

Abstract
AIM: Microsatellites are the repeated DNA sequences scattered widely within the genomes and closely linked with many important genes. This study was designed to characterize the changes of microsatellite DNA loss of heterozygosity (LOH) in esophageal carcinogenesis.
METHODS: Allelic deletions in 32 cases of matched precancerous, cancerous and normal tissues were examined by syringe microdissection under an anatomic microscope and microsatellite polymorphism analysis using 15 polymorphic markers on chromosomes 3p, 5q, 6p, 9p, 13q, 17p, 17q and 18q.
RESULTS: Microsatellite DNA LOH was observed in precancerous and cancerous tissues, except D9S1752. The rate of LOH increased remarkably with the lesions progressed from basal cell hyperplasia (BCH) to squamous cell carcinoma (SCC) (P<0.05). Three markers, D9S171, D13S260 and TP53, showed the highest incidence of LOH (>60%). LOH loci were different in precancerous and cancerous tissues. LOH in D3S1234 and TP53 was the common event in different lesions from the same patients.
CONCLUSION: Microsatellite DNA LOH occurs in early stage of human esophageal carcinogenesis, even in BCH. With the lesion progressed, gene instability increases, the accumulation of this change may be one of the important mechanisms driving precancerous lesions to cancer.
Keywords: Esophageal cancer, Precancerous lesion, LOH
INTRODUCTION
Esophageal carcinoma (EC) is one of the six most common malignant diseases in the world with a remarkable geographical distribution. The prognosis of EC is very poor, its 5-year survival rate is only about 10% for the patients at late or advanced stage. Linzhou city (formerly Linxian) and nearby counties in Henan Province, China, have been well recognized as the high incidence area in the world. In the past years, scientists from China and other countries have made much research work in this area and found that esophageal carcinogenesis was a multistep progressive process characterized by multiple genetic changes (accumulation and overlap). The early characteristic of the subjects predisposed to EC is the abnormal proliferation of epithelial cells, morphologically manifested as basal cell hyperplasia (BCH), dysplasia (DYS) and carcinoma in situ (CIS), which could be considered as precancerous lesions of EC. The precancerous lesions are instable, i.e., they can develop to cancer, or stay for a couple of years without any changes, or even return to normal. What is the most important factor to decide the precancerous lesions to develop in different directions, especially in those with a similar morphology? What is the key point to induce mild precancerous lesions to develop cancer? Our assumption is that there exist different molecular changes in precancerous lesions with a similar morphology. To characterize the molecular changes in carcinogenesis of EC, the mechanism of EC could be elucidated and the biomarkers for early diagnosis and mass survey of high-risk populations could be established[1].
Microsatellites are the repeated DNA sequences scattered widely within the genomes and closely linked with many important genes[2]. In recent years many researches have indicated that the alteration of microsatellite DNA is one of the important markers, which could induce normal cells to undergo immortal and neoplastic transformations. It was reported that an extensive loss of microsatellite DNA was discovered in many tumors such as colon cancer. Some microsatellite loci often exist in the hot spots of LOH at a high frequency in some specific malignancies. Tumor suppressor genes, which are associated with the development and progression of tumors, may harbor in the vicinity of these hot spots.
To characterize the changes of microsatellite DNA in esophageal carcinogenesis, loss of heterozygosity (LOH) in specific loci was analyzed in 32 surgically resected EC specimens using microsatellite polymorphic markers. Allelic deletions were examined using 15 polymorphic markers on chromosomes 3p, 5q, 6p, 9p, 13q, 17p, 17q and 18q.
MATERIALS AND METHODS
Precancerous and cancerous tissues
Thirty-two surgically resected squamous cell carcinoma (SCC) specimens were collected from Linxian Country, a high-incidence area of EC in Henan Province, China. Of the EC patients, 18 were males and 14 were females with an average age of 59 years (range 44-73 years). All the SCC patients were not treated by either chemotherapy or radiotherapy before operation. Surgically resected specimens were divided into two parts: One was fixed with 85% ethanol and paraffin embedded for histopathological diagnosis, and the other was stored in liquid nitrogen and then transferred in a -80 °C freezer for further use. Ten of thirty-two cases were taken from cancer and adjacent tissues. Various tissues from cancer and adjacent parts were frozen and cut into 5-μm thick sections and 30-60 slides were used for DNA extraction and LOH analysis. Five slides were stained with hematoxylin-eosin (HE) for histopathological diagnosis. According to cell morphologic changes, the esophageal epithelia were divided into BCH, DYS, CIS and SCC[3].
Syringe microdissection under anatomic microscope and DNA extraction
According to the distribution of cells in HE-stained sections, we dropped glycerol in matched spots, separated precancerous, cancerous and normal cells by a 5-mL syringe under an anatomy microscope, put them into 180 μL lysis buffer, added 20 μL proteinase K (50 μg/μL), and kept overnight at 56 °C. By this method, 90% purified cells could be collected[4]. DNA was extracted according to the protocols of the QIAGEN DNA Mini Kit.
LOH analysis
According to the results based on our previous work in esophageal carcinogenesis at high-incidence area for EC in Henan Province, China[5], we chose 15 microsatellite DNA loci for LOH analysis using a circulating p53-Rb system. These loci represented D3S966 (RASSF1A), D3S1234 and D3S1300 (FHIT), D5S82 (DP1), D5S346 (APC), D6S497 (HLA, Waf1), D9S1752 (INK4a), D9S171 (INK4b), D13S260 (BRCA2), D13S321 and D13S233 (Rb1), TP53 and D17S786 (p53), D17S855 (BRCA1) and D18S858 (DCC).
Microsatellite polymorphism analysis
Each locus was amplified from 40 to 50 ng of template DNA by PCR using paired primers, of which the forward primer was 5’ end-labeled with [γ-32P] ATP. Eight percent of nondenaturing polyacrylamide gel electrophoresis was performed at 1 650 V for 2-3 h. After electrophoresis, the gel was transferred to a 3 MM chromatography paper and dried in a vacuum gel dryer at 80 °C for 1 h. The dried gel was placed into an X-ray film cassette with an intensifying screen and exposed to a Kodak film at -70 °C or at room temperature for 2-48 h.
Result determination
Three individuals examined the relative intensities of polymorphic alleles in different lesions. LOH status was established if the intensity of one allele in the tumor was significantly reduced as compared with its corresponding allele in constitutive DNA. When the decision was not unanimous, the density of alleles was determined by quantitative densitometry. Reduction of 30% intensity of one allele in tumor DNA compared to its matching allele in the constitutive DNA was chosen as the criteria for LOH status. Individual results were classified into three categories: LOH (loss of one allele), heterozygosity retained (no allelic loss), and uninformative (homozygous alleles)[6].
Statistical analysis
The data were analyzed by SPSS10.0 statistical software. LOH frequency was performed by χ2 test. P<0.05 was considered statistically significant.
RESULTS
Relationship between LOH frequency and lesions (Figures 1 and 2, Table 1)
Figure 1.
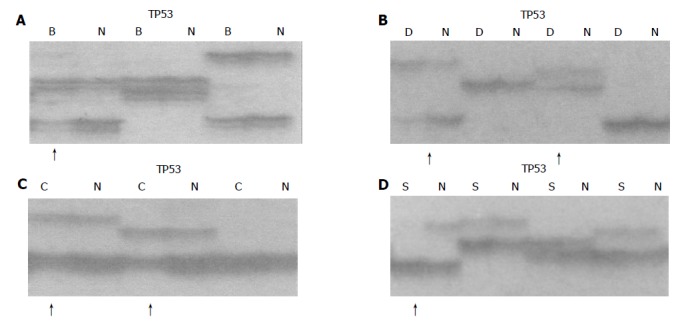
Allelic deletion patterns in TP53 microsatellite loci. Arrows indicate LOH in different lesions. B: basal cell hyperplasia (BCH), D: dysplasia (DYS), C: carcinoma in situ (CIS), S: squamous cell carcinoma(SCC), N: normal tissue, A: LOH in BCH; B: LOH in DYS; C: LOH in CIS; D: LOH in SCC.
Figure 2.
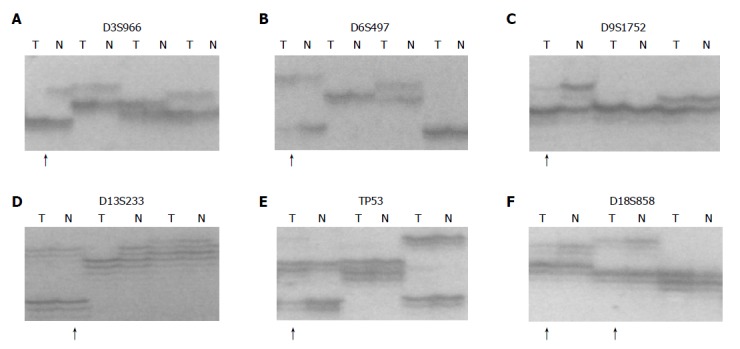
Allelic deletion patterns of various microsatellite markers in ESCC. Paired tumor DNA (T) and non-neoplastic DNA (N) were examined for each case. Arrows indicate LOH in tumor samples. A: D3S966 LOH; B: D6S497 LOH; C: D9S1752 LOH; D: D13S233 LOH; E: TP53 LOH; F: D18S858 LOH.
Table 1.
Comparison between frequency of microsatellite LOH in different lesions of esophagus.
| MSM |
LOH frequency n (%) |
P | |||
| BCH | DYS | CIS | SCC | ||
| D3S1234 | 1/14 (7.1) | 3/12 (25) | 5/15 (33.30) | 9/23 (39.1) | 0.042 |
| D3S1300 | 0/11 (0) | 2/10 (20) | 4/10 (40) | 10/26 (38.5) | 0.024 |
| D3S966 | 0/13 (0) | 3/13 (23.1) | 8/16 (50) | 11/20 (55.0) | 0.001 |
| D5S82 | 0/12 (0) | 2/12 (16.7) | 6/15 (40) | 8/20 (40.0) | 0.010 |
| D5S346 | 2/12 (16.7) | 3/12 (25) | 4/10 (40) | 14/25 (56.0) | 0.011 |
| D6S497 | 0/10 (0) | 0/13 (0) | 0/12 (0) | 9/26 (34.6) | 0.001 |
| D9S1752 | 1/15 (6.7) | 2/12 (16.7) | 3/15 (20) | 5/25 (20.0) | 0.315 |
| D9S171 | 0/13 (0) | 5/16 (31.3) | 7/14 (50) | 14/23 (60.8) | 0.000 |
| D13S233 | 0/11 (0) | 3/13 (23.1) | 6/16 (37.5) | 10/21 (47.6) | 0.006 |
| D13S321 | 0/14 (0) | 3/12 (25) | 6/14 (42.9) | 11/25 (44.0) | 0.006 |
| D13S260 | 0/16 (0) | 4/12 (33.3) | 7/14 (50) | 16/26 (61.5) | 0.000 |
| D17S855 | 2/13 (15.4) | 4/14 (28.6) | 6/15 (40) | 14/26 (53.8) | 0.014 |
| TP53 | 3/15 (20) | 4/13 (30.8) | 7/16 (43.8) | 15/22 (68.2) | 0.002 |
| D17S786 | 0/14 (0) | 3/12 (25) | 6/15 (40) | 12/22 (54.5) | 0.001 |
| D18S858 | 0/13 (0) | 3/16 (18.8) | 4/12 (33.3) | 10/18 (55.6) | 0.000 |
MSM: microsatellite marker; LOH: loss of heterozygosity; BCH: basal cell hyperplasia DYS: dysplasia; CIS: carcinoma in situ; SCC: squamous cell carcinoma -: not done.
Microsatellite DNA LOH was observed in precancerous and cancerous tissues, except D9S1752. The rate of LOH increased remarkably with the lesion developed from BCH to SCC (P<0.05). At least on LOH locus was 41% (7/17) in BCH, 82% (14/17) in DYS, 100% (17/17) in CIS and 97% (31/32) in SCC. Three samples from SCC and one sample from CIS were found to have deletions in 18 loci.
Distribution of microsatellite DNA LOH in different lesions (Figure 3)
Figure 3.

Distributions of microsatellite DNA-LOH in different precancerous lesions of esophagus FAL: fractional allelic loss for each tumor ○ retention of heterozygosity ■ loss of heterozygosity Ø uninformative, X not done.
LOH loci were different in precancerous and cancerous tissues. In BCH, DYS and SCC, the highest LOH loci were TP53 (20%), D13S260 (33.3%) and TP53 (68.2%) respectively. In CIS, the highest LOH markers had three loci, D13S260, D9S171 and D3S966 (50%). Three markers, D9S171, D13S260 and TP53, showed the highest incidence of LOH (>60%). LOH in D3S1234 and TP53 was the common event in different lesions of the same patients.
DISCUSSION
Inactivation of tumor suppressor genes appears to be one of the genetic mechanisms involved in the development of esophageal cancer. This process includes mutation of one allele, followed by a deletion of the remaining one (LOH) or homozygous deletion of both alleles. Allelic deletions detected as LOH have been proved useful for mapping regions of DNA that contain tumor suppressor genes[7]. LOH at specific chromosomal regions strongly suggested the existence of tumor suppressor genes at the relevant segments[8]. We performed deletion mapping analyses in 32 cases of matched precancerous, cancerous and normal tissues using 15 microsatellite markers on chromosomes 3p, 5q, 6p, 9p, 13q, 17p, 17q and 18q and found that microsatellite DNA LOH occurred in early precancerous stage and in BCH. With the development of the disease, the rate of LOH increased, indicating that the genetic changes occurred in the early stage of EC. With the lesion progressed, gene instability increased, the accumulation of this change may be one of the important mechanisms transforming precancerous lesions to cancer. LOH loci were different even at the same stages (such as DYS), indicating that there existed different molecular changes in precancerous lesions with a similar morphology. These changes might be the key factors to decide precancerous lesions developing in different directions, especially in those with a similar morphology. Esophageal carcinogenesis is a multistep progressive process characterized by multiple genetic changes (accumulation and overlap). The changes of p15INK4b, BRAC2 and p53 genes are common molecular events in esophageal carcinogenesis. LOH in D3S1234 and TP53 is the common event in different lesions of patients, indicating that the alternations of fragile histidine triad (FHIT) and p53 might be the key points to induce mild precancerous lesions to cancer. RASSF1A - one of the candidate TSGs - might be involved in the esophageal carcinogenesis in Henan Province, China.
The p15INK4b gene is an inhibitor of cyclin-dependent kinase 4, which has been identified in 95% genome sequence homogeneity with p16INK4a. They encode two important cyclin-dependent kinase inhibitors, which could negatively regulate G1-S transition of the proliferating cells by contributing to the maintenance of pRb in an active state[9]. Xing et al[10], reported that both p15INK4b and p16INK4a genes were frequently inactivated in EC, but the inactivation of p15INK4b and p16INK4a involved different mechanisms, with p16INK4a predominantly affected by aberrant methylation and p15INK4b by deletion. We analyzed the allelic loss of p15INK4b and p16INK4a using D9S1752 and D9S171 microsatellite markers, and found that the LOH frequency of p15INK4b was much higher than that of p16INK4a in precancerous and cancerous lesions. Our results support the speculation of Xing et al[10], and suggest that the deletion of p15INK4b gene might be involved in the multistage development of EC at the high-incidence area in Henan Province, China. LOH of p16INK4a might not be an important event in the esophageal carcinogenesis in the area.
BRCA1 and BRCA2 are tumor suppressor genes in familial breast-ovarian carcinoma syndrome and are located in different chromosomes. The BRCA2 gene is located on chromosome 13q12[11]. Extensive genetic and biochemical characterization has shown that BRCA2 is involved in the maintenance of chromosomal stability. It could serve as a critical mediator of DNA repair through direct interactions with Rad51 and might play an important role in recombination-mediated double-strand DNA break repair[12]. Harada et al[13], performed a fine deletion mapping on 13q by analyzing 60 EC patients with 18 polymorphic markers and found the frequent loss at D13S260 (43.7%). Up to now, to our knowledge, there are no other reports on the changes of BRCA2 in precancerous tissues. In our experiment, BRCA2 gene alternations were detected as 0% in BCH, 33% in DYS, 50% in CIS, and 61.5% in SCC, indicating that the deletion of BRCA2 might be involved in the development of EC and is one of the common molecular events in the esophageal carcinogenesis.
The p53 tumor suppressor gene is located on chromosome 17p13.1 and encodes a 53 ku nuclear phosphoprotein that binds to DNA and blocks the progression of the cell cycle in response to DNA damage and mediates apoptosis[14]. Allelic loss of p53 gene on chromosome 17p13.1 has been demonstrated to be one “hit” of inactivation of p53 gene. It was reported that the frequencies of LOH ranged from 65% to 83.8%, and often co-existed with mutations (“two hits”) in EC[15]. In our experiments, the deletion of p53 occurred in the early stage of BCH (20%), indicating that the alternations of p53 might be the key points to induce mild precancerous lesions to cancer.
Frequent allele loss has been observed on chromosome 3p in EC and its premalignant lesions, indicating that inactivation of putative tumor suppressor genes on 3p may be involved in early stages of esophageal carcinogenesis. It has been reported that FHIT gene is a novel tumor suppressor gene located on chromosome 3p14.2[16]. Highly frequent abnormal transcripts of FHIT gene have been found in a variety of human cancers, including cancers of the digestive tract, lung, breast, and head and neck[17,18]. Point mutations of the FHIT gene were also seen in gastric and breast carcinomas, but very rarely. A review of the literature showed that different results were obtained by different researches[19-21]. Zou et al[19], reported that the deletions of FHIT were involved in 11 of 50 (22%) EC samples. On the other hand, Mori et al[22], observed that the LOH frequency of the FHIT gene was 76%. In our experiment, loss of FHIT was detectable in 7% BCH, 20% DYS, 33% CIS and 39% SCC. Although the LOH frequency of FHIT gene was low in our results, the change occurred in each stage of esophageal carcinogenesis. Exposure to different environmental carcinogens might be one of the reasons why there existed differences in different areas.
RASSF1A is a novel tumor suppressor gene that was isolated recently from the lung tumor suppressor locus 3p21.3[23]. The presence of a Ras associated domain in RASSF1A suggested that this protein might function as an effector of Ras signaling in normal cells. Its protein structure also suggested that RASSF1A might participate in the DNA damage response or in DNA damage-induced regulation of other cell signaling events[24]. Chan et al[25], demonstrated the methylation status of RASSF1A and the frequency of LOH in 3p21.3 region in bladder cancer and found that the frequency of LOH and methylation of RASSF1A were 57.9% and 47.5% respectively, showing that RASSF1A might be inactivated in accordance with the two-hit inactivation model, involving deletion of one allele and hypermethylation of the other[26]. Up to now, much research work has been done in promoter hypermethylation of RASSF1A. In the present study, we observed that the LOH frequency of D3S966 was 23.1% in DYS, 50% in CIS and 55% in SCC, indicating that RASSF1A might be involved in the esophageal carcinogenesis at the high-incidence area in Henan Province, China. To our knowledge, this report is the first to identify allelic loss of RASSF1A during esophageal carcinogenesis in Henan Province, China.
Footnotes
Supported by National Outstanding Young Scientist Foundation of China, No. 30025016; State Basic Research Development Program of China, No. G1998051206; Foundation of Henan Education Committee, No. 1999125 and the US NIH Grant, No. CA65871
References
- 1.Wang LD, Zheng S. The mechanism of esophageal and gastric cardia carcinogenesis from the subjects at high-incidence area for esophageal cancer in Henan. Zhengzhou Daxue Xuebao. 2002;37:717–729. [Google Scholar]
- 2.Cullis CA. The use of DNA polymorphisms in genetic mapping. Genet Eng (N Y) 2002;24:179–189. doi: 10.1007/978-1-4615-0721-5_8. [DOI] [PubMed] [Google Scholar]
- 3.Wang LD, Shi ST, Zhou Q, Goldstein S, Hong JY, Shao P, Qiu SL, Yang CS. Changes in p53 and cyclin D1 protein levels and cell proliferation in different stages of human esophageal and gastric-cardia carcinogenesis. Int J Cancer. 1994;59:514–519. doi: 10.1002/ijc.2910590414. [DOI] [PubMed] [Google Scholar]
- 4.Wang LD, Zhou Q, Hong JY, Qiu SL, Yang CS. p53 protein accumulation and gene mutations in multifocal esophageal precancerous lesions from symptom free subjects in a high incidence area for esophageal carcinoma in Henan, China. Cancer. 1996;77:1244–1249. [PubMed] [Google Scholar]
- 5.Wang LD, Hong JY, Qiu SL, Gao H, Yang CS. Accumulation of p53 protein in human esophageal precancerous lesions: a possible early biomarker for carcinogenesis. Cancer Res. 1993;53:1783–1787. [PubMed] [Google Scholar]
- 6.Reed AL, Califano J, Cairns P, Westra WH, Jones RM, Koch W, Ahrendt S, Eby Y, Sewell D, Nawroz H, et al. High frequency of p16 (CDKN2/MTS-1/INK4A) inactivation in head and neck squamous cell carcinoma. Cancer Res. 1996;56:3630–3633. [PubMed] [Google Scholar]
- 7.Thiagalingam S, Foy RL, Cheng KH, Lee HJ, Thiagalingam A, Ponte JF. Loss of heterozygosity as a predictor to map tumor suppressor genes in cancer: molecular basis of its occurrence. Curr Opin Oncol. 2002;14:65–72. doi: 10.1097/00001622-200201000-00012. [DOI] [PubMed] [Google Scholar]
- 8.Kinzler KW, Nilbert MC, Vogelstein B, Bryan TM, Levy DB, Smith KJ, Preisinger AC, Hamilton SR, Hedge P, Markham A. Identification of a gene located at chromosome 5q21 that is mutated in colorectal cancers. Science. 1991;251:1366–1370. doi: 10.1126/science.1848370. [DOI] [PubMed] [Google Scholar]
- 9.Xing EP, Nie Y, Song Y, Yang GY, Cai YC, Wang LD, Yang CS. Mechanisms of inactivation of p14ARF, p15INK4b, and p16INK4a genes in human esophageal squamous cell carcinoma. Clin Cancer Res. 1999;5:2704–2713. [PubMed] [Google Scholar]
- 10.Xing EP, Nie Y, Wang LD, Yang GY, Yang CS. Aberrant methylation of p16INK4a and deletion of p15INK4b are frequent events in human esophageal cancer in Linxian, China. Carcinogenesis. 1999;20:77–84. doi: 10.1093/carcin/20.1.77. [DOI] [PubMed] [Google Scholar]
- 11.Wooster R, Neuhausen SL, Mangion J, Quirk Y, Ford D, Collins N, Nguyen K, Seal S, Tran T, Averill D. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13. Science. 1994;265:2088–2090. doi: 10.1126/science.8091231. [DOI] [PubMed] [Google Scholar]
- 12.Shamoo Y. Structural insights into BRCA2 function. Curr Opin Struct Biol. 2003;13:206–211. doi: 10.1016/s0959-440x(03)00033-2. [DOI] [PubMed] [Google Scholar]
- 13.Harada H, Tanaka H, Shimada Y, Shinoda M, Imamura M, Ishizaki K. Lymph node metastasis is associated with allelic loss on chromosome 13q12-13 in esophageal squamous cell carcinoma. Cancer Res. 1999;59:3724–3729. [PubMed] [Google Scholar]
- 14.Hu N, Huang J, Emmert-Buck MR, Tang ZZ, Roth MJ, Wang C, Dawsey SM, Li G, Li WJ, Wang QH, et al. Frequent inactivation of the TP53 gene in esophageal squamous cell carcinoma from a high-risk population in China. Clin Cancer Res. 2001;7:883–891. [PubMed] [Google Scholar]
- 15.Baker SJ, Kinzler KW, Vogelstein B. Knudson's hypothesis and the TP53 revolution. Genes Chromosomes Cancer. 2003;38:329. doi: 10.1002/gcc.10249. [DOI] [PubMed] [Google Scholar]
- 16.Pekarsky Y, Palamarchuk A, Huebner K, Croce CM. FHIT as tumor suppressor: mechanisms and therapeutic opportunities. Cancer Biol Ther. 2002;1:232–236. doi: 10.4161/cbt.73. [DOI] [PubMed] [Google Scholar]
- 17.Scully C, Field JK, Tanzawa H. Genetic aberrations in oral or head and neck squamous cell carcinoma 2: chromosomal aberrations. Oral Oncol. 2000;36:311–327. doi: 10.1016/s1368-8375(00)00021-x. [DOI] [PubMed] [Google Scholar]
- 18.Yang Q, Yoshimura G, Sakurai T, Kakudo K. The Fragile Histidine Triad gene and breast cancer. Med Sci Monit. 2002;8:RA140–RA144. [PubMed] [Google Scholar]
- 19.Zou TT, Lei J, Shi YQ, Yin J, Wang S, Souza RF, Kong D, Shimada Y, Smolinski KN, Greenwald BD, et al. FHIT gene alterations in esophageal cancer and ulcerative colitis (UC) Oncogene. 1997;15:101–105. doi: 10.1038/sj.onc.1201169. [DOI] [PubMed] [Google Scholar]
- 20.Mimori K, Inoue H, Shiraishi T, Matsuyama A, Mafune K, Tanaka Y, Mori M. Microsatellite instability is often observed in esophageal carcinoma patients with allelic loss in the FHIT/FRA3B locus. Oncology. 2003;64:275–279. doi: 10.1159/000069317. [DOI] [PubMed] [Google Scholar]
- 21.Roth MJ, Hu N, Emmert-Buck MR, Wang QH, Dawsey SM, Li G, Guo WJ, Zhang YZ, Taylor PR. Genetic progression and heterogeneity associated with the development of esophageal squamous cell carcinoma. Cancer Res. 2001;61:4098–4104. [PubMed] [Google Scholar]
- 22.Mori M, Mimori K, Shiraishi T, Alder H, Inoue H, Tanaka Y, Sugimachi K, Huebner K, Croce CM. Altered expression of Fhit in carcinoma and precarcinomatous lesions of the esophagus. Cancer Res. 2000;60:1177–1182. [PubMed] [Google Scholar]
- 23.Chow LS, Lo KW, Kwong J, To KF, Tsang KS, Lam CW, Dammann R, Huang DP. RASSF1A is a target tumor suppressor from 3p21.3 in nasopharyngeal carcinoma. Int J Cancer. 2004;109:839–847. doi: 10.1002/ijc.20079. [DOI] [PubMed] [Google Scholar]
- 24.Dammann R, Li C, Yoon JH, Chin PL, Bates S, Pfeifer GP. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat Genet. 2000;25:315–319. doi: 10.1038/77083. [DOI] [PubMed] [Google Scholar]
- 25.Chan MW, Chan LW, Tang NL, Lo KW, Tong JH, Chan AW, Cheung HY, Wong WS, Chan PS, Lai FM, et al. Frequent hypermethylation of promoter region of RASSF1A in tumor tissues and voided urine of urinary bladder cancer patients. Int J Cancer. 2003;104:611–616. doi: 10.1002/ijc.10971. [DOI] [PubMed] [Google Scholar]
- 26.Kuroki T, Trapasso F, Yendamuri S, Matsuyama A, Alder H, Mori M, Croce CM. Allele loss and promoter hypermethylation of VHL, RAR-beta, RASSF1A, and FHIT tumor suppressor genes on chromosome 3p in esophageal squamous cell carcinoma. Cancer Res. 2003;63:3724–3728. [PubMed] [Google Scholar]