

Abstract
Periventricular leukomalacia is the predominant injury in the preterm infant leading to cerebral palsy. Oxygen exposure may be an additional cause of brain injury in these infants. In this study, we investigated pathways of maturation-dependent oligodendrocyte (OL) death induced by hyperoxia in vitro and in vivo. Developing and mature OLs were subjected to 80% oxygen (0–24 h). Lactate dehydrogenase (LDH) assay was used to assess cell viability. Furthermore, 3-, 6-, and 10-d-old rat pups were subjected to 80% oxygen (24 h), and their brains were processed for myelin basic protein staining. Significant cell death was detected after 6–24 h incubation in 80% oxygen in pre-OLs (O4+,O1−), but not in mature OLs (MBP+). Cell death was executed by a caspase-dependent apoptotic pathway and could be blocked by the pan-caspase inhibitor zVAD-fmk. Overexpression of BCL2 (Homo sapiens B-cell chronic lymphocytic leukemia/lymphoma 2) significantly reduced apoptosis. Accumulation of superoxide and generation of reactive oxygen species (ROS) were detected after 2 h of oxygen exposure. Lipoxygenase inhibitors 2,3,5-trimethyl-6-(12-hydroxy-5-10-dodecadiynyl-1,4-benzoquinone and N-benzyl-N-hydroxy-5-phenylpentamide fully protected the cells from oxidative injury. Overexpression of superoxide dismutase (SOD1) dramatically increased injury to pre-OLs but not to mature OLs. We extended these studies by testing the effects of hyperoxia on neonatal white matter. Postnatal day 3 (P3) and P6 rats, but not P10 pups, showed bilateral reduction in MBP (myelin basic protein) expression with 24 h exposure to 80% oxygen. Hyperoxia causes oxidative stress and triggers maturation-dependent apoptosis in pre-OLs, which involves the generation of ROS and caspase activation, and leads to white matter injury in the neonatal rat brain. These observations may be relevant to white matter injury observed in premature infants.
Keywords: oxidative stress, periventricular leukomalacia, development, oligodendrocyte, apoptosis, lipoxygenase inhibitors
Introduction
In premature infants, periventricular leukomalacia (PVL) is a common type of injury of the cerebral white matter resulting in a chronic disturbance of myelination. It is the predominant underlying pathology of cerebral palsy and cognitive impairment in survivors of premature birth (Volpe, 2001a). PVL has its peak incidence during the period of extensive oligodendrocyte (OL) migration and maturation (Volpe, 2001b). In the human brain, the predominant stage of the oligodendrocyte lineage present during this period is the premyelinating oligodendrocyte (Back et al., 2001; Kinney and Armsrong, 2002). Mature oligodendrocytes become abundant after term. Two major causes are generally considered to be responsible for perinatal white matter injury: cerebral ischemia/reperfusion in the premature infant, combined with the propensity for impaired vascular autoregulation (Menke et al., 1997; Volpe, 1998) and bacterial infection in the mother and/or fetus that triggers a cytokine response in the fetal brain (Kinney et al., 2004; Leviton and Dammann, 2004). In the past 10 years, there has been no improvement in the neurodevelopmental outcome of premature infants (Hamrick et al., 2004).
Oxygen is widely used in resuscitation and in treatment of neonatal lung disease. Furthermore, premature infants are subjected much earlier to relative hyperoxia, because of a dramatic rise of oxygen tissue tension compared with intrauterine conditions. Hyperoxia has been implicated in the pathogenesis of bronchopulmonary dysplasia and retinopathy of prematurity (Saugstad, 2001; Chow et al., 2003). There is increasing evidence that hyperoxia may negatively influence brain maturation and development (Collins et al., 2001; Felderhoff-Mueser et al., 2004; Klinger et al., 2005; Gerstner et al., 2006). However, the mechanism of hyperoxia-induced injury and the identification of vulnerable cells in the human brain are still unknown.
It has been found recently that oxygen-induced cell death in vivo is age dependent (Felderhoff-Mueser et al., 2004). Hyperoxia increased apoptotic cell death in cells of 7-d-old Wistar rats and C57BL/6 mice in brain regions such as the caudate nucleus, layers II and IV of the frontal, parietal, cingulate, and retrosplenial cortices, as well as white matter tracts and the periventricular region. This cell death was associated with oxidative stress, decreased expression of neurotrophins, decreased activation of neurotrophin-regulated pathways, and increased levels of proinflammatory cytokines (Felderhoff-Mueser et al., 2004; Felderhoff-Mueser et al., 2005). In these studies, neuroanatomical localization of cell death was characterized, but cell type was not specified. Because the predominant brain injury in the preterm infant is damage to the cerebral white matter, we wanted to characterize the effect of oxygen on the oligodendrocyte lineage. In addition, we aimed to identify which reactive oxygen species (ROS) are involved in the toxicity of hyperoxia and to characterize the death pathway. Furthermore, we investigated the effects of hyperoxia on white matter in the immature rat brain.
Materials and Methods
Materials
DMEM, HBSS, Earle's balanced salt solution, fetal bovine serum, penicillin, and streptomycin were purchased from Invitrogen (Carlsbad, CA). Fetal bovine serum was obtained from HyClone (Logan, UT). Antibodies against recombinant human superoxide dismutase (SOD1) were obtained from Calbiochem (La Jolla, CA). The pan-caspase inhibitor zVAD-fmk (Promega, Madison, WI) was used in a final concentration of 40 μm. 2,3,5-Trimethyl-6-(12-hydroxy-5–10-dodecadiynyl)-1,4-benzoquinone (AA861) was purchased from Biomol (Plymouth Meeting, PA). Ebselen, a glutathione (GSH) peroxidase (GPx) mimic, was purchased from Calbiochem (San Diego, CA). BMD-122 (N-benzyl-N-hydroxy-5-phenylpentanamide) was a gift from Dr. Lawrence J. Marnett (School of Medicine, Vanderbilt University, Nashville, TN). Unless otherwise specified, all other chemicals [staurosporine (#S3939), superoxide dismutase (S-2515; lot #073K7667), and catalase (C-40; lot # 034K7064)] were obtained from Sigma (St. Louis, MO).
Oligodendrocyte primary cultures
Primary rat OLs were prepared from the cerebral hemispheres of Sprague Dawley rats at postnatal day 1 (P1) to P2 using a shaking method (Rosenberg et al., 2003) with modifications, as described previously (Li et al., 2003). Animals were killed by decapitation. Purified OLs were cultured in 24-well plates (500 μl/well) for 7–8 d in a serum-free basal-defined medium: DMEM (Invitrogen #11960–069), 0.1% bovine serum albumin (Sigma #A9647), 50 μg/ml human apo-transferrin (Sigma #T2252), 50 μg/ml insulin (Sigma #I6634), 30 nm sodium selenite (Sigma #S5261), 10 nm d-biotin (Sigma #B4501), 10 nm hydrocortisone (Sigma #H0888), 200 μm l-cystine, 10 ng/ml human recombinant platelet-derived growth factor (#100–13A; Peprotech, Rocky Hill, NJ), and 10 ng/ml human recombinant basic fibroblast growth factor (Peprotech #100–18B). At 7–8 d, the cultures were composed primarily of progenitors and pre-OLs (O4+, O1−, MBP−). After 7 d, culture medium was changed to serum-free basal-defined medium containing 10 ng/ml rat recombinant ciliary neurotrophic factor (Peprotech #450–50) and 15 nm 3,3′,5-triiodo-l-thyronine (Sigma #T6397) for 10 additional days until cells were differentiated into mature OLs (MBP+). The purity of OL cultures was consistently >95% OLs with <5% astrocyte contamination.
Infection of cultured OLs with recombinant herpes-simplex virus
Primary oligodendrocyte cells were maintained in culture for 6 d before being exposed to the herpes simplex viral (HSV) vector for overexpression of Homo sapiens B-cell chronic lymphocytic leukemia/lymphoma 2 (BCL2) or SOD1. Vector construction for HSV carrying BCL2 (NM_0006633), SOD1 (X02317), or green fluorescent protein (GFP) has been described previously (Neve and Geller, 1999). Cultures were exposed to 1 × 104 pfu/ml (1 × 105 particles/ml) HSV vector for 16 h in regular culture medium, followed by a complete change of the culture medium on day 7. Cells were cultured for an additional 24 h to allow for protein expression before any experiment or analysis was performed. Noninfected cells and cells exposed to GFP containing HSV vector (HSV-GFP) were included as controls. Infection rate was determined by counting the proportion of GFP-expressing cells (∼70%).
Oxygen exposure
At the beginning of the experiment, cultures were placed in the appropriate growth medium, at a volume of 250 μl per well and transferred to a humidified hyperoxia chamber. The chamber was maintained at 80% oxygen using a PRO:OX 110 oxygen controller (Biospherix, Redfield, NY) that was connected to a tank containing 95% O2 plus 5% CO2. Experiments were performed for selected periods of time, and the lid of the culture plate was propped 0.5 cm above the plate to facilitate diffusion. Control plates were kept under 21% oxygen, indicated as normoxic, plus 5% CO2 conditions at 37°C. Cell density was found to influence susceptibility to hyperoxia-induced cytotoxicity. Maximal vulnerability occurred at a density of ∼11,000 cells per well.
