

Abstract
The lateral habenula (LHb), a key regulator of monoaminergic brain regions, is activated by negatively-valenced events. Its hyperactivity is associated with depression. While enhanced excitatory input to the LHb has been linked to depression, little is known about inhibitory transmission. We discovered that GABA is co-released with its functional opponent, glutamate, from long-range basal ganglia inputs (which signal negative events) to limit LHb activity in rodents. At this synapse, the balance of GABA/glutamate signaling is shifted towards reduced GABA in a model of depression and increased GABA by antidepressant treatment. GABA and glutamate co-release therefore controls LHb activity, and regulation of this remarkable form of transmission may be important for determining the impact of negative life events on mood and behavior.
Excitatory transmission in the brain is generally balanced by feed-forward and feedback inhibitory actions of GABAergic interneurons (1). The LHb largely lacks GABAergic interneurons (2) and thus other mechanisms must operate to control hyperactivity. One potential mechanism is for input neurons to release both excitatory (e.g. glutamate) and inhibitory (e.g. GABA) transmitter ((3–8) but see (7, 9, 10)); balance could be achieved by regulating the relative amounts of transmitters released (11).
Hyperactivity in the LHb may contribute to depression (12–19), possibly by over-processing (20) negatively-valenced events (21). While enhanced excitatory input to the LHb contributes to depression-like behaviors in rodents (14, 15), little is known about LHb inhibitory transmission. Here we examine the nature of the inhibitory input to the LHb from the basal ganglia, as well as its regulation in conditions related to depression. This pathway is of particular interest because it is active during negatively-valenced events (22), which are thought to play a causal role in depression (23).
To examine the effects of GABAergic input on LHb output, we infected in vivo the basal ganglia output region, the entopeduncular nucleus (EP), of rodents with an adeno-associated virus (AAV) expressing channelrhodopsin-2 (ChR2) (24). Two to three weeks later, brain slices were prepared and whole-cell responses were obtained from neurons in the LHb (Fig. 1A). Trains of brief light pulses produced a few synaptically driven action potentials, indicating predominant excitation (25); bath application of the GABA-A receptor (−R) antagonist picrotoxin, increased the number of action potentials (Fig. 1B, C), indicating that the EP excitatory drive was balanced by inhibition. In voltage-clamp mode recordings, light-evoked synaptic responses displayed AMPA-R, GABA-A-R and NMDA-R components (Fig. 1D, E). In other brain slice experiments (Fig. 1F), pairs of light pulses elicited EP-LHb transmission and action potentials; the ratio of GABA-A-R to AMPA-R mediated EP-LHb synaptic transmission was inversely related to the generation of action potentials.
Fig. 1.
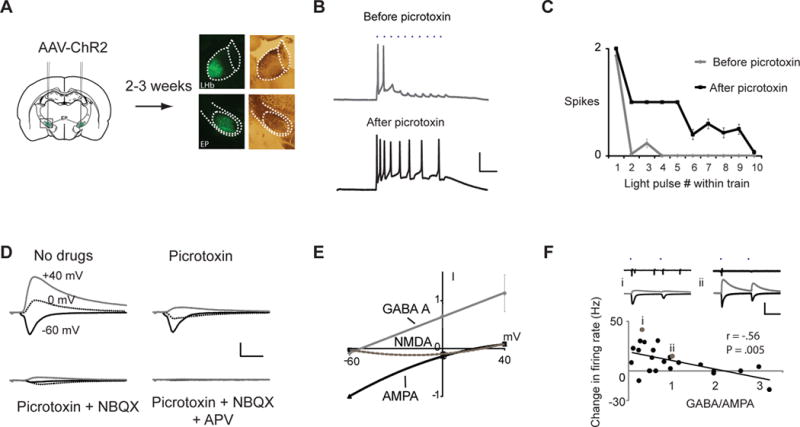
Mixed excitatory and inhibitory transmission from EP input controls persistent LHb activity. (A) Diagram of experimental protocol. Microscopic images depict example injection of AAV-ChR2 into WT rat; left, YFP fluorescence; right, bright-field illumination. (B) Trains of synaptic responses evoked by light pulses (50 Hz, blue dots) before (top) and after (bottom) bath application of picrotoxin recorded in whole-cell current clamp mode. Scale bars: 25 mV, 50 ms. (C) Summary of average responses for recording in (B). Similar results were obtained in 3 cells. (D) Whole-cell recordings in voltage-clamp mode of light-evoked EP-LHb transmission at different holding potentials (as indicated) and after application of drugs (as indicated). Scale bars: 100 pA, 10 ms. (E) Current-voltage plots for different EP-LHb synaptic conductances normalized to AMPA-R-mediated response at −60 mV (N= 9 cells). (F) Cell-attached (top) and subsequent whole-cell (middle) recordings at +20 mV (GABA, gray) and −50 mV (AMPA, black) holding potentials from two cells (i, ii) during synaptic responses evoked by pairs of light pulses (as indicated). Below, plot of change in firing rate (measured in cell-attached mode) versus GABA-A-R-mediated/AMPA-R-mediated synaptic response (measured in voltage-clamp mode; N = 24 cells); values for cells i and ii indicated by gray symbols. Scale bars: 400 pA, 50 ms. Error bars throughout indicate s.e.m.
