

Graphical abstract
Abbreviations: DsbA, thiol:disulfide interchange protein DsbA; GB1, immunoglobulin binding domain B1 of streptococcal protein G; eGFP, enhanced green fluorescent protein; GST, glutathione-S-transferase; HSQC, heteronuclear single quantum coherence; MBP, maltose-binding protein; OspC, outer surface protein C; Salp15, salivary protein of 15 kDa; Trx, thioredoxin
Keywords: Lyme borreliosis, Protein NMR, Saliva-assisted transmission, Solubility-enhanced fusion proteins, Tick saliva, Tick saliva antibodies
Highlights
-
•
Tick saliva proteins Salp15 and Iric-1 promote tick feeding and pathogen transmission.
-
•
We established the first bacterial expression system for soluble Salp15 and Iric-1.
-
•
Using this system we mapped monoclonal antibody epitopes on Salp15 and Iric-1.
-
•
We defined the interaction sites with Borrelia outer surface protein C (OspC).
-
•
We elucidated first secondary structure features in Iric-1 by NMR.
Abstract
Ticks transmit numerous pathogens, including borreliae, which cause Lyme disease. Tick saliva contains a complex mix of anti-host defense factors, including the immunosuppressive cysteine-rich secretory glycoprotein Salp15 from Ixodes scapularis ticks and orthologs like Iric-1 from Ixodesricinus. All tick-borne microbes benefit from the immunosuppression at the tick bite site; in addition, borreliae exploit the binding of Salp15 to their outer surface protein C (OspC) for enhanced transmission. Hence, Salp15 proteins are attractive targets for anti-tick vaccines that also target borreliae. However, recombinant Salp proteins are not accessible in sufficient quantity for either vaccine manufacturing or for structural characterization. As an alternative to low-yield eukaryotic systems, we investigated cytoplasmic expression in Escherichia coli, even though this would not result in glycosylation. His-tagged Salp15 was efficiently expressed but insoluble. Among the various solubility-enhancing protein tags tested, DsbA was superior, yielding milligram amounts of soluble, monomeric Salp15 and Iric-1 fusions. Easily accessible mutants enabled epitope mapping of two monoclonal antibodies that, importantly, cross-react with glycosylated Salp15, and revealed interaction sites with OspC. Free Salp15 and Iric-1 from protease-cleavable fusions, despite limited solubility, allowed the recording of 1H–15N 2D NMR spectra, suggesting partial folding of the wild-type proteins but not of Cys-free variants. Fusion to the NMR-compatible GB1 domain sufficiently enhanced solubility to reveal first secondary structure elements in 13C/15N double-labeled Iric-1. Together, E. coli expression of appropriately fused Salp15 proteins may be highly valuable for the molecular characterization of the function and eventually the 3D structure of these medically relevant tick proteins.
1. Introduction
Ticks are obligate ectoparasites that infest nearly all vertebrates. After mosquitos they comprise the second-largest group of arthropod vectors, transmitting numerous pathogenic viruses, bacteria and protozoa, with a high public health and economic impact [1]. Prominent examples for tick-borne human disease agents are tick-borne encephalits virus [2,3] and various Borrelia species causing Lyme disease [4–6]; the main agents are Borrelia burgdorferi sensu stricto in North-America, and B. burgdorferi plus Borrelia garinii and Borrelia afzelii in Eurasia. Each step of the tick life-cycle through the larval, nymphal and adult stages requires a bloodmeal. Ixodid (“hard”) ticks, including Ixodes scapularis and Ixodes pacificus from North-America and Ixodes ricinus from Eurasia as prime human disease-relevant species, take long-term (several days) bloodmeals. This exposes the engorged tick to numerous host defenses, such as vasoconstriction, complement activation, and innate and adaptive immune responses [6]. Tick saliva therefore contains a complex cocktail of counteracting factors, including immunosuppressants [7]. As yet few of these factors have been characterized; one of the best-studied is the salivary protein Salp15 from I. scapularis [8,9], a secretory cysteine-rich glycoprotein of about 15 kDa (in unglycosylated form). Salp15 binds to the T cell coreceptor CD4, compromising IL-2 production and thereby T cell activation and proliferation [8,10], and also dendritic cell-specific adhesion receptor DC-SIGN [11], further inhibiting cytokine expression. Less well characterized Salp 15 orthologs have been found in other Ixodes species (Fig. S1), including Iric-1 (Fig. 1) in I. ricinus which shares ∼76% sequence identity with Salp15 [12]. Salp15 and its orthologs are synthesized as about 135 aa precursors predicted to contain cleavable N terminal signal sequences of around 20 aa. The predicted cleavage sites vary slightly, depending on the specific ortholog and prediction algorithm.
Fig. 1.

Primary sequence features of I. scapularis Salp15 and I. ricinus Iric-1. The sequences represent the uncleaved precursor proteins. Amino acid identities in Iric-1 are indicated by dots, or where emphasizing a specific feature, by lower case lettering. Predicted signal sequences (Salp15: 1-20; Iric-1: 1–24) are shown in italics, N-glycosylation motifs in red. Cys residues are high-lighted by yellow background and consecutive numbering C1, C2 etc. Note that Iric-1 lacks one residue, by alignment at the position of Ser71 in Salp15. A more comprehensive alignment of Salp15 orthologs is provided in Fig. S1. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Tick-borne pathogens profit generally from the local immunosuppression at the tick feeding site which facilitates their transmission [13–15]. B. burgdorferi exploits on top the specific binding to Salp15 of its outer surface protein C (OspC) to provide itself with a protective coat against both innate and adaptive immune responses, thus enhancing dissemination in the host [16]. Similar binding of Iric-1 to the respective OspC proteins from the Eurasian Lyme disease-relevant borreliae suggests an analogous role [17].
Based on these data, Salp15, and by inference Iric-1, have gained interest as targets for anti-tick vaccines [18,19] which might reduce tick infestation and tick-borne pathogen transmission in general, and Borrelia transmission in particular. After withdrawal of a first-generation, lipidated B. burgdorferi OspA-based Lyme disease vaccine and with >100.000 confirmed new cases of Lyme disease per year in Europe and the US the need for improved vaccines is obvious; moreover, the actual number of Borrelia infections may be severely underdiagnosed [4,20]. An experimental Salp15-based vaccine has indeed shown some promise in a mouse model [21]. However, there is currently no expression system that would allow to produce recombinant Salp15 in sufficient quantity for vaccine manufacturing. Moreover, this scarcity of material is responsible for the complete lack of structural knowledge on Salp15 and other tick saliva proteins, including the location of potentially neutralizing epitopes. Furthermore, it prevents the rational design of advanced vaccines, e.g. presentation on immune-enhancing nanocarriers such as virus-like and virus capsid-like particles, respectively (e.g. [22]; Kolb, Nguyen, Walker and Nassal, Viral Nanotechnology, CRC Press; in press) or epitope-focused vaccines [23]. Conversely, immune-enhancement will be mandatory to overcome the immunosuppressive activity of Salp15 [8] and, likely, of its orthologs.
The major challenge for recombinant expression of Salp15 and related proteins is their presence in tick saliva as glycosylated proteins of unknown structure that contain seven cysteines of unknown disulfide status. Virtually all previous studies were restricted to the secretory expression of Salp15 in stably transfected Drosophila melanogaster S2 cells [8]. While likely producing a near-authentic tick protein, the yields from this system are very low. Explicit numbers have not been published, yet in our own experiments we hardly reached yields above 50 ng/ml of S2 cell culture (R.W. and J.H.; unpublished data). Furthermore, the use of stable cell clones greatly hampers generation of mutant Salp15 which would be instrumental for the identification of functional interaction sites. We also failed to achieve substantially higher yields using Salp15-encoding baculoviruses and SF9 insect cells, or transiently or stably Salp15 transfected Leishmania tarentolae (P.K., J.V., M.N.; unpublished data). Hence here we focused on Escherichia coli as expression host. Conventionally, eukaryotic secretory cysteine-rich proteins are expressed in the E. coli periplasm [24] to allow for disulfide-formation, however often at the cost of low yield [25]. Conversely, expression in the reducing cytoplasm suffers from the lack of disulfide formation, often accompanied by general folding problems resulting in insolubility [24]. This was indeed observed for a construct in which the N terminal 19 residues of full-length Salp15 (essentially the signal sequence) were replaced by an artificial 28 aa sequence providing an N terminal His6 tag (NH6-Salp15) which was well expressed but virtually insoluble. While this protein could be solubilized from inclusion bodies and be used to generate Salp15-specific mAbs (see below), its structural status remained unknown.
A potential alternative was to express Salp15 as a fusion with a solubility enhancing protein tag in an E. coli strain engineered to provide a more oxidizing cytosolic environment such as SHuffle Express (NEB). These cells express, in addition, a cytoplasmic version of the periplasmic disulfide isomerase DsbC; this may enable reshuffling of potentially improper disulfide bonds [26]. Various proteins, e.g. maltose-binding protein (MBP) and glutathione-S-transferase (GST), are in use as solubility enhancing tags [27,28]. Also thioredoxin (Trx) superfamily members like Trx itself, or cytosolic versions of DsbC or DsbA [29], a likewise periplasmic thiol oxidoreductase, have in some instances successfully been applied for the soluble expression of cysteine-containing proteins [24]. In fact, a Trx-Salp15 fusion protein was used to demonstrate the suppressive impact of the Salp15 part on the induction of Trx-specific antibodies [8], but yields and solubility were not reported.
As shown below, out of various combinations of fusion partners and E. coli expression strains, expression of Salp15 and Iric-1 as DsbA fusions in SHuffle Express cells resulted in milligram amounts of soluble proteins per liter of E. coli culture. The easy access to variants provided by this system enabled us to define the epitopes of two Salp15/Iric-1 cross-reactive mAbs and to map the OspC interaction sites to a predicted α-helical segment around aa 50 (numbering based on the uncleaved precursor proteins). Free Salp15 and Iric-1 from protease-cleavable DsbA fusions, and more so fusions with the NMR-compatible GB1 tag, allowed production of 15N and 13C/15N labeled samples for heteronuclear NMR spectroscopy, giving preliminary insights into secondary structure elements in Iric-1 and paving the way for determining the 3D structure of these medically relevant tick saliva proteins.
2. Results
2.1. DsbA as efficient solubility-enhancing fusion partner for Salp15
In a first series of experiments we inserted the open reading frame (ORF) of mature Salp15 (starting with residue A20, at the predicted signal peptide cleavage site; see Fig. 1) into bacteriophage T7 promoter-controlled vectors providing the ORFs for N terminally His6-tagged versions of the protein tags MBP, GST, Trx, and leaderless, i.e. cytoplasmic, DsbA (Fig. 2A). In DsbA, Cys30 and Cys33 in the catalytic C-X-X-C motif [30] were either replaced by Ser-residues (termed DsbA) to minimize potentially improper disulfide formation between DsbA and Salp15 [31], or were maintained (termed DsbA*) to possibly promote proper disulfide formation in the Salp15 part. Later on, the immunoglobulin-binding domain 1 from protein G (GB1), a small (56 aa) NMR-compatible tag [32], was employed as additional fusion partner.
Fig. 2.
