

Abstract
Calcium is of critical importance to mitochondrial and cell function, and calcium signaling is highly localized in the cell. When stimulated, mitochondria are capable of rapidly taking up calcium, affecting both matrix energetics within mitochondria and shaping the amplitude and frequency of cytosolic calcium “waves”. During pathological conditions a large increase in mitochondrial calcium levels is thought to activate the mitochondrial permeability transition pore, resulting in cell death. The protein responsible for mitochondrial calcium uptake, the mitochondrial calcium uniporter (MCU), was identified in 2011 and its molecular elucidation has stimulated and invigorated research in this area. MCU knockout mice have been created, a variety of other regulators have been identified, and a disease phenotype in humans has been attributed to the loss of a uniporter regulator. In the three years since its molecular elucidation, further research into the MCU has revealed a complex uniporter, and raised many questions about its physiologic and pathologic cell roles.
1. Mitochondria and Calcium Uptake-a History
The clinical significance of calcium has been appreciated for centuries, since Ringer first discovered in 1883 that the addition of the divalent ion could trigger contractions in cardiac myocytes [1]. Mitochondrial calcium uptake was first measured over 50 years ago, when studies in the 1960s showed that mitochondria were capable of rapidly taking up calcium [2,3]. When this occurred mitochondrial matrix concentrations of total calcium could rise by factors of 10 or more [2,4,5]. The ability of isolated mitochondria to accumulate calcium led to suggestions in the late 1970's that mitochondria might contribute to the regulation of cytosolic calcium. David Nicholls showed that because of the relative kinetics of mitochondrial uptake by the uniporter and release by the Na-H (or Na-Ca) exchanger that mitochondria could regulate extra-mitochondrial calcium to a “set point”: if extra-mitochondrial calcium was raised above the set point the mitochondria would accumulate calcium, but if extra-mitochondrial calcium was reduced below the set point, mitochondria would release calcium via the efflux pathway [6]. This concept that mitochondria regulate cytosolic calcium was challenged by studies in giant squid axon in which the cytosol could be loaded with calcium sensitive dyes such as arsenazo. These studies suggested that, under physiological conditions, cytosolic calcium did not rise to levels sufficient to support mitochondrial calcium uptake; others suggested that the role of mitochondrial calcium uptake was not to regulate cytosolic calcium, but rather to regulate mitochondrial matrix calcium and the activity of calcium sensitive mitochondrial dehydrogenases [7,8].
In the 1980s, the role of intracellular organelles in regulating cell calcium homeostasis turned away from the mitochondria and towards other organelles [9–11]. One reason for this was that baseline levels of mitochondrial calcium were found to be relatively low, and generally comparable to that of the cytosol (∼ 100 nM), suggesting that mitochondria do not serve as reservoirs for large amounts of calcium, at least at baseline cell conditions [8,12–16]. Even with agonist stimulation or at peak contractility bulk cytosolic calcium only rose to ∼1μM, and only very transiently [12,17].
Although it was well established that the sarcoplasmic reticulum was the intracellular organelle involved in calcium release and reuptake during calcium transients, the intracellular source of agonist-induced calcium release was unknown, and mitochondria were considered a possible source. In the 1980s, several groups found that agonists that led to the generation of inositol 1,4,5 triphosphate (IP3), caused calcium release from the endoplasmic reticulum [10,11]. Other research showed that the endoplasmic reticulum had a much higher affinity for calcium than the mitochondria [18]. It was general agreed that mitochondria did not have a major role in regulating cytosolic calcium homeostasis and that mitochondria only accumulated calcium under pathological cell death conditions associated with a massive increase in cytosolic calcium [19]. Accordingly, research turned to focus largely on the endoplasmic reticulum's role in cellular calcium handling.
Attention shifts back towards mitochondria
Attention returned to the mitochondria as a major player in cellular calcium in the 1990s, when the development of highly specific probes made it possible to demonstrate microdomains of high calcium near the mitochondria [20–22]. When channels on sarcoplasmic/endoplasmic reticulum (SR/ER) or plasma membrane opened, there was a sudden, local increase of calcium five to ten times the general cytosolic calcium concentration. Mitochondria near these microdomains of high calcium concentration were able to rapidly take up calcium. Therefore high levels of cytosolic calcium, sufficient to activate MCU did exist, in small focused areas often in close proximity to mitochondria, which were then able to accumulate calcium [23].
The concept emerged that calcium release from SR/ER exposed mitochondria to a much higher calcium concentration that what is typically present in the cytosol [24,25]. This picture also helped to reconcile the fact that mitochondrial calcium was essential for aerobic metabolism with its roles in propagating cell death: while an accumulation of calcium could cause cell death, a rapid and transient rise in calcium, the kind initiated by the brief appearance of these microdomains of high cytosolic calcium, could exist physiologically [24].
Mitochondria also appear to be docked to the ER/SR at designated signaling sites, ensuring their proximity and their ability to utilize these small, locally potent releases of calcium [26]. It was shown that the if the tethers between the ER/SR and mitochondria were tightened, mitochondria became more prone to calcium overload, mPTP opening and subsequent cell death, presumably because of their increased exposure to microdomains of high cytosolic calcium (see section 2b) [23,27].
2. (Patho)physiological roles of mitochondrial calcium
Balanced calcium uptake by the mitochondria is essential: at appropriate levels, it can stimulate important metabolic processes such as activation of mitochondrial dehydrogenases, but higher mitochondrial calcium can be detrimental for a cell, initiating cell death pathways such as apoptosis and necrosis. Mechanisms for altering mitochondrial calcium levels, and maintaining homeostasis, are therefore essential for both aerobic metabolism and cell survival [4].
a) Metabolism
Mitochondria are classically referred to as the powerhouse of the cell: provided with oxygen and reducing equivalents, respiring mitochondria are able to produce ATP and maintain a membrane potential. Three mitochondrial matrix dehydrogenases essential for ATP production are activated by calcium: pyruvate dehydrogenase, alpha-ketoglutarate, and isocitrate-dehydrogenase [28]. The stimulation of these dehydrogenases by calcium increases NADH availability, and therefore the flow of elections down the respiratory chain: mitochondrial calcium increases mitochondrial ATP production [29]. Calcium is also known to activate several complexes of electron transport [30,31].