Because the LHb contains few inhibitory neurons (2), we tested if the GABA-A response was due to long-range monosynaptic EP to LHb transmission. Light-evoked synaptic responses, measured at −15 mV holding potential (to allow simultaneous measurement of AMPA-R and GABA-A-R-mediated currents), displayed inward followed by outward currents (Fig. 2A). NBQX, an AMPA-R antagonist, blocked the inward current, leaving intact a picrotoxin-sensitive outward current (Fig. 2A, S1), indicating monosynaptic EP to LHb inhibition (see also Fig. S2). In some whole-cell recordings of LHb neurons voltage-clamped at −15 mV, we observed isolated spontaneous responses (in the absence of tetrodotoxin; i.e. action potentials not blocked) that were inward immediately followed by outward currents, indicating co-release of glutamate and GABA from individual axons onto the recorded cell (Fig. 2B, S3). Co-release from some individual vesicles could also be detected. In 4 of 6 experiments monitoring spontaneous miniature EPSCs (in tetrodotoxin), biphasic events occurred more often than could be accounted for by independent inward and outward responses (Fig. 2C; p< 0.05 for 4 experiments; see (26); see also Fig. S4).
Fig. 2.
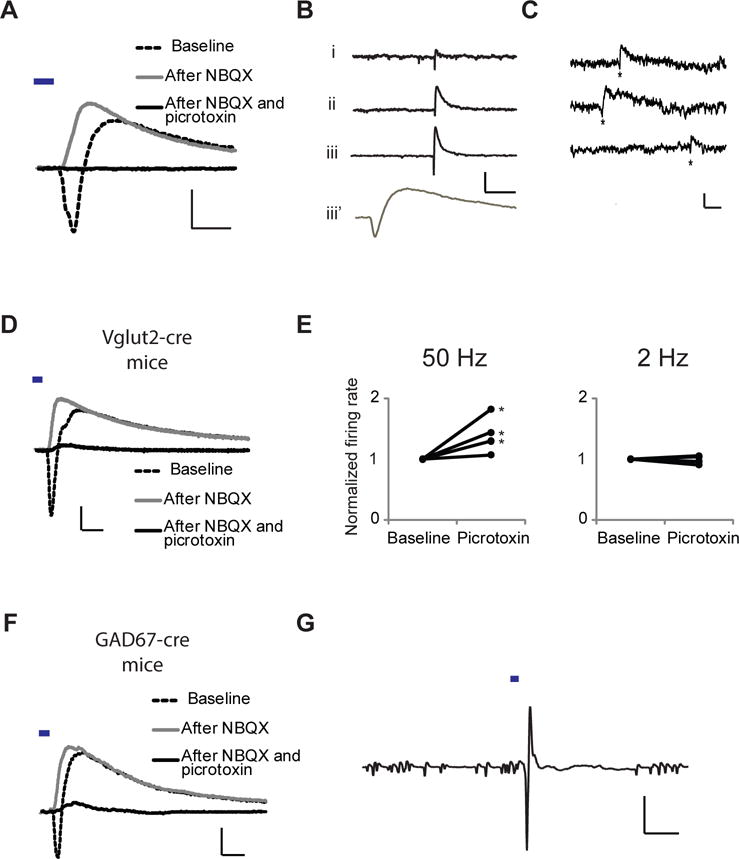
Co-release of GABA and glutamate from EP inputs to LHb: electrophysiological, optogenetic, and mouse genetic evidence. (A) Whole-cell recording at −15 mV holding potential of light-evoked EP-LHb transmission after bath application of indicated drugs. Scale bars: 60 pA, 10 ms. (B) Spontaneous responses from whole-cell recordings at −15 mV holding potential from 3 different LHb neurons (i, ii, iii) in adult WT mice. Lower trace (iii’) is the same as above (iii) at higher temporal resolution. Scale bars 50 pA, 200 ms (upper), 10 ms (lower). Biphasic events accounted for 2/3 of all large (> 15 pA) outward events in these cells (79, 56, and 67%; large, biphasic events were found in 3/12 recordings). (C) Spontaneous miniature biphasic events (*) from whole-cell recordings at −40 mV with 0.5 mM chloride internal solution. Scale bars, 10 pA, 20 ms. (D) Whole-cell recording from Vglut2-cre mouse injected with AAV-DIO-ChR2-YFP at −15 mV holding potential of light-evoked EP-LHb transmission with indicated drugs in bath. Both inward and outward currents found in 17/17 cells. Scale bars: 100 pA, 10 ms. (E) Plot of effect of light-evoked EP-LHb transmission from Vglut2+ axons of adult mice delivered at 50 Hz (left) or 2 Hz (right) on normalized LHb neuronal firing rate before and after bath application of picrotoxin (* P < .05). (F) Same as (D) for GAD67-cre mice. 11/13 cells, inward/outward; 2/13 cells, outward only. Scale bars: 40 pA, 10 ms. (G) cell-attached recording (average of 10 trials) from adult GAD67-cre mouse LHb neuron; light delivery indicated; scale bars: 5 mV, 20 ms.
To test specifically if EP neurons projecting to the LHb co-release glutamate and GABA, we first took a genetic approach. In Vglut2-Cre mice, only glutamatergic neurons expressing the vesicular glutamate transporter Vglut2 express Cre recombinase (27). We injected their EP with an AAV that expresses ChR2 only in cells expressing Cre (AAV-DIO-ChR2, 28); Fig. S5, S6), and prepared brain slices two to three weeks later. As in WT animals, light-evoked synaptic responses, measured at −15 mV holding potential, displayed a NBQX-sensitive inward current followed by a NBQX-insensitive, picrotoxin-sensitive outward current (Fig. 2D), indicating EP-LHb glutamatergic neurons release both glutamate and GABA. In cell-attached recordings, picrotoxin increased spikes evoked by 50 Hz, but not 2 Hz, stimulation of Vglut2-expressing EP axons (Fig. 2E, S7; NBQX blocked evoked spikes, Fig. S8, (26)), indicating that co-release of GABA is important for limiting LHb output during high-frequency EP input; the greater effect at higher frequency is likely due to the slower GABA-A-R kinetics permitting greater summation of GABA-A-R currents at 50 Hz.
In complementary experiments, we used GAD-67-Cre mice, in which only GABAergic neurons expressing glutamic acid decarboxylase type 67 express Cre recombinase (29–31). We injected their EP with AAV-DIO-ChR2 (Fig. S5, S6), and prepared brain slices two to three weeks later. As in WT and Vglut2-Cre mice, light-evoked synaptic responses, measured at −15 mV holding potential, displayed a NBQX-sensitive inward current followed by a NBQX-insensitive, picrotoxin-sensitive outward current (Fig. 2F), indicating EP-LHb GABAergic neurons release both glutamate and GABA. In cell-attached recordings, EP-LHb transmission evoked spikes, indicating that EP GABAergic neurons produce excitatory responses in LHb (Fig. 2G).
Co-release from EP to LHb was further supported by the following observations: a) the trial-to-trial variability in evoked EP-LHb transmission recorded at −15 mV displayed significantly greater co-variation of early inward and late outward currents than would be expected from independent release of glutamate and GABA (Fig. S9); b) evoked EP-LHb GABA-A-R- and AMPA-R-mediated transmission displayed similar presynaptic modulation by bath application of serotonin ((25), Fig. S10); c) evoked EP-LHb transmission recorded from different LHb neurons displayed similar GABA-A-R- and AMPA-R-mediated paired pulse response ratios (Fig. S11); and d) GABA-A-R- and AMPA-R-mediated transmission displayed similar response latencies and jitter (Fig. S12).
We next examined immunohistochemically the synapses made by EP neurons to the LHb. In adult mice the EP was injected with an AAV expressing green fluorescent protein (AAV-GFP; Fig. S6). Subsequently, 400 nm-thin sections of the LHb were prepared, immunolabeled for GFP, GAD and Vglut2, and imaged with confocal microscopy. GFP-expressing structures displayed GAD and Vglut2 (Fig. 3A; Fig. S13), indicating GABA and glutamate in individual EP-LHb terminals. A control region displayed little GAD and Vglut2 co-labeling (Fig. 3B). Quantification of labeling ((26), Fig. S14) in structures originating from the EP indicated that ~50% of Vglut2-positive structures showed GAD and ~ 80% that were GAD-positive had Vglut2; outside the LHb, Vglut2 showed little overlap with GAD (Fig. 3C, D; Fig. S15). Thus, in EP-LHb structures ~50% had both GAD and Vglut2, ~10% had only GAD and ~40% had only Vglut2. Consistent with co-release, glutamate receptors and GABA receptors displayed overlap in LHb (Fig. S16).
Fig. 3.
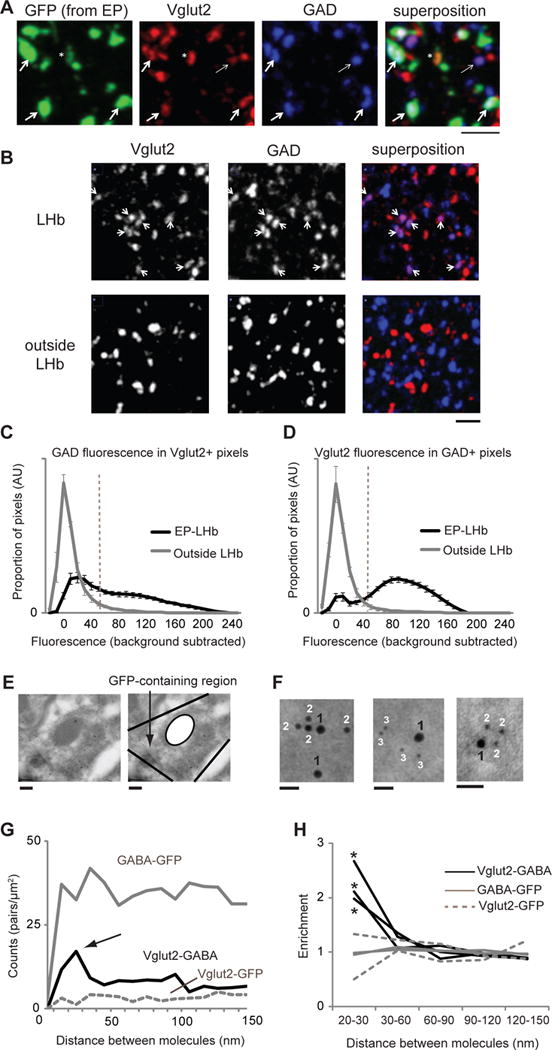
Co-localization of GABA-producing enzyme (GAD) and vesicular glutamate transporter (Vglut2) at EP neuron terminals in LHb. (A) Confocal fluorescent images of LHb sections from mouse injected with AAV-GFP into EP labeled for indicated proteins. Thick arrows indicate some of the sites of Vglut2, GAD and GFP colocalization. Thin arrow indicates Vglut2 and GAD colocalization (with no GFP). Asterisk indicates GFP-expressing structure showing only Vglut2 expression. Scale bar: 3 μm. (B) Confocal fluorescent images of sections from LHb (top) and outside LHb (bottom) labeled for indicated proteins. Arrows indicate structures displaying co-labeling for GAD and Vglut2. Scale bar: 3 μm. (C) Distribution of GAD fluorescence in pixels with Vglut2 intensity above Vglut2 threshold (dashed line in D; (26)), for images in LHb (for pixels displaying GFP, black line) and outside LHb (gray line). (D) Distribution of Vglut2 fluorescence in pixels with GAD intensity above GAD threshold (dashed line in C), as indicated (C, D, EP-LHb, N = 21 images, 3 mice; outside LHb, N = 5 images, 3 mice). (E) Electron micrograph showing a GFP-containing region in the LHb. Gold particles in the mitochondria were not analyzed. Scale bars: 200 nm. See Fig. S17, S18 for larger images of same region. (F) Left image: 18 nm gold labels Vglut2 (1), 12 nm gold labels GABA (2). Middle image: 18 nm gold labels Vglut2 (1), 6 nm gold labels GFP (3). Right image: 12 nm gold labels Vglut2 (1), 6 nm gold labels GABA (2). Scale bars: 30 nm. (G) Density of gold label pairs for distance intervals on x-axis for a single experiment. Black line, Vglut2-GABA distance; gray line, GABA-GFP; dotted gray line, Vglut2-GFP. N = 72 Vglut2 (18 nm) labels, 486 GABA (12 nm) labels, 416 GFP (6 nm) labels. Arrow indicates peak for Vglut2-GABA counts at 20–30 nm. (H) Increased enrichment (26) of Vglut2-GABA (black lines; 3 experiments) but not GABA-GFP (gray lines; 2 experiments) or Vglut2-GFP (dotted gray; 2 experiments) labels at 15–30 nm separation. N = 1624, 2556, and 1558 total Vglut2, GABA, and GFP labels, respectively. * > 4 standard deviations away from the mean.
To examine anatomically if GABA and glutamate can be released from individual vesicles at EP-LHb synapses, we used immunogold electron microscopy (EM). Two to three weeks after injecting adult mice with AAV-GFP in the EP, LHb sections were processed for EM and immunolabeled for GFP, Vglut2 and GABA with secondary antibodies linked to gold of three different sizes (26). Structures originating from the EP (Fig. 3E, Figs. S17, S18) showed gold particles of three clearly different sizes (Fig. 3F, Fig. S17). The frequency of distances between Vglut2 and GABA gold labels displayed a peak at ~20–30 nm (Fig. 3G), consistent with co-localization of Vglut2 and GABA at individual vesicles (32). The distribution of GFP-Vglut2 and GFP-GABA distances was flat indicating random distribution of particles. The number of gold particles less than 30 nm apart, compared to the number of gold particles between 30 and 100 nm apart was significantly higher for Vglut2-GABA (34/248) compared to Vglut2-GFP (4/92; p < 0.03, χ2-test) and GABA-GFP (64/999; p < 0.001, χ2-test). Increased enrichment of Vglut2-GABA gold pairs at less than 30 nm was obtained from three separate experiments using different antibodies and size gold (Fig. 3F, H; (26)). Vglut2 and the GABA vesicular transporter (VGAT), which are responsible for glutamatergic and GABAergic transmission, respectively (Fig. S19), also displayed enrichment at less than 30 nm (Fig. S20).
Activity of the LHb is elevated in animal models of human depression (12, 14, 33). Chronic serotonin-based antidepressant (e.g., citalopram (34)) treatment reduces LHb activity (35) and normalizes behaviors modeling human depression (36). We thus examined EP-LHb transmission as above in mice treated with citolapram or saline for two weeks. The ratio of GABA-A-R- to AMPA-R-mediated light-evoked synaptic responses was significantly higher in the citalopram- compared to saline-treated group (Fig. 4A, C, S21). This ratio was also reduced in cLH rats, a rat model of human depression (37), compared to control animals (Fig. 4B, D, S21). No differences in paired-pulse ratio, a measure of presynaptic release probability, were observed (Fig. S21, S22). These changes were matched immunohistochemically: in animals treated with citalopram, the ratio of GAD to Vglut2 was larger in EP-LHb boutons (Fig. 4E, S21) due to increased GAD labeling (Fig. 4G, I, S21); while in cLH animals, this ratio was reduced (Fig. 4F, S21), due to decreased GAD labeling (Fig. 4H, K, S21). Little change was seen in Vglut2 labeling (Fig. 4G, H, J, L, S21). Consistent with these results, chronic antidepressant treatment normalized the GAD to Vglut2 ratio in cLH animals (Fig. S23).
Fig. 4.
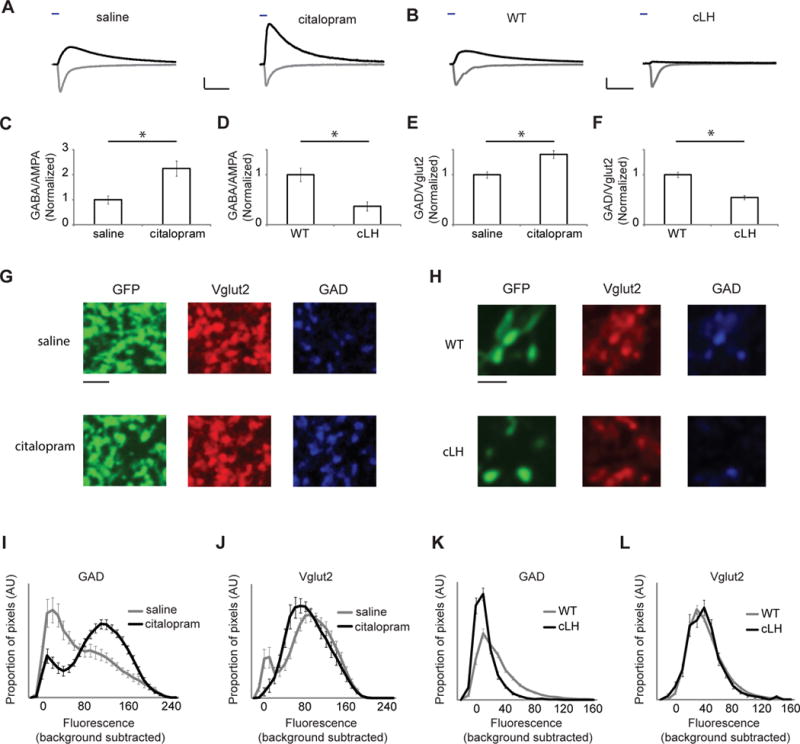
Altered co-release of GABA in conditions related to depression. (A) Sample whole-cell recordings of GABA-A-R- (black) and AMPA-R-mediated (gray) light-evoked EP-LHb responses from animals treated with saline or citalopram (10 mg/kg, i.p.). Scale bars: 200 pA, 20 ms. (B) Sample recordings, as in (A) in WT rats and cLH rats. Scale bars: 200 pA, 20 ms. (C) Bar graph of GABA-A-R-mediated/AMPA-R-mediated response at EP-LHb synapses for indicated groups normalized to average of saline ratios (N = 19 cells from 4 saline-treated mice and 24 cells from 4 citalopram-treated mice). (D) Bar graph of responses, as in (C) for WT and cLH rats normalized to average of WT ratios (N = 41 cells from 9 WT rats and 23 cells from 9 cLH rats). (E) Ratio of average background subtracted pixel intensities for GAD and Vglut2 in terminals originating from EP (26) for indicated groups (N = 24 images from 3 saline-treated mice, 21 images from 3 citalopram-treated mice). (F) Ratios, as in (E) for WT and cLH rats (N = 32 images from 7 WT rats, 21 images from 3 cLH rats). (G) Example confocal images from a saline and a citalopram-treated mouse. Scale bar: 3 μm. (H) Example confocal images from a WT rat and a cLH rat. Scale bar: 3 μm. (I) GAD fluorescence in pixels displaying Vglut2 and GFP labels in tissue from animals treated with citalopram or saline (% of Vglut2+, GFP+ pixels showing GAD fluorescence: saline, 54 ± 5%, 21 images; citalopram, 79 ± 2%, 24 images; P < 10−4). (J) Vglut2 fluorescence in pixels displaying GAD and GFP labels in tissue from animals treated with citalopram or saline (% of GAD+, GFP+ pixels showing Vglut2 fluorescence: saline, 80 ± 3%, 21 images; citalopram, 79 ± 3%, 24 images; P = 0.8). (K) GAD fluorescence, as in (I), for images from WT and cLH rats (WT, 50 ± 3%, 32 images; cLH, 19 ± 3%, 21 images; P < 10−7). (L) Vglut2 fluorescence, as in (J), for images from WT and cLH rats (WT, 61 ± 3%, 32 images; cLH, 61 ± 3%, 21 images; P = 0.9). * P < .002.
The LHb plays important roles in reward computations (21, 22), is activated during negatively-valenced events (21) via its basal ganglia inputs (22), and its hyperexcitation may contribute to human depression (16, 38). We found that co-release of GABA and glutamate occurs in the LHb and that the opponent actions of these neurotransmitters control the level of LHb activity by basal ganglia inputs. Our studies identify an unusual form of inhibition controlling the activity of this key brain region.
There is physiological evidence for co-release of GABA and glutamate (3) in the auditory system (5) and CA3 region of the hippocampus (4, 6–8), where it may contribute to refinement of neuronal connectivity during development (6, 39) and be induced by seizures in the adult hippocampus (11); GABA and glutamate co-release has not been universally accepted (10). Here we show using a combination of electrophysiological, optogenetic, mouse genetic, and fluorescent and EM immunohistochemical techniques that in the adult rodent brain a significant fraction of EP inputs to the LHb simultaneously release both glutamate and GABA and that these neurotransmitters have opposite effects on post-synaptic spike output. Co-release of glutamate and GABA can permit very local control, at the level of individual presynaptic terminals, in the balance of excitation and inhibition, a key mechanism controlling neuronal excitability (1).
The LHb receives long-range excitatory and inhibitory inputs from several sources (40–42). It remains to be determined if other inputs to the LHb co-release glutamate and GABA and the extent to which co-release of glutamate and GABA is used to regulate excitability in the rest of the nervous system (3). Co-expression of Vglut2 and GAD mRNA has been found in EP neurons that project to the motor thalamus, suggesting that co-release of glutamate and GABA may also occur in this pathway (43).
Given the proposed role of increased neuronal activity in the LHb in mood disorders (16), we examined the ratio of GABA to glutamate released at the EP-LHb synapse in conditions related to depression. In animals chronically treated with citalopram, a drug used to treat depression, evidence indicated enhanced GABA at EP-LHb synapses. In line with these data, EP-LHb synapses in cLH rats, which model aspects of human depression, showed evidence of reduced GABA, although additional changes (e.g., in receptor number) cannot be ruled out. Our results identify co-release of GABA and glutamate as a regulator of LHb output and potential determinant of the impact of negatively-valenced events on mood and behavior.
Supplementary Material
Acknowledgments
We thank H. Monyer and O. Kiehn for transgenic mice, R. Edwards for Vglut2 antibodies and DNA, K. Deisseroth for ChR2 DNA, J. Lin and R. Tsien for oChiEf DNA, T. Meerloo for electron microscopy assistance, and V. Joseph for histology assistance. This work was supported by an NIH grant to R.M. awarded to the UCSD Neuroscience Core (NS047101). C.D.P was supported by a postdoctoral award by the Instituts de Recherche en Santé du Canada.
Footnotes
References and Notes
- 1.Isaacson JS, Scanziani M. Neuron. 2011 Oct 20;72:231. doi: 10.1016/j.neuron.2011.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brinschwitz K, et al. Neuroscience. 2010 Jun 30;168:463. doi: 10.1016/j.neuroscience.2010.03.050. [DOI] [PubMed] [Google Scholar]
- 3.Seal RP, Edwards RH. Current opinion in pharmacology. 2006 Feb;6:114. doi: 10.1016/j.coph.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 4.Walker MC, Ruiz A, Kullmann DM. Neuron. 2001 Mar;29:703. doi: 10.1016/s0896-6273(01)00245-8. [DOI] [PubMed] [Google Scholar]
- 5.Gillespie DC, Kim G, Kandler K. Nat Neurosci. 2005 Mar;8:332. doi: 10.1038/nn1397. [DOI] [PubMed] [Google Scholar]
- 6.Beltran JQ, Gutierrez R. J Physiol. 2012 Oct 1;590:4789. doi: 10.1113/jphysiol.2012.236372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cabezas C, Irinopoulou T, Gauvain G, Poncer JC. J Neurosci. 2012 Aug 22;32:11835. doi: 10.1523/JNEUROSCI.5543-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Safiulina VF, Fattorini G, Conti F, Cherubini E. J Neurosci. 2006 Jan 11;26:597. doi: 10.1523/JNEUROSCI.4493-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caiati MD. J Neurosci. 2013 Jan 30;33:1755. doi: 10.1523/JNEUROSCI.5019-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uchigashima M, Fukaya M, Watanabe M, Kamiya H. J Neurosci. 2007 Jul 25;27:8088. doi: 10.1523/JNEUROSCI.0702-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gutierrez R. Trends Neurosci. 2005 Jun;28:297. doi: 10.1016/j.tins.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 12.Shumake J, Edwards E, Gonzalez-Lima F. Brain Res. 2003 Feb 14;963:274. doi: 10.1016/s0006-8993(02)04048-9. [DOI] [PubMed] [Google Scholar]
- 13.Caldecott-Hazard S, Mazziotta J, Phelps M. J Neurosci. 1988 Jun;8:1951. doi: 10.1523/JNEUROSCI.08-06-01951.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li B, et al. Nature. 2011 Feb 24;470:535. doi: 10.1038/nature09742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li K, et al. Science. 2013 Aug 30;341:1016. [Google Scholar]
- 16.Sartorius A, et al. Biol Psychiatry. 2010 Jan 15;67:e9. doi: 10.1016/j.biopsych.2009.08.027. [DOI] [PubMed] [Google Scholar]
- 17.Winter C, Vollmayr B, Djodari-Irani A, Klein J, Sartorius A. Behav Brain Res. 2011 Jan 1;216:463. doi: 10.1016/j.bbr.2010.07.034. [DOI] [PubMed] [Google Scholar]
- 18.Morris JS, Smith KA, Cowen PJ, Friston KJ, Dolan RJ. Neuroimage. 1999 Aug;10:163. doi: 10.1006/nimg.1999.0455. [DOI] [PubMed] [Google Scholar]
- 19.Amat J, et al. Brain Res. 2001 Oct 26;917:118. doi: 10.1016/s0006-8993(01)02934-1. [DOI] [PubMed] [Google Scholar]
- 20.Eshel N, Roiser JP. Biol Psychiatry. 2010 Jul 15;68:118. doi: 10.1016/j.biopsych.2010.01.027. [DOI] [PubMed] [Google Scholar]
- 21.Matsumoto M, Hikosaka O. Nature. 2007 Jun 28;447:1111. doi: 10.1038/nature05860. [DOI] [PubMed] [Google Scholar]
- 22.Hong S, Hikosaka O. Neuron. 2008 Nov 26;60:720. doi: 10.1016/j.neuron.2008.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patton GC, Coffey C, Posterino M, Carlin JB, Bowes G. Psychol Med. 2003 Oct;33:1203. doi: 10.1017/s0033291703008626. [DOI] [PubMed] [Google Scholar]
- 24.Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. Nat Neurosci. 2005 Sep;8:1263. doi: 10.1038/nn1525. [DOI] [PubMed] [Google Scholar]
- 25.Shabel SJ, Proulx CD, Trias A, Murphy RT, Malinow R. Neuron. 2012 May 10;74:475. doi: 10.1016/j.neuron.2012.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Materials and methods are available as supplementary material on Science Online.
- 27.Vong L, et al. Neuron. 2011 Jul 14;71:142. doi: 10.1016/j.neuron.2011.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsai HC, et al. Science. 2009 May 22;324:1080. doi: 10.1126/science.1168878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Melzer S, et al. Science. 2012 Mar 23;335:1506. doi: 10.1126/science.1217139. [DOI] [PubMed] [Google Scholar]
- 30.Fuchs EC, et al. Proc Natl Acad Sci U S A. 2001;98:3571. doi: 10.1073/pnas.051631898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tolu S, et al. FASEB J. 2010 Mar;24:723. doi: 10.1096/fj.09-139790. [DOI] [PubMed] [Google Scholar]
- 32.Tokuno H, Moriizumi T, Kudo M, Kitao Y, Nakamura Y. Brain Res. 1988 Dec 6;474:390. doi: 10.1016/0006-8993(88)90457-x. [DOI] [PubMed] [Google Scholar]
- 33.Caldecott-Hazard S. Exp Neurol. 1988 Jan;99:73. doi: 10.1016/0014-4886(88)90128-8. [DOI] [PubMed] [Google Scholar]
- 34.Hyttel J. Progress in neuro-psychopharmacology & biological psychiatry. 1982;6:277. doi: 10.1016/s0278-5846(82)80179-6. [DOI] [PubMed] [Google Scholar]
- 35.Freo U, Ori C, Dam M, Merico A, Pizzolato G. Brain Res. 2000 Jan 31;854:35. doi: 10.1016/s0006-8993(99)02261-1. [DOI] [PubMed] [Google Scholar]
- 36.Jarosik J, Legutko B, Unsicker K, von Bohlen Und Halbach O. Exp Neurol. 2007 Mar;204:20. doi: 10.1016/j.expneurol.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 37.Vollmayr B, et al. Behav Brain Res. 2004 Apr 2;150:217. doi: 10.1016/S0166-4328(03)00259-6. [DOI] [PubMed] [Google Scholar]
- 38.Hikosaka O. Nat Rev Neurosci. 2010 Jul;11:503. doi: 10.1038/nrn2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Noh J, Seal RP, Garver JA, Edwards RH, Kandler K. Nat Neurosci. 2010 Feb;13:232. doi: 10.1038/nn.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stamatakis AM, et al. Neuron. 2013 Nov 20;80:1039. doi: 10.1016/j.neuron.2013.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Herkenham M, Nauta WJ. J Comp Neurol. 1977 May 1;173:123. doi: 10.1002/cne.901730107. [DOI] [PubMed] [Google Scholar]
- 42.Gottesfeld Z, Massari VJ, Muth EA, Jacobowitz DM. Brain Res. 1977 Jul 8;130:184. doi: 10.1016/0006-8993(77)90856-3. [DOI] [PubMed] [Google Scholar]
- 43.Barroso-Chinea P, et al. Neurobiol Dis. 2008 Sep;31:422. doi: 10.1016/j.nbd.2008.05.019. [DOI] [PubMed] [Google Scholar]
- 44.Shumake J, Colorado RA, Barrett DW, Gonzalez-Lima F. Brain Res. 2010 Jul 9;1343:218. doi: 10.1016/j.brainres.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Micheva KD, Busse B, Weiler NC, O’Rourke N, Smith SJ. Neuron. 2010 Nov 18;68:639. doi: 10.1016/j.neuron.2010.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.