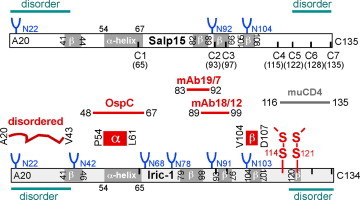
Impact of fusion partner and E. coli expression strain on Salp15/Iric-1 solubility. (A) General structure of the expression constructs used. The Salp15 and Iric-1 sequences starting with A20 were fused to various solubility-enhancing domains (SEDs) carrying an N terminal His6-tag; in some constructs, a TEV protease recognition sequence was inserted in-between. (B) Differential solubility enhancement by different protein tags. Total SDS-lysates versus the soluble fraction of native lysates from E. coli BL21*CP cultures expressing the indicated Salp15 fusion proteins were analyzed by SDS–PAGE followed by Coomassie Blue staining. (C) Impact of expression strain and Salp15 protein sequence. Total protein (T) in native lysates, and in the insoluble pellet (P) versus soluble supernatant (S) after centrifugation were analyzed by SDS–PAGE. Left panel: DsbA-Salp15 expressed in BL21*CP versus SHexpCP cells. Note the lower level of fusion protein in the insoluble pellet from SHexpCP cells. Right panel: DsbA-Salp15 versus DsbA-Iric-1 expressed in SHexpCP cells. Note the lower level of DsbA-Iric-1 in the insoluble pellet. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
In E. coli BL21* Codonplus (CP) RIL cells (Stratagene), providing IPTG-inducible T7 RNA polymerase plus rare E. coli tRNAs from the CP plasmid, IPTG induction led to visible overexpression of all fusion proteins; however, only the MBP and DsbA fusions (and in part the corresponding GB1 fusions, see below) were well soluble (Fig. 2B). The same was observed in T7 SHuffle Express cells carrying the CP plasmid (referred to as SHexpCP cells). However, the fraction of DsbA-Salp15 remaining in the insoluble fraction of the native bacterial lysates was reproducibly lower than in BL21*CP cells (Fig. 2C, left panel, lanes P), and the soluble fraction contained lower levels of smaller expression vector-related products migrating around the 25 kDa marker position. A comparably beneficial effect of the SHexpCP strain was observed for Salp15 fused to MBP and DsbA* (not shown), and to GB1 (see Fig. S5). Because of DsbA’s considerably smaller size (∼23 kDa) compared to MBP (∼40 kDa), and the lack of positive impact of the Cys-containing DsbA* versus Cys-less DsbA (see below; Fig. 10A), most subsequent constructs were based on DsbA and expressed in SHexpCP cells. This combination worked also well for Iric-1, which was obtained in almost completely soluble form (Fig. 2C, right panel). An intrinsically higher solubility of Iric-1 compared to Salp15 was confirmed in various subsequent experiments.
Fig. 10.
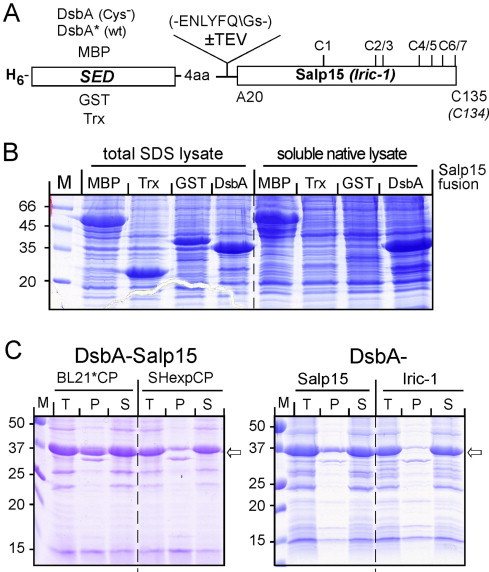
Basic structural features of free Salp15 and Iric-1. (A) Lack of impact of redox-active (DsbA*) versus inactive DsbA on Iric-1 disulfide status. Iric-1 was released by TEV protease as in Fig. 9B from analogous fusion proteins comprising DsbA* carrying the catalytic C-X-X-C motif, or the Cys-free DsbA variant used throughout this study. Free Iric-1 proteins were treated with excess iodoacetamide, then analyzed by SDS–PAGE under reducing (+DTT) versus non-reducing (−DTT) conditions. The dominant band in the non-reduced samples comigrating with the reduced samples plus a less intense faster migrating band are marked by arrows. The ∼24 kDa band marked by * was not affected by DTT and hence does not represent a disulfide-linked Iric-1 dimer. (B) Structural heterogeneity of solubilized NH6-Salp15. Solubilized NH6-Salp15 was treated with iodoacetamide and analyzed by SDS–PAGE under reducing versus non-reducing conditions as in (A). Note that nearly half the protein was present as disulfide-linked dimer. (C) Protease-released and solubilized Salp15 differ in quaternary structure. Both proteins were subjected to SEC on Superdex 75. The elution volume of protease-released Salp15 was consistent with a monomeric state, that of solubilized NH6-Salp15 with a dimeric state. (D, E) 1H–15N HSQC NMR spectra suggest a positive impact of the tick protein cysteines on folding. Wild-type and Cys-free Salp15 (D) and Iric-1 (E) were released from 15N-labeled DsbA-TEV fusion proteins as in Fig. 9. Spectra from the wild-type (wt; in black) and Cys-free (Cys−; in red) proteins were superimposed to emphasize the broader peak dispersion of the wild-type proteins. Larger versions of the spectra are shown in Fig. S4. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
2.2. Soluble Salp15 and Iric-1 DsbA fusion proteins are monomeric
For preparative purification, the soluble fractions of lysates from induced cultures of SHexpCP cells transformed with the DsbA-Salp15 and DsbA-Iric-1 vectors were first subjected to immobilized Ni2+ ion affinity chromatography (IMAC). Full-length (∼35.5 kDa) DsbA-Salp15 eluted between 50 and 300 mM imidazole, however accompanied by nearly equal amounts of smaller (∼25 kDa) products (Fig. 3A, left panel). As a protease inhibitor cocktail was present during the entire work-up, we suspected these products to arise from translation initiation at the DsbA-internal Met residue (M91 in the His-tagged protein) which would result in a ∼25 kDa protein; however, mutation of M91 to Leu did not significantly change this pattern (not shown), suggesting the fusion protein is highly sensitive to proteolytic cleavage. Depletion of the smaller products by subsequent size exclusion chromatography (SEC) on Superdex 75 (Fig. 3A, right panel) allowed the isolation of ∼30% of the total DsbA-Salp15 protein in >90% purity in a single run, as estimated from densitometric scanning of the Coomassie-Blue stained SDS–PAGE gel, in an overall yield of around 8 mg/L culture. By comparison with a set of molecular mass standards, the full-length fusion protein eluted at a volume corresponding to an apparent molecular mass of ∼42 kDa, consistent with a monomeric state. Similar results were obtained with the DsbA-Iric-1 fusion protein, except that the smaller products co-eluting from the IMAC column were much less abundant (Fig. 3B, left panel) and could be more efficiently separated from the full-length protein by SEC (Fig. 3B, right panel); this increased the yield of pure full-length DsbA-Iric-1 protein to about 30 mg/L culture. The elution volume of the Iric-1 protein was again in line with a monomeric state.
Fig. 3.
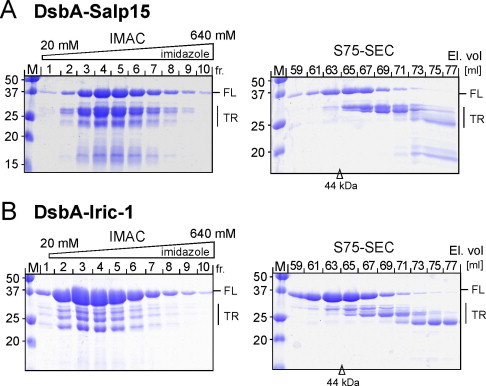
Representative purifications of the DsbA fusion proteins. (A) DsbA-Salp15. (B) DsbA-Iric-1. Cleared native lysates were first subjected to IMAC using a Ni-NTA column developed using a linear imidazole gradient. Peak fractions were concentrated and subjected to SEC on Superdex S75. Fractions were analyzed by SDS–PAGE followed by Coomassie Blue staining. The arrowhead marks the elution volume of a 44 kDa size marker protein. The positions of full-length (FL) and truncated (TR) fusion proteins are indicated. Note the lower level of truncated products for DsbA-Iric-1 compared to DsbA-Salp15. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Together these data demonstrated that highly enriched Salp15 and Iric-1 are accessible from E. coli in milligram amounts per liter of culture as soluble, monomeric DsbA fusion proteins.
2.3. Mapping of mAb epitopes using DsbA-Salp15 and DsbA-Iric-1 fusions
The solubilized NHis-Salp15 preparation, available before the above reported results were obtained, was used to generate monoclonal antibodies (mAbs) 19/7.4 and 18/12.1 (R.W. and J.H., unpublished data). In order to map their epitopes, we first generated a panel of DsbA-Salp15 proteins in which from one to all of the seven Cys-residues (Fig. 4A) were replaced by Ser-residues (termed Salp15Cx, where x specifies the replaced Cys-residue(s); variants lacking all cysteines were named Salp15Cys− and Iric-1Cys−). All were efficiently expressed in soluble form (Fig. 4B, top panel). We also included, as the first ones in a series (see below), two truncated Salp15 variants lacking the N terminal 44 aa (SaΔN44) or the C terminal aa from position 115 on (SaΔC115); these truncations appeared to enhance proteolytic sensitivity, as suggested by the reduced presence of full-length versus faster migrating products. When used as test antigens on Western blots, all proteins were similarly recognized by mAb 19/7.4 (Fig. 4B, middle panel), suggesting its epitope did not involve any of the Cys-residues and was contained between Salp15 residues 45 and 114. The latter was also true for mAb 18/12.1, however the Cys-variants were differentially recognized. The common feature of all non-reactive variants was the absence of C93 (C2) and C97 (C3). This was confirmed for Iric-1 in a side-by-side comparison of the wild-type versus Cys-free variants of Salp15 and Iric-1 DsbA fusion proteins (Fig. 4C).
Fig. 4.
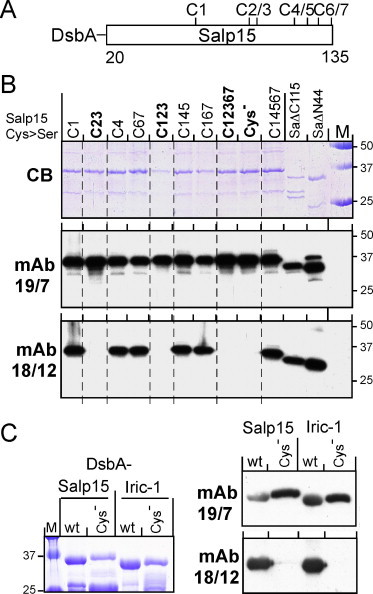
Impact of Cys-mutations on DsbA-Salp15 recognition by mAbs 19/7.4 and 18/12.1. (A) Cysteine-map of DsbA-Salp15. For reference to the explicit aa positions, see Fig. 1. DsbA-Salp15 proteins with one to all seven cysteines (Cys−) replaced by Ser-residues were expressed in SHexpCP cells; the number(s) in their names indicate the replaced cysteine(s). For comparison, an N terminal (SaΔN44) and a C terminal truncation variant (SaΔC115) were included. (B) SDS–PAGE analysis and immunoblots. Owing to the high expression levels, total SDS lysates could be used. Three SDS–PAGE gels were run in parallel. The first (upper panel) was stained with Coomassie Blue. The second and third, loaded with one fifth the amount of samples, were blotted and probed with the indicated mAbs, followed by visualization using a peroxidase-conjugated anti-mouse secondary antibody and a chemiluminescent substrate. Note the failure of mAb 18/12.1 to detect any of the proteins lacking C2 and C3. (C) Cross-reactivity of anti-Salp15 mAbs with Iric-1. DsbA fusions of wild-type Salp15 and Iric-1 and their Cys-free variants were separated by SDS–PAGE and immunoblotted as in (B). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
For further mapping, we employed additional terminal truncation variants (Fig. 5A). The shortest ones still recognized by both mAbs (Fig. 5B) lacked the first 59 aa (SaΔN59), or the C terminal residues from aa 100 on (SaΔC100). To fine-map the epitopes, we fused various Salp15 and Iric-1 sequences from within that region to DsbA. The fusion containing Salp15 residues I83-H99, which are strictly conserved in Iric-1 (Fig. 1), reacted with both mAbs. However, within that sequence mAb 19/7.4 recognized only the subsegment I83-N92, and mAb 18/12.1 only the subsegment D89-H99. Hence the epitopes are closely juxtaposed but distinct. Notably, the latter segment contains C93 (C2) and C97 (C3), which were also required for recognition by mAb18/12.1 in the context of full-length Salp15 (Fig. 4). Reactivity of both mAbs with DsbA-Salp15 and DsbA-Iric-1 full-length proteins was also confirmed in an ELISA format where the two proteins were coated onto the plate (data not shown; but see OspC binding below).
