

Abstract
AIM: To investigate the tumor-suppressive effect of the phosphatase and tensin homologue deleted from chromosome (PTEN) in human gastric cancer cells that were wild type for PTEN.
METHODS: Adenoviruses expressing PTEN or luciferase as a control were introduced into gastric cancer cells. The effect of exogenous PTEN gene on the growth and apoptosis of gastric cancer cells that are wtPTEN were examined in vitro and in vivo.
RESULTS: Adenovirus-mediated transfer of PTEN (Ad-PTEN) suppressed cell growth and induced apoptosis significantly in gastric cancer cells (MGC-803, SGC-7901) carrying wtPTEN in comparison with that in normal gastric epithelial cells (GES-1) carrying wtPTEN. This suppression was induced through downregulation of the Akt/PKB pathway, dephosphorylation of focal adhesion kinase and mitogen-activated protein kinase and cell-cycle arrest at the G2/M phase but not at the G1 phase. Furthermore, treatment of human gastric tumor xenografts (MGC-803, SGC-7901) with Ad-PTEN resulted in a significant (P<0.01) suppression of tumor growth.
CONCLUSION: These results indicate a significant tumor-suppressive effect of Ad-PTEN against human gastric cancer cells. Thus, Ad-PTEN may be used as a potential therapeutic strategy for treatment of gastric cancers.
Keywords: PTEN, Gastric cancer
INTRODUCTION
Phosphatase and tensin homolog deleted from chromosome 10 (PTEN) gene is a tumor-suppressor gene located on human chromosome 10q23.3[1,2]. Frequent deletions and somatic mutations of PTEN have been reported in glioblastoma, endometrial cancer, prostate cancer, and small cell lung cancer[3-7]. Furthermore, germline mutations of PTEN are the cause of Cowden disease and Bannayan-Zonana syndrome, which are autosomal dominant cancer predisposition disorders[8-10]. PTEN functions both as a protein and as a lipid phosphatase in cells that are genotypically wild type for PTEN[11,12]. PTEN also modulates the phosphatidylinositol 3-kinase (PI3K) pathway by catalyzing degradation of PtdIns (3, 4, 5) P3 generated by PI3K. This mechanism inhibits downstream functions mediated by the PI3K pathway, such as activation of Akt/protein kinase B (PKB), cell survival, and cell proliferation[13]. Thus, overexpression of PTEN in cancer cells carrying mutant- or deletion-type PTEN can inhibit cell proliferation and tumorigenicity via induction of cell-cycle arrest at G1 phase and apoptosis[14-17]. Although studies demonstrating the inhibitory function of PTEN in tumor cell mutants for PTEN exist, very little information is available on the effect of PTEN in tumor cells that are wild type for PTEN (wtPTEN). More recently, studies using melanoma cells, ovarian and thyroid cancer cells that are wtPTEN demonstrated ectopic expression of PTEN resulting in growth inhibition and cell death[18-20]. Based on these reports, we investigated the tumor-suppressive effect of PTEN on gastric cancer cells that were wtPTEN.
In the present study, we demonstrated that adenovirus mediated overexpression of PTEN (Ad-PTEN) in gastric cancer cells and in normal gastric epithelial cells that were wtPTEN resulted in selective induction of G2 cell-cycle arrest and apoptosis in tumor cells, but not in normal cells. Furthermore, treatment of gastric tumor xenografts with Ad-PTEN resulted in tumor inhibition. These results indicate that Ad-PTEN can be used as a gene therapeutic strategy for the treatment of gastric cancers.
MATERIALS AND METHODS
Cell lines and cell culture
Human gastric carcinoma metastatic lymph node cell line, SGC-7901, and normal gastric epithelial cell line, GES-1, were obtained from Cancer Research Institute of Beijing, China. Human gastric mucinous adenocarcinoma cell line, MGC-803, was obtained from the Cell Bank of Shanghai Institute of Cell Biology, Chinese Academy of Sciences (Shanghai, China). The derived cell lines were grown in IMDM medium supplemented with 10% heat-inactivated fetal calf serum, 50 U/mL penicillin and 50 µg/mL streptomycin. The cells were maintained at 37 °C in a humidified atmosphere containing 50 mL/L CO2. Viability of the cells used in these experiments was consistently more than 95% when evaluated by the trypan blue exclusion method.
Construction of recombinant adenoviral vector
To construct adenoviruses carrying PTEN (Ad-PTEN), cDNA of PTEN was cloned by RT-PCR using placental mRNA as a template and the following primers: 5’-CTTCA-GCCACAGGCTCCCA-3’ and 5’-TGGTGTTTTATCCC-TCTTGATA-3’. A 1.2-kb blunt-ended fragment of PTEN cDNA was inserted into the SwaI site of the cosmid pAxCAwt (TaKaRa) that contained the CAG promoter and an entire genome of type 5 adenoviruses except for E1 and E3 regions. This procedure generated pAxCAPTEN. The pAxCAPTEN was cotransfected with pJM17 (Microbix Biosystems Inc., Canada) into 293 cells to obtain Ad-PTEN viruses. As a control, Ad-Luc viruses were constructed. Viruses were propagated in the 293 cells lines and purified by two rounds of CsCl density centrifugation. Viral titers were measured in a limiting-dilution bioassay using the 293 cells. Recombinant adenovirus infections of the cell lines were carried out by dilution of viral stock to certain concentrations, addition of viral solution to cell monolayers, and incubation at room temperature for 30 min with agitation every 10 min. This was followed by addition of culture medium and the return of the infected cells to the 37 °C incubator.
Transduction and cell proliferation assay
All the cell lines were plated in six-well tissue culture plates at a density of 1×105 cells/well. Tumor cells and normal cells were then treated with Ad-PTEN or Ad-Luc, or treated with PBS as a mock control. Cells in each treatment group were plated in triplicate and cultured for 5 d. Then, at designated time points, cells were harvested and stained with 0.4% trypan blue (GIBCO BRL, Grand Island, NY, USA) to reveal dead cells. Viable cells were then counted using a hemocytometer.
Apoptotic staining
Cells were seeded in six-well tissue culture dishes at a density of 1×105 cells/well and treated with Ad-PTEN or Ad-Luc. At 72 h after treatment, cells were analyzed for apoptosis using Hoechst 33 258 staining (Sigma Chemicals, St. Louis, MO, USA). Apoptotic cells were determined via apoptotic body and/or chromosome condensation.
Cell-cycle analysis
Cells were seeded in 10-cm culture dishes (5-10×105 cells/dish) and treated with Ad-PTEN or Ad-Luc, or treated with PBS, 20 μmol/L LY294002 (Cell Signaling Technology, Beverly, MA, USA). At specific time points after treatment, cells were harvested, washed once with ice-cold PBS, fixed with 70% ethanol, and stored at -20 °C. Cells were then washed twice with ice-cold PBS and treated with RNase (for 30 min at 37 °C, 500 U/mL; Sigma Chemicals), and DNA was stained with propidium iodide (50 mg/mL; Boehringer Mannheim, Indianapolis, IN, USA). The cell cycle phase and apoptotic rate (cells at sub-G0/G1 phase) were analyzed using a FACScan (Beckman Coulter Inc., Fullerton, CA, USA).
Antibody and Western blotting analysis
Following antibodies were used as primary antibodies: PTEN (Santa Cruz, CA, USA), β-actin (Sigma Chemicals), p27Kip1, Akt, phospho-Akt, p44/42MAPK, and phospho-p44/42MAPK (Cell Signaling Technology Inc., Beverly, MA, USA), phospho-FAK (Pharmingen, San Diego, CA, USA). To perform Western blotting analysis, cells were harvested by centrifugation. After being washed with ice-cold PBS, cells were lysed with EBC buffer (120 mmol/L NaCl, 10 mmol/L Tris at pH 8.0, 0.1 mmol/L EDTA, 1 mmol/L DTT) containing 0.5%NP-40 (Sigma), and protease inhibitors (Roche, Indianapolis, IN, USA). Cell lysates were normalized before loading onto 4-20% SDS-PAGE gradient gels (Invitrogen, Carlsbad, CA, USA). Proteins were subsequently transferred to a nitrocellulose membrane (Invitrogen) and probed with primary and secondary antibodies. The proteins were visualized on an enhanced chemiluminescence (ECL) film (Amersham) using the ECL Western blot detection regent (Amersham).
In vivo tumor xenograft studies
Subcutaneous tumor xenografts (MGC803, SGC7901) were established in nude mice by injecting tumor cells (1×106 cells/animal) in the right dorsal flank. When the tumor reached a size of 50-100 mm3, animals were randomized into three groups (n = 9, animals/group) and treatment was initiated as follows. Group 1 received no treatment, group 2 received Ad-Luc (2×1010 vp/dose), and group 3 received Ad-PTEN (2×1010 vp/dose). All treatments were given on alternate days for a total of six doses. Intratumoral injections were performed under anesthesia.
Experiments were terminated when tumors showed signs of necrosis. The therapeutic effect was determined by statistical analysis using Student’s t -test.
RESULTS
Inhibition of cell proliferation in gastric cancer due to overexpression of PTEN
Tumor cells (MGC-803, SGC-7901) and normal cells (GES-1) were treated with PBS, Ad-Luc, or Ad-PTEN and examined for exogenous PTEN expression and cell proliferation at various time points. Treatment with Ad-PTEN resulted in exogenous PTEN expression in all the cell lines tested (Figure 1A). However, no endogenous PTEN protein could be detected by Western blotting in tumor cells, suggesting that additional epigenetic mechanisms might compromise the expression of PTEN, consistent with tumor suppressor role of PTEN. Endogenous PTEN expression was observed in normal cells, indicating that they were wild type for PTEN. Furthermore, analysis for cell viability daily on days 1-5 demonstrated significant inhibition of cell proliferation (P<0.01) in all the cancer cell lines treated with Ad-PTEN, when compared with that in control cells treated with Ad-Luc or PBS (Figure 1B). In normal cells (GES-1), Ad-PTEN treatment resulted in moderate inhibition of cell growth compared to cells treated with Ad-Luc and PBS (Figure 1B). However, Ad-PTEN-mediated inhibitory effect on tumor cell proliferation was greater and more pronounced than that observed in normal cells. Minimal to no inhibitory effect on cell proliferation was also observed in normal human fibroblast cell line when treated with Ad-PTEN (data not shown). These results suggested that Ad-PTEN might selectively and more effectively inhibit tumor cells than normal cells.
Figure 1.
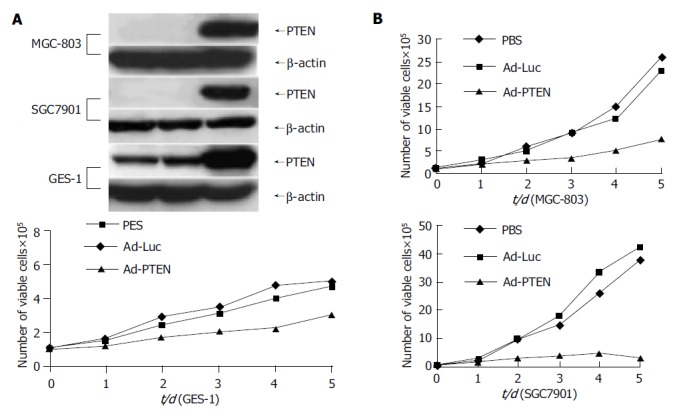
Inhibition of cell proliferation in gastric cancer cells due to overexpression of PTEN. A: Cells analyzed for PTEN protein expression by Western blot analysis; B: Measurement of cell proliferation as determined by viability assay on d 1, 2, 3, 4, and 5.
Induction of apoptosis in gastric cancer cells due to overexpression of PTEN
After treatment with Ad-PTEN, tumor cells (MGC-803, SGC-7901) and normal cells (GES-1) were analyzed for apoptotic changes using a fluorescence-activated cell sorter (FACS) and/or Hoechst 33258 staining. At 72 h after Ad-PTEN treatment, an increase in the number of cells in the sub-G0/G1 phase, an indicator of apoptotic changes, was observed in the two cancer cell lines by FACS analysis (Figure 2A). The percent increase in apoptotic cells was 20-30%, depending on the tumor cell type. However, no changes were observed in tumor cells treated with Ad-Luc or PBS. In contrast, no significant induction of apoptosis was observed in normal cells treated with Ad-PTEN compared to cells treated with PBS and Ad-Luc (Figure 2A). To confirm these results further, Hoechst 33258 staining was performed 72 h after treatment. Tumor cells underwent apoptosis following Ad-PTEN treatment, but normal cells did not. No changes were observed in any of the cells treated with Ad-Luc (Figure 2B).
Figure 2.
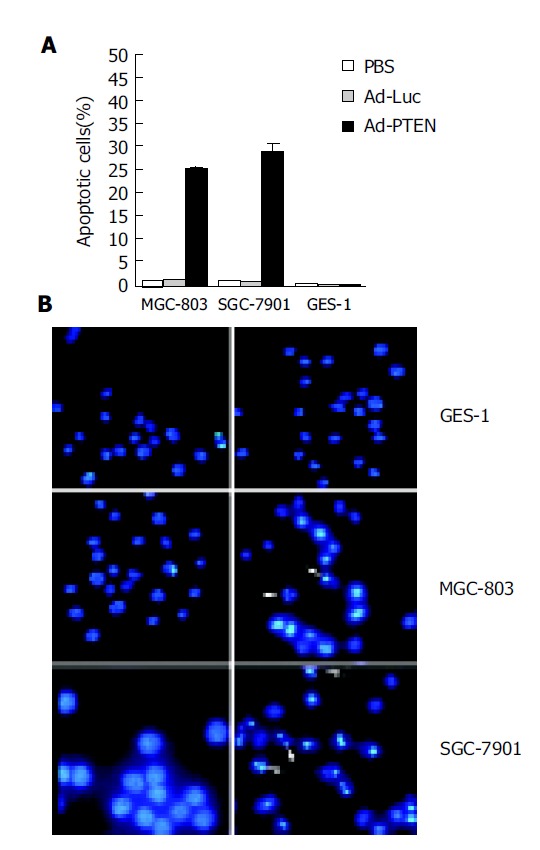
Induction of apoptosis due to overexpression of PTEN. A: Tumor cells (MGC-803, SGC-7901) and normal cells (GES-1) treated with PBS, Ad-Luc, or Ad-PTEN and harvested at 72 h after treatment; B: Analysis of tumor cells (MGC-803, SGC-7901) and normal cells (GES-1) by Hoechst 33258 staining 72 h after treatment with Ad-Luc and Ad-PTEN. Arrows indicate apoptotic cells. Magnification ×40 for all cell lines.
Induction of G2/M cell-cycle arrest due to overexpression of PTEN
To determine whether Ad-PTEN was capable of inducing G1 cell-cycle arrest as previously reported[19-22], cell-cycle phases were analyzed using a FACS. Cell-cycle analysis demonstrated an increase in the percentage (10-25%) of tumor cells (MGC-803, SGC-7901) in the G2/M population 72 h after treatment with Ad-PTEN (Figure 3) when compared to control cells treated with Ad-Luc, PBS, or exposed to 20 μmol/L LY294002. However, tumor cells treated with LY294002 were arrested at the G1 phase since LY294002 was a PI3K inhibitor. Although one of the PTEN functions was PI3K inhibition, like LY294002[15,20,23], Ad-PTEN did not induce G1 arrest in cancer cells carrying wtPTEN.
Figure 3.
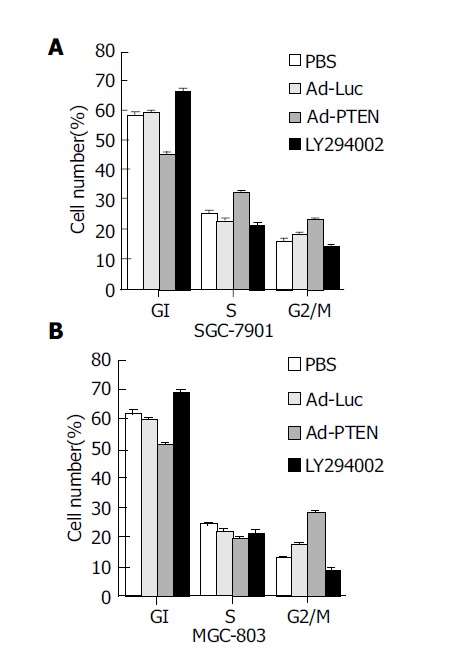
Induction of G2/M cell-cycle arrest due to overexpression of PTEN. (A: SGC-7901 cells; B: MGC-803 cells.) Bars denote standard error (SE).
Signaling pathways modulated by PTEN overexpression in gastric cancer cells
To investigate if PI3K downstream signaling was involved in PTEN-mediated growth suppression, we examined the expression of Akt which was downstream of the Akt kinase pathway and transcriptional regulators leading to apoptotic cell death[24], in tumor cells (SGC-7901) and normal cells (GES-1) by Western blot analysis (Figure 4). In SGC-7901 cells, phospho-Akt was strongly dephosphorylated by Ad-PTEN and LY294002 (PI3K inhibitor). The reduction in Akt phosphorylation signal was not due to the decrease of phosphorylated and unphosphorylated forms of Akt (Figure 4A). In contrast, no significant change in pAkt was observed in Ad-PTEN-treated GES-1 cells compared to control cells (Figure 4B). These results demonstrated that PTEN downregulated the Akt-kinase pathway and suppressed cell growth and proliferation in cancer cells, but not in normal cells carrying wtPTEN.
Figure 4.
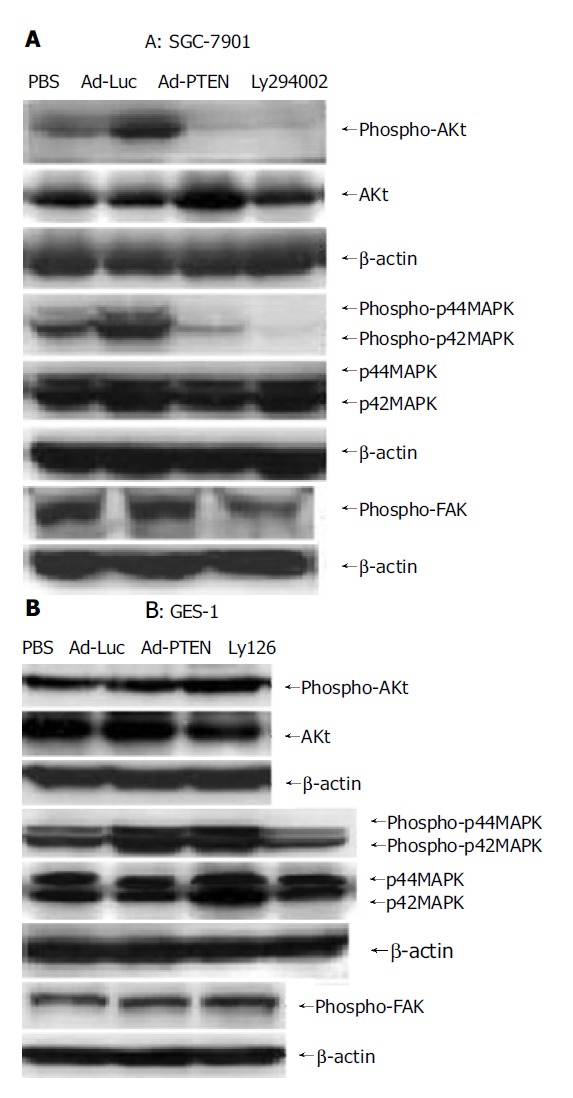
Signaling pathways regulated by PTEN overexpression. A: Downregulation of phospho-Akt, phospho-FAK, and inhibition of phospho-p44/42MAPK in SGC-7901 cells after Ad-PTEN treatment compared to Ad-Luc- and PBS-treated cells; B: No change in the expression levels of phospho-Akt, phospho-FAK, and phospho-p44/42MAPK observed in GES-1 cells after Ad-PTEN treatment compared to Ad-Luc- and PBS-treated cells.
We also examined the phosphorylation status of focal adhesion kinase (pFAK) and p44/42MAPK associated with cell migration and/or focal adhesions in SGC-7901 and GES-1 cells 72 h after infection[25-28]. In SGC-7901 cells, pFAK and p44/42MAPK were dephosphorylated by Ad-PTEN, but not by Ad-Luc or PBS (Figure 5A). Ly126, an MEK 1/2 inhibitor, also inhibited p44/42MAPK and served as a positive control. No changes in pFAK and p44/42MAPK expression were observed in Ad-PTEN-treated GES-1 cells compared to cells treated with Ad-Luc and PBS (Figure 5B). These results demonstrated that PTEN could selectively inhibit cell migration and adhesion through downregulation of the FAK and mitogen-activated protein kinase (MAPK) pathways in cancer cells carrying wtPTEN, but not in normal cells.
Figure 5.
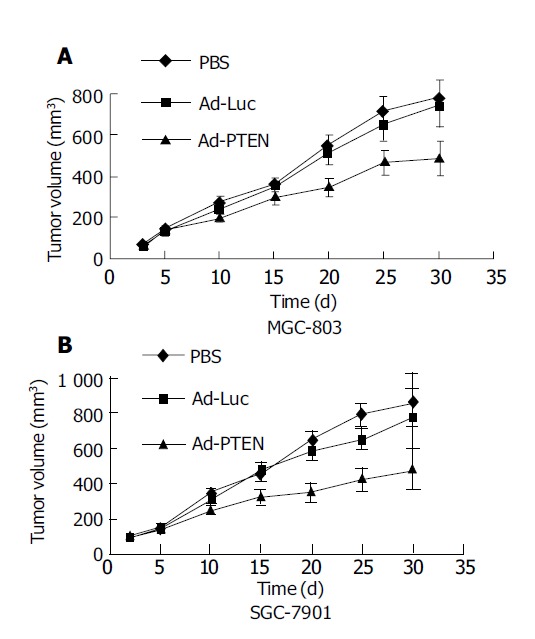
Therapeutic effect of Ad-PTEN on subcutaneous human gastric tumor xenografts. Bars represent standard error (SE). A: MGC-803 cells; B: SGC-7901 cells.
Inhibition of gastric tumor xenografts by PTEN
The ability of Ad-PTEN to inhibit subcutaneous tumor xenografts was evaluated next. Subcutaneous colorectal tumor xenografts established in nude mice were divided into groups (n = 9/group) and treated as follows. Group 1 received PBS, group 2 received Ad-Luc, and group 3 received Ad-PTEN. Intratumoral injections of Ad-PTEN on SGC-7901 and MGC-803 tumor xenografts resulted in a significant (P<0.01) growth inhibition compared to control tumors that were treated with PBS or treated with Ad-Luc (Figures 5A and 5B). However, no complete tumor regression and no treatment-related toxicity were observed during the course of the experiment.
DISCUSSION
In the present study, we investigated the effects of overexpressing PTEN proteins in gastric cancer cells that were wtPTEN. Prior to the start of the study, we examined the mutational status of PTEN gene in the cancer cell lines used in the study. Genetic alterations were determined by sequencing the coding region using exon-specific primers. Both cancer cell lines were sequenced and found to contain no mutations in this gene. Furthermore, we showed that overexpression of PTEN suppressed the growth of cancer cells and induced apoptosis in gastric cancer cell lines significantly compared to normal cells. These results are similar to the findings previously reported in ovarian and thyroid cancers that were wtPTEN[18,19]. However, one major difference observed in our results from others is that we observed that gastric tumor cells that were wtPTEN were arrested in G2/M phase after Ad-PTEN treatment. In contrast, previous studies reported that ectopic expression of PTEN in cancer cells resulted in G1 arrest that was associated with an increase in p27 expression[15,16,19,29-31]. Analysis of p27 expression in gastric cancer cells after Ad-PTEN treatment demonstrated no change in expression levels (data not shown). In support of our findings, the recent report by Stewart et al[20], demonstrated that enforced expression of PTEN inhibited growth of melanoma cells that were wtPTEN. The discrepancy in the results is not clear. However, one possibility is that the induction of cell-cycle arrest by PTEN may be cell-type-dependent. Alternatively, differences in the mutational changes in different cancers may contribute to the determination of cell-cycle phases. In fact, as demonstrated in the present study, overexpression of PTEN in gastric cancers that were wtPTEN underwent G2 arrest, while tumor cells that were mutants for PTEN underwent G1 arrest. These results indicate that the endogenous status of PTEN may play a role in the cell cycle arrest and warrants further investigation. Whatever the underlying mechanism, it is evident from the present study that the ability of PTEN to inhibit tumor growth is independent of the mutational status of PTEN in cancer cells and is in agreement with the findings previously reported by Li et al[32].
The mechanism by which PTEN suppresses cell growth and induces apoptosis appears to be dependent upon its phosphatase activity. Requirement of the PTEN phosphatase activity to downregulate the Akt, FAK, and p44/42MAPK pathways has been previously demonstrated in tumor cells that were mutants or null for PTEN[25,26,28,33-35]. The results from the present study agree with these reports. Treatment of gastric cancer cells with Ad-PTEN resulted in the inhibition of Akt, FAK, and p44/42MAPK pathways. However, Ad-PTEN treatment in normal cells did not result in the regulation of these pathways, which may explain their inability to undergo apoptotic cell death, thereby achieving tumor-selective killing. The ability of tumor cells, but not normal cells that are wtPTEN, to undergo apoptotic cell death is in agreement with the findings of Li et al[32], who demonstrated that colorectal cancer cells that were wtPTEN were sensitive to PTEN, but not normal cells. It is also possible that tumor cells have other signaling pathways that are defective, result in triggering of apoptotic cell death upon PTEN expression. An alternate explanation can be that the endogenous PTEN expressed in tumor cell lines, but not in normal cells, is not functional. To gain an insight into this possibility, further investigation is warranted.
Finally, the ability of Ad-PTEN to suppress the growth of xenograft tumors (MGC-803, SGC-7901) was investigated. Ad-PTEN treatment inhibited tumor growth significantly, supporting our in vitro findings presented here. The ability of Ad-PTEN to inhibit human gastric tumor growth that wtPTEN has not been previously documented. In conclusion, Ad-PTEN inhibits gastric cancer growth, both in vitro and in vivo. Thus, adenovirus-mediated transfer of the PTEN gene may be a promising therapeutic strategy for the treatment of patients with gastric cancer.
Footnotes
Supported by the National Natural Science Foundation of China, No. 39970229
References
- 1.Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 2.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 3.Rasheed BK, Stenzel TT, McLendon RE, Parsons R, Friedman AH, Friedman HS, Bigner DD, Bigner SH. PTEN gene mutations are seen in high-grade but not in low-grade gliomas. Cancer Res. 1997;57:4187–4190. [PubMed] [Google Scholar]
- 4.Wang SI, Puc J, Li J, Bruce JN, Cairns P, Sidransky D, Parsons R. Somatic mutations of PTEN in glioblastoma multiforme. Cancer Res. 1997;57:4183–4186. [PubMed] [Google Scholar]
- 5.Tashiro H, Blazes MS, Wu R, Cho KR, Bose S, Wang SI, Li J, Parsons R, Ellenson LH. Mutations in PTEN are frequent in endometrial carcinoma but rare in other common gynecological malignancies. Cancer Res. 1997;57:3935–3940. [PubMed] [Google Scholar]
- 6.Cairns P, Okami K, Halachmi S, Halachmi N, Esteller M, Herman JG, Jen J, Isaacs WB, Bova GS, Sidransky D. Frequent inactivation of PTEN/MMAC1 in primary prostate cancer. Cancer Res. 1997;57:4997–5000. [PubMed] [Google Scholar]
- 7.Yokomizo A, Tindall DJ, Drabkin H, Gemmill R, Franklin W, Yang P, Sugio K, Smith DI, Liu W. PTEN/MMAC1 mutations identified in small cell, but not in non-small cell lung cancers. Oncogene. 1998;17:475–479. doi: 10.1038/sj.onc.1201956. [DOI] [PubMed] [Google Scholar]
- 8.Nelen MR, van Staveren WC, Peeters EA, Hassel MB, Gorlin RJ, Hamm H, Lindboe CF, Fryns JP, Sijmons RH, Woods DG, et al. Germline mutations in the PTEN/MMAC1 gene in patients with Cowden disease. Hum Mol Genet. 1997;6:1383–1387. doi: 10.1093/hmg/6.8.1383. [DOI] [PubMed] [Google Scholar]
- 9.Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z, Bose S, Call KM, Tsou HC, Peacocke M, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. 1997;16:64–67. doi: 10.1038/ng0597-64. [DOI] [PubMed] [Google Scholar]
- 10.Marsh DJ, Dahia PL, Zheng Z, Liaw D, Parsons R, Gorlin RJ, Eng C. Germline mutations in PTEN are present in Bannayan-Zonana syndrome. Nat Genet. 1997;16:333–334. doi: 10.1038/ng0897-333. [DOI] [PubMed] [Google Scholar]
- 11.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 12.Myers MP, Stolarov JP, Eng C, Li J, Wang SI, Wigler MH, Parsons R, Tonks NK. P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc Natl Acad Sci USA. 1997;94:9052–9057. doi: 10.1073/pnas.94.17.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 14.Cheney IW, Johnson DE, Vaillancourt MT, Avanzini J, Morimoto A, Demers GW, Wills KN, Shabram PW, Bolen JB, Tavtigian SV, et al. Suppression of tumorigenicity of glioblastoma cells by adenovirus-mediated MMAC1/PTEN gene transfer. Cancer Res. 1998;58:2331–2334. [PubMed] [Google Scholar]
- 15.Li DM, Sun H. PTEN/MMAC1/TEP1 suppresses the tumorigenicity and induces G1 cell cycle arrest in human glioblastoma cells. Proc Natl Acad Sci USA. 1998;95:15406–15411. doi: 10.1073/pnas.95.26.15406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Furnari FB, Huang HJ, Cavenee WK. The phosphoinositol phosphatase activity of PTEN mediates a serum-sensitive G1 growth arrest in glioma cells. Cancer Res. 1998;58:5002–5008. [PubMed] [Google Scholar]
- 17.Davies MA, Lu Y, Sano T, Fang X, Tang P, LaPushin R, Koul D, Bookstein R, Stokoe D, Yung WK, et al. Adenoviral transgene expression of MMAC/PTEN in human glioma cells inhibits Akt activation and induces anoikis. Cancer Res. 1998;58:5285–5290. [PubMed] [Google Scholar]
- 18.Minaguchi T, Mori T, Kanamori Y, Matsushima M, Yoshikawa H, Taketani Y, Nakamura Y. Growth suppression of human ovarian cancer cells by adenovirus-mediated transfer of the PTEN gene. Cancer Res. 1999;59:6063–6067. [PubMed] [Google Scholar]
- 19.Weng LP, Gimm O, Kum JB, Smith WM, Zhou XP, Wynford-Thomas D, Leone G, Eng C. Transient ectopic expression of PTEN in thyroid cancer cell lines induces cell cycle arrest and cell type-dependent cell death. Hum Mol Genet. 2001;10:251–258. doi: 10.1093/hmg/10.3.251. [DOI] [PubMed] [Google Scholar]
- 20.Stewart AL, Mhashilkar AM, Yang XH, Ekmekcioglu S, Saito Y, Sieger K, Schrock R, Onishi E, Swanson X, Mumm JB, et al. PI3 kinase blockade by Ad-PTEN inhibits invasion and induces apoptosis in RGP and metastatic melanoma cells. Mol Med. 2002;8:451–461. [PMC free article] [PubMed] [Google Scholar]
- 21.Gottschalk AR, Basila D, Wong M, Dean NM, Brandts CH, Stokoe D, Haas-Kogan DA. p27Kip1 is required for PTEN-induced G1 growth arrest. Cancer Res. 2001;61:2105–2111. [PubMed] [Google Scholar]
- 22.Weng LP, Brown JL, Eng C. PTEN coordinates G(1) arrest by down-regulating cyclin D1 via its protein phosphatase activity and up-regulating p27 via its lipid phosphatase activity in a breast cancer model. Hum Mol Genet. 2001;10:599–604. doi: 10.1093/hmg/10.6.599. [DOI] [PubMed] [Google Scholar]
- 23.Taylor V, Wong M, Brandts C, Reilly L, Dean NM, Cowsert LM, Moodie S, Stokoe D. 5' phospholipid phosphatase SHIP-2 causes protein kinase B inactivation and cell cycle arrest in glioblastoma cells. Mol Cell Biol. 2000;20:6860–6871. doi: 10.1128/mcb.20.18.6860-6871.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu Y, Lin YZ, LaPushin R, Cuevas B, Fang X, Yu SX, Davies MA, Khan H, Furui T, Mao M, et al. The PTEN/MMAC1/TEP tumor suppressor gene decreases cell growth and induces apoptosis and anoikis in breast cancer cells. Oncogene. 1999;18:7034–7045. doi: 10.1038/sj.onc.1203183. [DOI] [PubMed] [Google Scholar]
- 25.Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science. 1998;280:1614–1617. doi: 10.1126/science.280.5369.1614. [DOI] [PubMed] [Google Scholar]
- 26.Gu J, Tamura M, Yamada KM. Tumor suppressor PTEN inhibits integrin- and growth factor-mediated mitogen-activated protein (MAP) kinase signaling pathways. J Cell Biol. 1998;143:1375–1383. doi: 10.1083/jcb.143.5.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tamura M, Gu J, Takino T, Yamada KM. Tumor suppressor PTEN inhibition of cell invasion, migration, and growth: differential involvement of focal adhesion kinase and p130Cas. Cancer Res. 1999;59:442–449. [PubMed] [Google Scholar]
- 28.Gu J, Tamura M, Pankov R, Danen EH, Takino T, Matsumoto K, Yamada KM. Shc and FAK differentially regulate cell motility and directionality modulated by PTEN. J Cell Biol. 1999;146:389–403. doi: 10.1083/jcb.146.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Furnari FB, Lin H, Huang HS, Cavenee WK. Growth suppression of glioma cells by PTEN requires a functional phosphatase catalytic domain. Proc Natl Acad Sci USA. 1997;94:12479–12484. doi: 10.1073/pnas.94.23.12479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robertson GP, Furnari FB, Miele ME, Glendening MJ, Welch DR, Fountain JW, Lugo TG, Huang HJ, Cavenee WK. In vitro loss of heterozygosity targets the PTEN/MMAC1 gene in melanoma. Proc Natl Acad Sci USA. 1998;95:9418–9423. doi: 10.1073/pnas.95.16.9418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheney IW, Neuteboom ST, Vaillancourt MT, Ramachandra M, Bookstein R. Adenovirus-mediated gene transfer of MMAC1/PTEN to glioblastoma cells inhibits S phase entry by the recruitment of p27Kip1 into cyclin E/CDK2 complexes. Cancer Res. 1999;59:2318–2323. [PubMed] [Google Scholar]
- 32.Li J, Simpson L, Takahashi M, Miliaresis C, Myers MP, Tonks N, Parsons R. The PTEN/MMAC1 tumor suppressor induces cell death that is rescued by the AKT/protein kinase B oncogene. Cancer Res. 1998;58:5667–5672. [PubMed] [Google Scholar]
- 33.Nakamura N, Ramaswamy S, Vazquez F, Signoretti S, Loda M, Sellers WR. Forkhead transcription factors are critical effectors of cell death and cell cycle arrest downstream of PTEN. Mol Cell Biol. 2000;20:8969–8982. doi: 10.1128/mcb.20.23.8969-8982.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dijkers PF, Medema RH, Pals C, Banerji L, Thomas NS, Lam EW, Burgering BM, Raaijmakers JA, Lammers JW, Koenderman L, et al. Forkhead transcription factor FKHR-L1 modulates cytokine-dependent transcriptional regulation of p27(KIP1) Mol Cell Biol. 2000;20:9138–9148. doi: 10.1128/mcb.20.24.9138-9148.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu Y, Lin YZ, LaPushin R, Cuevas B, Fang X, Yu SX, Davies MA, Khan H, Furui T, Mao M, et al. The PTEN/MMAC1/TEP tumor suppressor gene decreases cell growth and induces apoptosis and anoikis in breast cancer cells. Oncogene. 1999;18:7034–7045. doi: 10.1038/sj.onc.1203183. [DOI] [PubMed] [Google Scholar]