

Abstract
AIM: To investigate the functions of promoter hypermethylation of O6-methylguanine-DNA methyltransferase (MGMT) gene in colorectal tumorigenesis and progression.
METHODS: The promoter hypermethylation of MGMT gene was detected in 27 sporadic colorectal adenomas, 62 sporadic colorectal carcinomas and 20 normal colorectal mucosa tissues by methylation-specific PCR. At the same time, the expression of MGMT protein was carried out in the same samples using immunohistochemistry. Mutant-allele-specific amplification was used to detect K-ras G to A point mutation in codon 12.
RESULTS: None of the normal colorectal mucosa tissues showed methylated bands. Promoter hypermethylation was detected in 40.7% (11 of 27) of adenomas and 43.5% (27 of 62) of carcinomas. MGMT proteins were expressed in nucleus and cytoplasm of normal colorectal mucosa tissues. Loss of MGMT expression was found in 22.2% (6 of 27) of adenomas and 45.2% (28 of 62) of carcinomas. The difference between them was significant (P = 0.041). In the 6 adenomas and 28 carcinomas losing MGMT expression, 5 and 24 cases presented methylation, respectively (P = 0.027, P<0.001). Thirteen of the 19 colorectal tumors with K-ras G to A point mutation in codon 12 had methylated MGMT (P = 0.011). The frequencies of K-ras G to A point mutation were 35.3% (12 of 34) and 12.7% (7 of 55) in tumors losing MGMT expression and with normal expression, respectively.
CONCLUSION: Promoter hypermethylation and loss of expression of MGMT gene were common events in colorectal tumorigenesis, and loss of expression of MGMT occurs more frequently in carcinomas than in adenomas in sporadic patients. Hypermethylation of the CpG island of MGMT gene was associated with loss of MGMT expression and K-ras G to A point mutation in colorectal tumor. The frequency of K-ras G to A point mutation was increased in tumors losing MGMT expression. It suggests that epigenetic inactivation of MGMT plays an important role in colorectal neoplasia.
Keywords: O6-methylguanine-DNA methyltransferase, CpG island, DNA methylation, Epigenetic change, K-ras mutation
INTRODUCTION
O6-methylguanine-DNA methyltransferase (MGMT) is a ubiquitous DNA repair protein that removes mutagenic and cytotoxic adducts from the O6-guanine in DNA, the preferred point of attack of many carcinogens and alkylating chemotherapeutic agents[1,2]. Alkylating of DNA at the O6 position of guanine is an important step in the formation of mutations in cancer, primarily due to the tendency of the O6-methylguanine to pair with thymine during replication, resulting in a conversion of guanine-cytosine to adenine-thymine pairs in DNA[3]. Furthermore, the O6-alkylguanine-DNA adduct may cross-link with the opposite cytosine residues, blocking DNA replication[4-6]. MGMT expression is decreased in some tumor tissues, and lack of activity has been observed in some cell lines[7,8]. Loss of expression is rarely due to genetic changes of the MGMT gene, but methylation of discrete regions of the CpG island of MGMT has been associated with the silencing of the gene in cell lines[9,10]. K-ras mutation occurs in approximately one-half of colorectal carcinomas. Alteration of codon 12 GGT is the most common change, and often results from G to A transitions. In this study, we examined whether the epigenetic silencing of MGMT might be linked to the presence of K-ras mutations in human colorectal tumorigenesis.
MATERIALS AND METHODS
Materials
Twenty-seven sporadic colorectal adenomas and 62 sporadic carcinomas were obtained from surgical resection specimens of surgical patients at the Zhongnan Hospital of Wuhan University between May 2002 and June 2003. Twenty normal colorectal mucosa tissues were obtained from healthy volunteers examined by endoscopy in the clinic. Each sample was divided into two sections: one was stored at –70 °C, the other was fixed in formalin solution, embedded by paraffin until processing. No patient had received chemotherapy or radiation therapy prior to surgery. Genomic DNA was extracted by the phenol–chloroform standard methods.
Methods
Methylation-specific PCR (MSP) Bisulfite genomic DNA modification was performed as previously reported[11]. Genomic DNAs purified from tissues were denatured by NaOH and modified by sodium bisulfite, then purified using Wizard DNA purification resin (Promega), again treated with NaOH, precipitated with ethanol, and resuspended in water. PCR amplification was performed to determine the DNA methylation patterns in the CpG island of the MGMT. Primer sequences of MGMT were for the unmethylated reaction 5’-TTTGTGTTTTGATGTTTGTAGGTTTTTGT-3’ (sense primer), 5’-AACTCCACACTCTTCCAAAAACAAAACA-3’ (antisense primer) and for the methylated reaction 5’-TTTCGACGTTCGTAGGTTTTCGC-3’ (sense primer), 5’-GCACTCTTCCGAAAACGAAACG-3’ (antisense primer). Amplifications were performed in 50 μL of reaction mixture under the following conditions: 1 cycle at 95 °C for 360 s, 35 cycles at 94 °C for 45 s, 59 °C 45 s, and 72 °C for 60 s; and a final extension at 72 °C for 600 s. Amplified products were visualized by 8% polyacrylamide gel electrophoresis.
Mutant allele-specific amplification (MASA) G to A mutations at codon 12 (GGT) were detected by MASA[12]. The 3’-terminal bases of sense primers were set to the mutant base of K-ras codon 12; the sense-1 primer was for the first base G to A mutation and the sense-2 primer was for the second. The primer sequences were sense-1, 5’-ACTTGTGGTAGTTGGAGCTA-3’; sense-2, 5’-CTTGTGGTAGTTGGAGCTGA-3’. The antisense primer for both senses was 5’-CTCATGAAAATGGTCAGAGAAACC-3’. Reaction conditions were 1 cycle at 94 °C for 300 s, 35 cycles at 94 °C for 30 s, 63.5 °C for 90 s, and 72 °C for 90 s and a final extension at 72 °C for 300 s. Amplified products were visualized by 8% polyacrylamide gel electrophoresis.
Immunohistochemistry Formalin-fixed, paraffin-embedded samples were cut into 4-μm sections. Immunohistochemistry was carried out using a SP kit and mouse anti-MGMT monoclonal antibody. Nuclear staining was counted as positive. Those cases with lesser than 10% positively stained cells were regarded as having loss of expression. The stain was assessed by a pathologist.
Statistical analysis
Statistical analysis was performed using SPSS 11.0 software. Associations between the discrete variables were assessed using the two-sided Fisher’s exact test or Pearson’s χ2 tests. P value less than 0.05 was regarded as statistically significant.
RESULTS
MGMT promoter hypermethylation
DNA obtained from 89 colorectal tumors and 20 normal colorectal mucosa tissues was subjected to MGMT promoter methylation study using MSP. None of the normal tissues showed methylated bands (81 bp), but unmethylated bands (93 bp). Promoter hypermethylation was detected in 38 of 89 (42.7%) colorectal tumors (Figure 1). Among the adenomas and carcinomas tested, 11 of 27 (40.7%) and 27 of 62 (43.5%) had promoter hypermethylation, respectively. No statistical difference could be found between them (χ2 = 0.061, P = 0.806). But when we compared the promoter hypermethylation status of colorectal adenomas and carcinomas with normal colorectal mucosa tissues, the percentages of them were both higher than that of the normal tissues (Fisher’s exact test, P = 0.001, P<0.001). Aberrant MGMT promoter hypermethylation was frequent in both sporadic colorectal adenomas and carcinomas.
Figure 1.

Methylation-specific PCR result of MGMT in colorectal tumors and normal colorectal mucosa. Colorectal carcinomas (45 °C) showed both unmethylated (U) and methylated (M) bands, but normal tissues (N) showed only unmethylated bands.
Expression of MGMT
We analyzed MGMT expression by immunohistochemistry so that the expression in the neoplastic cells could be directly measured. MGMT protein was normally expressed in the nucleus of most parenchymal and stromal cells (Figure 2). All normal colorectal mucosa tissues examined had nuclear staining of MGMT. Among 89 colorectal tumors analyzed, 34 were judged to have loss of MGMT expression, whereas 55 tumors expressed MGMT (Figure 3). In these cases, 6 of 27 (22.2%) adenomas and 28 of 62 (45.2%) carcinomas showed loss of MGMT expression. Therefore, carcinoma significantly differs from adenoma in terms of MGMT expression (χ2 = 4.192, P = 0.041). And also colorectal adenomas and carcinomas both differ from normal colorectal mucosa tissues (Fisher’s exact test, P = 0.031, P<0.001).
Figure 2.
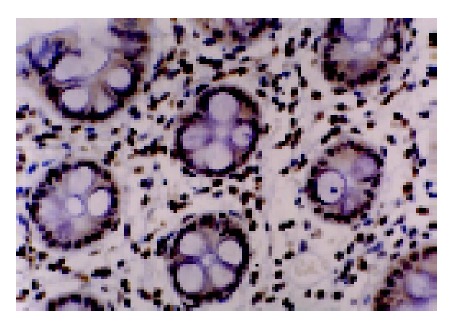
MGMT expressed in the nuclei of normal colorectal mucosa cells.
Figure 3.

Loss of MGMT expression in colorectal carcinomas.
When the expression data were matched to previous studies of the methylation of MGMT, 5 of 6 adenomas that lack MGMT expression showed MGMT promoter hypermethylation, whereas 15 of 21 adenomas that retained expression of MGMT were unmethylated at the MGMT CpG island (Fisher’s exact test, P = 0.027); 24 of 28 carcinomas lacking MGMT expression showed MGMT promoter hypermethylation, whereas 31 of 34 carcinomas that retained expression of MGMT were unmethylated at the CpG island (χ2 = 36.927, P<0.001). It suggests that the presence of aberrant hypermethylation in MGMT promoter was significantly associated with loss of MGMT protein.
K-ras G to A point mutation in codon 12
Among 89 colorectal tumors, 19 (21.3%) had K-ras G to A point mutation in codon 12 (Figure 4). This event was associated with the promoter hypermethylation of MGMT, for 13 (68.4%) of the 19 tumors with mutant K-ras had methylated MGMT, whereas only 25 (35.7%) of the 70 tumors without K-ras mutations had methylated MGMT (χ2 = 6.534, P = 0.011).
Figure 4.

MASA-PCR products for detection of K-ras point mutations in colorectal tumors. Set 1 and set 2 showed the first and second G to A mutation in K-ras codon 12 (GGT), respectively.
In the tumors that had loss of MGMT expression, the G to A point mutation rate of K-ras at codon 12 was 35.3% (12 of 34). This was 2.8 times greater than the rate 12.7% (7 of 55) in the tumors with normal MGMT expression (χ2 = 6.373, P = 0.012).
DISCUSSION
Gene function in cancer can be disrupted either through genetic alterations, which directly mutate or delete genes, or epigenetic alterations, which alter the heritable state of gene expression. It is increasingly apparent that, in human cancers, synergy between these two processes drives tumor progression from the earliest to latest stages. Methylation is the main form of epigenetic change in humans, and alterations in methylation patterns play an important role in tumorigenesis[1]. In this report, we provide an example of the interaction between epigenetic and genetic events in human cancer.
Our data showed that MGMT promoter hypermethylation and loss of MGMT expression were both frequent in sporadic colorectal tumors. This is conformed by the previous report[13]. Loss of expression is not commonly due to deletion, mutation or rearrangement of the MGMT gene, or mRNA instability, but due to methylation of the CpG island of MGMT[9,14]. In cell culture, a relationship between methylation of the MGMT CpG island and transcriptional silencing of the gene has been demonstrated[9]. Our study demonstrates that MGMT promoter hypermethylation associated with loss of expression is frequent in sporadic colorectal tumorigenesis as well. This tight correlation between MGMT CpG island methylation and loss of expression in these tumors provides an explanation for the loss of MGMT activity in earlier and our current reports.
We also found that MGMT promoter hypermethylation was present in equal frequency in adenomas and carcinomas, but the rate of loss of MGMT expression was higher in carcinomas than in adenomas, which suggests that MGMT methylation occurs early in colorectal neoplastic progression, and methylated MGMT, DNA repair gene in the adenoma stage resulted in the inactivation of MGMT, as it brings about the loss of MGMT protein expression when adenoma transforms into carcinoma. Therefore, MGMT methylation might play a key role in malignant transformation.
MGMT is a key enzyme in the DNA repair network. MGMT removes alkyl adducts from the O6 position of guanine, and then inactivates the proliferation of cells with damaged DNA. Therefore, it can protect cells from alkylating damages and maintain genomic integrity[15]. Our results suggest that the epigenetic alteration in MGMT promoter may confer an increased risk of alkylating agent-induced colorectal carcinogenesis.
Alkylating agents causing the promutagenic lesion may be provided from dietary nitrates reduced in the proximal colon by bacteria, by nitrosation of amines and amides derived of protein catabolism[16,17]. During DNA replication, O6-methylguanine can pair with thymine, resulting in conversion of guanine–cytosine to adenine–thymine pairs in DNA[3]. Supporting these data, the most common mutations caused by alkylating agents are G:C to A:T transitions. K-ras mutation occurs in approximately one-half of colorectal carcinomas. Ras oncogenes acquire transforming activity following single-point mutations within their coding sequences. Mutations in naturally occurring ras oncogenes have been localized in codons 12, 13, 59 and 61. Alteration of codon 12, GGT, is the most common change. Substitution of the wild-type glycine 12 by any other amino acid results in oncogenic activation of this molecule. Previous studies had suggested that loss of MGMT activity in the normal colorectal mucosa was found in patients with G to A mutations in K-ras[18,19]. However, our study suggests that epigenetic silencing of the DNA repair gene MGMT by hypermethylation is strongly associated with G to A mutations in K-ras in colorectal tumorigenesis. On the other hand, the absence of K-ras mutations is a common feature of sporadic human breast carcinomas. Concordant with these phenomena, MGMT is not inactivated by promoter hypermethylation in breast tumors[4]. These suggest that one potential consequence of loss of MGMT expression could be an increase in susceptibility of G to A mutation in K-ras gene.
In conclusion, our studies show loss of MGMT expression via hypermethylation in MGMT promoter leading to activation of K-ras gene by accumulation of G to A transition is a new potential pathway of colorectal tumorigenesis. Otherwise, our data concerns the fact that each guanine in the human genome may generate a promutagenic lesion in the absence of correct repair. Other genetic alterations and mutations in target genes in human cancer that might be induced by MGMT epigenetic inactivation should receive close attention, and be investigated in the future.
Footnotes
Supported by the Key Technologies R&D Program of Hubei Province, No. 2002AA301C84
Science Editor Guo SY Language Editor Elsevier HK
References
- 1.Esteller M, Herman JG. Generating mutations but providing chemosensitivity: the role of O6-methylguanine DNA methyltransferase in human cancer. Oncogene. 2004;23:1–8. doi: 10.1038/sj.onc.1207316. [DOI] [PubMed] [Google Scholar]
- 2.Esteller M. Cancer epigenetics: DNA methylation and chromatin alterations in human cancer. Adv Exp Med Biol. 2003;532:39–49. doi: 10.1007/978-1-4615-0081-0_5. [DOI] [PubMed] [Google Scholar]
- 3.Costello JF, Plass C. Methylation matters. J Med Genet. 2001;38:285–303. doi: 10.1136/jmg.38.5.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Esteller M, Hamilton SR, Burger PC, Baylin SB, Herman JG. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res. 1999;59:793–797. [PubMed] [Google Scholar]
- 5.Bhakat KK, Mitra S. CpG methylation-dependent repression of the human O6-methylguanine-DNA methyltransferase gene linked to chromatin structure alteration. Carcinogenesis. 2003;24:1337–1345. doi: 10.1093/carcin/bgg086. [DOI] [PubMed] [Google Scholar]
- 6.Kondo Y, Shen L, Issa JP. Critical role of histone methylation in tumor suppressor gene silencing in colorectal cancer. Mol Cell Biol. 2003;23:206–215. doi: 10.1128/MCB.23.1.206-215.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Esteller M, Toyota M, Sanchez-Cespedes M, Capella G, Peinado MA, Watkins DN, Issa JP, Sidransky D, Baylin SB, Herman JG. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is associated with G to A mutations in K-ras in colorectal tumorigenesis. Cancer Res. 2000;60:2368–2371. [PubMed] [Google Scholar]
- 8.Esteller M, Risques RA, Toyota M, Capella G, Moreno V, Peinado MA, Baylin SB, Herman JG. Promoter hypermethylation of the DNA repair gene O(6)-methylguanine-DNA methyltransferase is associated with the presence of G: C to A: T transition mutations in p53 in human colorectal tumorigenesis. Cancer Res. 2001;61:4689–4692. [PubMed] [Google Scholar]
- 9.Suzuki H, Gabrielson E, Chen W, Anbazhagan R, van Engeland M, Weijenberg MP, Herman JG, Baylin SB. A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nat Genet. 2002;31:141–149. doi: 10.1038/ng892. [DOI] [PubMed] [Google Scholar]
- 10.Wolf P, Hu YC, Doffek K, Sidransky D, Ahrendt SA. O(6)-Methylguanine-DNA methyltransferase promoter hypermethylation shifts the p53 mutational spectrum in non-small cell lung cancer. Cancer Res. 2001;61:8113–8117. [PubMed] [Google Scholar]
- 11.Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93:9821–9826. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watanabe H, Ha A, Hu YX, Ohtsubo K, Yamaguchi Y, Motoo Y, Okai T, Toya D, Tanaka N, Sawabu N. K-ras mutations in duodenal aspirate without secretin stimulation for screening of pancreatic and biliary tract carcinoma. Cancer. 1999;86:1441–1448. [PubMed] [Google Scholar]
- 13.Zaidi NH, Liu L, Gerson SL. Quantitative immunohistochemical estimates of O6-alkylguanine-DNA alkyltransferase expression in normal and malignant human colon. Clin Cancer Res. 1996;2:577–584. [PubMed] [Google Scholar]
- 14.Nakagawachi T, Soejima H, Urano T, Zhao W, Higashimoto K, Satoh Y, Matsukura S, Kudo S, Kitajima Y, Harada H, et al. Silencing effect of CpG island hypermethylation and histone modifications on O6-methylguanine-DNA methyltransferase (MGMT) gene expression in human cancer. Oncogene. 2003;22:8835–8844. doi: 10.1038/sj.onc.1207183. [DOI] [PubMed] [Google Scholar]
- 15.Teo AK, Oh HK, Ali RB, Li BF. The modified human DNA repair enzyme O(6)-methylguanine-DNA methyltransferase is a negative regulator of estrogen receptor-mediated transcription upon alkylation DNA damage. Mol Cell Biol. 2001;21:7105–7114. doi: 10.1128/MCB.21.20.7105-7114.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rowland IR, Granli T, Bøckman OC, Key PE, Massey RC. Endogenous N-nitrosation in man assessed by measurement of apparent total N-nitroso compounds in faeces. Carcinogenesis. 1991;12:1395–1401. doi: 10.1093/carcin/12.8.1395. [DOI] [PubMed] [Google Scholar]
- 17.Roos W, Baumgartner M, Kaina B. Apoptosis triggered by DNA damage O6-methylguanine in human lymphocytes requires DNA replication and is mediated by p53 and Fas/CD95/Apo-1. Oncogene. 2004;23:359–367. doi: 10.1038/sj.onc.1207080. [DOI] [PubMed] [Google Scholar]
- 18.Jackson PE, Hall CN, O'Connor PJ, Cooper DP, Margison GP, Povey AC. Low O6-alkylguanine DNA-alkyltransferase activity in normal colorectal tissue is associated with colorectal tumours containing a GC--& gt; AT transition in the K-ras oncogene. Carcinogenesis. 1997;18:1299–1302. doi: 10.1093/carcin/18.7.1299. [DOI] [PubMed] [Google Scholar]
- 19.Kohya N, Kitajima Y, Kitahara K, Miyazaki K. Mutation analysis of K-ras and beta-catenin genes related to O6-methylguanin-DNA methyltransferase and mismatch repair protein status in human gallbladder carcinoma. Int J Mol Med. 2003;11:65–69. [PubMed] [Google Scholar]