Fig. 5.
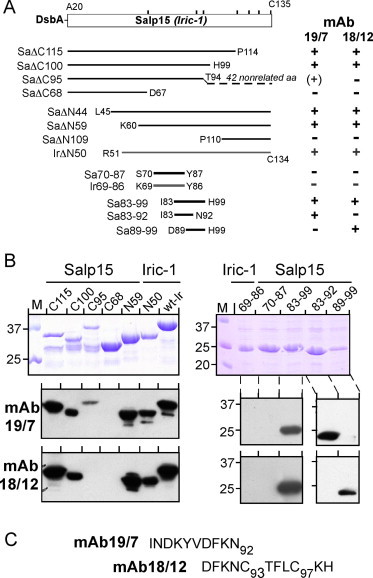
Mapping the epitopes of mAbs 19/7.4 and 18/12.1. (A) Schematic overview of the constructs used. Perpendicular marks on the box representing the Salp15 (Iric-1) ORF indicate the positions of the seven Cys-residues. Salp15 (Sa) derivatives are indicated as black lines, Iric-1 (Ir) derivatives as grey lines. Reactivities with the two mAbs derived from immunoblotting results are summarized on the right. (B) SDS–PAGE analysis and immunoblots. The indicated DsbA fusion proteins were analyzed as in Fig. 4B. Note that the Salp15 sequence 83-99, the smallest internal segment recognized by both mAbs, is identical to the Iric-1 sequence 82-98 (Fig. 1). (C) Salp15 primary sequences encompassing the epitopes of mAbs 19/7.4 and 18/12.1.
To test for reactivity of the mAbs with authentic Salp15 we investigated their ability to recognize glycosylated Salp15, in particular because in both Salp15 and Iric-1 one of the N-glycosylation motifs (N92CT in Salp15, N91CT in Iric1) is very close to or even overlaps with the epitopes (see Fig. 1). To this end, we transfected eukaryotic expression vectors encoding three potentially secreted variants of Salp15 (Fig. 6A), carrying the authentic Salp15 signal sequence (Salp-Sig-Salp15) or that of the E. coli OmpA protein (OmpA-Sig-Salp15), into human HEK293 cells. To account for potential non-reactivity with the mAbs, all constructs were provided with a C terminal His6 tag to allow detection with an anti-His antibody and enrichment by Ni-NTA affinity capture. To possibly increase detection sensitivity, a third construct (Salp-Sig-NH6-Salp15) contained an additional His6 tag after the predicted signal sequence cleavage site. Cells transfected with a vector for intracellular eGFP expression and non-transfected cells served as controls. Four days post transfection, equal aliquots of the culture supernatants were incubated with Ni-NTA agarose beads, and the affinity-captured material was used for immunoblotting, either directly or after prior treatment with peptide-N-glycosidase (PNGase) F for glycan removal. Detection with an anti-His-tag antibody (Fig. 6B, left panel) revealed for all three Salp15 proteins the presence of smeary bands above the 25 kDa marker position which collapsed into distinct ∼20 kDa bands after PNGase F treatment, in line with the secretion of heterogeneously glycosylated Salp15 from the cells. Equal aliquots of the Ni-NTA pulled-down material from the Salp-Sig-NH6-Salp15 transfected cells were then again subjected to immunoblotting, using either the anti-His-tag antibody or mAbs 19/7.4 and 18/12.1. All three antibodies reacted well the main product obtained after PNGase F treatment (Fig. 6B, right panels) yet, importantly, also with the glycosylated forms. Because glycan shielding of the epitopes might depend on 3D structure, we also tested the reactivity of both mAbs with native glycosylated Salp15 in an ELISA format. To this end, the secreted Salp15 proteins were affinity-captured on Ni-NTA HisSorb microtiter plates. Culture supernatants from cells transfected with the eGFP vector and from non-transfected cells served as negative controls, E. coli derived DsbA-Salp15 (100 ng and 10 ng per well) and solubilized NHis-Salp15 (10 ng per well) as positive controls. As shown in Fig. 6C, both mAbs generated signals with the Salp-Sig-NH6-Salp15 samples that clearly exceeded those of the negative controls. For mAb 18/12.1 the signal intensity (from 200 μl supernatant) was similar to that produced by 10 ng DsbA-Salp15 or, accounting for the DsbA part, by ∼3.5 ng recombinant Salp15. This translates into about 20 ng Salp15 per ml of culture supernatant and thus is in a similarly low range as seen in stably transfected insect cells. Together, these data imply that glycosylation does not shield the epitope of either mAb; hence both might be able to neutralize Salp15 from tick saliva.
Fig. 6.
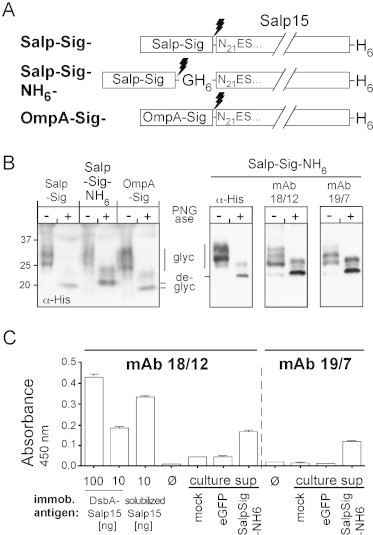
Recombinant Salp15 induced mAbs crossreact with secreted, glycosylated Salp15. (A) Schematic representation of the constructs used. All constructs contained a C terminal His6 tag to allow mAb-independent detection and affinity capture via Ni-NTA supports but differed regarding their N terminal signal sequences. Construct Salp-Sig comprises the original Salp15 precursor sequence; in Salp-Sig-NH6 a second His6-tag was inserted after the signal sequence; in OmpA-Sig the Salp15 signal sequence was replaced by the E. coli OmpA signal sequence. Lightning symbols, predicted signal peptide cleavage sites. (B) Immunoblot analysis. Constructs from (A) were transfected into HEK293 cells. Proteins in the supernatants captured using Ni-NTA beads were analyzed by immunoblotting without, or with prior deglycosylation using PNGase F. Left panel: Anti-His-tag immunoblot. Right panels: Detection of secreted Salp-Sig-NH6 proteins using an anti-His-tag mAb or mAbs 18/12.1 and 19/7.4. (C) ELISA. Wells of a His-Sorb microtiter plate were incubated, or not (ø), with the indicated recombinant Salp15 proteins, or 200 μl of culture supernatant from cells transfected with empty vector (mock) or vectors for eGFP or Salp-Sig-NH6, followed by incubation with the mAbs and detection by peroxidase-conjugated secondary antibody and TMB as substrate. All measurements were performed in quadruplicate; error bars indicate standard error of the mean (SEM).
2.4. DsbA-Salp15 and DsbA-Iric-1 are functional in OspC binding and allow mapping of the OspC binding sites
A distinct biological property of Salp15 and Iric-1 discovered using insect cell-derived proteins is their interaction with OspC [21,33]. To assess the ability of the E. coli expressed tick proteins to recapitulate this interaction we set up an ELISA in which four different recombinant OspC proteins (see Materials and Methods), OspCa and OspCb from B. burgdorferi strain ZS7 (with ∼96% sequence similarity), and B. afzelii NE [34] OspC A3 and OspC YU (with sequence similarities of about 75% to each other and to the ZS7 isolates), were coated on the plate (100 ng/well). Wells without OspC coating served as negative control. Next, the wells were incubated with excess (1 μg/well) DsbA-fusion proteins carrying wild-type or Cys-free Salp15 and Iric-1. Potentially bound tick proteins were detected using mAb 19/7.4. All combinations of OspC and Salp15/Iric-1, but not the negative controls, gave strong signals (Fig. 7). While the reason for the particularly intense signals with Cys-free Salp15 remains to be determined, this result indicated that OspC binding does not depend on the tick-protein Cys-residues. Regarding OspC specificity, the OspCs from B. burgdorferi were reproducibly better bound than those from B. afzelii. This ranking was corroborated by comparable results using mAb 18/12.1 for detection (Fig. S2); the results also confirmed in a non-denaturing ELISA setting that this mAb does not recognize the Cys-free variants. Specificity of the interaction between recombinant Salp15 and Iric-1 and OspC was further confirmed in a reverse setting where the DsbA-fusions were coated on the plate, followed by incubation with the various OspC proteins and their detection by the OspC-specific mAb LA97.1 [35]. For lack of reactivity with OspC A3 and OspC YU of this mAb, these data were restricted to the B. burgdorferi OspC proteins (not shown).
Fig. 7.
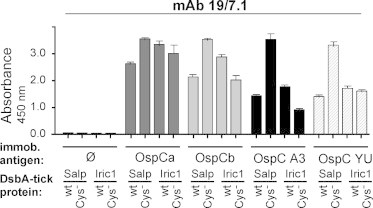
DsbA-Salp15 and DsbA-Iric-1 both interact specifically with different Borrelia OspC proteins. ELISA plate wells were coated with the indicated recombinant OspC proteins from B. burgdorferi (OspCa, Ospb) or B. afzelii (OspC A3, OspC YU), then incubated with DsbA-fused wt or Cys− Salp15 or Iric-1. Bound tick proteins were detected using mAb 19/7.4 as in Fig. 6C. All measurements were performed in quadruplicates; error bars indicate SEM. An analogous ELISA using mAb 18/12.1 is shown in Fig. S2.
Together, these data demonstrated the ability of E. coli derived Salp15 and Iric-1 to specifically interact with OspC proteins from the major Borrelia species in North-America and Eurasia, with implications for anti-tick saliva protein vaccines (see Section 3).
