

Abstract
Two azobenzene α-cyclodextrin based nanovalves are designed, synthesized and assembled on mesoporous silica nanoparticles. When in aqueous conditions, the cyclodextrin cap is tightly bound to the azobenzene moiety and capable of holding back loaded cargo molecules. Upon irradiation with a near-UV light laser, trans to cis- photoisomerization of azobenzene initiates a dethreading process, which causes the cyclodextrin cap to unbind followed by the release of cargo. The addition of a bulky stopper group to the end of the stalk allows this design to be reversible; complete dethreading of cyclodextrin as a result of unbinding with azobenzene is prevented as a consequence of steric interference. As a result, thermal relaxation of cis- to trans-azobenzene allows for the rebinding of cyclodextrin and resealing of the nanopores, a process which entraps the remaining cargo. Two stalks were designed with different lengths and tested with alizarin red S and propidium iodide. No cargo release was observed prior to light irradiation, and the system was capable of multiuse. On / off control was also demonstrated by monitoring the release of cargo when the light stimulus was applied and removed, respectively.
Introduction
Mesoporous silica has found its application in many fields of chemistry.1 A subset of mesoporous silica is the MCM-41 type mesoporous silica nanoparticle (MSN), which is characterized by its ordered, 2D hexagonal pore structure.2 Since its discovery by Mobil researchers several decades ago,3 MCM-41 has been an ideal choice as a drug delivery platform for its robust nature, high surface area to volume ratio, facile surface modification, low toxicity and high rates of cell internalization.4 To be an effective drug delivery system, MSNs must be able to retain cargo molecules while in storage and in circulation, but capable of delivering their payload upon arriving at its target. A variety of methods have been employed to prepare MSN to meet these criteria; researchers have exploited hydrophobicity as a method of passive cargo retention,5 and/or engineered nanomachines based on supramolecular nanovalves,6a–c snap-tops,6d metal nanocrystals,6e–f nanoimpellers,6g supported lipid bilayers,6h biomolecules,6i and reversible chelation6j towards the goal of retaining cargo inside the pores of MSN. These systems have been designed to deliver their payload in response to a variety of stimuli; some of which may already be present inside the cell, or is externally applied in the form of magnetic fields7 and light.8 The use of internal stimuli such as pH or enzyme activation allows for the creation of a self-contained delivery system, however, they can inevitably lead to higher rates of undesired activation. The use of external stimuli such as light9 can allow for an additional degree of control through selective irradiation of the target site. Current designs adopt an all-or-nothing type of release: once initiated by a stimulus, cargo is continuously released until the nanocarrier is emptied. While a complete delivery of cargo may sometimes be desired, an alternative approach is to create a nanocarrier capable of delivering its payload in controllable doses. In this way, a single treatment with such nanocarriers can deliver a range of therapeutic concentrations based on the external activating stimulus. To achieve this level of control, a system must be designed that is capable of resealing itself when a particular stimulus is removed. In the case of internal stimuli such as pH or enzyme activation, removing the stimulus is not feasible. Therefore, light, an external stimulus, was considered for its facile on / off manipulation.
We have previously reported a nanovalve based on an azobenzene derivative that is light-operated.8a Azobenzenes exhibit the property of photoisomerization, where its more stable trans- form can be converted into cis-azobenzene through light irradiation of the π -> π* band. Thermal relaxation of cis-azobenzene regenerates the trans- isomer.10 This transformation results in a large change in dipole moment; as a result, cis-azobenzene is relatively more hydrophilic than the trans- conformation. This property allows macrocycles which possess hydrophobic interior regions to selectively bind with trans-azobenzene. In the previously reported pseudorotaxane design, light irradiation of an α-cyclodextrin / azobenzene adduct causes the more stable trans-azobenzene to isomerize into cis-azobenzene. Due to the low binding affinity of α-cyclodextrin towards cis-azobenzene, the α-cyclodextrin unthreads from the stalk and allows the cargo to be released. Thermal-relaxation of cis-azobenzene back to trans- theoretically allows α-cyclodextrin to rebind, but is highly improbable due to the low cap concentration in the media. Therefore, to hold the α-cyclodextrin in proximity to the particle even after it dissociates from the azobenzene, a full rotaxane design was utilized. This allows for recomplexation after thermal conversion back to trans-azobenzene, producing a resealing system upon removal of light. Building on our previous azobenzene / β-cyclodextrin design, herein we report the (i) design and synthesis of two new azobenzene stalks 3a and 3b (Fig. 1) capable of binding α-cyclodextrin, (ii) the assembly of the pseudorotaxane, FRS1-NHS or EXT2-NHS, onto MSNs to synthesize the full rotaxane nanovalve, FRS1-MSN or EXT2-MSN), (iii) the delivery of three different sized fluorescent dyes (alizarin red S (ARS), propidium iodide (PI) and Hoechst 33342) as a result of near-UV light irradiation which allows α-cyclodextrin to slide away from the pore openings, and (iv) the capability of this design to reseal their cargo after the light irradiation is turned off, which allows for dosage control.
Fig. 1.

Schematic overview of the synthesis of FRS1-MSN and EXT2-MSN; p-nitrobenzoic acid is reacted with glucose to give 1. Reaction with N-hydroxysucciminide yields 2. Monosubstitution of 2 with one equivalent of 2a or 2b yields 3a or 3b, respectively. Complexation with α-cyclodextrin forms the α-cyclodextrin/azobenzenepseudorotaxanes FRS1-NHS or FRS1-NHS. Reaction of the pseudorotaxanes with APTES-MSNs yields FRS1- MSN or EXT2-MSN.