Immunocytochemistry and immunofluorescence microscopy
Cells were fixed with 4% paraformaldehyde in PBS for 10 min at room temperature, washed three times with PBS, and blocked with TBST (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, and 0.1% Triton X-100) for the myelin basic protein (MBP) labeling, and TBS for the O1 and O4 labeling containing 5% goat serum, for 1 h at room temperature. The coverslips were incubated with mouse monoclonal antibodies O4, O1 (1:200), and MBP (1:400) overnight at 4°C. The following day, after three to four washes with PBS for 5 min each, the appropriate secondary antibody conjugated with Alexa-red goat anti-mouse IgM for O4 and O1, or with Alexa-red goat anti-mouse IgG for MBP (Invitrogen, Carlsbad, CA) were added to the coverslips and incubated for 1 h at RT. After extensive washes with TBS, nuclei were stained by adding Hoechst 33258 at a final concentration of 2 μg/ml for 1 min. After three more washes, the coverslips were mounted onto glass slides with FluoroMount (Southern Biotech, Birmingham, AL) and kept in the dark at 4°C. Cell images were captured with a fluorescence microscope (Eclipse E800; Nikon, Tokyo, Japan) equipped with a Spot RT digital camera (Diagnostic Instruments, Sterling Heights, MI).
Immunoblotting
Cells were lysed in 1% SDS buffer [pH 7.6, 20 mm HEPES, 1 protease inhibitor mixture tablet per 10 ml (Roche, Mannheim, Germany)], and lysates were collected and sonicated for 12 s. Protein concentrations were determined using the Bio-Rad (Hercules, CA) Dc Protein assay. Protein extracts (5–20 μg per sample) and a biotinylated molecular weight marker (Cell Signaling Technology, Beverly, MA) were denaturated in Laemli's sample loading buffer at 95°C, separated by 4–20% PAGE and electrotransferred in transfer buffer to a polyvinylidene fluoride membrane (0.2 μm pore; Bio-Rad). The membrane was rinsed with 0.1% Tween 20-containing Tris-buffered saline (TBST) and treated with blocking solution (5% nonfat dry milk in TBST). Equal loading and transfer of proteins was confirmed by staining the membranes with Ponceau S solution (Fluka, Buchs, Switzerland). The membrane was incubated overnight at 4°C with rabbit polyclonal anti-BCL2 antibody (1:200, 1 μg/ml; Santa Cruz Biotechnology, Santa Cruz, CA), and polyclonal rabbit anti-actin antibody (1:10,000; Sigma). Secondary incubations were performed with horseradish peroxidase-linked anti-mouse (1:2000; Bio-Rad) or anti-rabbit (1:7500; GE Healthcare, Bucks, UK) antibodies. Positive signals were visualized using enhanced chemiluminescence (ECL; PerkinElmer, Boston, MA), and serial exposures were made to radiographic film (Premium Autoradiography Film; Denville Scientific, Metuchen, NJ).
Survival assay
Lactate dehydrogenase release.
After exposure to hyperoxia, cell culture media were collected and centrifuged, and lactate dehydrogenase (LDH) activity was quantified in 50 μl samples of medium supernatant, using a colorimetric cytotoxicity assay kit according to the manufacturer directions (Roche). Absorbance data were obtained using a 96-well plate reader (Molecular Devices) with a 450 nm filter, and 650 nm as reference wavelength. Maximum LDH release was determined by cell lysis of one well of untreated cells by cell culture media containing 1% Triton X-100.
Detection of mitochondrial superoxide production
Mitochondrial superoxide production in premyelinating OLs was evaluated using MitoSOX red mitochondrial superoxide indicator for live-cell imaging (Invitrogen). MitoSOX red mitochondrial superoxide indicator is a nonratiometric assay using a fluorogenic dye and derivate of ethidium bromide for highly selective detection of superoxide in the mitochondria of live cells. Once in the mitochondria, MitoSOX reagent is oxidized by superoxide and exhibits red fluorescence after binding to nucleic acids. After cells were treated with 80% oxygen for 2–12 h, they were loaded with 5 μm MitoSOX for 10 min at 37°C, protected from light. For nucleic acid stain, Hoechst 33342 was added at a final concentration of 1 μm during the last 5 min of MitoSOX incubation. The fluorescence of the cells loaded with MitoSOX was measured using confocal microscopy (Zeiss, Thornwood, NY) (LSM 510 META, excitation λ, 510 nm/emission λ, 580 nm for MitoSOX; excitation λ, 350 nm/emission λ, 461 nm for Hoechst 33342 nuleic acid stain). The fluorescence intensity of the MitoSOX-loaded cells exposed to normoxia for 2–12 h served as controls.
Detection of reactive oxygen species accumulation
The generation of reactive oxygen species in the cytoplasm of premyelinating OLs was evaluated using 5-(and-6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (carboxy-H2DCFDA, Image-iT; Invitrogen). Oxidation of H2DCFDA to the fluorescent product 2′,7′-dichlorofluorescein (DCF) serves as an indicator of the overall degree of intracellular oxidative stress (Barja, 2002). The nonratiometric assay is based on carboxy-H2DCFDA, a reliable fluorogenic marker for ROS in live cells. In addition, the common inducer of ROS production tert-butyl hydroperoxide at 100 μm for 60–90 min served as a positive control. The blue-fluorescent, cell-permeant nucleic acid stain Hoechst 33342 was used for detection of the cell nuclei. After cells were treated with 80% oxygen for 2–12 h, they were loaded with 25 μm carboxy-H2DCFDA for 30 min at 37°C, protected from light. The fluorescence of the cells loaded with carboxy-H2DCFDA was measured using confocal microscopy (Zeiss) (LSM 510 META: excitation λ, 495 nm/emission λ, 529 nm for ROS detection; excitation λ, 350 nm/emission λ, 461 nm for Hoechst 33342 nuleic stain).
Animal studies
All animal experiments were performed in accordance with the guidelines of the Humboldt University (Berlin, Germany). P3, P6, and P10 Wistar rat pups (BgVV, Berlin, Germany) were placed together with their mothers in an oxygen chamber containing 80% oxygen for 24 h. Animals were killed at P11 by receiving an overdose of chloral hydrate and were transcardially perfused with heparinized 0.1 m PBS, and with 4% paraformaldehyde. Brains were postfixed for 24 h with 4% paraformaldehyde and then cryoprotected in 30% sucrose in PBS.
Immunocytochemistry and histological analysis
OL maturation was evaluated by immunocytochemistry (ICC) with antibodies to the specific OL marker MBP in rats killed at P11. Histological and ICC analysis on rodent tissue were performed on serial 16 μm coronal sections, as detailed previously (Follett et al., 2000). Sections were blocked and incubated overnight with anti-MBP (1:1000; SMI-99; Sternberger Monoclonals, Baltimore, MD), rinsed, and then incubated with the appropriate secondary Ab (Invitrogen, Eugene, OR) for 1 h at room temperature. MBP was used as a marker for white matter injury. Lesion size was assessed by a blinded observer, with reproducibility demonstrated by a second blinded observer, using a semiquantitative scale of 1–5 (corresponding to 100–0%): 0, no abnormality; 1, any loss of cortical processes; 2, some loss of processes, without any capsule involvement; 3, loss of processes and fragmenting or thinning of capsule; 4, ragged appearing capsule and thinning; 5, discontinuity in capsule, caused by massive thinning, full thickness loss in areas (Follett et al., 2000, 2004).
Statistical analysis
Data were analyzed using GraphPad Prism version 4.0 for Windows (GraphPad Software, San Diego, CA). Unless otherwise indicated, the results shown are one experiment representative of three to five separate experiments that were performed. Either Student's t test, one-way ANOVA with the Tukey-Kramer post hoc analysis, or two-way ANOVA were performed for statistical analyses. Statistical significance was assumed for p < 0.05.
Results
Hyperoxia reduces oligodendrocyte viability in a time-dependent manner
To estimate the time course of oxygen toxicity, pre-OLs were incubated with 80% oxygen for 0, 6, 12, and 24 h. Cell viability decreased with time of exposure (Fig. 1). After 12 h of incubation, only ∼25% of cells survived, and after 24 h, almost no cells were viable. Control cells were kept in 21% oxygen for 0–24 h and displayed a constant amount (8–10%) of cell death for each time period (data not shown).
Figure 1.

Hyperoxia causes cell death in pre-OLs. Primary oligodendrocyte cytotoxicity, as measured by LDH release, after 0, 6, 12, and 24 h incubation with 80% oxygen (means ± SEM of 3 independent experiments). *p < 0.05, ***p < 0.001 in one-way ANOVA followed by Tukey–Kramer test; comparison between 0 and 6, 12, and 24 h of 80% oxygen exposure.
Hyperoxia-induced reduction of oligodendrocyte viability is maturation dependent
When pre-OLs were exposed to 80% oxygen for 12 h, cell viability significantly decreased (Fig. 2A,B). In contrast, mature OLs, incubated under the same conditions, did not exhibit vulnerability to hyperoxia (Fig. 2D). Assessment of LDH release demonstrated that the toxicity of 80% oxygen was found only in pre-OLs but not in mature OLs (Fig. 2E).
Figure 2.