To map the OspC interaction site(s) on Salp15 and Iric-1, OspCa coated ELISA plates were incubated with the terminally truncated (but mAb 19/7.4 reactive) DsbA-Salp15 and DsbA-Iric1 fusion proteins (see Fig. 5), followed by detection of potentially bound tick proteins by mAb 19/7.4. The most pertinent results are shown in Fig. 8A. Accordingly, C terminal deletions after aa 100 (SaΔC100) and N terminal deletion before aa 44 (SaΔN44) had no negative effect. However, deleting 50 aa from the N terminus of Iric-1 (IrΔN50) and 59 aa from the N terminus of Salp15 (SaΔN59) substantially decreased signal strength, suggesting the OspC interaction site(s) to involve residues downstream of aa 44. For final confirmation, we fused the sequence from P48 to D67 of Salp15, or P48 to N67 of Iric-1, to an N terminally His6-tagged eGFP (Fig. 8B); the same eGFP protein without fused peptide served as control. All eGFP proteins were well and solubly expressed in E. coli (Fig. 8B, bottom panel). After enrichment by IMAC, they were incubated with plate-immobilized OspCa (Fig. 8C, “OspCa-mediated binding”); binding was detected using an anti-GFP mAb (Chromotek); functionality of the assay was confirmed by direct immobilization of the peptide-fused GFP proteins to the ELISA plate (Fig. 8C, “direct immob.”). Strong signals in the OspCa-mediated format were detected for both peptide-fused eGFP proteins, but not the eGFP-only control. Specificity was further corroborated by the absence of signal when the OspCa coated wells were incubated with the DsbA-SaΔN59 protein. Hence the sequences comprising residues P48-D67 of Salp15 and P48 to N67 of Iric-1 were necessary and sufficient for specific OspC binding.
Fig. 8.
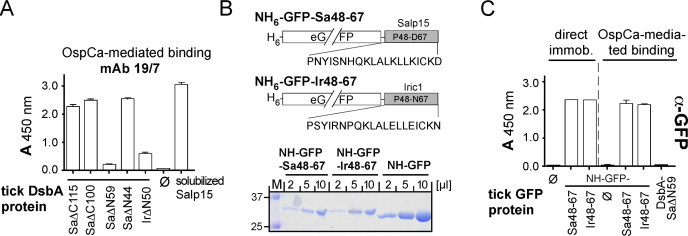
Mapping of the OspC interaction sites on Salp15 and Iric-1. (A) OspC binding ELISA. ELISA plate wells were coated with recombinant OspCa as in Fig. 7, then incubated, or not (ø), with the indicated DsbA-fused deletion variants (see Fig. 5A) of Salp15 or Iric-1. Bound tick proteins were detected using mAb 19/7.4 as in Fig. 6C. (B) Expression of internal Salp15 and Iric-1 fragments 48-67 as C terminal fusions to N terminally His-tagged GFP. Upper panel: Schematic representation of the constructs. Lower panel: SDS–PAGE analysis of the peak fractions after IMAC purification of the GFP proteins expressed in E. coli. Material from these fractions was used in the ELISA shown in (C). (C) Selective binding to OspC of the tick protein fragment-bearing GFP fusions. For the assays to the left of the dashed line (“direct immob.”), the ELISA plate wells were directly coated, or not (ø), with the GFP fusion proteins. For the remaining assays, the wells were coated with OspCa as in (A), then incubated with NH6-GFP (ø) or the tick protein fragment bearing variants, or DsbA-SaΔN59 which did not bind OspCa. Bound GFP proteins were detected using an anti-GFP mAb. Assays were performed in quadruplicates; error bars represent SEM.
2.5. E. coli derived Salp15 and Iric-1 proteins for NMR structural analyses
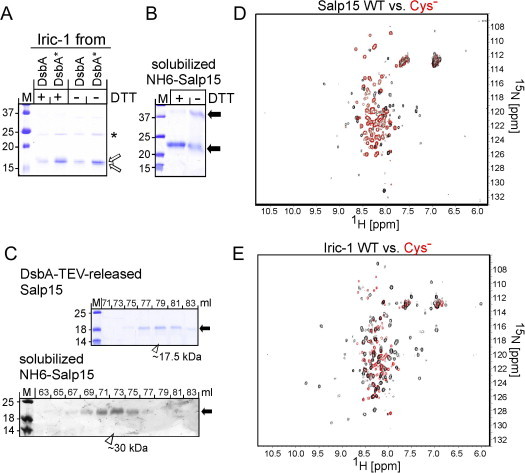
The high level soluble expression in E. coli plus their functionality in antibody and OspC binding suggested the DsbA fusions with Salp15 and/or Iric-1 as a source of proteins suitable for direct structural analyses, e.g. by NMR spectroscopy. 3D structure determination by NMR typically requires up to ∼0.5 ml of a ⩾0.1 mM protein solution (1.5 mg/ml for a 15 kDa protein) and access to 15N and 13C isotope labeled derivatives. With about 100 aa in length, the tick proteins fall well into the range of protein NMR [36,37]; however, the larger DsbA parts in the fusion protein would convolute the spectra. To obtain DsbA-free Salp15 and Iric-1, we therefore introduced a TEV protease recognition sequence between DsbA and Salp15, Iric-1, or their Cys-free variants, respectively (see Fig. 2A). All constructs were as highly expressed in SHexpCP cells as the parental fusion proteins, were mostly soluble, and could be enriched by sequential IMAC and Superdex 75 SEC where they eluted at a volume indicative of a monomeric state; also their recognition by mAb 19/7.4 (all) and 18/12.1 (only the wild-type proteins) was the same as for the parental DsbA fusions (data not shown).
Next, the purified fusion proteins were subjected to cleavage by His6-tagged TEV protease. For the wild-type Salp15 fusion this produced a distinct SDS–PAGE gel band running at about 27 kDa that corresponded to the DsbA part (nominally 24 kDa) and probably the TEV protease, plus a weakly Coomassie Blue staining band of around 20 kDa (Fig. 9A, left panel) whose identity as free Salp15 was confirmed by immunoblotting with mAb 19/7.4 (not shown). Taking advantage of the His6-tags on the TEV protease and DsbA, free Salp15 was obtained in the flow-through of a Ni-NTA column whereas the bulk of uncleaved precursor, cleaved DsbA and TEV protease eluted, as expected, only at higher imidazole concentration (Fig. 9A, right panel). Attempts to concentrate the protein from the flow-through by ultrafiltration indicated an upper solubility limit of about 0.1 mg/ml, thus limiting its suitability for further characterization (see below). Salp15Cys− analogously released from a DsbA-TEV fusion protein was more well-behaved (Fig. S3). This also held for free Iric-1 (Fig. 9B, left panel) and its Cys-free variant (Fig. 9B, right panel) which could both be concentrated to up to ∼0.5 mg/ml. Notably, both released tick proteins, independent from their Cys-content, migrated more slowly during SDS–PAGE than expected from their nominal molecular mass of ∼13 kDa; Salp15 comigrated with a 18 kDa marker protein, Iric-1 had a slightly higher mobility (see Fig. 9B, left panel, for a direct comparison). Probably this reflects their high content (25%) of charged amino acids [38].
Fig. 9.
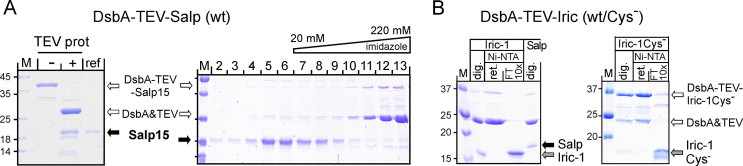
Free Salp15 and Iric-1 by TEV protease-mediated release from DsbA-TEV fusions. Wild-type (wt) and Cys-free (Cys−) forms of Salp15 and Iric-1 were expressed as DsbA fusions carrying a TEV protease recognition site between the two domains (see Fig. 2A) and enriched by Ni-NTA IMAC. Material from the peak fractions was used as protease substrate. (A) Salp15. Left panel: SDS–PAGE comparison of untreated versus His-tagged TEV protease-treated DsbA-TEV-Salp15. Salp15 from a prior pilot scale experiment served as reference (ref). Right panel: Depletion of uncleaved fusion protein, DsbA and TEV protease by IMAC. Free Salp15 was collected in the flow-through; the other protein components were retained due to their His-tags. Data for Cys-free Salp15 are shown in Fig. S3. (B) Iric-1. Wild-type (left panel) and Cys-free (right panel) DsbA-TEV-Iric-1 proteins were treated as in (A). Aliquots analyzed by SDS–PAGE are labeled as follows: dig., TEV protease reaction; Ni-NTA ret., retained on Ni-NTA column; FT 10×; flow-through of Ni-NTA column, concentrated 10-fold by ultrafiltration. Note the mobility difference between protease-released Iric-1 and Salp15.
To test for a potentially beneficial effect on solubility of the released tick proteins we also expressed them as fusions with cysteine-containing, i.e. potentially redox-active, DsbA (DsbA*). In all aspects of expression level, solubility, purification, and TEV-protease cleavage no significant differences to the fusions with Cys-free DsbA were observed (data not shown). To reveal potential differences in disulfide status, Iric-1 released from the DsbA versus the DsbA* fusion protein were subjected, after blocking free SH groups by iodoacetamide, to SDS–PAGE under non-reducing versus reducing conditions (Fig. 10A). In the absence of reducing agent, only trace amounts (<5% as determined by densitometry) of a band with a mobility expected for dimeric Iric-1 was seen in both preparations; at the monomer position one major band comigrating with the reduced samples plus a weaker, faster migrating band were observed, possibly reflecting a species with more extensive intramolecular disulfide bonding. However, the overall similar band patterns revealed no advantage of the DsbA* versus the DsbA Iric-1 fusion protein, and neither for the corresponding Salp15 fusions (data not shown).
As a potential alternative for structural studies the solubilized NH6-Salp15 protein was subjected to the same type of analysis (Fig. 10B). This revealed about half of the protein being present as disulfide-linked dimer. To compare the native quaternary structures of the DsbA-released Salp15 versus the solubilized NH6-Salp15, both proteins were also subjected to analytical SEC (Fig. 10C). The released Salp15 eluted around 79 ml, corresponding to a molecular mass of 17.5 kDa. In contrast, the solubilized NH6-Salp15 eluted at 73 ml, corresponding to about 30 kDa, i.e. twice its nominal molecular mass of 15.6 kDa. Hence unless refolding conditions can be improved so as to direct the protein into a single major conformation, the renatured Salp15 does not appear suitable for structural work.
Despite the limited solubility of the DsbA released tick proteins, the procedure could be adapted to generate uniformly 15N labeled derivatives of Salp15 and Iric-1 plus their Cys-free variants (see Materials and Methods) in sufficient quantity to record 2D 1H–15N heteronuclear single quantum coherence (HSQC) spectra. These provide information about NH groups in the protein backbone and on nitrogen-containing aa side chains. A wide peak dispersion indicates a stable tertiary structure because the NMR-active nuclei experience a variety of distinct three-dimensional environments; a narrow peak dispersion is indicative of a lack of stable tertiary structure and typically seen with intrinsically disordered proteins or random-coil peptides [37]. DsbA-released Salp15 showed a modest peak dispersion which was drastically reduced in its Cys-free variant (Fig. 10D; see Fig. S4A for higher magnification). Hence Cys-free Salp15 is largely unstructured and/or highly flexible, whereas the Cys-residues in the wild-type protein contribute to holding at least parts of the protein in a stable conformation. This was confirmed, better visible due to the higher protein concentrations, for Iric-1 versus its Cys-free variant (Fig. 10E; see Fig. S4B for higher magnification). However, 3D structure determination requires, besides 13C/15N double-labeling, higher sample concentrations than were accessible for the free tick proteins.