2. Experimental
2.1. General Methods
All of the chemicals used in this study were used as purchased from major suppliers such as Sigma-Aldrich and Fisher Scientific. Release profiles were generated using a time-resolved fluorescence spectroscopy setup (Fig. S12). A Coherent 377 nm CUBE laser was used as the probe beam (1–15 mW, 1 mm diameter), and the emission spectra were monitored using a Princeton Instruments Roper CCD detector cooled to −127 K. The pump beam used was a Coherent 403 nm CUBE laser (1–100 mW, 1 mm diameter) with a manually operated shutter. Powder X-ray diffraction measurements were made using a Panalytical X'Pert Pro diffractometer. All 1H NMR was performed on a Bruker ARX400 spectrometer in chloroform-d or DMSO-d solvents (400 MHz). Chemical shift is listed in ppm. The solvent signal was used as an internal standard. TEM images were acquired on a JEM1200-EX microscope. TOF-ESI+ spectra were collected on a Waters LCT Premier XE Time of Flight instrument. DLS readings were collected on a ZetaPals DLS and zeta potential instrument.
2.2. Synthesis of MCM-41 Nanoparticles
Cetyltrimethylammonium bromide (CTAB, 0.25 g, 0.7 mmol) was dissolved in deionized water (120 mL) with heat and stirring. The stir speed and temperature (80 °C) was allowed to stabilize for 1 hour, and then NaOH (2 M, 870 μL, 1.7 mmol) was added to the mixture. To this mixture, triethoxysilane (TEOS, 1.2 mL, 5.4 mmol) was then added drop wise under vigorous stirring (800 RPM) and reacted for 2 hours at 80 °C. The particle suspension was cooled to room temperature, and collected by centrifugation (7830 RPM, 15 min). The collected nanoparticles were washed with methanol until the effluent was a pH of 7. Synthesized nanoparticles showed a monodisperse DLS reading and low PDI (224 nm, PDI 0.01). Pore structure and particle morphology were verified using TEM and XRD, respectively (Fig. S1, S2).
2.3. Synthesis of 3-Aminopropyltriethoxysilane Modified MSN
Synthesized MSNs (50 mg) were washed twice with toluene before being resuspended in anhydrous toluene (10 mL). After purging a round bottom flask with dry argon, the reaction vessel was charged with the MSN / toluene slurry and 3-aminopropyltriethoxysilane (30 μL, 0.13 μmol) was added. The mixture was refluxed under dry argon atmosphere for 12 hours, before isolating the nanoparticles via centrifugation (5000 RPM, 5 min). The modified nanoparticles were washed twice with toluene and with DMF. Successful modification of MSN was verified after surfactant extraction with FTIR spectroscopy, as shown in the appearance of two primary amine stretching peaks at 3200 cm−1, 3400 cm−1 and the appearance of sp3 hybridized C–H stretching at 2980 cm−1.
2.4. Extraction of Templating Agent
MSNs (120 mg) were suspended in a solution of ammonium nitrate in ethanol (133 mg in 50 mL). The mixture was stirred under reflux for 2 hours. The particles were recollected by centrifugation (7830 RPM, 15 min) and washed with methanol (5 × 2 mL). To verify complete extraction of CTAB from the pores, FTIR spectroscopy was performed on the isolated particles (Fig. S3). DLS readings after surfactant extraction indicated a monodispersed MSNs (180 nm, PDI 0.005).
2.5. Synthesis of 4,4′-Azobenzenecarboxylic Acid
P-nitrobenzoic acid (15 g, 9 mmols) was dissolved in an aqueous solution of sodium hydroxide (50 g in 225 mL). In a separate reaction vessel, a glucose solution was prepared (100 g, 55 mmols) in water (150 mL) by heating until homogenous. After cooling to room temperature, the glucose solution was added dropwise to the dissolved p-nitrobenzoic acid at a rate in order to maintain a temperature of 60 °C and stirred at room temperature for 12 hours under atmospheric oxygen. After the allotted reaction time, the dark brown colored mixture was aged for four hours before filtering. The bright orange precipitate was dissolved in water (50 mL) and acidified with acetic acid (10%, 10 mL) producing a light orange precipitate. After filtering, the product was washed with portions of purified water (3 × 50 mL) and dried under vacuum to yield 4,4'-azobenzenedicarboxylic acid. 1H NMR (DMSO-d6): δ 7.99 (d, 4H), δ 8.13 (d, 4H).
2.6. Synthesis of 2
4,4′-Azobenzenedicarboxylic acid (300 mg, 1.1 mmols) was dissolved in anhydrous DMF (30 mL) with heating. After cooling to room temperature, N-hydroxysuccinimide (300 mg, 2.6 mmols) was added to the reaction mixture along with 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC, 500 mg, 3.2 mmols). A catalytic amount of 4-dimethylaminopyridine (DMAP) was added to the mixture and reacted for 12 hours at room temperature under dry nitrogen. After complete reaction, water (100 mL) was added to the reaction vessel, precipitating out a red solid. After filtering, the product was washed with portions of water (3 × 50 mL) and methanol (30 mL) and vacuum dried to yield a dark red powder. 1H NMR (DMSO-d6): δ 8.32 (d, 4H), δ 8.14 (d, 4H), δ 2.89 (s, 8H).
2.7. Synthesis of 1-Adamantanecarbonyl Chloride
1-Adamantanecarboxylic acid (2 g, 11 mmol) was dissolved in DCM (50 mL) and a catalytic amount of DMF was added before cooling to 0 °C. To this mixture, oxalyl chloride (1.5 mL, 18 mmol) was introduced dropwise. After the dropwise addition, the reaction was allowed to warm to room temperature and reacted for an additional two hours. Completion of the reaction was verified by TLC, and the solvent was removed under reduced pressure to isolate adamantanecarbonyl chloride. The white solid was used without further purification. 1H NMR (DMSO-d6): δ 2.09 (s, 3H), δ 1.98 (d, 6H), δ 1.78 (q, 6H).