b) Cell Death
Mitochondria are capable of rapidly taking up calcium, but at very high levels, their ability to buffer that calcium can be overwhelmed. When this occurs, pathological calcium concentrations are reached, and a large conductance channel known as the mitochondrial permeability transition pore (mPTP) opens in the inner mitochondrial membrane [32–34] First formally described by Haworth and Hunter in 1976, this pore has since been implicated in a multitude of cell death pathways, including cardiac and neuronal cell death, hepatotoxicity, and nervous and muscular dystrophies [35] this pore has since been implicated in a multitude of cell death pathways, including cardiac and neuronal cell death, hepatotoxicity, and nervous and muscular dystrophies. The process appears to begin with an oxidative stress and/or ATP depletion, which is followed by mitochondrial calcium loading to pathologically high levels, inducing the mPTP to open [36]. When the mPTP opens there follows a collapse of the mitochondrial membrane potential and a subsequent bioenergetic crisis. MPT-dependent mitochondrial swelling occurs, and cell death rapidly ensues [37]. Opening of the mPTP appears to play a fundamental role in reperfusion injury in the heart [32–34,38]. The low pH during ischemia is known to inhibit the mPTP, but as cytosolic pH is restored on reperfusion the mPTP opens [38]. In both I/R injury and other forms of mPTP induced cell death such as neuronal glutamate toxicity, blocking of either the mPTP or the reduction of mitochondrial calcium uptake appears to be protective, suggesting that mitochondrial calcium uptake may be a potential site for therapeutic intervention [39,40].
3. MCU Identified
Although it had been clear for decades that mitochondrial calcium levels were involved in the regulation of processes ranging from aerobic metabolism to cell death, the actual protein responsible for calcium uptake into the mitochondria had not been identified. Because the outer membrane of the mitochondria has channels such as the voltage dependent anion channel (VDAC) that render it freely permeable to calcium, the MCU was proposed to be on the inner membrane of the mitochondria, but its molecular identity was unknown. Evidence suggested that the MCU would be i.) highly selective ii.) sensitive to ruthenium red (RuR) and iii.) have low affinity for the cation [41,42]. The driving force for calcium uptake by the MCU was established to be the steep negative membrane potential established by the respiratory chain [43]. Furthermore, measurements in mitoplasts had shown that the MCU was a calcium-specific ion channel [44].
Despite the well-described nature of the MCU, it took 5 decades to elucidate the molecular structure of this putative membrane channel. In 2010 Mootha's group identified Mitochondrial Calcium Uptake 1 (MICU1), a mitochondrial protein that bound calcium by examining MitoCarta. They identified a 54 kDa protein that they named MICU1, that, when silenced, abrogated mitochondrial calcium uptake. They correctly hypothesized that this protein was more likely to be a regulator than the core component of the uniporter, because it had maximally only one predicted transmembrane domain, while a pore would need at least two. It did however have two EF hands that could bind calcium, making it potentially capable of regulating a partner channel [45,46].
With an associated protein identified, the path to pursue the putative uniporter became somewhat more straightforward, and two groups simultaneously identified MCU over the next year, using the MitoCarta database. Both groups identified CCDC109A, now called MCU, as a universally expressed protein, present in mitochondria, with two transmembrane domains that oligomerizes to form a larger complex [46,47].
In 2010, Mootha's group examined RNA and protein expression profiles to identify proteins that were co-expressed in proportion to MICU1 [46]. They discovered a protein that was not similar in sequence with MICU1, but whose expression varied proportionately with MICU1: they were co-expressed or co-absent in 495 of 500 organisms. Rizzuto's group searched the MitoCarta database for a protein expressed in most organisms, but specifically Trpanosomatida, which was known to have RuR-sensitive calcium uptake, but not present in S. cerevisiae, which was known to lack RuR-sensitive mitochondrial uptake. They further limited their search to proteins that had the two transmembrane domains necessary to be an ion channel [47]. Both groups found that CCDC109A fit their criteria. They both further showed, in a variety of cell types, that MCU over and under-expression led to the expected changes in mitochondrial calcium uptake. With the addition of histamine, an inositol 1,4,5-triphosphate-generating agonist that causes calcium release from the endoplasmic reticulum, MCU overexpression doubled calcium concentrations in the mitochondrial matrix. In these cells, mitochondria took calcium up more quickly and reached a higher concentration of calcium. Furthermore, these cells were more sensitized to apoptotic stimuli, consistent with previous hypothesis that the MCU was highly participatory in cell death pathways [47].
Both groups also found that silencing the MCU with siRNA abrogated mitochondrial calcium uptake proportional to the decrease in MCU mRNA, with no effect on mitochondrial respiration, membrane potential, or basic morphology [46]. Interestingly, Mootha's group found that when they silenced MCU in mice, their liver cells were physiologically intact and still capable of undergoing normal respiratory state changes, despite being unable to take up calcium into the mitochondria [46,47].
a) Topology
MCU has two transmembrane domains, and is localized to the inner membrane. Though there was originally some dissent on the orientation of the N and C-termini of the MCU, the development and use of ascorbate peroxidase, or APEX, which functions as an electron microscopy tag, identified the mitochondrial matrix as the site of both the N and C-termini [48]. MCU appears to oligomerize within mitochondrial inner membrane as part of a larger molecular weight complex, consistent with the presence of MICU1 and MICU2, its major regulators. MICU2 appears to associate with MCU via MICU1, though whether MICU1 is directly bound to MCU or attached via another regulatory protein known as Essential MCU Regulators (EMRE, see section 3b, iii) remains unknown in metazoans, as does the exact orientation of EMRE [49]. A short stretch of amino acids facing the intermembrane space appear critical for calcium transport: ruthenium red and the related compound Ru360 are known potent inhibitors of the MCU, but mutations in residues in this stretch of amino acids and in the intermembrane carboxy terminus both conferred a marked resistance to both calcium uptake and the inhibitory effect of Ru360, strongly suggesting that this is the pore-forming unit of the uniporter [46].
b) Regulators
Following the identification of MCU, several other regulators and a dominant-negative paralog of MCU have been identified. This has lead to the understanding that MCU exist as a multiprotein complex and that these regulators modulate MCU activity. Though their exact stoichiometry, role and interaction within the uniporter are still under investigation, what has become clear is that this is a complex ion channel. Furthermore, it appears that the relative ratios of these regulators, as well as the ratio MCU to its paralog, may vary from tissue to tissue in proportion to levels of mitochondrial calcium uptake. This exacting level of regulation is consistent with the important and complex role that mitochondrial calcium, and the MCU play in cell physiology [50].
i) MCUb
MCUb is a 33 kDa protein that looks very similar to the MCU in structure and orientation but that acts as a dominant-negative paralog of the MCU due to substitutions in the previously discussed loop region of the channel. This channel does not allow the movement of calcium ions across the inner mitochondrial membranes, but instead appears to reduce the movement of calcium into the mitochondria when it oligomerizes with the MCU. Perhaps most intriguingly, the ratio of MCU and MCUb mRNA expression varies from 40:1 to 3:1 across different tissue types, suggesting a mechanism to differentially regulate mitochondrial calcium uptake in different tissues. Cardiomyocytes, for instance, take up dramatically less calcium into their mitochondria, and have a much lower ratio (3:1) of MCU: MCUb than skeletal muscle, which has been observed to take up 28 times more calcium, and which has an MCU:MCUb at a 40:1 ratio [51].
ii) MICU1 and MICU2
With the MCU and one of its regulatory units MICU1 identified, research began to identify the capacities in which MICU1 regulated MCU. Results were almost immediately confusing: Overexpression of MICU1 clearly led to higher levels of mitochondrial calcium, especially at higher levels of cytosolic calcium, but silencing of MICU1 also unexpectedly led to higher levels of mitochondrial calcium, even at basal cytosolic calcium levels. This led to the understandable, but ultimately incorrect assessment that MICU1 had different roles at different calcium concentrations [45,52,53].
In 2014, the picture was greatly clarified by the discovery of a 45-kDa homolog of MICU1 called MICU2. Both proteins are exclusively found in the mitochondrial membrane, and have EF hands, but appear to play very different roles. MICU1 appears to primarily act to respond to high cytosolic calcium levels by stimulating MCU to uptake calcium, whereas MICU2 appears to inhibit the function of the MCU at lower cytosolic calcium levels. When MICU2 was specifically silenced, there was an increase in mitochondrial calcium levels. When MICU2 was overexpressed, there was also a small, though statistically significant, decrease in mitochondrial calcium concentration. MICU2 therefore effectively sets a threshold for MCU function and for the calcium concentration of the mitochondria thereby preventing calcium overload. It now appeared MICU1 and MICU2 work in balanced opposition to finely tune mitochondrial calcium intake [52]. This model also explains the sigmoidal response of the mitochondria, and the MCU, to calcium: at lower concentrations, cytosolic calcium fluctuations may go largely ignored by the MCU—but at higher concentrations, there can be a large and immediate uptake of calcium into the mitochondria via the MCU.
Furthermore, research found that while MICU1 is capable of homodimerizing, that MICU2 necessarily heterodimerizes with MICU1 via a disulfide bond: MICU2 appears to associate with the MCU via the MICU1. The MICU2 protein physically connects to the MICU1, which then binds to the MCU, and so a cell with silenced or absent MICU1 has similarly compromised levels functional MICU2 protein (though not mRNA). This explained some of the confusing results initially found by labs, specifically that cells without or with less MICU1 actually had higher levels of calcium within their mitochondrial matrices. It was now obvious that without MICU1 to stabilize it, MICU2 levels were also reduced, and the MCU lost not only its stimulatory regulator (MICU1) but also by extension its inhibitory regulator (MICU2). The loss of MICU2 then contributed to the higher observed baseline mitochondrial calcium levels [52,54].
Mootha's group did find that MICU2 had some ability to stabilize or potentially act as a MICU1 paralog: when MICU2 was administered to MICU1 silenced cells, MICU1 levels were restored to half of wild-type levels, suggesting that there was further complexity to the interaction of MICU1 with MICU2, which has not yet been fully described. A third paralog, MICU3, was also identified, but present almost exclusively skeletal muscle and the nervous system [52]. Overall, ratios of each of the three MICU proteins were found to vary from tissue to tissue, suggesting a very complex mechanism for cytosolic and mitochondrial calcium handling that differs from tissue to tissue.
The control of these two paralogs both appears to be mediated by calcium binding to their EF hands: when cytosolic calcium levels rise, the EF hands of MICU1 and MICU2 are bound, simultaneously relieving the inhibitory MICU2 and stimulating the activating MICU1. Mootha's group made several insightful observations that underscored the essential nature of these EF hands. First, they found that mutating the EF hands of either MICU1 or MICU2 so that they were unable to bind calcium significantly reduced calcium uptake by the MCU. This effect was most obvious with the mutation of MICU2, consistent with its identified role as an inhibitor, and the release of MICU2 inhibition via calcium binding to its EF hands. EF mutation of MICU1 also reduced overall calcium uptake, though it appeared to be most significant at higher levels of cytosolic calcium. This is consistent with Rizzuto's finding that MICU1 stimulates calcium uptake: at higher levels of calcium, binding of its EF hands promotes calcium uptake by the MCU [52,53]. It was also found that MICU2 can stabilize or potentially act as a MICU1 paralog: when MICU2 was administered to MICU1 silenced cells, MICU1 levels were restored to half of wild-type levels, suggesting that there was further complexity to the interaction of MICU1 with MICU2, which has not yet been fully described. A third paralog, MICU3, was also identified; it is present almost exclusively in skeletal muscle and the nervous system [52]. Overall, ratios of each of the three MICU proteins were found to vary from tissue to tissue, suggesting a very complex mechanism for cytosolic and mitochondrial calcium handling that differs from tissue to tissue. Given the role of mitochondrial calcium in both pathologic and physiologic cell processes, the need multiple regulators is expected.
iii) Essential MCU Regulator
In 2013, Mootha's lab identified, using SILAC, a 10 kDa single-pass transmembrane regulator that appears essential to MCU function, which they called Essential MCU Regulator (EMRE). MCU and MICU1/2 are exquisitely well preserved across phylum, which suggests that they were present even in early mitochondria. In contrast, metazoans appear to be the only lineage with EMRE: it has no equivalent in plants, fungi, or protozoa, suggesting that it is a fairly recent adaptation. Within mammals, however, it appears to be widely expressed, and essential for mitochondrial calcium transport. In both isolated mouse cells and organelles, EMRE silencing rendered the MCU completely ineffective. Thus far, research has suggested that while MICU1 and MICU2 are important MCU regulators, they are not essential for MCU function: EMRE therefore represents the first identified regulator essential for uniporter activity. The observation that EMRE is essential for uniporter activity appears to be in opposition to findings in in vitro bilayer studies, which found that purified MCU was sufficient for mitochondrial calcium transport [47,55]. However, Mootha and coworkers point out that the electrophysiological properties of MCU in bilayers were different from that in mitoplast and suggest that regulation might be different in mitochondria versus bilayers.
Interestingly, when MCU was silenced, EMRE expression was reduced, but normal levels of EMRE mRNA were present, suggesting the loss of EMRE was posttranslational, and possibly due loss of MCU destabilization. Furthermore, co-precipitation experiments have suggested that EMRE is responsible for the interaction between the MICU1/MICU2 heterodimer and MCU. The orientation of EMRE has not yet been fully identified, however it currently appears that EMRE may interact with both MICU1 and MICU2 in the inner membrane space and with the MCU within the inner membrane itself [49,55].
iv) SLC25A23
SLC25A23 is EF-hand containing mitochondrial carrier that exchanges Mg-ATP/Pi. Hoffman et al have reported that silencing SLC25A23 reduces, but does not eliminate, mitochondrial calcium uptake [56]. Hela cells with knockdown of SLC25A23 exhibit a smaller decrease in mitochondrial membrane potential following mitochondrial calcium uptake. It is suggested that SLC25A23 might regulate MCU by calcium activation of the phosphate anion transporter, but it has also been suggested that SLC25A23 interacts with MCU. It should be noted that SLC25A23 was not found in the MCU complex in the SILAC study by Mootha and coworkers. Silencing of SLC25A23 also appeared to be potentially protective in cell death [56]. In apparent contrast to the results of Hoffman et al, Amigo et al report that loss of SLC25A23 reduces mitochondrial calcium retention capacity, consistent with enhanced mPTP opening [57].
v) MCUR1
Mitochondrial Calcium Uniporter Regulator 1, or MCUR1, is a 40-kDa protein with two transmembrane domains and one coiled-coil region, which, like MCU, has N and C-termini that face the intermembrane space. Cells without MCUR1 appear similar to cells with MCU knockdown, and have no obvious calcium uptake. Cells also exhibit increased autophagy and increased resistance to apoptotic and necrotic cell death [58]. Overexpression of MCUR1 results in increased mitochondrial calcium uptake, but only when MCU is expressed. This suggests that calcium is indeed transported through the MCU. Furthermore, silencing of MCUR1 led to an increase in MCU mRNA and protein levels. Perhaps most interestingly, an MCUR1 equivalent has been identified in Saccharomyces cerevisie, or yeast, which has demonstrable mitochondrial calcium uptake, but no MCU equivalent [59,60].
Other regulators
A variety of other proteins, including mitochondrial sodium calcium exchanger (NCLX), uncoupling proteins (UPC) 2 and 3, leucine-zipper EF-hand containing transmembrane protein 1 (LETM1), EPR57, and transient receptor potential 3 (TRCP3), also appear important for mitochondrial calcium physiology, but their exact relationships, both physical and biochemical, have yet to be fully elucidated [43,55].
c) Knockout Mice
Once the molecular identity of the MCU was described, the Finkel lab created knockout mice to further elucidate the role of the MCU in biologic processes, with some perplexing results. The mice showed no evidence of calcium uptake into the mitochondria. Although mitochondrial calcium has been proposed to play a role in responding to changing energy demands, surprisingly when they analyzed oxygen consumption of MCU−/− MEFs under basal conditions, there were no detectable differences from WT MEFs using a variety of potential metabolic substrates. In addition there was no change in total body basal oxygen consumption between the WT and MCU-/- mice. Thus it was concluded that “that basal metabolism is not markedly altered in the absence of MCU expression.” Only in skeletal muscle, under conditions of maximum work, was there a difference between WT and MCU-KO mice; the MCU knockout mice had reduced abilities to generate maximal power.
One surprising finding was that it was possible to create MCU KO mice at all. Given the important role of mitochondrial calcium in a variety of cell processes, it was very possible that an MCU KO phenotype would be lethal. Though MCU-KO mice were viable in an outbred background, they were born in non-Mendelian ratio, suggesting that there was some embryonic lethality. Furthermore, the MCU-KO mice were embryonic lethal in a C57 background (See Ref 68 for further discussion).
Surprisingly, while the KO mice were smaller than their WT counterparts, they appeared to be physiologically very similar on a gross level: their organ weights were proportional to their body size, and there were no changes in overall body composition between KO and WT mice. Furthermore, examination by electron microscopy revealed no defects in mitochondrial population or morphology.
Mitochondria from MCU-KO mice, mouse embryo fibroblasts (MEFs), and isolated adult cardiac myocytes were unable to take up calcium. In contrast, WT mitochondria were able to quickly and efficiently take up calcium, unless they were treated with ruthenium red or Ru360 to inhibit the MCU, which reduced their mitochondrial calcium levels to levels comparable to that observed in the KO mice. As had been observed in MCU silenced and knockdown models, the loss of the ability to take up calcium had no obvious effects on aerobic respiration in the purified mitochondria from KO mice [61].
While there was a significant reduction in total mitochondrial matrix calcium, there was not a complete absence of mitochondrial matrix calcium in KO mice: in skeletal muscle it was reduced to about 25% of WT levels. The presence of mitochondrial calcium in the KO mice might argue for alternative mechanisms for calcium entry, although the data from the MCU mice suggest that if there are alternative calcium uptake pathways they must be very slow, as no calcium uptake occurred over a 10 to 20 minute time scale. It is also work noting that mitochondria have a sodium-calcium exchanger that normally functions to extrude calcium from the mitochondria. However this exchanger is in electrochemical equilibrium with sodium (and possibly the membrane potential if it is electrogenic) [62]. This exchanger can run in reverse and could allow a mechanism for calcium entry into the mitochondria [63,64].
Because mPTP opening and subsequent cell death is generally initiated by a large influx of calcium into the mitochondria via the MCU, there was a great deal of interest in whether the MCU KO mice would be protected from mPTP opening and resultant cell death following ischemia and reperfusion. When exposed to pathologic levels of calcium, mitochondria from WT mice experience mPTP opening, and are protected by cyclosporine A (CsA), which reduces the mPTP opening, and ruthenium red, known to reduce mitochondrial calcium influx [65]. Mitochondria or permeabilized MEFs from KO mice exhibited no signs of mPTP opening when exposed to high calcium levels that resulted in mPTP opening in WT mitochondria. However, there was no evidence that the MCU-KO MEFs were protected from cell death. MCU-KO MEFs died as quickly and in similar numbers to WT MEFs and had similar levels of caspase-3 activation and mitochondrial swelling. Furthermore, perfused hearts from MCU-KO mice subjected to ischemia and reperfusion showed no difference in infarct size or in recovery of contractile function compared to WT hearts. It should be noted that while there was no mPTP opening in MCU KO mitochondria exposed to large amounts of calcium, this does not preclude mPTP opening by ROS, independent of calcium, in cells following ischemia-reperfusion. Thus it is possible that the mPTP is activated in the MCU-KO hearts in a calcium independent manner. This would be consistent with suggestions that ROS is the primary activator of mPTP in vivo [37,66,67]. If mPTP activation occurs in MCU-KO heart, then one would expect that CsA, an inhibitor of the mPTP, should provide protection in the MCU-KO hearts. This was tested by Pan et al in the MCU knockout mice. They found that following ischemia and reperfusion that WT hearts were protected from cell death by the administration of CsA, but that MCU-KO hearts were not [61]. These data suggest that in contrast to the WT hearts, inhibition of the mPTP is not protective in the MCU-KO hearts. There are several possible explanations for this surprising finding. One explanation for the comparable rates of cell death is an up-regulation of a different cell death pathway, one that does not involve mPTP opening. Another possible explanation is that mPTP opening is occurring in the MCU-KO hearts, but it is not inhibited by CsA, perhaps because the stimulus for mPTP opening is greater in MCU-KO hearts than in the WT hearts. It is well established that mPTP opening is only facilitated by CyP-D and can still occur in its absence or in the presence of CsA if the stimulus is sufficiently great. Finally, the observation that MCU knockout mice are not cardioprotected is also consistent with the hypothesis that cytosolic calcium, rather than mitochondrial calcium is the key determinant of cell death [68].
4) Other Channels
Recent observations by Graier's lab further suggest that there are alternative mitochondrial pathways for calcium uptake. In one of their recent studies they used electrophysiology to identify up to 5 different calcium currents in mitoplasts [69,70]. Another lab has found that the ryanodine receptor, previously known to be one of the major channels for SR calcium release, is also present on the mitochondrial inner membrane, and serves as an additional mitochondrial uptake mechanism in cardiomyocytes and neurons. Like the MCU, they found the ryanodine receptor responds to increases in cytosolic calcium by increasing mitochondrial calcium uptake [71]. These data suggest that there are alternative mitochondrial calcium uptake pathways. However, the MCU-KO mitochondria do not take up any measurable calcium over a 10 to 20 minute time period, making it unlikely that any of these alterative mitochondrial calcium uptake mechanisms can function on rapid time scale. It is also possible that during mitoplast isolation that ER/SR or plasma membrane might incorporate into the mitoplast, introducing additional currents. Future work will be needed to address this issue.
5) Human Disease
In 2014, a human disease process attributable to the uniporter was reported. They found that individuals with mutations in MICU1 had a clinical phenotype that included proximal myopathy, a progressive extrapyramidal movement disorder and learning disabilities. In these individuals, mitochondrial uptake at low cytosolic calcium levels was increased (likely due in part to the destabilization of MICU2) and cytosolic calcium signals occurred at a lower amplitude and frequency. Interestingly, they found that under basal conditions, fibroblast from diseased individuals had normal cellular metabolic function. Given this, and their progressive disease course, the researchers theorized that affected individuals were able to compensate to a certain degree, after which the chronic activation of the MCU channel and mitochondrial calcium overload stressed cells enough to create the observed phenotype. The discovery of an MCU disease also re-emphasized the importance of the uniporter, and its regulator, on both mitochondrial calcium uptake and, more broadly, on human health [72].
Figure 1. Impact of MICU1 or MICU2 loss on MCU activity.
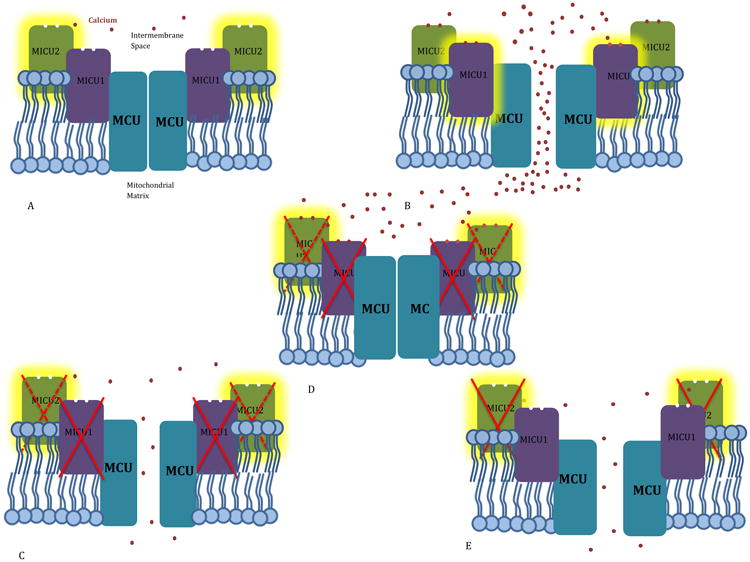
Highlighting indicates the dominant regulator at a given calcium concentration A. At low calcium levels, MICU2's EF hands are unbound, and it inhibits the MCU, keeping it closed and preventing calcium entry into the mitochondria. B. At high calcium levels, the EF hands of both MICU2 and MICU1 are bound, quieting inhibitory MICU2 and activating stimulatory MICU1. The MCU opens, and calcium is taken up rapidly into the mitochondria. C. With MICU1 silenced, MICU2 loses its physical connection to the MCU, and is unable to set a threshold for calcium uptake. Calcium therefore “leaks” into the mitochondria, even at low cytosolic calcium levels. D When MICU1 has been silenced, the stimulatory regulator is unable to encourage MCU to open, even at high cytosolic calcium levels, and calcium uptake is greatly reduced. E. With MICU2 silenced, the MCU loses its primary threshold for calcium uptake. Calcium therefore “leaks” into the mitochondria, even at low cytosolic calcium levels.
Figure 2. Ratio of MCU to MCUb in Uniporter Affects Calcium Uptake.

Relative amounts of calcium uptake by the MCU tetramer (represented by green arrows) vary based on the ration of MCU to MCUb, the negative paralog of MCU. MCU tetramers with lower ratios of MCU:MCUb have less calcium uptake. The ratio of MCU:MCUb has been noted to vary from tissue to tissue, suggesting that mitochondrial calcium handling varies by site.
The MCU: Questions Answered and Unanswered.
In the three years since the discovery of the MCU, it has become clear that the uniporter exists as a complex, with a large number of regulatory proteins. Though a variety of regulators and a paralog of MCU have been identified, their exact roles and stoichiometric variance from tissue to tissue are still not fully understood. The creation of MCU KO mice has established the importance of the uniporter in generating maximal power, but also suggested that the MCU may not be essential, or even important, at baseline oxygen consumption or basal metabolism. Additionally, lack of protection from I/R injury in MCU-KO hearts, in spite of the apparent lack of calcium active mPTP, might lead to a re-examination of the role of mPTP in ischemia-reperfusion cell death. Finally, the identification of a progressive human disease characterized by MICU1 deficiency has underscored the essential, though not yet fully characterized, nature of this complex.
MCU Highlights.
The mitochondrial calcium uniporter (MCU) was recently identified.
Regulators, including MICU1, MICU2, EMRE and a null paralog, have been identified
Mitochondria from MCU KO mice do not take up calcium
Mitochondria from MCU KO mice do not undergo calcium activated mPTP.
Hearts from MCU KO mice were not protected from ischemic/reperfusion injury
Acknowledgments
EM was supported by the NHLBI Intramural Research Program. Support for JH was made possible through the National Institutes of Health (NIH) Medical Research Scholars Program, a public-private partnership supported jointly by the NIH and generous contributions to the Foundation for the NIH from Pfizer Inc, The Doris Duke Charitable Foundation, The Newport Foundation, The American Association for Dental Research, The Howard Hughes Medical Institute, and the Colgate-Palmolive Company, as well as other private donors. For a complete list, please visit the Foundation website at: http://fnih.org/work/education-training-0/medical-research-scholars-program
Abbreviations
- EMRE
Essential MCU Regulator
- ER
endoplasmic reticulum
- IP3
inositol 1,4,5 triphosphate
- LETM1
leucine-zipper EF-hand containing transmembrane protein 1
- MCU
Mitochondrial Calcium Uniporter
- MCUR1
Mitochondiral Calcium Uniporter Regulator 1
- MICU1
Mitochondrial Calcium Uptake 1
- mPTP
mitochondrial permeability transition pore
- NCLX
mitochondrial sodium calcium exchanger
- RuR
ruthenium red
- UPC
uncoupling proteins
- SR
sarcoplasmic reticulum
- VDAC
voltage dependent anion channel
Footnotes
Disclosures: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ringer S. A third contribution regarding the Influence of the Inorganic Constituents of the Blood on the Ventricular Contraction. J Physiol. 1883 Aug 1;4(2-3):222–5. doi: 10.1113/jphysiol.1883.sp000127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeLuca HF, Engstrom GW. Calcium Uptake by Rat Kidney Mitochondria*. Proc Natl Acad Sci U S A. 1961 Nov;47(11):1744–50. doi: 10.1073/pnas.47.11.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rossi CS, Lehninger AL. Stoichiometry of Respiratory Stimulation, Accumulation of Ca++ and Phosphate, and Oxidative Phosphorylation in Rat Liver Mitochondria. J Biol Chem. 1964 Nov 1;239(11):3971–80. [PubMed] [Google Scholar]
- 4.Jouaville LS, Pinton P, Bastianutto C, Rutter GA, Rizzuto R. Regulation of mitochondrial ATP synthesis by calcium: Evidence for a long-term metabolic priming. Proc Natl Acad Sci. 1999 Nov 23;96(24):13807–12. doi: 10.1073/pnas.96.24.13807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Denton RM, McCormack JG. On the role of the calcium transport cycle in heart and other mammalian mitochondria. FEBS Lett. 1980 Sep 22;119(1):1–8. doi: 10.1016/0014-5793(80)80986-0. [DOI] [PubMed] [Google Scholar]
- 6.Nicholls DG. The regulation of extramitochondrial free calcium ion concentration by rat liver mitochondria. Biochem J. 1978 Nov 15;176(2):463–74. doi: 10.1042/bj1760463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Denton RM, McCormack JG, Edgell NJ. Role of calcium ions in the regulation of intramitochondrial metabolism. Effects of Na+, Mg2+ and ruthenium red on the Ca2+-stimulated oxidation of oxoglutarate and on pyruvate dehydrogenase activity in intact rat heart mitochondria. Biochem J. 1980 Jul 15;190(1):107–17. doi: 10.1042/bj1900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brinley FJ, Tiffert T, Scarpa A, Mullins LJ. Intracellular calcium buffering capacity in isolated squid axons. J Gen Physiol. 1977 Sep;70(3):355–84. doi: 10.1085/jgp.70.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tarasov AI, Griffiths EJ, Rutter GA. Regulation of ATP production by mitochondrial Ca2+ Cell Calcium. 2012 Jul;52(1):28–35. doi: 10.1016/j.ceca.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Joseph SK, Thomas AP, Williams RJ, Irvine RF, Williamson JR. myo-Inositol 1,4,5-trisphosphate. A second messenger for the hormonal mobilization of intracellular Ca2+ in liver. J Biol Chem. 1984 Mar 10;259(5):3077–81. [PubMed] [Google Scholar]
- 11.Streb H, Irvine RF, Berridge MJ, Schulz I. Release of Ca2+ from a nonmitochondrial intracellular store in pancreatic acinar cells by inositol-1,4,5-trisphosphate. Nature. 1983 Nov 3;306(5938):67–9. doi: 10.1038/306067a0. [DOI] [PubMed] [Google Scholar]
- 12.Tsien RY, Pozzan T, Rink TJ. Calcium homeostasis in intact lymphocytes: cytoplasmic free calcium monitored with a new, intracellularly trapped fluorescent indicator. J Cell Biol. 1982 Aug;94(2):325–34. doi: 10.1083/jcb.94.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O'Rourke B, Blatter LA. Mitochondrial Ca2+ uptake: tortoise or hare? J Mol Cell Cardiol. 2009 Jun;46(6):767–74. doi: 10.1016/j.yjmcc.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dedkova EN, Blatter LA. Calcium signaling in cardiac mitochondria. J Mol Cell Cardiol. 2013 May;58:125–33. doi: 10.1016/j.yjmcc.2012.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sheu SS, Sharma VK. Rapid report: a novel technique for quantitative measurement of free Ca2+ concentration in rat heart mitochondria. J Physiol. 1999 Jul 15;518(Pt 2):577–84. doi: 10.1111/j.1469-7793.1999.0577p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marban E, Rink TJ, Tsien RW, Tsien RY. Free calcium in heart muscle at rest and during contraction measured with Ca2+ -sensitive microelectrodes. Nature. 1980 Aug 28;286(5776):845–50. doi: 10.1038/286845a0. [DOI] [PubMed] [Google Scholar]
- 17.Allen DG, Blinks JR. Calcium transients in aequorin-injected frog cardiac muscle. Nature. 1978 Jun 15;273(5663):509–13. doi: 10.1038/273509a0. [DOI] [PubMed] [Google Scholar]
- 18.Becker GL, Fiskum G, Lehninger AL. Regulation of free Ca2+ by liver mitochondria and endoplasmic reticulum. J Biol Chem. 1980 Oct 10;255(19):9009–12. [PubMed] [Google Scholar]
- 19.Horikawa Y, Goel A, Somlyo AP, Somlyo AV. Mitochondrial Calcium in Relaxed and Tetanized Myocardium. Biophys J. 1998 Mar;74(3):1579–90. doi: 10.1016/S0006-3495(98)77869-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Drummond RM, Mix TCH, Tuft RA, Walsh JV, Fay FS. Mitochondrial Ca2+ homeostasis during Ca2+ influx and Ca2+ release in gastric myocytes from Bufo marinus. J Physiol. 2000 Feb 1;522(3):375–90. doi: 10.1111/j.1469-7793.2000.t01-2-00375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hajnóczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell. 1995 Aug 11;82(3):415–24. doi: 10.1016/0092-8674(95)90430-1. [DOI] [PubMed] [Google Scholar]
- 22.Rudolf R, Mongillo M, Magalhães PJ, Pozzan T. In vivo monitoring of Ca(2+) uptake into mitochondria of mouse skeletal muscle during contraction. J Cell Biol. 2004 Aug 16;166(4):527–36. doi: 10.1083/jcb.200403102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Csordas G, Renken C, Varnai P, Walter L, Weaver D, Buttle KF, et al. Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol. 2006 Sep 25;174(7):915–21. doi: 10.1083/jcb.200604016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rizzuto R, Brini M, Murgia M, Pozzan T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science. 1993 Oct 29;262(5134):744–7. doi: 10.1126/science.8235595. [DOI] [PubMed] [Google Scholar]
- 25.Marsault R, Murgia M, Pozzan T, Rizzuto R. Domains of high Ca2+ beneath the plasma membrane of living A7r5 cells. EMBO J. 1997 Apr 1;16(7):1575–81. doi: 10.1093/emboj/16.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008 Dec 4;456(7222):605–10. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- 27.Dorn GW, Scorrano L. Two close, too close: sarcoplasmic reticulum-mitochondrial crosstalk and cardiomyocyte fate. Circ Res. 2010 Sep 17;107(6):689–99. doi: 10.1161/CIRCRESAHA.110.225714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Denton RM. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta. 2009 Nov;1787(11):1309–16. doi: 10.1016/j.bbabio.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 29.Territo PR, Mootha VK, French SA, Balaban RS. Ca(2+) activation of heart mitochondrial oxidative phosphorylation: role of the F(0)/F(1)-ATPase. Am J Physiol Cell Physiol. 2000 Feb;278(2):C423–435. doi: 10.1152/ajpcell.2000.278.2.C423. [DOI] [PubMed] [Google Scholar]
- 30.Glancy B, Balaban RS. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry. 2012 Apr 10;51(14):2959–73. doi: 10.1021/bi2018909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Balaban RS. Domestication of the cardiac mitochondrion for energy conversion. J Mol Cell Cardiol. 2009 Jun;46(6):832–41. doi: 10.1016/j.yjmcc.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baines CP. The molecular composition of the mitochondrial permeability transition pore. J Mol Cell Cardiol. 2009 Jun;46(6):850–7. doi: 10.1016/j.yjmcc.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Halestrap AP. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol. 2009 Jun;46(6):821–31. doi: 10.1016/j.yjmcc.2009.02.021. [DOI] [PubMed] [Google Scholar]
- 34.Di Lisa F, Carpi A, Giorgio V, Bernardi P. The mitochondrial permeability transition pore and cyclophilin D in cardioprotection. Biochim Biophys Acta. 2011 Jul;1813(7):1316–22. doi: 10.1016/j.bbamcr.2011.01.031. [DOI] [PubMed] [Google Scholar]
- 35.Hunter DR, Haworth RA, Southard JH. Relationship between configuration, function, and permeability in calcium-treated mitochondria. J Biol Chem. 1976 Aug 25;251(16):5069–77. [PubMed] [Google Scholar]
- 36.Szabadkai G, Duchen MR. Mitochondria: The Hub of Cellular Ca2+ Signaling. Physiology. 2008 Apr 1;23(2):84–94. doi: 10.1152/physiol.00046.2007. [DOI] [PubMed] [Google Scholar]
- 37.Juhaszova M, Wang S, Zorov DB, Nuss HB, Gleichmann M, Mattson MP, et al. The identity and regulation of the mitochondrial permeability transition pore: where the known meets the unknown. Ann N Y Acad Sci. 2008 Mar;1123:197–212. doi: 10.1196/annals.1420.023. [DOI] [PubMed] [Google Scholar]
- 38.Halestrap AP. Calcium, mitochondria and reperfusion injury: a pore way to die. Biochem Soc Trans. 2006 Apr;34(Pt 2):232–7. doi: 10.1042/BST20060232. [DOI] [PubMed] [Google Scholar]
- 39.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005 Mar 31;434(7033):658–62. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 40.Griffiths EJ, Halestrap AP. Protection by Cyclosporin A of ischemia/reperfusion-induced damage in isolated rat hearts. J Mol Cell Cardiol. 1993 Dec;25(12):1461–9. doi: 10.1006/jmcc.1993.1162. [DOI] [PubMed] [Google Scholar]
- 41.Reed KC, Bygrave FL. The inhibition of mitochondrial calcium transport by lanthanides and ruthenium red. Biochem J. 1974 May;140(2):143–55. doi: 10.1042/bj1400143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Szabó I, Zoratti M. The giant channel of the inner mitochondrial membrane is inhibited by cyclosporin A. J Biol Chem. 1991 Feb 25;266(6):3376–9. [PubMed] [Google Scholar]
- 43.Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol. 2012 Sep;13(9):566–78. doi: 10.1038/nrm3412. [DOI] [PubMed] [Google Scholar]
- 44.Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004 Jan 22;427(6972):360–4. doi: 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- 45.Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, et al. MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature. 2010 Sep 16;467(7313):291–6. doi: 10.1038/nature09358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011 Jun 19;476(7360):341–5. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.De Stefani D, Raffaello A, Teardo E, Szabò I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011 Aug 18;476(7360):336–40. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marchi S, Pinton P. The mitochondrial calcium uniporter complex: molecular components, structure and physiopathological implications. J Physiol. 2013 Dec 23; doi: 10.1113/jphysiol.2013.268235. jphysiol 2013 268235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kamer KJ, Sancak Y, Mootha VK. The uniporter: From newly identified parts to function. Biochem Biophys Res Commun. 2014 Jul 11;449(4):370–2. doi: 10.1016/j.bbrc.2014.04.143. [DOI] [PubMed] [Google Scholar]
- 50.Pendin D, Greotti E, Pozzan T. The elusive importance of being a mitochondrial Ca2+ uniporter. Cell Calcium. 2014 Mar;55(3):139–45. doi: 10.1016/j.ceca.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 51.Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, et al. MICU2, a Paralog of MICU1, Resides within the Mitochondrial Uniporter Complex to Regulate Calcium Handling. [cited 2014 Aug 27];PLoS ONE. 2013 Feb 7;8(2) doi: 10.1371/journal.pone.0055785. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M, et al. MICU1 and MICU2 Finely Tune the Mitochondrial Ca2+ Uniporter by Exerting Opposite Effects on MCU Activity. Mol Cell. 2014 Mar 6;53(5):726–37. doi: 10.1016/j.molcel.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Csordas G, Golenar T, Seifert EL, Kamer KJ, Sancak Y, Perocchi F, et al. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca2+ uniporter. Cell Metab. 2013 Jun 4;17(6):976–87. doi: 10.1016/j.cmet.2013.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kamer KJ, Mootha VK. MICU1 and MICU2 play nonredundant roles in the regulation of the mitochondrial calcium uniporter. EMBO Rep. 2014 Mar 1;15(3):299–307. doi: 10.1002/embr.201337946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sancak Y, Markhard AL, Kitami T, Kovács-Bogdán E, Kamer KJ, Udeshi ND, et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science. 2013 Dec 13;342(6164):1379–82. doi: 10.1126/science.1242993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hoffman NE, Chandramoorthy HC, Shanmughapriya S, Zhang XQ, Vallem S, Doonan PJ, et al. SLC25A23 augments mitochondrial Ca2+ uptake, interacts with MCU, and induces oxidative stress-mediated cell death. Mol Biol Cell. 2014 Mar 15;25(6):936–47. doi: 10.1091/mbc.E13-08-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Amigo I, Traba J, Gonzalez-Barroso MM, Rueda CB, Fernandez M, Rial E, et al. Glucagon regulation of oxidative phosphorylation requires an increase in matrix adenine nucleotide content through Ca2+-activation of the mitochondrial ATP-Mg/Pi carrier SCaMC-3. J Biol Chem. 2013 Jan 23; doi: 10.1074/jbc.M112.409144. jbc M112 409144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mallilankaraman K, Cardenas C, Doonan P, Chandramoorthy HC, Irrinki KM, Golenar T, et al. MCUR1 is an Essential Component of Mitochondrial Ca2+ Uptake that Regulates Cellular Metabolism. Nat Cell Biol. 2012 Dec;14(12):1336–43. doi: 10.1038/ncb2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kovács-Bogdán E, Sancak Y, Kamer KJ, Plovanich M, Jambhekar A, Huber RJ, et al. Reconstitution of the mitochondrial calcium uniporter in yeast. Proc Natl Acad Sci U S A. 2014 Jun 17;111(24):8985–90. doi: 10.1073/pnas.1400514111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Uribe S, Rangel P, Pardo JP. Interactions of calcium with yeast mitochondria. Cell Calcium. 1992 Apr;13(4):211–7. doi: 10.1016/0143-4160(92)90009-h. [DOI] [PubMed] [Google Scholar]
- 61.Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter (MCU) [cited 2014 Aug 26];Nat Cell Biol. 2013 Dec;15(12) doi: 10.1038/ncb2868. Internet. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3852190/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boyman L, Williams GSB, Khananshvili D, Sekler I, Lederer WJ. NCLX: the mitochondrial sodium calcium exchanger. J Mol Cell Cardiol. 2013 Jun;59:205–13. doi: 10.1016/j.yjmcc.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Murphy E, Eisner DA. Regulation of intracellular and mitochondrial sodium in health and disease. Circ Res. 2009 Feb 13;104(3):292–303. doi: 10.1161/CIRCRESAHA.108.189050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Griffiths EJ. Reversal of mitochondrial Na/Ca exchange during metabolic inhibition in rat cardiomyocytes. FEBS Lett. 1999 Jun 25;453(3):400–4. doi: 10.1016/s0014-5793(99)00726-7. [DOI] [PubMed] [Google Scholar]
- 65.Moore CL. Specific inhibition of mitochondrial Ca++ transport by ruthenium red. Biochem Biophys Res Commun. 1971 Jan 22;42(2):298–305. doi: 10.1016/0006-291x(71)90102-1. [DOI] [PubMed] [Google Scholar]
- 66.Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL. Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta. 2009 Nov;1787(11):1395–401. doi: 10.1016/j.bbabio.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014 Jul;94(3):909–50. doi: 10.1152/physrev.00026.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Murphy E, Pan X, Nguyen T, Liu J, Holmström KM, Finkel T. Unresolved questions from the analysis of mice lacking MCU expression. Biochem Biophys Res Commun. 2014 Jul 11;449(4):384–5. doi: 10.1016/j.bbrc.2014.04.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bondarenko AI, Jean-Quartier C, Malli R, Graier WF. Characterization of distinct single-channel properties of Ca2+ inward currents in mitochondria. Pflüg Arch - Eur J Physiol. 2013 Jul 1;465(7):997–1010. doi: 10.1007/s00424-013-1224-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bondarenko AI, Jean-Quartier C, Parichatikanond W, Alam MR, Waldeck-Weiermair M, Malli R, et al. Mitochondrial Ca2+ uniporter (MCU)-dependent and MCU-independent Ca2+ channels coexist in the inner mitochondrial membrane. Pflugers Arch. 2014;466:1411–20. doi: 10.1007/s00424-013-1383-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jakob R, Beutner G, Sharma VK, Duan Y, Gross RA, Hurst S, et al. Molecular and functional identification of a mitochondrial ryanodine receptor in neurons. Neurosci Lett. 2014 Jul 11;575:7–12. doi: 10.1016/j.neulet.2014.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Logan CV, Szabadkai G, Sharpe JA, Parry DA, Torelli S, Childs AM, et al. Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat Genet. 2014 Feb;46(2):188–93. doi: 10.1038/ng.2851. [DOI] [PubMed] [Google Scholar]