We therefore generated constructs carrying non-cleavable GB1 as fusion partner. N-terminal GB1 can increase solubility of the fused partner protein, and due to its small size and stable fold does not require removal prior to NMR analysis [32,39]. Under analogous conditions as described above for the DsbA fusions, the GB1-fusion proteins were well expressed in SHexpCP cells. Total expression was again even higher in BL21*CP cells, however at the cost of a lower fraction of soluble protein and an increased content in truncated products (Fig. S5). While even in SHexpCP cells GB1 was a less powerful solubility enhancer than DsbA (about 50% remaining in the insoluble fraction of the lysates; Fig. 2C versus Fig. S5), GB1-Salp15 and GB1-Iric were easily enriched by IMAC (data not shown). Removal of the bulk of truncated products from GB1-Salp15 required SEC (shown in Fig. 11A, left panel, for 15N labeled protein) and some of the protein was lost during subsequent concentration by ultrafiltration. Still, 1H–15N HSQC spectra revealed the same significant peak dispersion for the Salp15 part as in free wild-type Salp15 (Fig. 11A, right panel; red spectrum: free Salp15, blue spectrum: GB1-Salp15; a higher magnification is shown in Fig. S6A) although the spectrum was dominated by peaks from the GB1 part (peak assignment was done using the known chemical shifts of GB1; [40]). For GB1-Iric-1, the lesser content in smaller products after IMAC allowed to replace the SEC step by simple ultrafiltration; a larger contaminant (marked with * in Fig. 11B, left panel) was depleted by using a 50 kDa molecular weight cut-off (MWCO) filter membrane, and the GB1-Iric-1 protein in the flow-through could be concentrated, without substantial losses, by subsequent ultrafiltration using a 10 kDa MWCO membrane (Fig. 11B, left panel). In this way, 1 ml of GB1-Iric-1 at about 1.5 mg/ml was obtained from 250 ml of E. coli culture. 1H–15N HSQC spectra revealed a large number of widely dispersed Iric-1 specific peaks (Fig. 11B, right panel; a higher magnification is shown in Fig. S6B), many of which overlapped with the peaks seen for DsbA released free Iric-1. Hence substantial parts of Iric-1 in the GB1 fusion protein appear to adopt the same stable fold as in the absence of GB1 part.
Fig. 11.
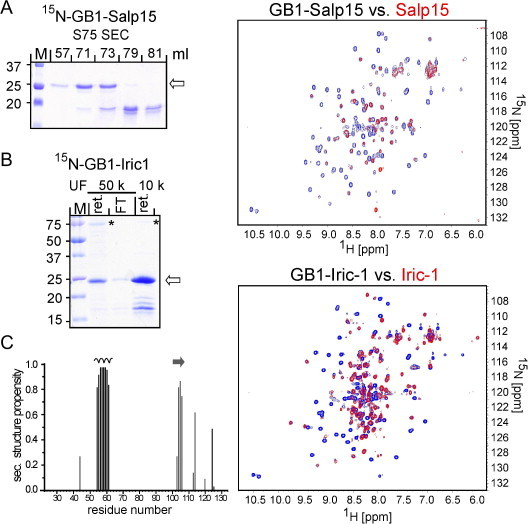
GB1 tick protein fusions enable higher sample concentration and more detailed structural analysis by NMR. GB1-Salp15 and GB1-Iric-1, including in 15N-labeled form, were expressed and enriched by IMAC analogously to the DsbA fusions. (A) Salp15. Like the unlabeled protein, 15N-GB1-Salp15 contained substantial amounts of truncated products, requiring further purification by SEC (left panel). Peak fractions containing the full-length fusion protein were concentrated and used to record 1H–15N spectra (upper right panel; GB1-Salp15 peaks in blue; superimposed red peaks from free Salp15). (B) Iric-1. 15N-GB1-Iric-1 contained fewer truncated products after IMAC, allowing to omit the SEC step. Instead, ultrafiltration (UF) through a 50 kDa MWCO membrane (50 k) depleted a larger contaminant (*; retained, ret.). GB1-Iric-1 in the flow-through (FT) was then concentrated ∼16-fold using a 10 kDa MWCO membrane (10 k) and used to record 1H–15N HSQC spectra (lower right panel). Larger versions of the spectra are shown in Fig. S6. (C) Direct evidence for secondary structure of Iric-1 in the GB1-Iric-1 fusion protein. TALOS-N was used to predict the secondary structure of the assigned regions of Iric-1 from the chemical shift values. The height of the bars indicates the probability to form α-helix (black bars) or β-strand (grey bars). Strip plots of the α-helix and β-strand relevant cross peaks (regions in the full-length sequence context marked with a helix symbol and an arrow) are shown in Fig. S7. The assigned N-terminal region of Iric-1 (N22-L45) does not contain any secondary structure up to residue V43 whereas T44 and L45 might be involved in a β-strand according to TALOS-N. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Using the same protocol, we obtained a first sample ∼0.08 mM sample of uniformly 13C/15N double-labeled GB1-Iric-1. Despite the low protein concentration and considerable broadening of a number of backbone amide resonances standard 3D triple-resonance spectra allowed up to now the backbone assignment of 54 Iric-1 residues in the fusion protein. Evaluation of the assigned chemical shifts using TALOS-N [41] and analysis of 15N-edited 3D NOESY spectra show an unstructured, flexible N-terminus (V21-L45), an α-helix encompassing residues P54QKLALEL61 and a β-strand involving residues V104 to D107, as graphically summarized in Fig. 11C. Pertinent sections of the NOESY spectra for the latter elements are shown in Fig. S7. In addition, the Cβ shifts (39.5 ppm and 42.2 ppm, respectively) of C114 (C4) and C121 (C5) clearly indicate their involvement in disulfide bridges. These data experimentally support sequence-based secondary structure predictions (see Section 3) and are in line with the loss of structure in the Cys-free Iric-1 and Salp15 proteins. Hence for future determination of the complete 3D structure of the tick Salp15 ortholog family GB1-Iric-1 appears as a most promising candidate.
3. Discussion
The current lack of efficient expression systems for tick salivary proteins severely hampers progress in better understanding the molecular interplay between vector, pathogen and host in general, and the development of anti-tick vaccines in particular. While secretory expression in insect cells yields (near) authentic recombinant forms of Salp15 and Iric-1, E. coli based systems provide numerous advantages, including easy handling and access to mutant proteins, potentially in milligram amounts. However, such bacterially expressed tick proteins would only be useful if they mimic important features of their natural, glycosylated counterparts. The results of this study show that fusion of wild-type Salp15 and Iric-1 to the DsbA partner enables generation of preparative amounts of highly soluble, monomeric fusion proteins retaining the ability to bind to a proven biological interaction partner, i.e. Borrelia OspC; that the DsbA fusion proteins, especially in combination with site-directed mutants, are useful for epitope mapping studies; that from slightly modified DsbA fusions free Salp15 and Iric-1 can be generated in amounts approaching those required for structural studies, including in isotope-labeled form; and that the wild-type forms of the recombinant tick proteins are folded to a substantial extent in a cysteine-dependent fashion. While fully establishing the similarities and potential differences to the genuine tick saliva proteins will require further evaluation, altogether our results should greatly facilitate future investigations into the biology of tick saliva proteins and the determination of their 3D structure.
3.1. Impact of the fusion partner and expression strain upon Salp15/Iric-1 solubility in E. coli
All fusion partners tested, including a simple N terminal His6-tag, led to high level expression yet only MBP, both forms of DsbA (Fig. 2, Fig. 10) and, less pronouncedly, GB1 (Fig. S5) mediated substantial solubility. Regarding the expression strain, total fusion protein levels were higher in E. coli BL21*CP than in SHexpCP cells, however, the levels of soluble protein per culture volume of SHexpCP cells were comparable and accompanied by lower levels of degradation products, thus simplifying purification (Fig. 3). This was corroborated by the efficient soluble expression of numerous Salp15 and Iric-1 cysteine (Fig. 4) and truncation variants (Fig. 5), as well as analogous TEV protease cleavable fusion proteins (Fig. 9). Plausibly, the more reducing intracellular environment in the SHexpCP versus BL21*CP cells favors formation of (an) intracellular disulfide bond(s), as is supported by the presence of the protease-released proteins in apparently one dominant form regarding cysteine-status (Fig. 10A); the more stable fold would reduce sensitivity towards proteolysis. Hence for most downstream applications using DsbA Salp15 or Iric fusions in SHexpCP cells appears as the combination of choice. A potential DsbA alternative might be DsbC [42]; in pilot experiments DsbC-Salp15 was well expressed and soluble yet a final statement will require a quantitative comparison with the DsbA fusions. GB1 did not perform quite as well in terms of solubility enhancement (Fig. S5), however for NMR its small size and the concomitant dispensability of further processing provide a distinct advantage.
3.2. MAbs raised against E. coli expressed Salp15 are cross-reactive with Iric-1 and secreted, glycosylated Salp15
As yet no monoclonal antibodies specific for Salp15 or its orthologs have been described. While the solubilized NHis-Salp15 protein used as immunogen turned out to be structurally heterogeneous (Fig. 10B), both mouse mAbs specifically recognized the DsbA-Salp15 and DsbA-Iric-1 fusions protein in denaturing (Western blot; Figs. 4 and 5) and non-denaturing (ELISA; Fig. 7) formats. Epitope mapping, enabled by numerous DsbA-fused variants (Figs. 4 and 5), showed that mAbs 19/7.4 and 18/12.1 recognize closely juxtaposed but distinct sequences (I83-N92, and D88-H99; see summary in Fig. 12). Recognition by mAb 18/12.1, but not mAb 19/7.4, was strictly dependent on the presence of cysteine C3. Because mAb 18/12.1 binding occurred after SDS–PAGE under reducing conditions, it is likely that C3 per se, rather than its involvement in a specific disulfide bond, contributes to the epitope. The epitope region is completely conserved in Iric-1 and, with few chemically similar replacements, in other orthologs (see alignment in Fig. S1) as well as in recent isolates from Chinese ticks [43]. Its potential immuno-dominance in the genuine tick saliva proteins remains to be determined; however, in a PepScan assay [21] overlapping 20-mer peptides representing Salp15 residues 72-91 (there called P6) and 82-101 (P7) were the only ones, in addition to the most C terminal peptides (P10, P11), that were recognized by polyclonal anti-Salp15 antisera, suggesting a strong immunogenicity. Hence mAbs 19/7.4 and 18/12.1 with their distinct binding sites in this region appear as valuable new tools for further basic research into the functions of Salp15 proteins. Furthermore, if the mapped mAb epitopes were protective, their conservation amongst different orthologs suggests that vaccination with a saliva protein from one tick species may cross-protect against other ticks. Most important for potential vaccination approaches, both mAbs also recognized glycosylated Salp15 (Fig. 6), indicating that their epitopes are not shielded by the glycan moieties. Hence pursuing E. coli produced, non-glycosylated Salp15- or Iric-1 as a basis for anti-tick vaccines is a very viable option.
Fig. 12.