2.8. Synthesis of 2b
Potassium carbonate (600 mg) was vacuum dried with heating for 30 minutes and purged with dry argon before adding anhydrous DCM (20 mL) and 2,2′-(ethylenedioxy)bis(ethylamine) (1.5 mL, 10 mmol) (2,2'-EDBEA). The reaction vessel was sealed before cooling to 0 °C in an ice bath. In a separate container, adamantanecarbonyl chloride (1 g, 5 mmol) was dissolved in anhydrous DCM (10 mL) and cooled to 0°C before adding dropwise to the first mixture. After the complete addition, the reaction mixture was allowed to warm to room temperature and stirred for an additional 2 hours, where TLC analysis confirmed reaction completion. The mixture was filtered to remove excess potassium carbonate, washed once with water (10 mL) and then acidified with hydrochloric acid (10%, 2 mL) before extracting the organic phase with water (3 × 15 mL). The combined aqueous layers were washed with DCM (3 × 10 mL) before adjusting to pH 8 with NaOH (10%). The aqueous layer was then extracted with DCM (5 × 15 mL), and the combined organic layers washed with brine, and dried under anhydrous sodium sulfate before concentrating under rotary evaporator to yield compound 2b as a viscous, clear liquid. 1H NMR (chloroform-d): δ 6.13 (s, 1H), δ 3.53 (s, 4H), δ 3.42 (m, 4H), δ 3.29 (q, 2H), δ 2.74 (t, 2H), δ 1.90 (s, 3H), δ 1.72 (s, 6H), δ 1.57 (t, 6H). TOF-ESI+ MS (calculated at 310.2): m/z 311.2 (M+H+).
2.9. Synthesis of 3a
In a reaction vessel, compound 2 (100 mg, 0.22 mmols) was dissolved in DMF (20 mL) with gentle heating and cooled to room temperature. Separately, adamantylamine (50 mg, 0.33 mmols) was dissolved in anhydrous DCM (5 mL) and cooled to 0 °C. The adamantylamine mixture was added to the mixture containing compound 2 dropwise over 2 hours, and allowed to react overnight at room temperature. After completion, the mixture was diluted with water (50 mL) and extracted with ethyl acetate (3 × 30 mL). The organic phases were combined and washed with water (5 × 20 mL) to remove the dissolved DMF, washed with brine and dried with anhydrous sodium sulfate. After concentrating the solution under reduced pressure, the crude product was purified with column chromatography on a silica stationary phase (EtOAc : hexanes = 1 : 2), and the second band isolated to yield quantitative amounts of 3a. 1H NMR (DMSO-d6): δ 8.29 (d, 2H), δ 8.09 (d, 2H), δ 7.95 (m, 4H), δ 2.89 (s, 4H), δ 2.24 (t, 3H), δ 2.05 (d, 6H), δ 1.64 (s, 6H). TOF-ESI+ MS (calculated at 500.2): m/z 501.2 (M+H+).
2.10. Synthesis of 3b
In a round bottom flask, compound 2 (100 mg, 0.22 mmols) was dissolved in DMF (20 mL) with gentle heating and allowed to cool to room temperature. In a separate vessel, compound 2b (100 mg, 0.32 mmols) was dissolved in DCM (5 mL) before adding to the solution of compound 2 over a period of 2 hours. After the complete addition, the mixture was allowed to react overnight at room temperature before diluting with water (30 mL) and extracting with ethyl acetate (5 × 10 mL). The combined organic layers were washed with water (5 × 15 mL), once with brine and dried over anhydrous sodium sulfate before concentrating on a rotary evaporator. The crude product was purified with column chromatography on a silica stationary phase, and eluted with ethyl acetate. The second band was isolated as a dark red solution, and the solvent evaporated under reduced pressure to isolate 3b as a dark red powder. 1H NMR (chloroform-d): δ 8.31 (d, 2H), δ 7.97–8.05 (m, 6H), δ 6.87 (s, 1H), δ 5.97 (s, 1H), δ 3.58–3.72 (m, 8H), δ 3.57 (t, 2H), δ 3.44 (q, 2H), δ 2.94 (s, 4H), δ 2.01 (s, 3H), δ 1.82 (d, 6H), δ 1.67 (s, 6H). TOF-ESI+ MS (calculated at 659.3): m/z 660.3 (M+H+).
2.11. Synthesis of FRS1-NHS
3a (6 mg, 1.1 μmol) and α-cyclodextrin (17 mg, 2.3 μmols) were dissolved in anhydrous DMF (1 mL). The reaction mixture was stirred at room temperature for an additional 2 hours to form pseudorotaxane FRS1-NHS.
2.12. Synthesis of EXT2-NHS
3b (2 mg, 0.3 μmols) and α-cyclodextrin (6 mg, 0.7 μmols) were added and dissolved in anhydrous DMF (1 mL). The reaction mixture was also stirred at room temperature for an additional 2 hours to allow α-cyclodextrin to thread onto 3b to synthesize pseudorotaxane FRS1-NHS.
2.13. Synthesis of FRS1-MSN
APTES-modified MSNs (20 mg) were washed twice with DMF before being resuspended in a mixture of FRS1-NHS (23 mg in 1 mL DMF). The reaction vessel was sealed under dry argon atmosphere and allowed to react for 12 hours at room temperature, before collecting through centrifugation (15000 RPM, 3 min). The stalk modified nanoparticles were washed with two portions of methanol and resuspended in ethanol. Successful attachment of the stalk was verified through UV-Vis spectroscopy of a suspension of FRS1-MSN (Fig. S10).
2.14. Synthesis of EXT2-MSN
A sealed reaction vessel purged with dry argon gas was charged with APTES-modified MSN (20 mg) that have been washed twice with anhydrous DMF. To this, a solution of EXT2-NHS (8 mg in 1 mL DMF) was added and allowed to react at room temperature for 12 hours. The product was collected through centrifugation (15000 RPM, 3 min), washed twice with methanol and suspended in ethanol. Successful attachment of the stalk was verified through UV-Vis spectroscopy of a suspension of EXT2-MSN (Fig. S11)
2.15. Loading of Fluorescent Dye and Sealing in Cargo
A sample of FRS1-MSN or EXT2-MSN was suspended in a concentrated dye solution. For the ARS loaded samples, the nanoparticles were suspended in an ethanoic ARS dye solution (2 mM, 1 mL). For Hoechst 33342 samples, the nanoparticles were suspended in a solution of Hoechst in ethanol:acetone = 1:1 (1 mmol, 1 mL). For PI loaded samples, EXT2-MSNs were suspended in an ethanoic PI dye solution (1 mM, 1 mL). Samples were stirred at room temperature for 24 hours before collecting with centrifugation (15000 RPM, 10 min). To seal in cargo and wash off excess surface-adsorbed dye, samples were washed with water until the supernatant exhibited no more fluorescence under 365 nm light and dried under vacuum prior to testing.