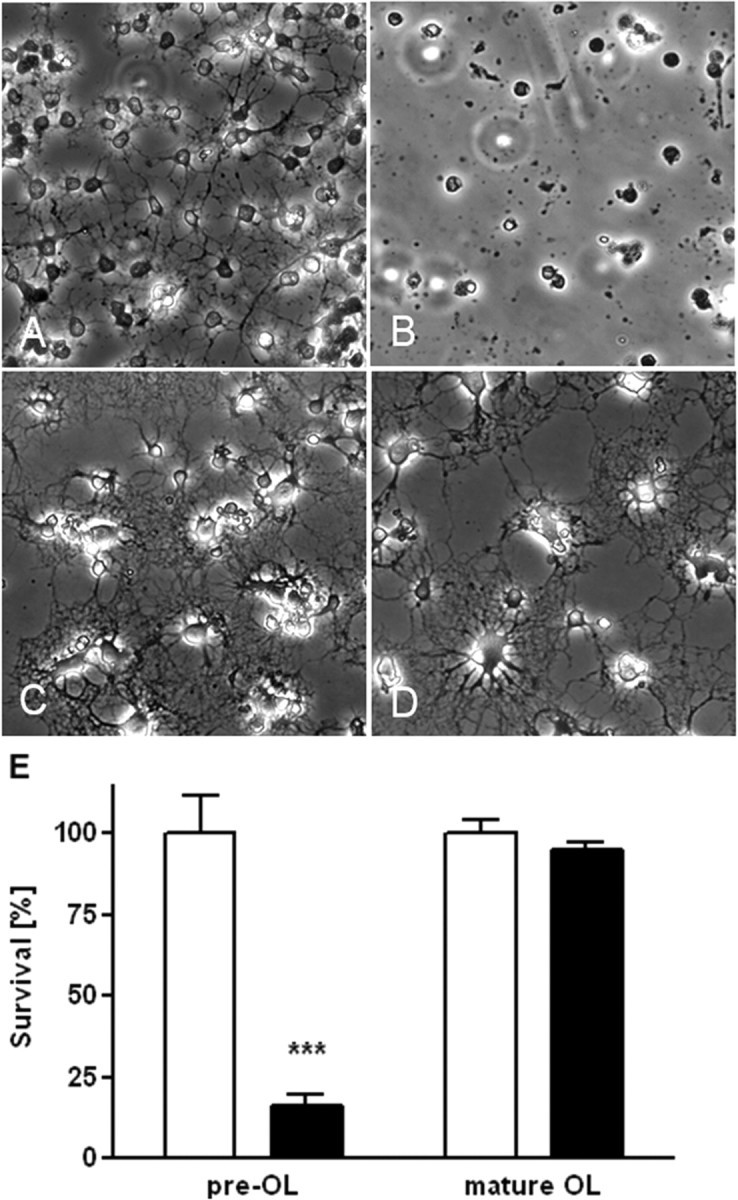
Representative phase contrast photomicrographs of primary oligodendrocytes after 12 h of incubation with either 21 or 80% oxygen. A–D, After incubation with 21% oxygen, pre-OLs (A) and mature OLs (C) are intact without signs of apoptosis. Hyperoxia-treated pre-OLs (B) show complete loss of processes, cell shrinkage, plasma membrane blebbing, and nuclear condensation. Mature OLs do not show signs of apoptosis after incubation in 80% oxygen for 12 h (D). E, Pre-OL and mature OL cell viability, as measured by LDH release, after 12 h of incubation with 80% oxygen (means ± SEM of 3 independent experiments). ***p < 0.001, Student's t test; comparison of hyperoxia group (black bar) versus the normoxia group (white bar) for each maturation stage.
Hyperoxia-induced pre-OL cell death is attenuated by the pan-caspase inhibitor zVAD-fmk
To assess whether inhibition of caspases could block apoptotic cell death induced by 80% oxygen, the pan-caspase inhibitor zVAD-fmk (40 μm) was added to the culture medium after transfer to the oxygen incubator (Fig. 3A–G). Pre-OLs that were kept in 80% oxygen for 12 h exhibited signs of apoptosis: cell shrinkage, plasma membrane blebbing, and condensation of cytoplasm (Fig. 3B). In contrast, cells that were treated with zVAD-fmk showed significantly less cell death (Fig. 3D,G). Staurosporine (1 μm), a nonselective protein kinase inhibitor and apoptosis inducer, was used as positive control (Fig. 3E–G).
Figure 3.
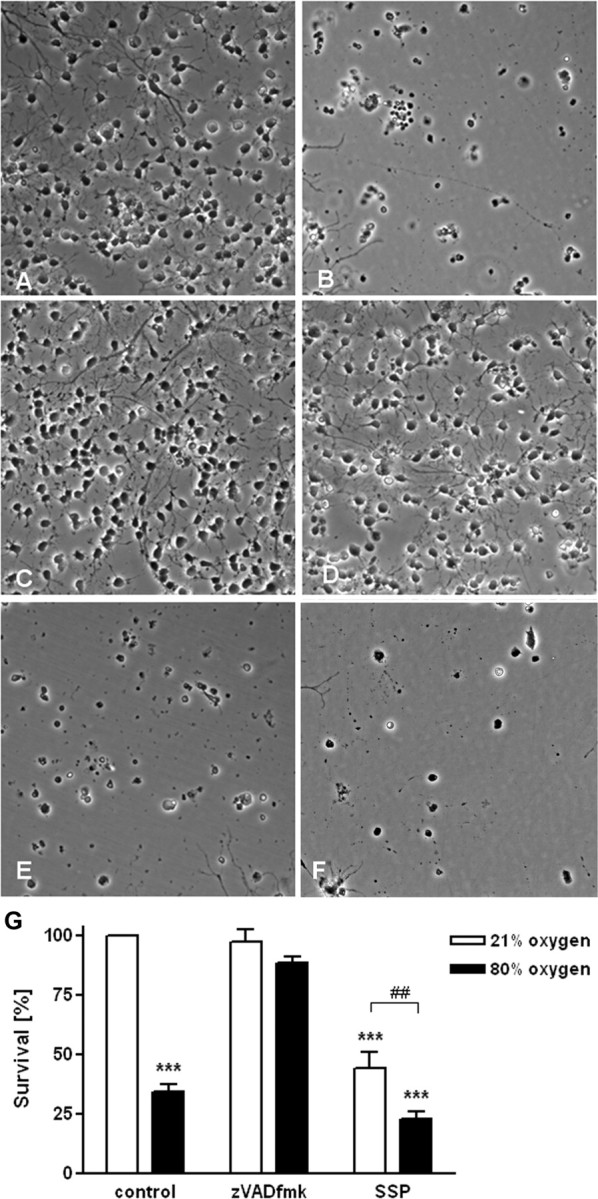
Involvement of caspases in cell toxicity by hyperoxia. A–F, Representative phase contrast photomicrographs of pre-OLs after 12 h of incubation with either 21% (A, C, E) or 80% oxygen (B, D, F). After incubation with 21% oxygen, pre-OLs (A) or cells treated with zVAD-fmk (40 μm) (C) do not display signs of apoptosis. E, Staurosporine (1 μm) was used as a positive control for inducing apoptosis in pre-OLs. B, After 12 h of 80% oxygen exposure, pre-OLs (B) showed signs of apoptosis, whereas zVAD-fmk (40 μm)-treated cells (D) were protected. E, F, Staurosporine-induced apoptosis in pre-OLs was increased after incubation with hyperoxia. G, Pre-OLs were treated with hyperoxia and/or with the pan-caspase inhibitor zVAD-fmk (40 μm), and/or with staurosporine (1 μm) for induction of apoptosis. Cells were incubated under normoxic (21% O2, white bars) or hyperoxic (80% O2, black bars) conditions for 12 h, and cell viability was determined via LDH release assay (bars represent mean ± SEM). ***p < 0.001 in two-way ANOVA, comparison between 21 and 80% oxygen-treated cells. ##p ≤ 0.01, Student's t test; comparison of staurosporine (1 μm)-treated pre-OLs between 21 and 80% oxygen.
Overexpression of BCL2 increased resistance of pre-OLs to hyperoxia
Oligodendroglial lineage cells vary in susceptibility to both necrosis and apoptosis depending on their developmental stages, which is hypothesized to be regulated by differential expression of BCL2-related genes (Itoh et al., 2003). To demonstrate the biological significance of the developmental regulation of BCL2 observed across the OL lineage, we next overexpressed BCL2 in pre-OLs via an HSV-derived vector containing BCL2. Pre-OLs were exposed to 104 pfu/ml of HSV- BCL2 or to HSV containing the reporter gene GFP (HSV-GFP). Infection rate usually ranged between 60 and 80% of total cells as assessed by counting GFP-positive cells. Infection with HSV-BCL2 strongly increased BCL2 protein expression in pre-OLs as assessed by Western blot analysis (Fig. 4A) without change in morphological appearance. Viral infection was associated with <10–15% cell death, as assessed by LDH release (data not shown). Overexpression of BCL2 in pre-OLs significantly increased their resistance to hyperoxia (Fig. 4C), compared with HSV-GFP-treated pre-OLs (Fig. 4B). In four separate experiments, BCL2-infected cells displayed significantly increased cell survival when exposed to 80% oxygen for 16 h (Fig. 4D) compared with cells infected with HSV-GFP.
Figure 4.