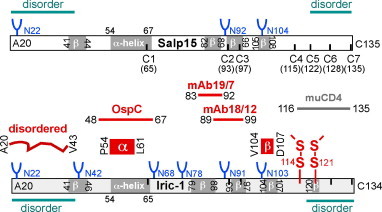
Schematic summary of functional interfaces and structural features in Salp15 and Iric-1. The bars represent, to scale, the primary sequences of signal sequence-less Salp15 and Iric-1. Y-shaped objects symbolize N-glycosylation motifs, upward and downward pointing marks indicate the positions of the seven Cys-residues. Secondary structure motifs (α-helix, β-strand) as predicted by Phyre2 are depicted inside the bars, disorder predictions above and below the bars. Interaction sites defined in this study (applying to both Salp15 and Iric-1), and secondary structure elements defined by NMR of GB1-Iric-1 are shown in red; binding to murine CD4 has previously been mapped to the C terminal 20 aa of Salp15 [44]. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.3. A charged α-helix containing region within Salp15 and Iric-1 mediates interaction with OspC variants from different borreliae
B. burgdorferi transmission enhancement by Salp15 is thought to rely at least in part on its recruitment to OspC [21], a rather variable protein on the spirochete surface. OspC binding has also been shown for insect-cell derived Iric-1 [33]. In vitro affinities to OspC variants from B. burgdorferi, B. garinii and B. afzelii were all in the low micromolar range, although protection against antibody-mediated killing of the bacteria was observed only for B. burgdorferi sensu stricto. Our immobilized OspC-binding ELISA using the E. coli-derived DsbA fused wild-type Salp15 and Iric-1 gave largely congruent results (Fig. 7; Fig. S2), except that the two B. burgdorferi OspCs reacted more strongly than their B. afzelii and B. garinii counterparts. The high reactivity of Cys-free Salp15, but not Iric-1, with the latter two OspCs may relate to better accessibility of the OspC binding region (see below) due to the lack of global structure (see below) and the primary sequence differences between Salp15 and Iric-1 in this region. Clearly, however, OspC binding required neither cysteines nor a complex 3D structure context because specific binding was recapitulated by GFP fusions carrying just residues 48–67 of either Salp15 or Iric-1 (Fig. 8). This agrees well with recent PepScan data according to which peptide P4 (residues 52-71 in our numbering system) is involved in OspC binding [21]; however, we found no evidence for the proposed contribution of C terminal residues, inferred from positive signals with peptide P11 (aa 116-135) which previously was reported to mediate CD4 binding [44]. Notably, within the sequence 48–67 residues 54–64 of both Salp15 and Iric-1 are predicted, e.g. by Phyre2 [45], to form an α-helix (the only one in the whole protein; see Fig. 12) which for residues 54–61 in GB1-Iric-1 is directly confirmed by our NMR data (see below). Remarkably, the basic residues K60 and K63 in Salp15 are both replaced by acidic residues in Iric-1 (E60, E63) yet both proteins interact equally well with OspC. A plausible explanation comes from the structure of OspC. OspC forms a mushroom-like symmetrical dimer [46,47], and a cavity in the dimer-interface in the stalk comprising a central patch of charged residues (K60, E61, E63; plus the homologous residues from the second subunit) lined by hydrophobic aa is proposed to act as ligand-binding domain [48]. As the KEXE motif is conserved in virtually all known OspC isolates, using slightly different electrostatic and hydrophobic interactions Salp15 and Iric-1 could still bind to the same target region on different OspC orthologs. The easy access to mutants provided by our E. coli expression system lends itself to further define the sequence and structure requirements for this interaction. Given the importance of Salp15/Iric-1 residues 48–67 for OspC binding this region also represents a prime target for neutralizing antibodies. Hence it will be important to evaluate polyclonal antisera against Salp15 and Iric-1 for their ability to recognize this region; if confirmed, the α-helix would also lend itself for epitope-focused vaccines in which a structured epitope is stabilized by its presentation on a matching protein scaffold [23].
3.4. Towards the 3D structure of Salp15 family proteins
Understanding the molecular interactions of the tick saliva proteins with the host as well as with tick-vectored pathogens will eventually require detailed structural knowledge. Our E. coli based system for soluble expression of free Salp15/Iric-1 by post-expression proteolytic cleavage and as fusions to the small NMR compatible GB1 tag represent the first approach with good prospects for such direct structural studies. The biological relevance of these proteins as models for the genuine tick saliva proteins is supported, though not proven, by their ability to induce mAbs that recognize glycosylated Salp15, and to specifically interact with various OspC proteins. Practically, our data demonstrate the suitability of the expression and purification protocols to obtain 15N and 13C/15N labeled samples for NMR analyses. The 2D spectra of the protease-released tick proteins (Fig. 10D, E; Fig. S4A, B), limited in sensitivity by the low solubility of free wild-type Iric-1 and particularly Salp15, still revealed a negative impact on general structural stability by replacement of the endogenous cysteines. This is not unexpected given that Cys-containing secretory proteins commonly contain disulfide bonds. Hence the better soluble Cys-free variants seem not suitable for structural work. However, the enhanced solubility of the GB1 fusions with the wild-type proteins and the preliminary NMR data obtained with the 13C/15N double-labeled GB1-Iric-1 appear most promising.
Primary sequence-based structure predictions have become powerful tools, especially if they involve template-based homology modeling such as Phyre2 [45] or Porter+[49]. Both predict for Salp15 and Iric-1 an α-helix from residues 54–66, and some regions with β-strand propensity (Fig. 12); Phyre2 also implies disorder for the N terminal region up to position 40, and the C terminal region downstream of position 117. However, the lack of homologs of known structure substantially limits reliability of the predictions. Notably, our preliminary NMR data for GB1-Iric-1 indicate indeed an α-helix for residues P54-L61 (Fig. S7), lending credit to the predictions. Interestingly, the charged residues K56 and E60 (plus E63 if the helix was slightly longer) are on one face, neighbored by hydrophobic residues, and the same arrangement of charged versus hydrophobic residues would hold for Salp15. Its relevance for the interaction with OspC (and possibly other, host-encoded proteins) might now easily be tested by mutations. The NMR data also reveal a β-strand involving at least V104-D107 (Fig. S7), plus the participation in disulfide bridges of C114 and C121, possibly between these two residues; neither of the above prediction programs provides information on disulfide bridges. While as yet giving only a glimpse into the structural features of Iric-1, solving the complete 3D structure of Salp15 family tick proteins appears now in reach. This will enable the rational design of vaccines that overcome their immunosuppressive properties and induce neutralizing antibodies. Our study also implies E. coli as a suitable expression host for manufacturing such vaccines in the required amounts.
4. Materials and methods
4.1. Prokaryotic expression vectors
pET28-derived vectors encoding N terminally His6-tagged DsbA, GB1, GST, MBP, or Trx fused to eGFP were kindly provided by Jerome Basquin (EMBL, Heidelberg, Germany). Synthetic, E. coli codon usage-optimized genes for Salp15 (Genbank: AAK97817.1) and Iric-1 (Genbank: ABU93613.1) comprising terminal Nco I and Hind III sites (obtained from Entelechon and GeneArt, respectively) were then used to replace the eGFP ORF flanked by the same restriction sites. Most variants were constructed using conventional mutagenic PCR. Vector pET28-NHis-GFP was obtained from the pET28-GB1-eGFP vector via deletion of the GB1-internal Hind III–Nco I fragment so as to leave only the His6-tag plus five GB1N terminal residues upstream of eGFP. GFP vectors encoding C terminal fusions with the Salp15 segment P48-D67 and the Iric-1 segment P48-N67 were obtained by cloning appropriate synthetic oligonucleotide duplexes between the 3′ proximal BsrG I site in the eGFP ORF and the Kpn I site immediately following the eGFP stop codon. The construct for NH6-Salp15 was obtained by inserting a leader-less PCR-amplified Salp15 cDNA into the BamH I site of pQE-30 Xa (Qiagen), such that the mature Salp15 sequence starting with V19 was preceded by the aa sequence MRGSHHHHHHGSGSGSGIEGRPYNGTGS; the factor Xa recognition sequence is underlined. For expression of the B. afzelii NE [34] OspC variants A3 (Genbank: AFA85556.1) and YU (Genbank: AIK19145.1), leader-less versions of the corresponding cDNAs were similarly cloned into pQE-30 Xa. pGEX-2T vectors (GE Healthcare) encoding GST-tagged leader-less B. burgdorferi ZS7 OspCa (Genbank: AAM19715.1) and OspCb (containing the following exchanges compared to OspCa: D87G, N88D, R92H, A133E, T135S, D136T, N140D, E141N, S144Q, K147I, K148Q, D149G, A150V, D153E, D154E) were previously described [50]. All constructs were verified by DNA sequencing.
4.2. Recombinant tick protein and OspC expression in E. coli
The respective Salp15 and Iric-1 expression vectors were transformed into E. coli BL21*CP (Stratagene) or T7 SHuffle express (NEB) cells carrying the CP rare tRNA plasmid. Expression was induced by addition of IPTG to a final concentration of 1 mM, and the cultures (usually 250 ml Luria broth containing appropriate antibiotics) were shaken in baffled flasks for 16 h at 22 °C. Cleared lysates were obtained via treatment, in the presence of protease inhibitor cocktail without EDTA (Roche), with Triton X-100 and lysozyme plus benzonase, sonication and removal of insoluble material by centrifugation as previously described [51]; in addition, the supernatants were passed through 0.22 μm sterile filters before further work-up. OspCa and OspCb were expressed as GST fusions in E. coli JM109 cells via induction with 1 mM IPTG for 3 h at 37 °C and affinity purification on glutathione sepharose 4B (GE Healthcare), followed by release from GST via factor Xa cleavage as described [52]. OspC A3 and OspC YU were expressed in E. coli B834(DE3) cells (Novagen) without IPTG addition, and purified by manual Ni2+ IMAC on Ni-NTA agarose beads (Qiagen).
4.3. Purification of Salp15 and Iric-1 fusion proteins from E. coli
For Ni2+-IMAC cleared bacterial lysates from 250 ml cultures were passed through a 1 ml Ni-NTA Superflow cartridge (Qiagen) mounted on an ÄKTA FPLC system (GE Healthcare, Unicorn software) and equilibrated in TN 150 buffer (25 mM Tris/HCl, 150 mM NaCl, pH 7.5) supplemented with 20 mM imidazole, followed by application of a linear gradient from 20 mM to 640 mM imidazole. His6-tagged proteins usually eluted between 100 mM to 300 mM imidazole and were collected in 1 ml fractions. Where appropriate, the peak fractions (usually around 4 ml) were subjected to SEC on a Superdex 75 16/60 column (GE Healthcare) equilibrated in TN150 buffer. After elution of 0.3 column volumes, 2 ml fractions were collected and analyzed by SDS–PAGE. Apparent molecular masses were determined from the elution volumes of a set of marker proteins of known sizes (BioRad gel filtration standard). If desired, proteins were concentrated via ultrafiltration through Amicon Ultra centrifugal filters (EMD Millipore) with appropriate molecular weight cut offs.
4.4. TEV protease-mediated release of Salp15 and Iric-1 from DsbA-TEV fusion proteins
The S129V variant of His6-tagged TEV protease was expressed from plasmid pRK793 plasmid essentially as described [53]. After Ni2+-IMAC, the protein was dialysed against TN50 containing 5 mM DTT plus 2 mM EDTA and concentrated using a 10 MWCO Amicon centrifugal filter to 1.5 mg/ml. Stock solutions containing 10% glycerol were stored at −20 °C. Digestions were usually performed at 4 °C over night or 4 h at 16 °C using a final concentration of 25 μg/ml TEV protease and a 50-fold to 100-fold mass excess of DsbA-TEV fusion protein substrate. Released Salp15 was obtained by removal of His-tagged uncleaved fusion protein, released DsbA and TEV protease by collecting the flow-through from a Ni-NTA superflow column as described above. For released Iric-1, the undesired His-tagged components were removed using Ni-NTA agarose in a batch procedure.