2.16. Assessment of Light activated Cargo Release
FRS1-MSN or EXT2-MSN (5 mg) loaded with an appropriate dye were confined to the corner of a glass cuvette. Water (4 mL) was gently added to the particles at a rate to prevent particles from being resuspended into solution. A 377 nm excitation laser (1 mW) was aimed through the solution supernatant. Emission of dye molecules released from each sample in the solution supernatant was monitored in real time by focusing the emitted light through a collecting lens, a 450 nm cutoff filter, and a monochromator (300 nm window) before integrating on a CCD detector (Fig. S12). To track release of ARS, the monochromator was centered on 550 nm, and the fluorescence integrated from 550–600 nm. PI release was monitored by centering the window at 600 nm and tracking dye emission from 600–620 nm. A baseline was collected over 1.5 hours to allow solution suspended particles to settle before a 403 nm pump beam (85 mW) was focused directly on the sample to stimulate cargo release. For the on/off experiments, the pump beam was turned off at timed intervals.
2.17. Reloading and Reuse Assessment
After acquiring a complete release profile, the sample was heated to 60 °C in ethanol for 12 hours in order to release the cargo from any particles that may not have been exposed to the irradiating laser. The solution was centrifuged (15000 RPM, 15 min) to remove suspended nanoparticles and analyzed under UV-Vis spectroscopy in order to verify the identity of the released cargo, and the sample pellet reloaded in an appropriate dye solution before washing away the unloaded excess. The reloaded samples were subjected to a second release profile test, and the corresponding time resolved fluorescence spectra collected in a similar fashion as described above.
3. Results and Discussion
3.1. Design of the Nanovalves
For adequate functioning of the nanovalve, a stalk must be designed that exhibits regional, reversible binding with a nanocap. After threading the nanocap with the stalk and assembling the 1:1 complex on the surface of the nanoparticle, the “closed” state of the stalk, the region of highest affinity, brings the nanocap close enough to the particle surface to prevent escape of the cargo. When binding is no longer favorable, the nanocap slide along the stalk away from the surface of the nanoparticle to its “open” state before reaching an end-terminating stopper group. This allows the cargo molecules to diffuse from the pores, but prevents the nanocap from unthreading completely. To achieve light responsiveness, the binding affinity of the capping agent must also be light dependent. For this reason, building on the previously designed pseudorotaxane azobenzene-cyclodextrin motif, an azobenzene moiety was chosen.
Similarly to the pseudorotaxane design,8a in the dark, the α-cyclodextrin can bind and form a supramolecular complex with the trans-azobenzene moiety, which is driven by favorable interactions between the hydrophobic α-cyclodextrin cavity and the azobenzene core.11 While all of the cyclodextrins exhibit a respectable binding affinity with trans-azobenzene, α-cyclodextrin was chosen for its ideal size in having an inner cavity diameter of 0.47 nm on the primary face and 0.53 nm on the secondary face.12 Irradiating with light causes trans-azobenzene to isomerize into its cis- form, which, due to its increased hydrophilicity, no longer has a favorable binding affinity with α-cyclodextrin and results in translation of the nanocap along the stalk. Although the α-cyclodextrin is able to slide along the stalk, after irradiation, complete dethreading is prevented by incorporating a stalk-terminating stopper group.
Several requirements for the choice of a stopper group were considered. The stopper must simultaneously be bulky enough to prevent the α-cyclodextrin from unthreading, but also have a lower binding affinity with α-cyclodextrin than trans-azobenzene in order to limit competitive binding with the stopper. For these reasons, an adamantane functional group was selected as the stopper. This molecule, with a diameter of 0.5 nm, is large enough to prevent α-cyclodextrin from unthreading, while exhibiting a low binding affinity for the nanocap. While other members of the cyclodextrin family –namely, β- and γ-cyclodextrin–have binding constants of 7000 M−1 and 3500 M−1 with adamantane,13a respectively, the strength of the α-cyclodextrin / adamantane adduct is considerably weaker by an order of magnitude (100 M−1).13 In this way, α-cyclodextrin will preferentially bind trans-azobenzene, but in the case when only cis-azobenzene forms by photoexcitation, the nanocap forms a metastable adamantane / α-cyclodextrin state that holds the nanocap away from the pore openings to allow release of the cargo. To ensure an adequate spatial clearance for cargo movement during this event, the distance between the azobenzene moiety and end stopper must be carefully tailored.
Because the size of many chemotherapy drugs falls between 1 – 2.5 nm, we designed two stalks of different lengths in order to load and deliver both 1 nm and 2 nm sized cargo molecules. To illustrate this, three fluorescent dyes: alizarin red S (ARS), propidium iodide (PI), and Hoechst 33342 with diameters 1.2 nm, 1.8 nm and 2.0 nm, respectively, were chosen as the cargo molecules (Fig. 2a). FRS1-MSN was tailored so that its “open” state could to allow for movement of cargo with sizes up to 1.6 nm, whereas in its “closed” state, the clearance is reduced to 0.8 nm (Fig. 2b-left). Therefore, ARS was selected as a candidate to test FRS1-MSN since it is 1.2 nm in size. EXT2-MSN was designed with a 2.8 nm “open” distance when fully extended (Fig. 2b-right). Similarly to FRS1-MSN, in its closed conformation, EXT2-MSN reduces the α-cyclodextrin-to-pore distance to 0.8 nm in order to seal the mesopores. As such, stalk FRS1-MSN is expected to load and deliver ARS but fail with PI or Hoechst, and stalk EXT2-MSN is expected to be compatible with PI.
Fig. 2.