BCL2 overexpression decreased cell death in pre-OLs. A, Western blot analysis of pre-OLs (O4) overexpressing BCL2 showing that BCL2 protein was detectable with HSV-BCL2 (BCL2) but not with control reporter gene HSV-GFP (GFP) infection. B, C, Pre-OLs, either infected with virus containing the reporter gene GFP (HSV-GFP) (B) or BCL2 (HSV- BCL2) (C) were cultured for 16 h under hyperoxic (80% oxygen) conditions and then assayed for cell survival using LDH release. D, HSV-BCL2-infected OLs demonstrated an increased resistance to hyperoxia-induced oxidative stress compared with HSV-GFP-infected or control cells. Values shown are mean ± SEM from three independent experiments that were performed. *p ≤ 0.05, ***p ≤ 0.001, in two-way ANOVA; comparison of hyperoxia group (black bar) versus the normoxia group (white bar) for each group. ###p ≤ 0.01; comparison of hyperoxia-treated HSV-GFP-infected OLs versus HSV-BCL2-infected OLs.
Hyperoxia induces superoxide and ROS generation in pre-OLs
Mitochondria are a major source of superoxide in the cell (Staniek et al., 2002). Next, we asked whether hyperoxia might be associated with superoxide generation. First, we assayed intracellular mitochondrial superoxide accumulation and generation of ROS in pre-OLs subjected to either 21 or 80% oxygen for 6 h (Fig. 5) (Abramov et al., 2007). We used a derivate of ethidium bromide as a specific dye for superoxide and found an increase in rate of superoxide production in cells subjected to hyperoxia compared with control cells kept under normoxic conditions (Fig. 5A,B). Furthermore, we surveyed pre-OLs for the generation of reactive oxygen species using carboxy-H2DCFDA and found an increase in ROS production (Fig. 5C,D) after 6 h oxygen exposure compared with cells that were kept under normoxic conditions. Confocal microscopy revealed the evidence for superoxide production in the mitochondria (Fig. 5E) and the detection of ROS in the cytoplasm of premyelinating oligodendrocytes (Fig. 5F) after only 2 h of oxygen exposure.
Figure 5.
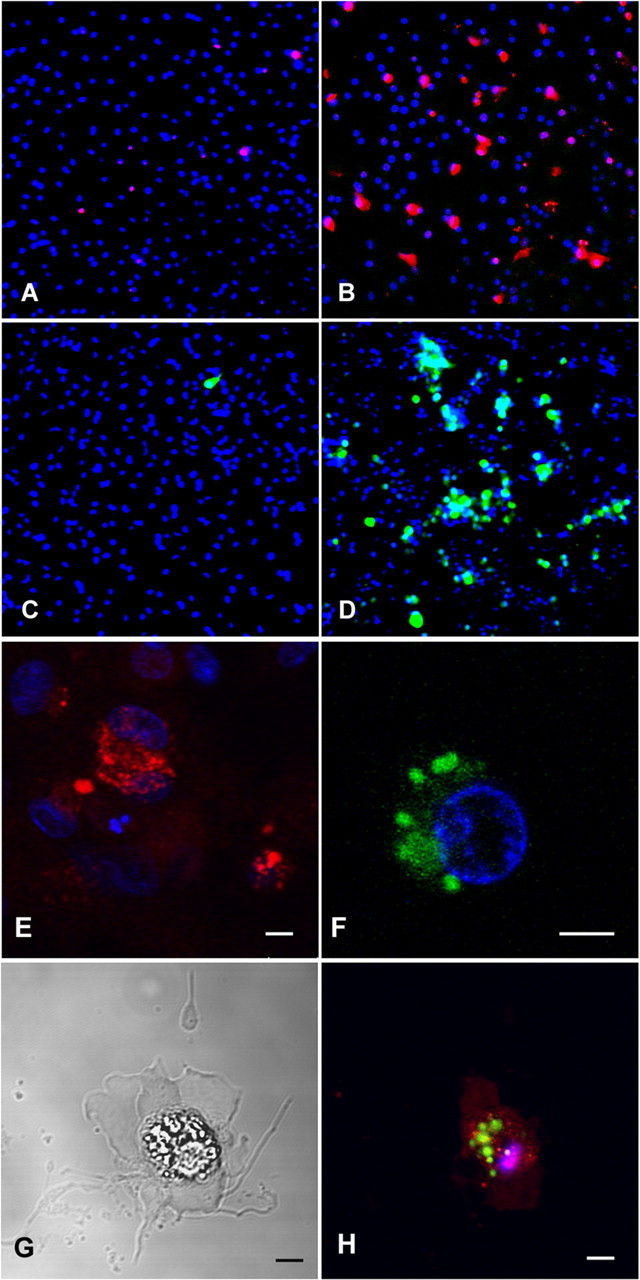
A, B, Mitochondrial dysfunction during hyperoxia in pre-OLs. Pre-OLs were cultured for 6 h in normoxic (21% oxygen) or hyperoxic (80% oxygen) conditions and then stained using MitoSOX red, a mitochondrial superoxide indicator and derivate of ethidium bromide. Once in the mitochondria, MitoSOX reagent is oxidized by superoxide and exhibits red fluorescence after binding to nucleic acids. If pre-OLs were kept under normoxic conditions, only a small number of cells displayed red fluorescence staining (A), whereas pre-OLs that were exposed to 80% oxygen showed an increased number of cells with red labeling of mitochondria indicating increased superoxide production (B). C, D, Generation of ROS in the cytoplasm of premyelinating OLs. Pre-OLs were cultured for 6 h in normoxic (21% oxygen) (C) or hyperoxic (80% oxygen) (D) conditions and then stained using carboxy-H2DCFDA, which serves as an indicator of the overall degree of intracellular oxidative stress. If pre-OLs were kept under normoxic conditions, only a small number of cells showed green fluorescence staining (C), whereas pre-OL cultures that were exposed to 80% oxygen showed an increased number of cells with detection of ROS in the cytoplasm (D). E–H, Higher magnification with confocal microscopy (60×) showed the distribution of the red fluorescent-stained mitochondria (E) and the distribution of the green fluorescent-stained ROS in the cytoplasm (F, H) of O4 cells exposed to 80% oxygen for 2 h. G and H show a combined light and phase contrast photomicrograph of an O4 cell that was exposed to 80% oxygen for 2 h and then stained with red MitoSOX, green carboxy-H2DCFDA, and blue Hoechst nuclear stain (H). Note the typical morphological signs of apoptotic cell death: cell shrinkage, loss of processes, nuclear condensation, and plasma membrane blebbing. Scale bar, 5 μm.
Inhibition of hyperoxia-induced oxidative stress by administration of extracellular superoxide dismutase, catalase, and the GPx mimic ebselen
The best studied cellular antioxidants are the enzymes superoxide dismutase, catalase, and glutathione peroxidase. There is a developmental mismatch in GPx and catalase expression and activity during oligodendrocyte maturation. GPx is upregulated with a twofold to threefold increase in its expression and activity in mature OLs versus pre-OLs (Baud et al., 2004b). Exposure to hydrogen peroxide causes downregulation of catalase activity and death in pre-OLs. If pre-OLs are preincubated with ebselen, a GPx mimic, OLs are fully protected against H2O2-induced decrease in catalase activity and cell death. We hypothesized that extracellular administration of SOD, catalase, or the GPx mimic ebselen should be able to protect pre-OLs from hyperoxia induced cell death. All three were effective in attenuating hyperoxia-mediated cell death in a dose-dependent manner (Fig. 6). Coadministration of SOD and catalase did not further increase the protection produced by SOD alone (Fig. 6).
Figure 6.

Inhibition of hyperoxia-induced oxidative stress by extracellular superoxide dismutase, catalase, and the GPx mimic ebselen. Pre-OLs were exposed to 80% oxygen for 12 h and treated with or without SOD (100 U/ml, 1000 U/ml), catalase (100 U/ml, 1000 U/ml), SOD/catalase (1000 U/ml each), or the GPx mimic ebselen (10 μm). A second plate with the same treatments was kept under normoxic conditions and used as control. There was decreased cell death within the treatment groups compared with untreated cells under normoxic conditions. Values shown are means ± SEM from three independent experiments (***p ≤ 0.001 in one-way ANOVA followed by Tukey–Kramer test; comparison of drug-treated vs vehicle-treated cells).
Overexpression of SOD1 increased cell death in pre-OLs
After finding that extracellular SOD administration was protective in hyperoxia-mediated toxicity in pre-OLs, we next tested whether overexpression of SOD1 within the cells would also protect pre-OLs against hyperoxia-induced cell death. Recently, it has been found that cystine deprivation toxicity leading to glutathione depletion was associated with mitochondrial dysfunction and intracellular superoxide accumulation in pre-OLs, similar to our findings in hyperoxia-induced cell death (Baud et al., 2004c). SOD1 protein expression and enzyme activity was similar along the OL lineage (Baud et al., 2004c). Pre-OLs were exposed to 104 pfu/ml of HSV-SOD1 or to HSV containing the reporter gene GFP (HSV-GFP). Infection rate usually ranged between 60 and 70% of total cells as assessed by counting GFP-positive cells. SOD1 expression was measured by immunoblot and was found to be upregulated in HSV-SOD1-infected cells (data not shown). Surprisingly, we obtained the opposite result from what we predicted: overexpression of SOD1 in pre-OLs (Fig. 7C) increased cell death even under normoxic conditions (Fig. 7A) compared with HSV-GFP-treated pre-OLs (Fig. 7B). Treatment with hyperoxia increased toxicity to SOD1-overexpressing pre-OLs, however, to the same extent as in HSV-GFP-infected pre-OLs. Although pre-OLs were susceptible to the apparent toxicity of SOD1 overexpression, MBP+ cells were not (Fig. 7D).
Figure 7.
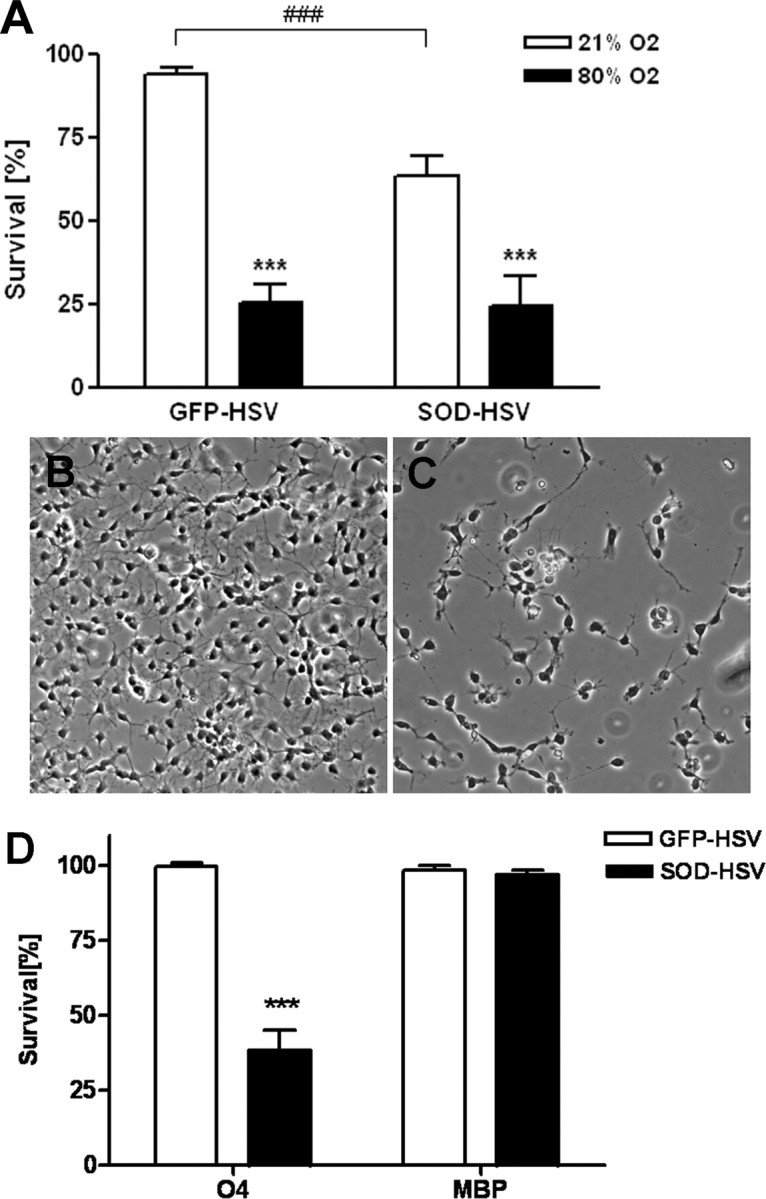
SOD1 overexpression increased cell death in pre-OLs. A, HSV-SOD1-infected pre-OLs demonstrated increased toxicity compared with HSV-GFP-infected control cells, when cells were kept under room air conditions (###p ≤ 0.001, Student's t test; comparison of HSV-GFP-infected cells vs HSV-SOD1-infected pre-OLs). After exposure to 80% oxygen, cell death increased significantly in HSV-GFP- or HSV-SOD1-infected pre-OLs. Values shown are means ± SEM from six independent experiments that were performed [**p < 0.01, ***p ≤ 0.001, in two-way ANOVA; comparison of hyperoxia group (black bar) vs the normoxia group (white bar)]. B, C Pre-OLs, either infected with virus containing the reporter gene GFP (HSV-GFP) (B) or SOD1 (HSV-SOD1) (C) were cultured for 24 h under normoxic conditions and then assayed for cell survival using LDH release. D, Toxicity from SOD1 overexpression was found in pre-OLs but not mature OLs. Values shown are mean ± SEM from three independent experiments that were performed [***p ≤ 0.001, Student's t test; comparison of hyperoxia group (black bar) vs the normoxia group (white bar) for each maturation stage].
12-Lipoxygenase activation is involved in hyperoxia-induced toxicity to pre-OLs
Cystine deprivation is a reliable paradigm for producing OL death resulting from intracellular GSH depletion (Back et al., 1998; Wang et al., 2004). 12-Lipoxygenase (LOX) inhibitors block cystine deprivation-induced cell death (Wang et al., 2004). To test the hypothesis that 12-LOX may also be involved in the hyperoxia-induced cell death, we tested the effects of 12-LOX inhibitors AA-861 (Nakadate et al., 1985; Li et al., 1997) and BMD-122 (Honn et al., 1994; Nie et al., 2000) against oxygen. Both 12-LOX inhibitors were very effective in blocking hyperoxia-induced cell death at 5 μm (n = 4) (Fig. 8). We also tested the LOX inhibitor baicalein, which also has antioxidant properties (Dailey and Imming, 1999), and found a protective effect against hyperoxia-mediated OL cell death (Fig. 8).
Figure 8.

The 12-LOX inhibitors, AA-861, BMD-122, and baicalein, all at 5 μm, protected against hyperoxia-induced toxicity in pre-OLs. The graph represents means ± SEM of three independent experiments. There was no difference between the three treatments. ***p < 0.001, one-way ANOVA; comparison of drug-treated versus vehicle-treated cells.
Oxygen induces white matter injury in neonatal rats
To determine the vulnerability of hyperoxia-induced injury to white matter in vivo, 3-, 6-, and 10-d-old rat pups were subjected to 80% oxygen over a 24 h period and killed at P11, and P10 animals additionally at P14. To investigate whether oligodendrocyte maturation is disturbed after oxygen exposure, brain sections were stained for MBP, an indicator of OL maturation. Injury was assessed on serial sections by two blinded observers for degree of MBP loss in cerebral white matter and with hematoxylin and eosin histology (data not shown). There was a significant increase in white matter damage in P11 rats treated with 80% oxygen at P3 or P6 (Fig. 9A–D,I): profound loss of processes, thinning of the capsule, and partial disruption of white matter. In contrast, for P11 and P14 rats subjected to hyperoxia at P10, no significant damage was noted (Fig. 9E–H, I). There was no significant difference in mortality or weight gain among any of the treatment groups.
Figure 9.
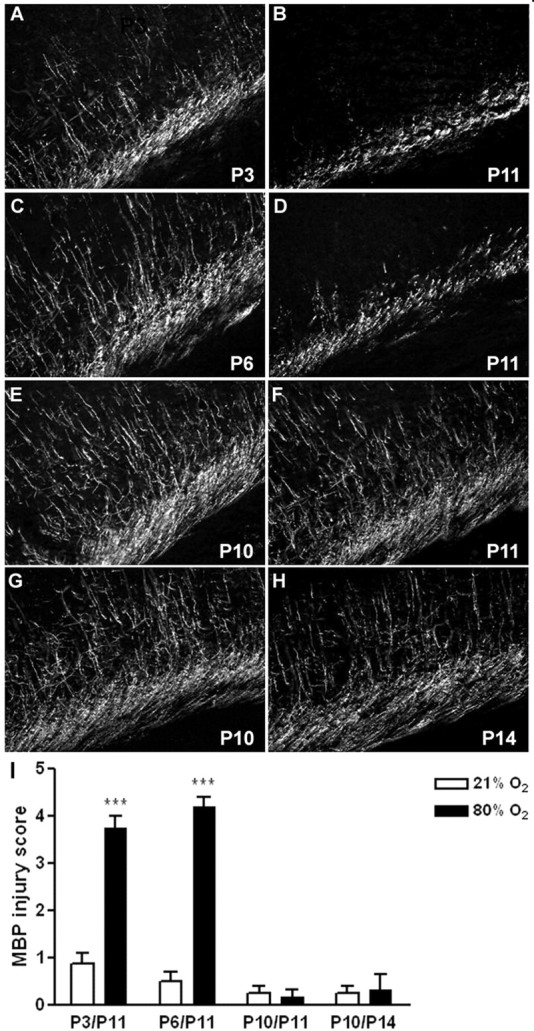
Hyperoxia caused decrease in MBP expression in the developing rat brain. A, C, E, Normal expression of MBP in the white matter tracts of P11 rats that were kept under room air conditions at P3, P6, and P10 (controls; n = 4 each group). B, D, I, Loss of MBP expression is seen in the P11 pup in the medial white matter, after 24 h of oxygen exposure (80%) at P3 (n = 4) (B) or P6 (n = 5) (D). E–I, No difference in MBP expression was detected at P11 (n = 4) (F) nor at P14 (n = 4) (H) if the animal was subjected to 80% oxygen at P10. ***p < 0.001, using the nonparametric Wilcoxon test.
Discussion
The present study demonstrates that exposure of oligodendrocytes to high concentrations of oxygen resulted in apoptotic cell death during a specific period of development. Furthermore, P3 and P6 rat pups exposed to hyperoxia displayed bilateral reduction in MBP expression, indicating disruption of myelination processes. In contrast, hyperoxia-exposed P10 rats were less vulnerable, because MBP expression was equal to room air-treated control animals.
Recently, we found that hyperoxia triggers maturation-dependent apoptosis in immature (O4+O1+MBP−) and pre-OLs (O4+O1−MBP−), which is dependent on caspase activation (Gerstner et al., 2006). This study used the oligodendroglial cell line OLN-93 (Richter-Landsberg and Heinrich, 1996). Previously studied examples of maturation-dependent toxicity in OLs have not been found to be caspase dependent (Back et al., 1998; Rosenberg et al., 2003; Baud et al., 2004b). The goal of the present study was to examine whether this maturation and caspase-dependent vulnerability could also be observed in primary OLs and also to characterize the oxidative stress that is involved upstream of the apoptotic pathway. First, we found differences in susceptibility toward oxygen exposure between primary OLs and the OL cell line OLN-93 as assessed by different viability assays. As described previously, after 24 h of oxygen incubation, OLN-93 cell viability is reduced to 80%, compared with 10–20% in primary OLs, and only after 96 h oxygen exposure the viability of OLN-93 cells is reduced to <80% (Gerstner et al., 2006). Therefore, primary pre-OLs are even more susceptible to oxygen exposure than the cell line. Second, we confirmed the involvement of caspases in hyperoxia-induced primary OL cell death. Hyperoxia-induced pre-OL death was blocked by the pan-caspase inhibitor zVAD-fmk. Furthermore, overexpression of the antiapoptotic protein BCL2 significantly protected pre-OLs from hyperoxia-induced cell death. Third, we demonstrated qualitatively that hyperoxia induces superoxide production in mitochondria and the generation of ROS in the cytoplasm. In an attempt to characterize measures that will counteract hyperoxia injury to developing white matter, we examined the effect of extracellular administration of superoxide dismutase, catalase, and ebselen, which all attenuated cell death. Overexpression of SOD1 in pre-OLs, but not in mature OLs, increased cell death under normoxic conditions, consistent with previous work demonstrating that an imbalance of SOD and peroxidase activity leads to cell death (Ditelberg et al., 1996; Baud et al., 2004c; McLean et al., 2005). 12-LOX inhibitors were very efficient in blocking hyperoxia-induced cell death.
Hyperoxia induces widespread cell death in brains of 3- to 7-d-old rats and mice in both gray and white matter regions (Felderhoff-Mueser et al., 2004). Oligodendrocytes were not specifically studied in this report. Vulnerability to oxygen-induced cell death was found to be age dependent with a maximum during the first week of life, consistent with our findings in primary OL cultures and in the white matter of neonatal rats. Hyperoxic brain injury also leads to nitrative stress, ensuing microvascular degeneration and diminished brain mass and function in immature rat pups (Sirinyan et al., 2006). Additionally, a role for reactive nitrogen species in hyperoxic death is suggested by the observation that hyperoxia causes upregulation of inducible nitric oxide synthase mRNA and protein in microglial cells and formation of nitrotyrosine in neurons of the immature rat brain (Hoehn et al., 2003). Baud et al. (2004a) demonstrated that nitric oxide is more toxic to developing OLs than to mature OLs and that neither peroxynitrite formation nor caspase activation was involved in its toxicity. Before 32 weeks gestation, ∼90% of oligodendrocytes are in the early stage of development referred to as the pre-oligodendrocyte (Back et al., 2001). These precursor cells are very sensitive to free radicals (Back et al., 2002), and we found these reactive oxygen species present as early as 2 h after exposure to hyperoxia.
Maturation-dependent vulnerability in an oligodendrocyte lineage was found by Back et al. (2002) using a neonatal rat model of hypoxic-ischemic injury. Late OL progenitors (O4+O1−MBP−) and immature OLs (O4+O1+MBP−), both premyelinating stages in the lineage, were the OL stages killed by hypoxia/ischemia, whereas more mature OLs were highly resistant (Back et al., 2002). This maturation-dependent vulnerability of pre-OLs to oxidative stress has been confirmed in several paradigms. Thus, oxidative stress produced by glutathione depletion results in marked cell death in pre-OLs, whereas mature OLs are resistant (Oka et al., 1993; Yonezawa et al., 1996; Back et al., 1998). This vulnerability appears to be related to accumulation of free radicals in pre-OLs but not in mature cells, suggesting a defect in the ability of immature OLs to remove ROS. This inability to remove ROS was shown to be caused by relative deficiencies of antioxidant enzymes (Baud et al., 2004b,c). Our results demonstrate that SOD and catalase, two of the most important cellular antioxidant enzymes, were effective in attenuating hyperoxia-mediated OL cell death when added to the extracellular medium (Fig. 6). In contrast to our expectations, overexpression of SOD1 in pre-OLs did not lead to protection from hyperoxia-induced cell death and instead significantly increased cell death in pre-OLs under normoxic conditions. Overexpression of SOD1 was not toxic to mature OLs under normoxic conditions. The failure of SOD1 overexpression to confer protection as well as its toxicity may be related to excess intracellular H2O2 production exceeding the capacity of peroxidase activity in pre-OLs. Previously reported GPx upregulation in mature OLs is likely to account for the increased resistance of mature OLs to H2O2 (Baud et al., 2004b). A precedent for an imbalance between SOD and peroxidase activity resulting in cell death has been reported in transgenic mice overexpressing SOD. In these animals, a paradoxical increase in brain injury was observed after neonatal hypoxia-ischemia versus adult hypoxia-ischemia, and this increase in injury has been attributed to excessive accumulation of H2O2 in the immature cells (Ditelberg et al., 1996; Fullerton et al., 1998; Ferriero, 2001; Lafemina et al., 2006).
We found that the LOX inhibitors AA-861 and BMD-122 fully protected pre-OLs from GSH depletion caused by cystine deprivation (Wang et al., 2004). The present study demonstrates that 12-LOX also plays a key role in oxidative injury caused by hyperoxia in pre-OLs. Inhibiting this enzyme completely protected OLs from hyperoxia-induced cell death (Fig. 8). Lipoxygenases are a family of enzymes that incorporate one molecule of oxygen stereo-specifically into arachidonic acid and docosahexaenoic acid to produce fatty acid hydroperoxides and their derivatives (Brash, 1999). The activity of 12-LOX is regulated by the redox status of the cell (Shornick and Holtzman, 1993; Li et al., 1997). Glutathione, the principal antioxidant in the cell, has been shown to modulate the activity of the enzyme and can directly inhibit it in concentrations of GSH from 1 to 10 mm (Hagmann et al., 1993). Depleting GSH by cystine deprivation induced an increase in activity of 12-LOX in pre-OLs (Wang et al., 2004). In addition, we showed previously that peroxynitrite-induced neuronal apoptosis and peroxynitrite induced necrosis of mature OLs (Zhang et al., 2004) was blocked by inhibitors of 12-LOX. Future research will need to determine the pathways that are involved in hyperoxia-induced apoptosis in pre-OLs. Inhibition of 12-LOX may provide an important therapeutic strategy for protecting developing OLs from hyperoxic injury or injury induced by oxidative stress.
Multiple stimuli can lead to the activation of the apoptotic cascade in oligodendrocytes, including oxidative stress. It is well known that free radicals can lead to direct DNA damage as well as damage of mitochondrial membranes, release of cytochrome c into the cytoplasm, and activation of caspases (Yuan and Yankner, 2000; Filomeni et al., 2003). However, glutathione depletion-induced pre-OL death was also found to be caspase independent (Back et al., 1998; Wang et al., 2004). Similarly, nitric oxide toxicity to pre-OLs was associated with translocation of apoptosis-inducing factor, but this event was not related to caspase activation (Baud et al., 2004a).
Here, we demonstrate a novel instance of an oxidative insult inducing apoptosis in oligodendrocytes that could be blocked by the pan-caspase inhibitor zVAD-fmk, or overexpression of the antiapoptotic molecule BCL2. Others have shown that BCL2 expression is associated with protection of OLs from different cell-death inducers (Burgmaier et al., 2000; FitzGerald et al., 2003). Recently, it was shown that the overexpression of BCL2 was able to prevent hyperoxia-induced cell death in a murine fibrosarcoma cell line, by affecting mitochondria-dependent apoptotic pathways and increasing intracellular antioxidant compounds (Metrailler-Ruchonnet et al., 2007). The present in vitro study in conjunction with our previous in vivo findings suggest that the principal mechanism of cell death in oligodendrocytes and neurons in response to hyperoxia is apoptosis (Felderhoff-Mueser et al., 2004).
Oxygen toxicity has been implicated in the pathogenesis of other diseases particularly associated with preterm birth such as bronchopulmonary dysplasia and retinopathy of prematurity (Saugstad, 2001). Recent studies have demonstrated that a change in clinical practice to avoid hyperoxia is associated with significant decrease in neonatal morbidity but does not have a detrimental effect on developmental outcome at 18 months (Deulofeut et al., 2006). Moreover, another study identified hyperoxia as one clinical risk factor for abnormal neurologic outcome of preterm infants (Collins et al., 2001). This finding is consistent with the observation that the duration of oxygen therapy in premature infants was predictive of the cognitive outcome at 8 years (Short et al., 2003). Interestingly, the clinically evident decline in cystic PVL rates in recent years (Hamrick et al., 2004) has been accompanied by a decrease in days of mechanical ventilation. However, this decline was not associated with improved developmental outcome, indicating that other forms of cerebral injury contribute to abnormal neurodevelopmental outcomes in premature infants (Hamrick et al., 2004).
We hypothesize that increased apoptotic death of developing OLs in response to hyperoxia might lead to pre-OL death and thereby impaired myelination. Our present in vivo results of disrupted myelin expression in P3 and P6 rat pups support this hypothesis. However, long-term white matter development and its influence on gray matter survival in these animals needs to be investigated further. Our results may be relevant to the concerns regarding the use of high oxygen levels for resuscitation and for treatment of lung disease in premature infants. Furthermore, our results raise the possibility that antioxidants or 12-LOX inhibitors might be suitable for specific treatment of premature infants who need to be exposed to oxygen. More data are urgently needed concerning the safety and efficacy of potential drugs in the neonatal period and the long-term neurodevelopmental outcome of their use.
Footnotes
This work was supported by grants from the Ernst Schering Research Foundation (Berlin, Germany), the German Federal Department of Education and Research (BMBF #01 ZZ 0101), Bonn, and the European Union (NEOBRAIN 036534, 6th framework 4th call), and by National Institutes of Health Grants NS38475 and HD18655. We thank Dr. Felicia M. Trachtenberg (New England Research Institute, Watertown, MA) for advice regarding statistical analysis. We are grateful to Drs. Yumin Zhang, Wenbin Deng, and Frances E. Jensen for helpful discussion and support.
References
- Abramov AY, Scorziello A, Duchen MR. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J Neurosci. 2007;27:1129–1138. doi: 10.1523/JNEUROSCI.4468-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Gan X, Li Y, Rosenberg PA, Volpe JJ. Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induced death caused by glutathione depletion. J Neurosci. 1998;18:6241–6253. doi: 10.1523/JNEUROSCI.18-16-06241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Luo NL, Borenstein NS, Levine JM, Volpe JJ, Kinney HC. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J Neurosci. 2001;21:1302–1312. doi: 10.1523/JNEUROSCI.21-04-01302.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Han BH, Luo NL, Chricton CA, Xanthoudakis S, Tam J, Arvin KL, Holtzman DM. Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J Neurosci. 2002;22:455–463. doi: 10.1523/JNEUROSCI.22-02-00455.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barja G. The quantitative measurement of H2O2 generation in isolated mitochondria. J Bioenerg Biomembr. 2002;34:227–233. doi: 10.1023/a:1016039604958. [DOI] [PubMed] [Google Scholar]
- Baud O, Li J, Zhang Y, Neve RL, Volpe JJ, Rosenberg PA. Nitric oxide-induced cell death in developing oligodendrocytes is associated with mitochondrial dysfunction and apoptosis-inducing factor translocation. Eur J Neurosci. 2004a;20:1713–1726. doi: 10.1111/j.1460-9568.2004.03616.x. [DOI] [PubMed] [Google Scholar]
- Baud O, Greene AE, Li J, Wang H, Volpe JJ, Rosenberg PA. Glutathione peroxidase-catalase cooperativity is required for resistance to hydrogen peroxide by mature rat oligodendrocytes. J Neurosci. 2004b;24:1531–1540. doi: 10.1523/JNEUROSCI.3989-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud O, Haynes RF, Wang H, Folkerth RD, Li J, Volpe JJ, Rosenberg PA. Developmental up-regulation of MnSOD in rat oligodendrocytes confers protection against oxidative injury. Eur J Neurosci. 2004c;20:29–40. doi: 10.1111/j.0953-816X.2004.03451.x. [DOI] [PubMed] [Google Scholar]
- Brash AR. Lipoxygenases: occurrence, functions, catalysis, and acquisition of substrate. J Biol Chem. 1999;274:23679–23682. doi: 10.1074/jbc.274.34.23679. [DOI] [PubMed] [Google Scholar]
- Burgmaier G, Schonrock LM, Kuhlmann T, Richter-Landsberg C, Bruck W. Association of increased bcl-2 expression with rescue from tumor necrosis factor-alpha-induced cell death in the oligodendrocyte cell line OLN-93. J Neurochem. 2000;75:2270–2276. doi: 10.1046/j.1471-4159.2000.0752270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow LC, Wright KW, Sola A. Can changes in clinical practice decrease the incidence of severe retinopathy of prematurity in very low birth weight infants? Pediatrics. 2003;111:339–345. doi: 10.1542/peds.111.2.339. [DOI] [PubMed] [Google Scholar]
- Collins MP, Lorenz JM, Jetton JR, Paneth N. Hypocapnia and other ventilation-related risk factors for cerebral palsy in low birth weight infants. Pediatr Res. 2001;50:712–719. doi: 10.1203/00006450-200112000-00014. [DOI] [PubMed] [Google Scholar]
- Dailey LA, Imming P. 12-Lipoxygenase: classification, possible therapeutic benefits from inhibition, and inhibitors. Curr Med Chem. 1999;6:389–398. [PubMed] [Google Scholar]
- Deulofeut R, Critz A, Adams-Chapman I, Sola A. Avoiding hyperoxia in infants < or = 1250 g is associated with improved short- and long-term outcomes. J Perinatol. 2006;26:700–705. doi: 10.1038/sj.jp.7211608. [DOI] [PubMed] [Google Scholar]
- Ditelberg JS, Sheldon RA, Epstein CJ, Ferriero DM. Brain injury after perinatal hypoxia-ischemia is exacerbated in copper/zinc superoxide dismutase transgenic mice. Pediatr Res. 1996;39:204–208. doi: 10.1203/00006450-199602000-00003. [DOI] [PubMed] [Google Scholar]
- Felderhoff-Mueser U, Bittigau P, Sifringer M, Jarosz B, Korobowicz E, Mahler L, Piening T, Moysich A, Grune T, Thor F, Heumann R, Bührer C, Ikonomidou C. Oxygen causes cell death in the developing brain. Neurobiol Dis. 2004;17:273–282. doi: 10.1016/j.nbd.2004.07.019. [DOI] [PubMed] [Google Scholar]
- Felderhoff-Mueser U, Sifringer M, Polley O, Dzietko M, Leineweber B, Mahler L, Baier M, Bittigau P, Obladen M, Ikonomidou C, Bührer C. Caspase-1-processed interleukins in hyperoxia-induced cell death in the developing brain. Ann Neurol. 2005;57:50–59. doi: 10.1002/ana.20322. [DOI] [PubMed] [Google Scholar]
- Ferriero DM. Oxidant mechanisms in neonatal hypoxia-ischemia. Dev Neurosci. 2001;23:198–202. doi: 10.1159/000046143. [DOI] [PubMed] [Google Scholar]
- Filomeni G, Rotilio G, Ciriolo MR. Glutathione disulfide induces apoptosis in U937 cells by a redox-mediated p38 MAP kinase pathway. FASEB J. 2003;17:64–66. doi: 10.1096/fj.02-0105fje. [DOI] [PubMed] [Google Scholar]
- FitzGerald UF, Gilbey T, Brodie S, Barnett SC. Transcription factor expression and cellular redox in immature oligodendrocyte cell death: effect of Bcl-2. Mol Cell Neurosci. 2003;22:516–529. doi: 10.1016/s1044-7431(02)00040-4. [DOI] [PubMed] [Google Scholar]
- Follett PL, Rosenberg PA, Volpe JJ, Jensen FE. NBQX attenuates excitotoxic injury in developing white matter. J Neurosci. 2000;20:9235–9241. doi: 10.1523/JNEUROSCI.20-24-09235.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follett PL, Deng W, Dai W, Talos DM, Massillon LJ, Rosenberg PA, Volpe JJ, Jensen FE. Glutamate receptor-mediated oligodendrocyte toxicity in periventricular leukomalacia: a protective role for topiramate. J Neurosci. 2004;24:4412–4420. doi: 10.1523/JNEUROSCI.0477-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullerton HJ, Ditelberg JS, Chen SF, Sarco DP, Chan PH, Epstein CJ, Ferriero DM. Copper/zinc superoxide dismutase transgenic brain accumulates hydrogen peroxide after perinatal hypoxia ischemia. Ann Neurol. 1998;44:357–364. doi: 10.1002/ana.410440311. [DOI] [PubMed] [Google Scholar]
- Gerstner B, Bührer C, Rheinländer C, Polley O, Schüller A, Berns M, Obladen M, Felderhoff-Mueser U. Maturation-dependent oligodendrocyte apoptosis caused by hyperoxia. J Neurosci Res. 2006;84:306–315. doi: 10.1002/jnr.20880. [DOI] [PubMed] [Google Scholar]
- Hagmann W, Kagawa D, Renaud C, Honn KV. Activity and protein distribution of 12-lipoxygenase in HEL cells: induction of membrane-association by phorbol ester TPA, modulation of activity by glutathione and 13-HPODE, and Ca(2+)-dependent translocation to membranes. Prostaglandins. 1993;46:471–477. doi: 10.1016/0090-6980(93)90066-g. [DOI] [PubMed] [Google Scholar]
- Hamrick SE, Miller SP, Leonard C, Glidden DV, Goldstein R, Ramaswamy V, Piecuch R, Ferriero DM. Trends in severe brain injury and neurodevelopmental outcome in premature newborn infants: the role of cystic periventricular leukomalacia. J Pediatr. 2004;145:593–599. doi: 10.1016/j.jpeds.2004.05.042. [DOI] [PubMed] [Google Scholar]
- Hoehn T, Felderhoff-Mueser U, Maschewski K, Stadelmann C, Sifringer M, Bittigau P, Koehne P, Hoppenz M, Obladen M, Bührer C. Hyperoxia causes inducible nitric oxide synthase-mediated cellular damage to the immature rat brain. Pediatr Res. 2003;54:179–184. doi: 10.1203/01.PDR.0000075220.17631.F1. [DOI] [PubMed] [Google Scholar]
- Honn KV, Tang DG, Grossi IM, Renaud C, Duniec ZM, Johnson CR, Diglio CA. Enhanced endothelial cell retraction mediated by 12(S)-HETE: a proposed mechanism for the role of platelets in tumor cell metastasis. Exp Cell Res. 1994;210:1–9. doi: 10.1006/excr.1994.1001. [DOI] [PubMed] [Google Scholar]
- Itoh T, Itoh A, Pleasure D. Bcl-2-related protein family gene expression during oligodendroglial differentiation. J Neurochem. 2003;85:1500–1512. doi: 10.1046/j.1471-4159.2003.01795.x. [DOI] [PubMed] [Google Scholar]
- Kinney HC, Armstrong DD. Perinatal neuropathology. In: Graham DI, Lantos PL, editors. Greenfield's neuropathology. Ed 7. London: Arnold; 2002. pp. 519–606. [Google Scholar]
- Kinney HC, Haynes RL, Folkerth RD. Pathology and genetics acquired and inherited diseases of the developing nervous system. Basel: ISN Neuropathology; 2004. White matter lesions in the perinatal period. [Google Scholar]
- Klinger G, Beyene J, Shah P, Perlman M. Do hyperoxaemia and hypocapnia add to the risk of brain injury after intrapartum asphyxia? Arch Dis Child Fetal Neonatal Ed. 2005;90:F49–F52. doi: 10.1136/adc.2003.048785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafemina MJ, Sheldon RA, Ferriero DM. Acute hypoxia-ischemia results in hydrogen peroxide accumulation in neonatal but not adult mouse brain. Pediatr Res. 2006;59:680–683. doi: 10.1203/01.pdr.0000214891.35363.6a. [DOI] [PubMed] [Google Scholar]
- Leviton A, Dammann O. Coagulation, inflammation, and the risk of neonatal white matter damage. Pediatr Res. 2004;55:541–545. doi: 10.1203/01.PDR.0000121197.24154.82. [DOI] [PubMed] [Google Scholar]
- Li J, Lin JC, Wang H, Peterson JW, Furie BC, Furie B, Booth SL, Volpe JJ, Rosenberg PA. Novel role of vitamin k in preventing oxidative injury to developing oligodendrocytes and neurons. J Neurosci. 2003;23:5816–5826. doi: 10.1523/JNEUROSCI.23-13-05816.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Maher P, Schubert D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron. 1997;19:453–463. doi: 10.1016/s0896-6273(00)80953-8. [DOI] [PubMed] [Google Scholar]
- McLean CW, Mirochnitchenko O, Claus CP, Noble-Haeusslein LJ, Ferriero DM. Overexpression of glutathione peroxidase protects immature murine neurons from oxidative stress. Dev Neurosci. 2005;27:169–175. doi: 10.1159/000085989. [DOI] [PubMed] [Google Scholar]
- Menke J, Michel E, Hillebrand S, von Twickel J, Jorch G. Cross-spectral analysis of cerebral autoregulation dynamics in high risk preterm infants during the perinatal period. Pediatr Res. 1997;42:690–699. doi: 10.1203/00006450-199711000-00023. [DOI] [PubMed] [Google Scholar]
- Metrailler-Ruchonnet I, Pagano A, Carnesecchi S, Ody C, Donati Y, Barazzone Argiroffo C. Bcl-2 protects against hyperoxia-induced apoptosis through inhibition of the mitochondria-dependent pathway. Free Radic Biol Med. 2007;42:1062–1074. doi: 10.1016/j.freeradbiomed.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Nakadate T, Yamamoto S, Aizu E, Kato R. Inhibition of mouse epidermal 12-lipoxygenase by 2,3,4-trimethyl-6-(12-hydroxy-5,10-dodecadiynyl)-1,4-benzoquinon e (AA861) J Pharm Pharmacol. 1985;37:71–73. doi: 10.1111/j.2042-7158.1985.tb04938.x. [DOI] [PubMed] [Google Scholar]
- Neve RL, Geller AI. Genetic analysis of neuronal physiology with defective herpes simplex virus vectors. Adv Neurol. 1999;79:1027–1032. [PubMed] [Google Scholar]
- Nie D, Tang K, Diglio C, Honn KV. Eicosanoid regulation of angiogenesis: role of endothelial arachidonate 12-lipoxygenase. Blood. 2000;95:2304–2311. [PubMed] [Google Scholar]
- Oka A, Belliveau MJ, Rosenberg PA, Volpe JJ. Vulnerability of oligodendroglia to glutamate: pharmacology, mechanisms, and prevention. J Neurosci. 1993;13:1441–1453. doi: 10.1523/JNEUROSCI.13-04-01441.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter-Landsberg C, Heinrich M. OLN-93: a new permanent oligodendroglia cell line derived from primary rat brain glial cultures. J Neurosci Res. 1996;45:161–173. doi: 10.1002/(SICI)1097-4547(19960715)45:2<161::AID-JNR8>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Rosenberg PA, Dai W, Gan XD, Ali S, Fu J, Back SA, Sanchez RM, Segal MM, Follett PL, Jensen FE, Volpe JJ. Mature myelin basic protein-expressing oligodendrocytes are insensitive to kainate toxicity. J Neurosci Res. 2003;71:237–245. doi: 10.1002/jnr.10472. [DOI] [PubMed] [Google Scholar]
- Saugstad OD. Chronic lung disease: oxygen dogma revisited. Acta Paediatr. 2001;90:113–115. doi: 10.1080/080352501300049127. [DOI] [PubMed] [Google Scholar]
- Shornick LP, Holtzman MJ. A cryptic, microsomal-type arachidonate 12-lipoxygenase is tonically inactivated by oxidation-reduction conditions in cultured epithelial cells. J Biol Chem. 1993;268:371–376. [PubMed] [Google Scholar]
- Short EJ, Klein NK, Lewis BA, Fulton S, Eisengart S, Kercsmar C, Baley J, Singer LT. Cognitive and academic consequences of bronchopulmonary dysplasia and very low birth weight: 8-year old outcomes. Pediatrics. 2003;112:359. doi: 10.1542/peds.112.5.e359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirinyan M, Sennlaub F, Dorfman A, Sapieha P, Gobeil F, Jr, Hardy P, Lachapelle P, Chemtob S. Hyperoxic exposure leads to nitrative stress and ensuing microvascular degeneration and diminished brain mass and function in the immature subject. Stroke. 2006;37:2807–2815. doi: 10.1161/01.STR.0000245082.19294.ff. [DOI] [PubMed] [Google Scholar]
- Staniek K, Gille L, Kozlov AV, Nohl H. Mitochondrial superoxide radical formation is controlled by electron bifurcation to the high and low potential pathways. Free Radic Res. 2002;36:381–387. doi: 10.1080/10715760290021225. [DOI] [PubMed] [Google Scholar]
- Volpe JJ. Brain injury in the premature infant: overview of clinical aspects, neuropathology, and pathogenesis. Semin Pediatr Neurol. 1998;5:135–151. doi: 10.1016/s1071-9091(98)80030-2. [DOI] [PubMed] [Google Scholar]
- Volpe JJ. Neurobiology of periventricular leukomalacia in the premature infant. Pediatr Res. 2001a;50:553–562. doi: 10.1203/00006450-200111000-00003. [DOI] [PubMed] [Google Scholar]
- Volpe JJ. Philadelphia: W. B. Saunders; 2001b. Neurology of the newborn, Ed 4 Unit III, pp 215–394. [Google Scholar]
- Wang H, Li J, Follett PL, Zhang Y, Cotanche DA, Jensen FE, Volpe JJ, Rosenberg PA. 12-Lipoxygenase plays a key role in cell death caused by glutathione depletion and arachidonic acid in rat oligodendrocytes. Eur J Neurosci. 2004;20:2049–2058. doi: 10.1111/j.1460-9568.2004.03650.x. [DOI] [PubMed] [Google Scholar]
- Yonezawa M, Back SA, Gan X, Rosenberg PA, Volpe JJ. Cystine deprivation induces oligodendroglial death: rescue by free radical scavengers and by a diffusible glial factor. J Neurochem. 1996;67:566–573. doi: 10.1046/j.1471-4159.1996.67020566.x. [DOI] [PubMed] [Google Scholar]
- Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. 2000;407:802–809. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wang H, Li J, Jimenez DA, Levitan ES, Aizenman E, Rosenberg PA. Peroxynitrite-induced neuronal apoptosis is mediated by intracellular zinc release and 12-lipoxygenase activation. J Neurosci. 2004;24:10616–10627. doi: 10.1523/JNEUROSCI.2469-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]