4.5. Solubilization of NH6-Salp15 from inclusion bodies
Cells from a 2 L culture of induced E. coli JM109 cells transformed with the pQE-30 Xa-NHis-Salp vector were incubated on ice in 50 mL BugBuster reagent (Novagen) for 30 min, followed by centrifugation at 20,000g. The insoluble pellet containing the bulk of Salp15 was washed twice in 50 mL MT-PBS buffer (150 mM NaCl, 16 mM Na2HPO4, 4 mM NaH2PO4; pH 7.4) and dissolved in 10 ml TPI-5 buffer (10 mM Tris/HCl, 100 mM NaH2PO4, 5 mM imidazole, pH 8.0) containing 8 M urea. Insoluble material was removed by centrifugation (20,000g, 10 min, 23 °C) and the supernatant was passed through a 5 ml (gel bed) Ni2+-NTA Sepharose (Qiagen) column. The column was washed with 2 gel bed volumes of 8 M urea in TPI-20 buffer (like TPI-5 but containing 20 mM imidazole) and bound proteins were eluted with 2 gel bed volumes of 8 M urea in TPI-500 buffer (as TPI-5 but containing 500 mM imidazole). The eluate was transferred into a dialysis tubing (6–8 kDa MWCO; Spectrapor) and initially dialysed against 200 ml of 8 M urea in MT-PBS buffer. The urea concentration was gradually reduced to ∼0.3 M by dropwise addition (4 ml/min) of a total of 5 L MT-PBS; thereafter dialysis was continued against MT-PBS without urea for 4 h. Insoluble material was removed by centrifugation at 10,000g for 10 min. Soluble protein in the supernatant was concentrated by ultrafiltration.
4.6. 15N and 13C/15N labeling
For labeling with NMR-active isotopes, DsbA-TEV Salp15 and Iric-1 proteins were expressed as described above except that 2 h prior to induction with IPTG, precultures grown in LB medium were transferred into minimal M9 medium [54] containing 15NH4Cl (Sigma), or 15NH4Cl plus 13C glucose (Deutero) as the only nitrogen and carbon sources, respectively.
4.7. Eukaryotic secretory Salp15 expression
The hGFP coding sequence in the CMV-IE promoter driven vector pTR-UF5 [55] was replaced by coding sequences for C terminally His6-tagged Salp15 precursor, ie. containing the authentic Salp15 signal sequence (pTRUF-SalpSig-Salp15H6), the same containing in addition a GH6 sequence between Salp15 aa 21 and aa 22 (pTRUF-SalpSigNH6-Salp15H6), or the mature Salp15 sequence aa 20-135 preceded by the E. coli OmpA signal peptide MKKTAIAIAVALAGFATVA plus an extra Met residue (pTRUF-OmpA-Salp15H6). The vectors were transfected into HEK293 cells grown in 10 cm diameter dishes in DMEM (PAA Laboratories) supplemented with 10% fetal calf serum, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37 °C and 5% CO2 using Mirus Transit LT1 reagent as recommended by the manufacturer. Cell culture supernatants were harvested on day 4 post transfection. For ELISA-based assays, 200 μl supernatant per well from the transfected cells and, for control, from cells transfected with a GFP vector or non-transfected cells, were incubated on Ni-NTA HisSorb (Qiagen) plates as described below.
For immunoblotting, 5 ml of the respective supernatants were adsorbed to 100 μl (gel-bed) Ni-NTA agarose beads (Qiagen), and washed 3× with 1 ml of TN150 buffer. Then 20 μl each of the beads were either directly boiled with 30 μl of SDS sample, or treated with PNGase F (NEB) as recommended by the manufacturer. After washing, the beads were likewise analyzed by SDS–PAGE and immuno-blotting.
4.8. Generation of anti-Salp15 mAbs
MAbs 18/12.1 and 19/7.4 were generated by immunization of Balb/c mice with solubilized NHis-Salp15 protein in monophosphoryl-lipid A (MPL) adjuvant. Spleen cell fusion with myeloma cells and hybridoma selection were performed as previously described [35]. Salp15 specific hybridomas were identified via immunoblotting for recognition of the immunization antigen. All animal research was approved in advance by the Laboratory Animal Committee of the University of Heidelberg (RP Karlsruhe 35-9185.82/A-43/10) and performed in accordance with the European Communities Council Directive of 24 November, 1986 (86/609/EEC).
4.9. Immuno-blotting and ELISA
For immunoblotting, proteins were separated by SDS–PAGE, electro-blotted to polyvinylidenfluoride (PVDF) membranes (Millipore) and probed as previously described [51]. For detection of His-tagged proteins, a Tetra-His mouse mAb (Qiagen) was used; Salp15 proteins were detected using mAbs 19/7.4 or 18/12.1. Bound mAbs were visualized using peroxidase- (PO-) conjugated anti-mouse antibody (Dianova) and ECL reagent (GE Healthcare), followed by exposure to X-ray film (BioMax MR, Kodak).
For ELISA, the desired proteins were immobilized on 96 well NUNC Maxisorp microtiter plates in coating buffer (50 mM Na2CO3, pH 9.6). Plates were washed three times in PBST buffer (phosphate buffer saline containing 0.05 % Tween-20) and blocked for 1 h at RT in blocking buffer (PBS containing 1% BSA). All additional incubations were performed in binding buffer (PBS containing 0.1% BSA) for 1 h at RT. For detection of secreted His-tagged Salp15 derivatives, pre-blocked Ni-NTA HisSorb plates (Qiagen) plates were employed as recommended by the manufacturer. Bound antibodies were detected using appropriate secondary antibody-PO conjugates (Dianova) with tetramethylbenzidine (TMB; Sigma) as substrate. Absorbance at 450 nm (A450nm) was monitored using a Genios Pro plate reader (Tecan). All assays were performed in quadruplicate. Mean values and standard error of the mean (SEM) were calculated using Graphpad Prism.
4.10. NMR spectroscopy
All NMR spectra were collected at 298 or 304 K on a Bruker Avance 600 MHz instrument equipped with a 5 mm TXI cryoprobe. 1H, 15N, and 13C chemical shift assignments of GB1-Iric-1 in 100 mM sodium phosphate (pH 6) and 10% D2O (v/v) were obtained using standard strategies based on 1H–15N HSQC spectra and the following 3D experiments: HNCA, HN(CO)CA, CBCA(CO)NH, CBCANH, HNCO, HN(CA)CO, HBHA(CO)NH and NOESY-1H–15N-HSQC (100 ms mixing time). All chemical shifts were referenced to internal 2,2-dimethyl-2-silapentane-5-sulfonic acid (DSS). NMR data were processed with Bruker’s Topspin 2.1 software and analyzed with CARA [56]. The TALOS-N [41] web server (http://spin.niddk.nih.gov/bax/nmrserver/talosn/) was used for chemical shift based identification of secondary structure elements.
Author contributions
P.K., D.B., R.W. and M.N. designed research; P.K., D.B., J.V., J.H., and M.N. performed research; P.K., D.B., R.W. and M.N. analyzed data; and P.K., D.B., R.W. and M.N. wrote the paper.
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft through grants NA 154/11-1 to M.N. and WA 533/8-1 to R.W.
D.B. acknowledges continued support by Dr. B. Fakler (University of Freiburg).
Appendix A. Supplementary data
Supplementary Figures.
References
- 1.Brossard M., Wikel S.K. Tick immunobiology. Parasitology. 2004;129(Suppl.):S161–S176. doi: 10.1017/s0031182004004834. [DOI] [PubMed] [Google Scholar]
- 2.Charrel R.N., Attoui H., Butenko A.M., Clegg J.C., Deubel V., Frolova T.V., Gould E.A., Gritsun T.S., Heinz F.X., Labuda M., Lashkevich V.A., Loktev V., Lundkvist A., Lvov D.V., Mandl C.W., Niedrig M., Papa A., Petrov V.S., Plyusnin A., Randolph S., Suss J., Zlobin V.I., de Lamballerie X. Tick-borne virus diseases of human interest in Europe. Clin. Microbiol. Infect. 2004;10:1040–1055. doi: 10.1111/j.1469-0691.2004.01022.x. [DOI] [PubMed] [Google Scholar]
- 3.Labuda M., Nuttall P.A. Tick-borne viruses. Parasitology. 2004;129(Suppl.):S221–S245. doi: 10.1017/s0031182004005220. [DOI] [PubMed] [Google Scholar]
- 4.Perronne C. Lyme and associated tick-borne diseases: global challenges in the context of a public health threat. Front. Cell. Infect. Microbiol. 2014;4:74. doi: 10.3389/fcimb.2014.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stanek G., Wormser G.P., Gray J., Strle F. Lyme borreliosis. Lancet. 2012;379:461–473. doi: 10.1016/S0140-6736(11)60103-7. [DOI] [PubMed] [Google Scholar]
- 6.Radolf J.D., Caimano M.J., Stevenson B., Hu L.T. Of ticks, mice and men: understanding the dual-host lifestyle of Lyme disease spirochaetes. Nat. Rev. Microbiol. 2012;10:87–99. doi: 10.1038/nrmicro2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stibraniova I., Lahova M., Bartikova P. Immunomodulators in tick saliva and their benefits. Acta Virol. 2013;57:200–216. doi: 10.4149/av_2013_02_200. [DOI] [PubMed] [Google Scholar]
- 8.Anguita J., Ramamoorthi N., Hovius J.W., Das S., Thomas V., Persinski R., Conze D., Askenase P.W., Rincon M., Kantor F.S., Fikrig E. Salp15, an ixodes scapularis salivary protein, inhibits CD4(+) T cell activation. Immunity. 2002;16:849–859. doi: 10.1016/s1074-7613(02)00325-4. [DOI] [PubMed] [Google Scholar]
- 9.Juncadella I.J., Anguita J. The immunosuppresive tick salivary protein, Salp15. Adv. Exp. Med. Biol. 2009;666:121–131. doi: 10.1007/978-1-4419-1601-3_10. [DOI] [PubMed] [Google Scholar]
- 10.Juncadella I.J., Garg R., Ananthnarayanan S.K., Yengo C.M., Anguita J. T-cell signaling pathways inhibited by the tick saliva immunosuppressor, Salp15. FEMS Immunol. Med. Microbiol. 2007;49:433–438. doi: 10.1111/j.1574-695X.2007.00223.x. [DOI] [PubMed] [Google Scholar]
- 11.Mason L.M., Veerman C.C., Geijtenbeek T.B., Hovius J.W. Menage a trois: Borrelia, dendritic cells, and tick saliva interactions. Trends Parasitol. 2014;30:95–103. doi: 10.1016/j.pt.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 12.Hovius J.W., Ramamoorthi N., Van’t Veer C., de Groot K.A., Nijhof A.M., Jongejan F., van Dam A.P., Fikrig E. Identification of Salp15 homologues in Ixodes ricinus ticks. Vector Borne Zoonotic Dis. 2007;7:296–303. doi: 10.1089/vbz.2006.0624. [DOI] [PubMed] [Google Scholar]
- 13.Nuttall P.A., Labuda M. Tick-host interactions: saliva-activated transmission. Parasitology. 2004;129(Suppl.):S177–S189. doi: 10.1017/s0031182004005633. [DOI] [PubMed] [Google Scholar]