(a) Size illustration of various cargo molecules in reference to (b) azobenzene stalks FRS1-MSN and EXT2-MSN. Because of the shorter stalk length, FRS1-MSN is limited to loading cargo molecules < 2 nm. Hoechst 33342 and propidium iodide dyes remain excluded from FRS1-MSN as a result of their size. However, EXT2-MSN is observed to load all three of the listed fluorophores. (c) Schematic illustration of the fully assembled rotaxane nanovalve and operation. (1) After synthesis, the nanovalves are suspended in an organic solvent, which destabilizes hydrophobic interactions between the α-cyclodextrin and azobenzene moiety and allows for cargo loading. (2) Upon solvent exchange to water, rebinding of the cyclodextrin to azobenzene seals in the loaded cargo. (3) Irradiation with light induces isomerization to cis-azobenzene, forcing cyclodextrin to move to the end of the stalk, and allowing cargo to release. (4) Upon removal of the light stimulus, thermal relaxation of cis-azobenzene to its more stable trans isomer allows rebinding of the cyclodextrin and seals in the remaining cargo.
3.2. Synthesis of MSNs
MSNs were synthesized utilizing a surfactant-templated sol-gel process.14 The resulting spherical nanoparticles were ~100 nm in diameter. After synthesis, MSN was directly modified with 3-aminopropyltriethoxysilane (APTES) by a condensation reaction with the surface silanol groups in order to bias modification to the nanoparticle surface and prepare for subsequent stalk attachment. After modification with APTES, removal of the templating agent resulted in nanoparticles with hexagonally arranged mesopores about 2.2 nm in diameter (Fig. S1–S3).
3.3. Synthesis of the Stalks
Since α-cyclodextrin / azobenzene binding is dependent on hydrophobic interactions, considerations in the synthesis of both stalks were made to ensure that the final attachment steps onto MSN could be made in a polar solvent. Because of this, a NHS ester functional group was chosen as the reactive end for its specificity and compatibility with non-aqueous, polar solvents. In the synthesis of both stalks, p-nitrobenzoic acid was reacted in basic conditions with glucose to form compound 1. To generate the di-substituted NHS ester product, 1 was reacted with N-hydroxysuccimide to form 2. To prepare the short stalk, 2 was directly reacted with adamantylamine (2a) under dilute conditions and purified via column choromatography to isolate the monosubstituted product 3a. To prepare the long stalk, first, adamantanecarboxylic acid was reacted with oxalyl chloride to generate adamantane carbonyl chloride. Then, nucleophilic substitution of the acid chloride with 2,2'-(ethylenedioxy)bis(ethylamine) afforded the monoalkylated product 2b. 2b was reacted with 2 under dilute conditions and purified under column chromatography to yield the longer stalk 3b. To synthesize the pseudorotaxane, compounds 3a or 3b were mixed with α-cyclodextrin in DMF to allow α-cyclodextrin to complex with azobenzene and form the pseudorotaxanes FRS1-NHS and EXT2-NHS, respectively. Attachment to MSNs was accomplished by first modifying MSNs with 3-aminopropyltriethoxysilane and extracting the templating agent before a direct reaction with FRS1-NHS or EXT2-NHS to complete the synthesis of FRS1-MSN or EXT2-MSN.
3.4. Loading and Sealing of Cargo
After grafting each of the pseudorotaxane stalks onto MSNs, the assembled machines were washed with methanol to remove unreacted excess material. In order to load cargo into the pores, FRS1-MSN or EXT2-MSN were suspended in an organic solvent under UV light irradiation to break the hydrophobic interactions between α-cyclodextrin and azobenzene to allow α-cyclodextrin to slide toward the adamantane end and allow for pore access (Fig. 2c–3). Suspending and stirring the nanoparticles in an organic solution of fluorescent dye allowed cargo to slowly load inside the pores. In order to seal the nanocap, the loaded nanoparticles were solvent exchanged with water, where the exclusion of solvent from the cyclodextrin cavity is favored, allowing the α-cyclodextrin cap to rebind with azobenzene and reseal tightly the loaded cargo (Fig. 2c–4). Excess, surface-adsorbed cargo was removed with subsequent water washes until the eluent no longer exhibited fluorescence.
Fig. 3.
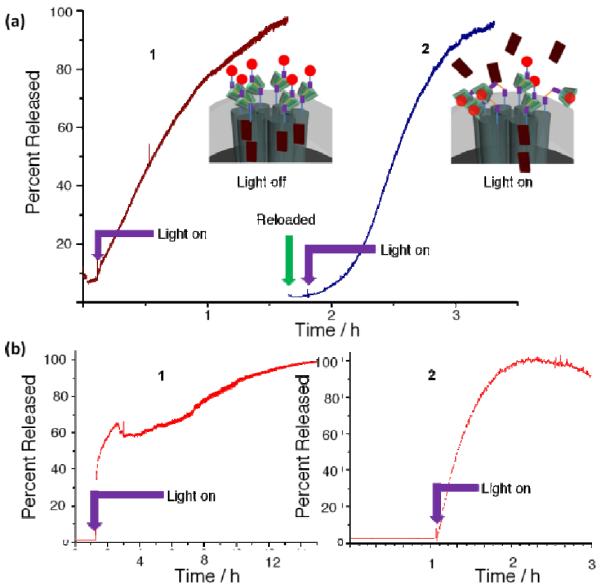
(a) Time resolved fluorescence spectra showing the light activated (403 nm) release of alizarin red S (ARS) dye from FRS1-MSN. Initial loading of the particles results in minimal release prior to irradiation (trace 1). When the pump beam is activated, ARS fluorescence steadily increases in the supernatant. After the release is complete, FRS1-MSN were reloaded with ARS and washed before reinitiating cargo release. Trace 2 shows these reloaded FRS1-MSN releases ARS equally as well. An identical experiment was performed on the EXT2-MSN system (b) using PI dye instead. EXT2-MSN exhibits the same behavior as FRS1-MSN with the larger fluorophore. These results demonstrate that the full rotaxane systems are reversible, reusable and can bedesigned to release different sized cargos.
Fig. 4.
Time resolved fluorescence spectra demonstrating the on-off properties of FRS1-MSN. (a) Stimuli-response release profile; light stimuli causes the release of the ARS. When the light is turned off, the fluorescence intensity levels out until the stimulus is reapplied. (b) Release from the particles modified by 3a rather than FRS1-NHS. The stalk, lacking the cyclodextrin nanocap, shows a continuous release of ARS without application of light. Therefore, the α-cyclodextrin is required to form the complete nanovalve system and prevent leakage of cargo.
3.5. Characterization
The MSN quality was determined through TEM imaging (Fig. S1), which confirmed an average diameter of 100 nm and a 2D-hexagonally packed pore structure with pore diameters of 2–2.4 nm. Powder XRD measurements (Fig. S2) were used to confirm the hexagonal mesopore phase, evident in the higher order Bragg diffraction peaks indexed as the (100), (110), and (200) planes with a lattice spacing of 4 nm. Complete removal of CTAB was verified by subjecting bare MSN to the extraction procedure and performing FTIR spectroscopy (Fig. S3) on the resulting material, which showed a complete removal of organic material with the disappearance of sp3 C-H stretching peaks present around 3000 cm−1. Successful amine modification of MSN was verified through FTIR spectroscopy, with the appearance of the dual primary amine stretches around 3200 cm−1 and sp3 C-H stretching peaks at 3400 cm–1 after CTAB extraction. To verify the success of each step in the synthesis of 3a and 3b1H NMR and ESI-TOF mass spectrometry were employed (Fig. S4–S8) on each of the intermediates in the stalk syntheses. The attachment efficiency of both FRS1-NHS and EXT2-NHS pseudorotaxanes to MSN was verified through UV-Vis spectroscopy,. FRS1-MSN and EXT2-MSN showed similar absorption peaks centered at 334 and 336 nm respectively, which can be attributed to a successful attachment of the azobenzene stalks (Fig. S9–S10). After isolating the modified nanoparticles through centrifugation, absorption spectroscopy of the corresponding supernatant showed a negligible absorbance in this region, which indicates the attachment process was highly efficient.
3.6. Light Activated Cargo Delivery
To verify the operation of the nanomachine, continuous monitoring by fluorescence spectroscopy was employed. In a corner of a quartz cuvette, a dried sample of FRS1-MSN or EXT2-MSN was confined and water gently added in order to prevent suspending the nanoparticles into solution. To monitor the release of cargo, a low power probe beam (377 nm, 1 mW) was aimed through the solution supernatant in order to excite the fluorescent dye molecules released from the sample. The corresponding emission was filtered through a cut-off filter and selected using a monochromator before focusing onto a CCD detector. Integrating over a specific range of wavelengths in real-time allowed for the generation of a time-resolved release profile. In order to irradiate the sample and induce cargo release, a pump beam (403 nm, 85 mW, 1 mm diameter), was directly focused onto the sample.
3.7. FRS1-MSN Light Activated Release
Using a time resolved spectroscopy technique (Fig. S12), an initial baseline reading of the FRS1-MSN sample was collected for two hours. Fig. 3a shows no dye release prior to activation of the pump beam, illustrating the strength of the α-cyclodextrin / trans-azobenzene complexation in sealing off the cargo. When irradiated, an increasing ARS fluorescence is observed (Fig. 3a–1), indicating the release of cargo. The curved shape illustrates an initially high release rate as dye molecules diffuse from the highly concentrated pore interiors into the solution. After several hours, the slope of the curve flattens out as fewer dye molecules remain inside the pores. To demonstrate the reusability of the rotaxane valve, after a completed release, the sample was washed with ethanol to remove any remaining dye, reloaded with ARS, and washed in water to remove surface adsorbed-dye and reseal the pores. This reloaded sample was subjected to the same release profile experiment to ensure that α-cyclodextrin is still present on the rotaxane stalk and capable of sealing in cargo. The baseline in Fig. 3a–2 demonstrates that this reloaded sample remains tightly sealed prior to light irradiation, indicating that the nanocap has not been removed from the rotaxane stalk. After irradiation with the pump laser, an increase in ARS fluorescence is observed, showing that the system is still light responsive, and suggesting the reusability of FRS1-MSN. To confirm the observed fluorescence change is due to ARS, the supernatant was analyzed (Fig. S11) after each release profile using UV-Vis spectroscopy and compared to a standard solution of ARS.
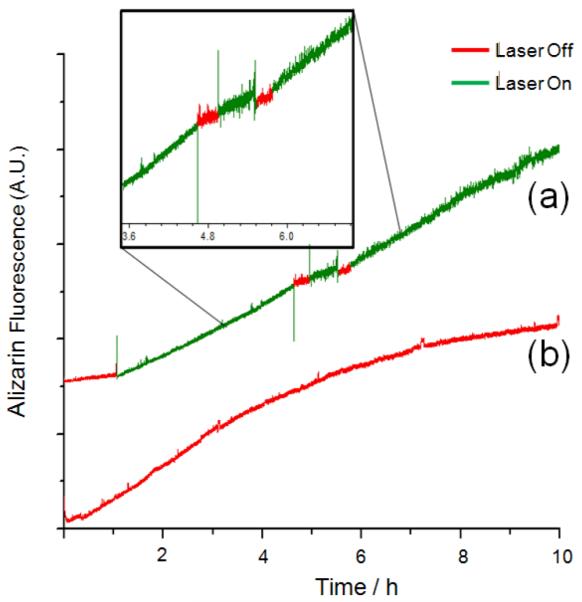
In order to also verify the reversibility of the rotaxane nanovalve, FRS1-MSN was subjected to a series of “on” / “off” tests. In the same experimental setup as described above, cargo release from a FRS1-MSN sample loaded with ARS was tracked while the pump beam was turned off for 30 minute intervals (Fig. 4a). At first, when irradiated, FRS1-MSN exhibits the presumed exponential shape indicative of ARS release, but flattens out when the pump beam is turned off. This can be attributed to a thermal relaxation of cis-azobenzene into trans-azobenzene in the dark, which allows α-cyclodextrin to rebind and prevent additional cargo release. Reintroducing the pump beam to the sample causes ARS fluorescence to increase once more, indicating the reversibility of this system and its potential for dosage control.
In order to ensure the necessity of α-cyclodextrin in the rotaxane design, a control experiment was performed in which a sample of 3a was assembled without first threading on α-cyclodextrin to produce FRS1-MSN. This sample was then loaded with ARS and washed until the supernatant was colorless. The corresponding fluorescence release profile was also acquired with this sample, which showed increasing ARS fluorescence prior to pump beam irradiation. This suggests that without a cyclodextrin cap, the azobenzene stalk alone is unable to retain cargo molecules (Fig. 4b), and thus demonstrates the necessity of the α-cyclodextrin nanocap for successfully sealing in ARS cargo.
Although FRS1-MSN is able to load and release ARS, the dimensions of these nanoparticles in their closed and open states indicate that it is unable able to load cargo greater than 1.5 nm. To test this, FRS1-MSN was assembled and loaded with Hoechst 33342 (diameter =2 nm). After the loading and washing steps, the loaded nanoparticles were not fluorescent under a UV-lamp, indicating that Hoechst was not able to access the pores of the FRS1-MSN. To corroborate this, the sample was tested in a release profile experiment while monitoring the solution for Hoechst fluorescence. After collecting a baseline, irradiating the sample with the pump laser caused no change in fluorescence over the background noise (Fig. S13). Because of its length, FRS1-MSNs are unable to load a larger sized cargo molecule like Hoechst. The longer stalk length of EXT2 allows cargo up to 2.2 nm to be loaded inside of the pores. To assess whether this longer design can achieve the goal of delivering larger cargo molecules, stalk EXT2 was tested similarly with PI as the fluorescent cargo molecule.
3.8. EXT2-MSN Light Activated Release
Using the same laser based time-resolved spectroscopy techniques, EXT2-MSN was tested as a delivery system for PI cargo. In order to detect PI emission, the monochromator was centered at 600 nm with a 300 nm window. Similarly, a baseline reading was collected before activating the pump beam and stimulating cargo release. Prior to activating the pump beam, the initial flat baseline reading for EXT2-MSN (Fig. 3b–1) shows no observable PI fluorescence, showing that this rotaxane stalk is capable of sealing in the PI cargo. When the pump beam is turned on, an increase in fluorescence is observed, indicating that PI is being released from the nanoparticles and into the solution. The solution fluorescence of PI increases gradually over several hours before approaching completion, when the dye has been completely released from EXT2-MSN. This experiment indicates that EXT2-MSN is capable of releasing PI dye with light stimulation.
In order to verify that light irradiation did not destroy the stalk, or cause α-cyclodextrin to unthread altogether, after a completed release the sample was washed to remove any remaining PI cargo before reloading in a fresh solution of PI. After washing off the excess, surface adsorbed dye with water, a second release profile was generated under the same conditions as used previously (Fig. 3b–2). The initial collection of data shows a flat baseline prior to pump beam activation, indicating that the reloaded EXT2-MSN system is still capable of sealing in PI. Upon irradiation with the pump beam, an increase in solution fluorescence is observed, suggesting that PI cargo is again diffusing from the nanoparticle pores and into the solution. Because the reloaded cargo releases only upon irradiation, EXT2-MSN is observed to be a reusable, light-responsive delivery system for PI.
4. Conclusions
In summary, two reversible, reusable nanovalves were designed based on the azobenzene / α-cyclodextrin recognition motif that is capable of the controlled release of both small cargo (ARS dye) and larger dye molecules (PI). In aqueous environments, both nanovalves remain tightly closed due to the strong binding between α-cyclodextrin and the trans-azobenzene moiety, but light irradiation causes the photoisomerization of trans- to cis-azobenzene, unbinding the α-cylodextrin cap and allowing it to slide to the adamantane end of the stalk, which results in the release of the cargo. Because this binding is reversible, once the light is turned off, thermal relaxation of cis-azobenzene regenerates trans-azobenzene, which allows the cyclodextrin to rebind and seal in the remaining cargo molecules. By tuning the length of the azobenzene / adamantane stalk, cargo size-selectivity can be achieved. Currently, this system is being tested in-vitro with the goal of developing an externally light-activated nanovalve to deliver anti-cancer drugs to cancerous cells.
Supplementary Material
Acknowledgements
The project described was supported by Grant Numbers S10RR025631 from the National Center for Research Resources and CA133697 from the National Institutes of Health. Support from the U.S. Department of Defense and from the Northwestern University International Institute for Nanotechnology is also acknowledged.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/b000000x/
Notes and references
- 1.(a) Lu Y, Ganguli R, Drewien CA, Anderson MT, Brinker CJ, Gong W, Guo Y, Soyez H, Dunn B, Huang MH, Zink JI. Nature. 1997;389:364–368. [Google Scholar]; (b) Slowing II, Trewyn GB, Giri S, Lin SV. Adv. Funct. Mater. 2007;17:1225–1236. [Google Scholar]; (c) Zhao D, Feng J, Huo Q, Melosh N, Fredrickson GH, Chmelka BF. Science. 1998;279:548–552. doi: 10.1126/science.279.5350.548. [DOI] [PubMed] [Google Scholar]; (d) Lin Y-S, Hurley KR, Haynes CL. J. Phys. Chem. Lett. 2012;3:364–374. doi: 10.1021/jz2013837. [DOI] [PubMed] [Google Scholar]; (e) Li Z, Barnes JC, Bosoy A, Stoddart JF, Zink JI. Chem. Soc. Rev. 2012;41:2590–2605. doi: 10.1039/c1cs15246g. [DOI] [PubMed] [Google Scholar]
- 2.Kresge CT, Leonowicz ME, Roth WJ, Vartuli JC, Beck JS. Nature. 1992;359:710–712. [Google Scholar]
- 3.Beck JS, Vartuli JC, Roth WJ, Leonowicz ME, Kresge CT, Schmitt KD, Chu CTW, Olson DH, Sheppard EW. J. Am. Chem. Soc. 1992;114:10834–10843. [Google Scholar]
- 4.(a) Liong M, Lu J, Kovochich M, Xia T, Ruehm SG, Nel AE, Tamanoi F, Zink JI. ACS Nano. 2008;2:889–896. doi: 10.1021/nn800072t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ambrogio MW, Thomas CR, Zhao Y-L, Zink JI, Stoddart JF. Acc. Chem. Res. 2011;44:903–913. doi: 10.1021/ar200018x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Trewyn BG, Slowing II, Giri S, Chen H-T, Lin VSY. Acc. Chem. Res. 2007;40:846–853. doi: 10.1021/ar600032u. [DOI] [PubMed] [Google Scholar]; (d) Zhang H, Dunphy DR, Jiang X, Meng H, Sun B, Tarn D, Xue M, Wang X, Lin S, Ji Z, Li R, Garcia FL, Yang J, Kirk M, Xia T, Zink JI, Nel AE, Brinker CJ. J. Am. Chem. Soc. 2012;134:15790–15804. doi: 10.1021/ja304907c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Yanes RE, Tarn D, Hwang AA, Ferris DP, Sherman SP, Thomas CR, Lu J, Pyle AD, Zink JI, Tamanoi F. Small. 2013;9:697–704. doi: 10.1002/smll.201201811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu J, Liong M, Zink JI, Tamanoi F. Small. 2007;3:1341–1346. doi: 10.1002/smll.200700005. [DOI] [PubMed] [Google Scholar]
- 6.(a) Meng H, Xue M, Xia T, Zhao Y-L, Tamanoi F, Stoddart JF, Zink JI, Nel AE. J. Am. Chem. Soc. 2010;132:12690–12697. doi: 10.1021/ja104501a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Angelos S, Yang Y-W, Khashab NM, Stoddart JF, Zink JI. J. Am. Chem. Soc. 2009;131:11344–11346. doi: 10.1021/ja9042752. [DOI] [PubMed] [Google Scholar]; (c) Li Z, Nyalosaso JL, Hwang AA, Ferris DP, Yang S, Derrien G, Charnay C, Durand J-O, Zink JI. J. Phys. Chem. C. 2011;115:19496–19506. doi: 10.1021/jp2047147. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Coti KK, Belowich ME, Liong M, Ambrogio MW, Lau TA, Khatib HA, Zink JI, Khashab NM, Stoddart JF. Nanoscale. 2009;1:16–39. doi: 10.1039/b9nr00162j. [DOI] [PubMed] [Google Scholar]; (e) Vivero-Escoto JL, Slowing II, Trewyn BG, Lin VS-Y. Small. 2010;6:1952–1967. doi: 10.1002/smll.200901789. [DOI] [PubMed] [Google Scholar]; (f) Lai C-Y, Trewyn BG, Jeftinija DM, Jeftinija K, Xu S, Jeftinija S, Lin VS-Y. J. Am. Chem. Soc. 2003;125:4451–4459. doi: 10.1021/ja028650l. [DOI] [PubMed] [Google Scholar]; (g) Lu J, Choi E, Tamanoi F, Zink JI. Small. 2008;4:421–426. doi: 10.1002/smll.200700903. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Liu JW, Stace-Naughton A, Jiang XM, Brinker CJ. J. Am. Chem. Soc. 2009;131:1354–1355. doi: 10.1021/ja808018y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Ferris DP, Lu J, Gothard C, Yanes R, Thomas CR, Olsen J-C, Stoddart JF, Tamanoi F, Zink JI. Small. 2011;7:1816–1826. doi: 10.1002/smll.201002300. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Tarn D, Xue M, Zink JI. Inorg. Chem. 2013;52:2044–2049. doi: 10.1021/ic3024265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomas CR, Ferris DP, Lee J-H, Choi E, Cho MH, Kim ES, Stoddart JF, Shin J-S, Cheon J, Zink JI. J. Am. Chem. Soc. 2010;132:10623–10625. doi: 10.1021/ja1022267. [DOI] [PubMed] [Google Scholar]
- 8.(a) Ferris DP, Zhao Y-L, Khashab NM, Khatib HA, Stoddard JF, Zink JI. J. Am. Chem. Soc. 2009;131:1686–1688. doi: 10.1021/ja807798g. [DOI] [PubMed] [Google Scholar]; (b) Nguyen TD, Leung KC-F, Liong M, Liu Y, Stoddart JF, Zink JI. Adv. Funct. Mater. 2007;17:2101–2110. [Google Scholar]
- 9.(a) Angelos S, Choi E, Vögtle F, De Cola L, Zink JI. J. Phys. Chem. C. 2007;111:6589–6592. [Google Scholar]; (b) Aznar E, Casasús R, García-Acosta B, Marcos MD, Martínez-Mánaz R, Sancenón F, Soto J, Amorós P. Adv. Mater. 2007;19:2228–2231. [Google Scholar]
- 10.Rabek JF. Photochemistry and Photophysics. Volume II. CRC Press, Inc; Boca Raton, Florida: 1990. [Google Scholar]
- 11.Murakami H, Kawabuchi A, Kotoo K, Kunitake M, Nakashima NJ. J. Am. Chem. Soc. 1997;119:7605–7606. [Google Scholar]
- 12.Del Valle EMM. Process Biochem. 2004;39:1033–1046. [Google Scholar]
- 13.(a) Falvey P, Lim CW, Darcy R, Revermann T, Karst U, Giesbers M, Marcelis ATM, Lazar A, Coleman AW, Reinhoudt DN, Ravoo BJ. Chem. Eur. J. 2005;11:1171–1180. doi: 10.1002/chem.200400905. [DOI] [PubMed] [Google Scholar]; (b) Cromwell WC, Bystrom K, Eftink MR. J. Phys. Chem. 1985;89:326–332. [Google Scholar]
- 14.Angelos S, Liong M, Choi E, Zink JI. Chem. Eng. J. 2008;137:4–13. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.