- 14.Wikel S. Ticks and tick-borne pathogens at the cutaneous interface. host defenses, tick countermeasures, and a suitable environment for pathogen establishment. Front. Microbiol. 2013;4:337. doi: 10.3389/fmicb.2013.00337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu X.Y., Bonnet S.I. Hard tick factors implicated in pathogen transmission. PLoS Negl. Trop. Dis. 2014;8:e2566. doi: 10.1371/journal.pntd.0002566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramamoorthi N., Narasimhan S., Pal U., Bao F., Yang X.F., Fish D., Anguita J., Norgard M.V., Kantor F.S., Anderson J.F., Koski R.A., Fikrig E. The Lyme disease agent exploits a tick protein to infect the mammalian host. Nature. 2005;436:573–577. doi: 10.1038/nature03812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schuijt T.J., Hovius J.W., van Burgel N.D., Ramamoorthi N., Fikrig E., van Dam A.P. The tick salivary protein Salp15 inhibits the killing of serum-sensitive Borrelia burgdorferi sensu lato isolates. Infect. Immun. 2008;76:2888–2894. doi: 10.1128/IAI.00232-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schuijt T.J., Hovius J.W., van der Poll T., van Dam A.P., Fikrig E. Lyme borreliosis vaccination: the facts, the challenge, the future. Trends Parasitol. 2011;27:40–47. doi: 10.1016/j.pt.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 19.Embers M.E., Narasimhan S. Vaccination against Lyme disease: past, present, and future. Front. Cell. Infect. Microbiol. 2013;3:6. doi: 10.3389/fcimb.2013.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stricker R.B., Johnson L. Lyme disease: call for a “Manhattan Project” to combat the epidemic. PLoS Pathog. 2014;10:e1003796. doi: 10.1371/journal.ppat.1003796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dai J., Wang P., Adusumilli S., Booth C.J., Narasimhan S., Anguita J., Fikrig E. Antibodies against a tick protein, Salp15, protect mice from the Lyme disease agent. Cell Host Microbe. 2009;6:482–492. doi: 10.1016/j.chom.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walker A., Skamel C., Nassal M. SplitCore: an exceptionally versatile viral nanoparticle for native whole protein display regardless of 3D structure. Sci. Rep. 2011;1:5. doi: 10.1038/srep00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Correia B.E., Bates J.T., Loomis R.J., Baneyx G., Carrico C., Jardine J.G., Rupert P., Correnti C., Kalyuzhniy O., Vittal V., Connell M.J., Stevens E., Schroeter A., Chen M., Macpherson S., Serra A.M., Adachi Y., Holmes M.A., Li Y., Klevit R.E., Graham B.S., Wyatt R.T., Baker D., Strong R.K., Crowe J.E., Jr., Johnson P.R., Schief W.R. Proof of principle for epitope-focused vaccine design. Nature. 2014;507:201–206. doi: 10.1038/nature12966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Marco A. Strategies for successful recombinant expression of disulfide bond-dependent proteins in Escherichia coli. Microb. Cell Factors. 2009;8:26. doi: 10.1186/1475-2859-8-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lebendiker M., Danieli T. Production of prone-to-aggregate proteins. FEBS Lett. 2014;588:236–246. doi: 10.1016/j.febslet.2013.10.044. [DOI] [PubMed] [Google Scholar]
- 26.Lobstein J., Emrich C.A., Jeans C., Faulkner M., Riggs P., Berkmen M. SHuffle, a novel Escherichia coli protein expression strain capable of correctly folding disulfide bonded proteins in its cytoplasm. Microb. Cell Factors. 2012;11:56. doi: 10.1186/1475-2859-11-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Waugh D.S. Making the most of affinity tags. Trends Biotechnol. 2005;23:316–320. doi: 10.1016/j.tibtech.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 28.Young C.L., Britton Z.T., Robinson A.S. Recombinant protein expression and purification: a comprehensive review of affinity tags and microbial applications. Biotechnol. J. 2012;7:620–634. doi: 10.1002/biot.201100155. [DOI] [PubMed] [Google Scholar]
- 29.Ito K., Inaba K. The disulfide bond formation (Dsb) system. Curr. Opin. Struct. Biol. 2008;18:450–458. doi: 10.1016/j.sbi.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 30.Zapun A., Cooper L., Creighton T.E. Replacement of the active-site cysteine residues of DsbA, a protein required for disulfide bond formation in vivo. Biochemistry. 1994;33:1907–1914. doi: 10.1021/bi00173a038. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Y., Olsen D.R., Nguyen K.B., Olson P.S., Rhodes E.T., Mascarenhas D. Expression of eukaryotic proteins in soluble form in Escherichia coli. Protein Expr. Purif. 1998;12:159–165. doi: 10.1006/prep.1997.0834. [DOI] [PubMed] [Google Scholar]
- 32.Zhou P., Lugovskoy A.A., Wagner G. A solubility-enhancement tag (SET) for NMR studies of poorly behaving proteins. J. Biomol. NMR. 2001;20:11–14. doi: 10.1023/a:1011258906244. [DOI] [PubMed] [Google Scholar]
- 33.Hovius J.W., Schuijt T.J., de Groot K.A., Roelofs J.J., Oei G.A., Marquart J.A., de Beer R., van‘t Veer C., van der Poll T., Ramamoorthi N., Fikrig E., van Dam A.P. Preferential protection of Borrelia burgdorferi sensu stricto by a Salp15 homologue in Ixodes ricinus saliva. J. Infect. Dis. 2008;198:1189–1197. doi: 10.1086/591917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perez D., Kneubuhler Y., Rais O., Jouda F., Gern L. Borrelia afzelii ospC genotype diversity in Ixodes ricinus questing ticks and ticks from rodents in two Lyme borreliosis endemic areas: contribution of co-feeding ticks. Ticks Tick Borne Dis. 2011;2:137–142. doi: 10.1016/j.ttbdis.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 35.Kramer M.D., Schaible U.E., Wallich R., Moter S.E., Petzoldt D., Simon M.M. Characterization of Borrelia burgdorferi associated antigens by monoclonal antibodies. Immunobiology. 1990;181:357–366. doi: 10.1016/S0171-2985(11)80504-8. [DOI] [PubMed] [Google Scholar]
- 36.Shin J., Lee W. Structural proteomics by NMR spectroscopy. Expert Rev. Proteomics. 2008;5:589–601. doi: 10.1586/14789450.5.4.589. [DOI] [PubMed] [Google Scholar]
- 37.Kwan A.H., Mobli M., Gooley P.R., King G.F., Mackay J.P. Macromolecular NMR spectroscopy for the non-spectroscopist. FEBS J. 2011;278:687–703. doi: 10.1111/j.1742-4658.2011.08004.x. [DOI] [PubMed] [Google Scholar]
- 38.Rath A., Glibowicka M., Nadeau V.G., Chen G., Deber C.M. Detergent binding explains anomalous SDS–PAGE migration of membrane proteins. Proc. Natl. Acad. Sci. USA. 2009;106:1760–1765. doi: 10.1073/pnas.0813167106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou P., Wagner G. Overcoming the solubility limit with solubility-enhancement tags: successful applications in biomolecular NMR studies. J. Biomol. NMR. 2010;46:23–31. doi: 10.1007/s10858-009-9371-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gronenborn A.M., Filpula D.R., Essig N.Z., Achari A., Whitlow M., Wingfield P.T., Clore G.M. A novel, highly stable fold of the immunoglobulin binding domain of streptococcal protein G. Science. 1991;253:657–661. doi: 10.1126/science.1871600. [DOI] [PubMed] [Google Scholar]
- 41.Shen Y., Bax A. Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR. 2013;56:227–241. doi: 10.1007/s10858-013-9741-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nozach H., Fruchart-Gaillard C., Fenaille F., Beau F., Ramos O.H., Douzi B., Saez N.J., Moutiez M., Servent D., Gondry M., Thai R., Cuniasse P., Vincentelli R., Dive V. High throughput screening identifies disulfide isomerase DsbC as a very efficient partner for recombinant expression of small disulfide-rich proteins in E. coli. Microb. Cell Factors. 2013;12:37. doi: 10.1186/1475-2859-12-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang X., Huang Y., Niu S.B., Jiang B.G., Jia N., van der Geest L., Ni X.B., Sun Y., Cao W.C. Genetic diversity of Salp15 in the Ixodes ricinus complex (Acari: Ixodidae) PLoS ONE. 2014;9:e94131. doi: 10.1371/journal.pone.0094131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garg R., Juncadella I.J., Ramamoorthi N., Ashish Ananthanarayanan.S.K., Thomas V., Rincon M., Krueger J.K., Fikrig E., Yengo C.M., Anguita J. Cutting edge: CD4 is the receptor for the tick saliva immunosuppressor, Salp15. J. Immunol. 2006;177:6579–6583. doi: 10.4049/jimmunol.177.10.6579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kelley L.A., Sternberg M.J. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 46.Kumaran D., Eswaramoorthy S., Luft B.J., Koide S., Dunn J.J., Lawson C.L., Swaminathan S. Crystal structure of outer surface protein C (OspC) from the Lyme disease spirochete, Borrelia burgdorferi. EMBO J. 2001;20:971–978. doi: 10.1093/emboj/20.5.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eicken C., Sharma V., Klabunde T., Owens R.T., Pikas D.S., Höök M., Sacchettini J.C. Crystal structure of Lyme disease antigen outer surface protein C from Borrelia burgdorferi. J. Biol. Chem. 2001;276:10010–10015. doi: 10.1074/jbc.M010062200. [DOI] [PubMed] [Google Scholar]
- 48.Earnhart C.G., Leblanc D.V., Alix K.E., Desrosiers D.C., Radolf J.D., Marconi R.T. Identification of residues within ligand-binding domain 1 (LBD1) of the Borrelia burgdorferi OspC protein required for function in the mammalian environment. Mol. Microbiol. 2010;76:393–408. doi: 10.1111/j.1365-2958.2010.07103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mooney C., Vullo A., Pollastri G. Protein structural motif prediction in multidimensional phi-psi space leads to improved secondary structure prediction. J. Comput. Biol. 2006;13:1489–1502. doi: 10.1089/cmb.2006.13.1489. [DOI] [PubMed] [Google Scholar]
- 50.Skamel C., Ploss M., Böttcher B., Stehle T., Wallich R., Simon M.M., Nassal M. Hepatitis B virus capsid-like particles can display the complete, dimeric outer surface protein C and stimulate production of protective antibody responses against Borrelia burgdorferi infection. J. Biol. Chem. 2006;281:17474–17481. doi: 10.1074/jbc.M513571200. [DOI] [PubMed] [Google Scholar]
- 51.Vogel M., Vorreiter J., Nassal M. Quaternary structure is critical for protein display on capsid-like particles (CLPs): efficient generation of hepatitis B virus CLPs presenting monomeric but not dimeric and tetrameric fluorescent proteins. Proteins. 2005;58:478–488. doi: 10.1002/prot.20312. [DOI] [PubMed] [Google Scholar]
- 52.Rossmann E., Kitiratschky V., Hofmann H., Kraiczy P., Simon M.M., Wallich R. Borrelia burgdorferi complement regulator-acquiring surface protein 1 of the Lyme disease spirochetes is expressed in humans and induces antibody responses restricted to nondenatured structural determinants. Infect. Immun. 2006;74:7024–7028. doi: 10.1128/IAI.01028-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kapust R.B., Tozser J., Fox J.D., Anderson D.E., Cherry S., Copeland T.D., Waugh D.S. Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 2001;14:993–1000. doi: 10.1093/protein/14.12.993. [DOI] [PubMed] [Google Scholar]
- 54.Sambrook J., Fritsch E.F., Maniatis T. second ed. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. Molecular Cloning - A Laboratory Manual. [Google Scholar]
- 55.Zolotukhin S., Potter M., Hauswirth W.W., Guy J., Muzyczka N. A “humanized” green fluorescent protein cDNA adapted for high-level expression in mammalian cells. J. Virol. 1996;70:4646–4654. doi: 10.1128/jvi.70.7.4646-4654.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Keller R. CANTINA Verlag; Goldau: 2004. The Computer Aided Resonance Assignment Tutorial. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures.