

Abstract
AIM: The mechanism of decreased vascular reactivity to vasoconstrictors in portal hypertension is still unclear. In addition to nitric oxide, defects in post-receptor signal transduction pathway have been suggested to play a role. However, substantial evidences observed equivocal changes of vascular reactivity following different agonists that challenged the hypothesis of the post-receptor defect. The current study was to evaluate the vascular reactivity to different agonists and the inositol trisphosphate (IP3) changes in signal transduction cascade from cirrhotic rats with portal hypertension.
METHODS: The endothelial denuded aortic rings from cirrhotic and sham-operated rats were obtained for ex vivo tension study and measurement of the corresponding [3H] IP3 formation following different receptor and nonreceptor-mediated agonists’ stimulation. Additionally, iNOS protein expression was measured in thoracic aorta. The contractile response curves to phenylephrine were performed in endothelial denuded aortic rings with and without preincubation with a specific iNOS inhibitor (L-N(6)-(1-iminoethyl)-lysine, L-NIL).
RESULTS: In endothelial denuded aortic rings of cirrhotic rats, the vascular responses were reduced with phenylephrine and arginine vasopressin (AVP) stimulation but were normal with U-46619, NaF/AlCl3, and phorbol esterdibutyrate (PdBU) stimulation. Compared to the corresponding control groups, the degree of the increment of [3H] IP3 formation from basal level was also decreased with phenylephrine and AVP stimulation, but was normal with U-46619 and NaF/AlCl3 stimulation. The preincubation with L-NIL did not modify the hyporesponsiveness to phenylephrine. Additionally, the iNOS protein expression in thoracic aorta was not different in cirrhotic and sham-operated rats.
CONCLUSION: Without the influence of nitric oxide, vascular hyporeactivity to vasoconstrictors persisted in cirrhotic rats with portal hypertension. However, the decreased vascular reactivity is an agonist-specific phenomenon. In addition, G-protein and phospholipase C pathway associated with the IP3 productions may be intact in cirrhotic rats with portal hypertension.
Keywords: Cirrhosis, Portal hypertension, Inositol trisphosphate, Vascular reactivity, Protein kinase C
INTRODUCTION
Peripheral arterial vasodilatation is a characteristic hemodynamic derangement observed in cirrhosis with portal hypertension[1-4]. Increased circulating vasodilators and endothelial-related vasodilatory activity have been suggested to play a role for the development of arterial vasodilatation[5-11]. Additionally, the decreased vascular reactivity to vasoconstrictors also contributes to the arterial vasodilatation in portal hypertension[12-18]. The vasoconstrictors used in those studies to evaluate vascular reactivity were mainly α1-adrenoceptor agonists, arginine vasopressin (AVP), and angiotensin II. Although increased nitric oxide (NO) production has been shown to play a role, the mechanisms of decreased vascular reactivity have not been completely established and the data on vascular reactivity in portal hypertensive humans and animals are conflicting (for reviews see Hadoke and Hayes[19]).
It has been hypothesized that the presence of a defect in the post-receptor signal transduction cascade plays, in part, a role for the vascular hyporeactivity[14,16,20,21]. Theoretically, if the post-receptor signal transduction cascade is generally impaired in portal hypertension, any vasoconstrictor that leads to a receptor and G-protein-linked stimulation of vascular contraction should lead to a decreased vascular contractility. However, two recent studies in portal hypertensive patients and animals showed a normal vascular reactivity to the agonist of thromboxane A2 (TXA2) receptor[22,23]. In addition, an increased vascular response to 5-hydroxytryptamine stimulation was found in cirrhotic patients[24]. The results from these studies do not support the presence of a defect in the post-receptor signal transduction cascade in portal hypertension. Therefore, the current study was undertaken in cirrhotic rats with portal hypertension to measure the vascular contractile response, without the influence of NO, following administration of different agonists. The inositol trisphosphate (IP3) formations following agonists’ stimulation and the responses to protein kinase C (PKC) activator were also measured to evaluate the post-receptor mechanism of vascular reactivity in cirrhotic portal hypertension.
MATERIALS AND METHODS
Animals
Adult male Sprague-Dawley rats (250-350 g) were used in all experiments. Cirrhosis with portal hypertension was produced by common bile duct ligation (CBL), as previously described[25]. Sham-operated rats had their bile duct exposed but not ligated. All rats were caged at 24 °C, with a 12-h light-dark cycle, and allowed free access to food and water. Animal studies were approved by the Animal Experiment Committee of the University and conducted humanely.
Hemodynamic measurements
Four to five weeks after surgery, rats were anesthetized with ketamine, 150 mg/kg. The first set of eight cirrhotic and six sham-operated rats were included in this experiment. The ileocolic vein was cannulated with PE-10 tubing for measuring portal venous pressure (PP) and the femoral artery was cannulated with PE-50 tubing for arterial blood pressure (MAP). Pressure and heart rate were monitored by a polygraph (RS 3400, Gould, Valley View, OH, USA) via strain-gauge transducers (P23XL, Viggo-Spectramed, Oxnard, CA, USA). Cardiac output was measured by thermodilution (Columbus Instruments, OH, USA), and cardiac index (CI) and systemic vascular resistance (SVR) were calculated as previously described[26,27].
Ex vivo tension experiments
The second set of CBL and sham-operated rats were used in this study. Rats were killed by an overdose of sodium pentobarbital. Then the thoracic aorta (above the diaphragm and below the aortic arch) was excised and put into aerated (mixture of 95% O2 5% and CO2) kreb’s Ringer bicarbonate solution (KRBS) and cleaned at room temperature. The composition of the KRBS was in millimolar: NaCl, 118.3; KCl, 4.7; CaCl2, 2.5; MgSO4, 1.2; KH2PO4, 1.2; NaHCO3, 25; and glucose, 11.1. The thoracic aorta was cut into 4-mm segments. In order to avoid the effect of endothelium on vascular contractility, the endothelium was removed by gently rubbing the intimal surface of the vessels. Endothelial cell removal was confirmed by the inability of rings pre-contracted with 0.1 μmol/L phenylephrine (PEP) to relax in response to 1 μmol/L acetylcholine. The rings were then suspended horizontally between two stainless steel wires in individual organ chambers filled with 10 mL of KRBS. The solution was continuously bubbled with a mixture of 95% O2 5% and CO2 and maintained at 37 °C with an outer water jacket and circulating heat pump. One wire was fixed to the chamber and the other was attached to a force displacement transducer (FT 03, Grass Instrument Co., Quincy, MA, USA) with a basic tension of 1.8 g. Tension was recorded on a physiograph (RS 3400, Gould, Valley View, OH, USA). The readiness of tissue indicated by consistent responses induced by three consecutive tests with KCl (30-90 mmol/L) in each group was obtained. The tissue was then rinsed and allowed to recover for 45 min. Segments of aorta from each animal were allocated into five subsets. Then, the cumulative concentration-response curves to PEP (10-10 to 10-4 mol/L), AVP (10-10 to 10-4 mol/L), synthetic TXA2 analog U-46619 (10-9 to 10-5 mol/L), receptor-independent G-protein activator by NaF (10-3 to 1 mol/L)/AlCl3 (30 μmol/L), direct activation of PKC by phorbol esterdibutyrate (PdBU) (10-8 to 3×10-5 mol/L) were obtained, respectively.
[3H]1,4,5 Inositol trisphosphate assay
The third set of endothelial-denuded aortic segments were weighted (4 mm in length) and incubated in 0.5 mL KRBS containing LiCl (20 mmol/L) and imipramine (4 μmol/L) at 37 °C under a mixture of 95% O2 5% and CO2 for 10 min. Then, agonists [PEP (10-7 to 10-5 mol/L), AVP (10-7 to 10-5 mol/L), U-46619 (10-7 and 10-6 mol/L), NaF/AlCl3 (0.1 and 1 mol/L)] or vehicle (0.1% ascorbic acid) were added into the tissue bath for 20 min. These concentrations of agonists were chosen according to the concentration-response contractile curve. We used the highest two or three concentrations of agonist to assess their corresponding IP3 formations under the same experimental condition. The reaction was stopped with 2 mL of CH3OH-CHCl3-HCl (40:20:1, v/v). The assay method was essentially the same as previously reported[28-30]. To extract inositol phosphates, the tissue was sonicated for 45 min and a mixture of 1.26 mL of H2O and 0.63 mL of CHCl3 was added to separate the organic and aqueous phases. Tubes were centrifuged at 2500 r/min for 10 min. The supernatant (aqueous phase containing extracted inositol phosphates) was removed, neutralized to pH 6.8 and 7.2. The neutralized supernatant was added to 300-500 μL of a 1:1 (v/v) mixture of Freon (1, 1, 2-trichlorotrifluoroethane) and tri-n-octylamine, and vortex-mix the separate phases. This neutralized mixture was centrifuged for 10 min at 2000 r/min. Then three separated phases can be found. These three phases are upper phase: neutralized sample plus water soluble components; middle phase: tri-n-octylamine perchlorate; lower phase: Freon plus unrecalled tri-n-octylamine. Then the upper phase was removed and stored in -70 °C for subsequent analysis. The IP3 formations with or without stimulation of agonist were measured in all samples by the commercial kit of radioimmunoassay (RIA) (PerkinElmerTM Life Sciences, Inc., Boston, MA, USA). The radioactivity of these samples after RIA was counted in a β-counter (LS 6500, Beckman Instruments, Fullerton, CA, USA).
Vascular reactivity with and without [L-N(6)-(1-iminoethyl)-lysine, L-NIL] preincubation and iNOS protein measurement
In order to clarify the role of iNOS on the decreased vascular reactivity, the fourth set CBL and sham-operated rats were killed by an overdose of sodium pentobarbital. The endothelial-denuded aorta segments were prepared as described above. Then, the cumulative concentration-response curves to PEP (10-10 to 10-4 mol/L) were obtained with and without preincubation with L-NIL (10-6 mol/L) in CBL and sham-operated rats. In addition, the iNOS protein expression was measured in the thoracic aorta from CBL and sham-operated rats by Western blot analysis.
Chemicals
All substances were purchased from Sigma Chemical Co. (St. Louis, MO, USA) and added into the organ bath in a volume of 100 μL. PEP was prepared in 0.1% ascorbic acid. U-46619 was dissolved in 10% of methanol. PdBU was dissolved in 10% dimethylsulfoxide. The NaF/AlCl3 acetylcholine chloride, L-NIL (L-N(6)-(1-iminoethyl)-lysine) and AVP were dissolved in distilled water. All these solvents had proved not to affect vascular tone in the organ bath.
Statistical analysis
The data are given as mean±SE. Contractile force is expressed in grams (g). Cumulative concentration-response curves to PEP, AVP, U-46619, NaF/AlCl3, and PdBU were obtained. pEC50 (negative logarithm of the concentration producing the half maximum effect) values were calculated from the sigmoid logistic curves in each vessel preparation. Statistical analysis was performed by two-way ANOVA. Additionally, values for maximal contraction (Rmax) were calculated in absolute values. Statistical analysis of the differences between Rmax of the aortic rings from rats with cirrhosis and normal were performed with unpaired Student’s t test. Dose-response curves were analyzed by repeat measures ANOVA followed by post hoc test (Games and Howell variant of the Tukey and t test) to compare the groups at each dose separately. The tissue (wet) weight of each segment was weighed in each experiment. The signal intensity (integral volume) of the appropriate iNOS protein bands on the autoradiogram was analyzed by use of the ImageQuant software package (Biosoft, Indianapolis, IN, USA). Basal IP3 formation was calculated as counts per minute per mg wet weight of tissue (cpm/mg). Agonist-stimulated IP3 formation was calculated as a percentage of cpm in the presence of agonist divided by cpm without agonist (basal formation). For each assay, both basal and agonist-stimulated tubes were prepared in duplicates and the means of duplicates were used for analysis. A P value <0.05 was considered statistically significant. Statistical analysis of the differences was performed with paired or unpaired Student’s t test when appropriate.
RESULTS
Hemodynamic studies
About 4 wk after CBL, cirrhotic rats showed jaundice, splenomegaly, mesenteric edema and variable amount of ascites. In Table 1, CBL rats had significantly lower SVR associated with higher CI and PP than sham-operated rats.
Table 1.
Hemodynamic values in sham-operated (sham) and common bile duct-ligated (CBL) rats.
| Sham (n = 6) | CBL (n = 8) | |
| Cardiac index (mL·min/100 g body wt) | 29.4±0.6 | 46.7±0.7a |
| Mean arterial pressure (mmHg) | 107±5 | 97±10 |
| Systemic vascular resistance | 289±9 | 167±7a |
| (dyn·s/cm5×103/100 g body wt) | ||
| Heart rate (beats/min) | 315±6 | 327±10 |
| Portal pressure (mmHg) | 7.9±1.3 | 19.8±0.6a |
Values are mean±SE.
P<0.05 vs sham rats.
Ex vivo contractile responses in thoracic aorta
Both PEP (10-10 to 10-4 mol/L) and AVP (10-10 to 10-4 mol/L) induced concentration-dependent contractions in the aorta from CBL and sham-operated rats. Our results demonstrated that the Rmax, but not pEC50, to PEP and AVP was significantly decreased in the aorta of CBL rats compared to those of sham-operated rats (Table 2). These results showed a decreased vascular reactivity and a normal vascular sensitivity to PEP and AVP of aorta in CBL rats (Figure 1 and Table 2). Similarly, U-46619, NaF/AlCl3, and PdBU also induced the concentration-dependent contractions curve in both study groups (Figure 2). However, neither the Rmax nor its pEC50 values differed between the two vessel groups (Table 2).
Table 2.
Contractile response of different agents to the endothelial-denuded aortic rings from sham and CBL rats.
| Vasopressor | Parameter | Sham | CBL |
| PEP | pEC50 | 6.41±0.1 | 6.53±0.8 |
| Rmax (g) (10-5 mol/L) | 3.29±0.37 | 1.99±0.21a | |
| AVP | pEC50 | 6.51±0.2 | 7.08±0.9 |
| Rmax (g) (10-5 mol/L) | 3.45±0.28 | 2.31±0.49a | |
| U-46619 | pEC50 | 7.43±0.13 | 7.81±0.26 |
| Rmax (g) (10-6 mol/L) | 3.18±0.27 | 3.19±0.17 | |
| NaF/AlCl3 | pEC50 | 0.83±0.43 | 1.01±0.01 |
| Rmax (g) (1 mol/L) | 3.68±0.13 | 3.42±0.25 | |
| PdBU | pEC50 | 7.05±0.4 | 7.05±0.7 |
| Rmax (g) (10-6 mol/L) | 3.87±0.14 | 3.92±0.22 |
Values are mean±SE; sham: sham-operated rats; CBL: common bile duct-ligation rats, PEP: phenylephrine, n = 10, in each group with different agonist stimulation,
P<0.05 vs sham rats.
Figure 1.
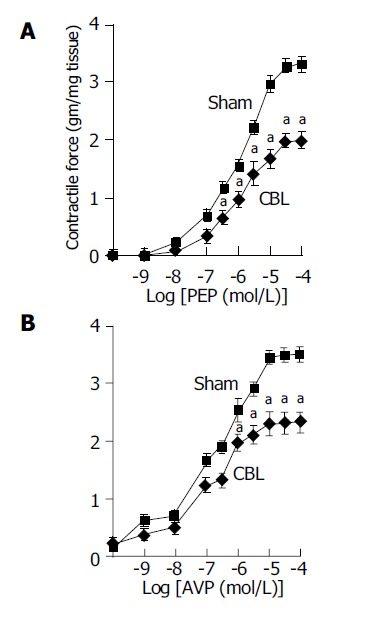
Maximum cumulative concentration-response curves to (A) phenylephrine (PEP) (10-10 to 10-4 mol/L), (B) AVP (10-10 to 10-4 mol/L) in thoracic aorta from CBL (◆) and sham (□) rats. aP<0.05 vs sham rats (n = 12 in each group with different agonist stimulation). Sham: sham-operated rats; CBL: common bile duct-ligated rats.
Figure 2.
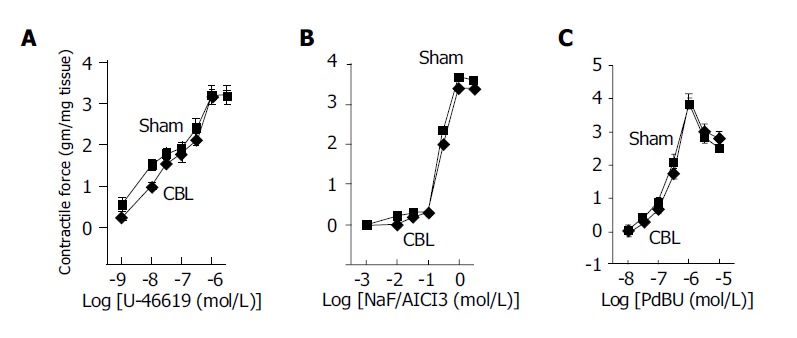
Maximum cumulative concentration-response curves to (A) synthetic TXA2 analog U-46619 (10-9 to 10-5 mol/L), (B) receptor-independent G-protein stimulus NaF (10-3-1 mol/L)/AlCl3 (30 μmol/L), (C) direct activation of protein kinase C by PdBU (10-8 to 3×10-5 mol/L) in thoracic aorta from CBL (◆) and sham-operated (□) rats, n = 10 in each group with different agonist stimulation.
[3H]1,4,5 Inositol trisphosphate (IP3) formation in aortic rings
The basal IP3 formation in aortic rings was similar between the CBL and sham-operated rats (385±6 and 402±10 cpm/mg tissue weight, n = 8 in each group with different agonist stimulation). Both PEP (10-7 to 10-5 mol/L) and AVP (10-7 and 10-5 mol/L) induced dose-dependent increases of [3H] IP3 in both groups. In the presence of PEP and AVP, the increments of [3H] IP3 (% of basal levels) in the CBL rats were significantly lower than that in the sham-operated rats at the concentration of 10-6 and 10-5 mol/L of PEP and 10-7 to 10-5 mol/L of AVP, respectively (Figure 3). In contrast, the percentage of increases after U-46619 (10-7 and 10-6 mol/L) and NaF/AlCl3 (0.1 and 1 mol/L) stimulation were similar between the two groups (Figure 4).
Figure 3.
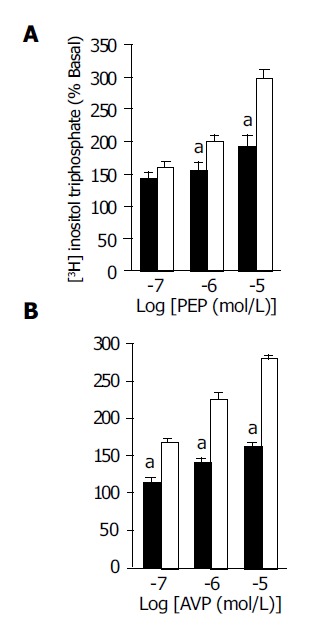
(A) Phenylephrine (PEP) (10-7 to 10-5 mol/L) and (B) AVP (10-7 to 10-5 mol/L)-induced [3H] IP3 formation in the aortic ring from CBL (black bars) and sham (white bars) rats. [3H] IP3 formation was expressed as the percentage of counts (cpm) per minute in the presence of agonist divided by the counts without agonist (basal formation). The data are expressed as mean±SE. aP<0.05 vs sham rats, n = 8 in each group with different agonist stimulation. Sham: sham-operated rats; CBL: common bile duct-ligated rats.
Figure 4.
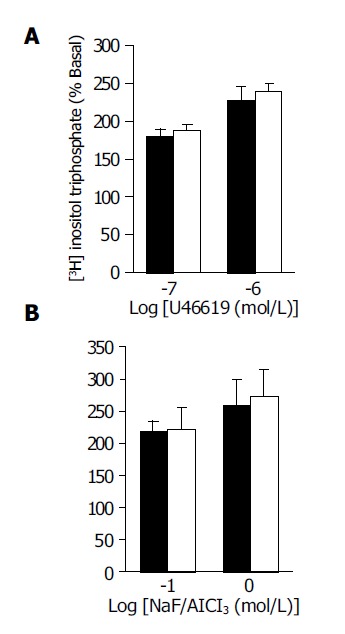
(A) U-46619, a synthetic TXA2 analog (10-7 and 10-6 mol/L), (B) NaF/AlCl3 (0.1 and 1 mol/L)-induced [3H] IP3 formation in the aortic ring from CBL (black bars) and sham (white bars) rats. [3H] IP3 formation was expressed as the percentage of counts (cpm) per minute in the presence of agonist divided by the counts without agonist (basal formation). The data are expressed as mean±SE, n = 8 in each group with different agonist stimulation.
Vascular reactivity with and without L-NIL preincubation and iNOS protein measurement
The effects of selective iNOS inhibition on vascular responses to PEP are shown in Figure 5A. In cirrhotic aortic rings, the vascular hyporeactivity to PEP stimulation still persisted after preincubation with L-NIL. In addition, the iNOS protein expression in the thoracic aorta of BDL rats was not different from sham-operated rats (Figure 5B). These results indicated that iNOS did not play a role in the decreased vascular contractility in portal hypertension.
Figure 5.
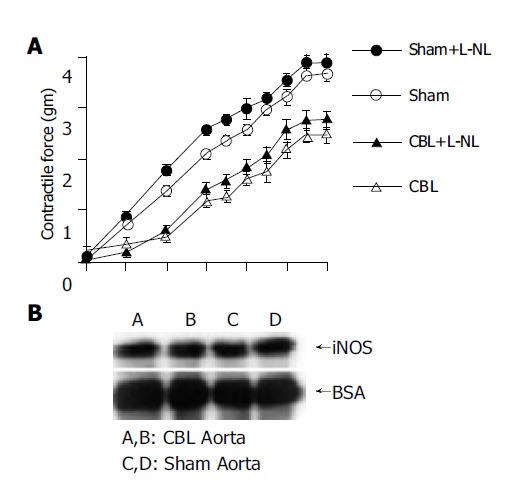
(A) Maximum cumulative concentration-response curves to phenylephrine (PEP) (10-10 to 10-4 mol/L) in thoracic aorta from CBL (◆) and sham (□) rats with and without preincubation with L-NIL (L-N(6)-(1-iminoethyl)-lysine, a selective iNOS inhibitor). n = 10 in each group with different agonist stimulation. CBL: common bile duct-ligated rats; sham: sham-operated rats. (B) Western blot analysis of inducible nitric oxide synthase (iNOS) protein expression from thoracic aorta in CDL and sham-operated rats. Lanes A and B: CBL rats; lanes C and D: sham rats.
DISCUSSION
The mechanism of agonists-induced contraction of vascular smooth muscle cells involves both membrane and intracellular changes[31,32]. Agonists bind to surface G-protein-linked transmembrane receptors may activate phospholipase C (PLC). This enzyme hydrolyzes the lipid precursor phosphatidylinositol (4,5)-biphosphate stored in the plasma membrane to give IP3 and 1,2-diacylglycerol. The IP3 binds to its receptor located in the membrane of the sarcoplasmic reticulum that lead to open the calcium channels and increases the cytosolic calcium concentration. The agonists have also been shown to open L-type calcium channel located on the plasma membrane to increase cytosolic calcium concentration. On the other hand, diacylglycerol activates PKC with regulation of intracellular calcium concentration. The cytosolic free calcium then binds to calmodulim to form calcium-calmodulim complex, which in turn interact with the contractile proteins that lead to smooth muscle cell contraction. Therefore, any alteration in the G-proteins receptor coupling, transmembrane receptor, or the signal transduction downstream from the transmembrane receptor may play a role in the vascular hyporesponsiveness in portal hypertension.
The mechanisms responsible for the decreased vascular reactivity in cirrhosis are not completely understood. In experimental portal hypertension, the increased nitric oxide production has been suggested to play an important role for the vascular hyporeactivity[7,10,26,33]. Although Vallance and Moncada proposed that the increased activity of iNOS led to NO overproduction and subsequent hyperdynamic circulation[34], substantial studies showed a lack of iNOS expression in the vessels from portal hypertensive animals despite the enhanced NO production[35,36]. It is now generally accepted that eNOS rather than iNOS is the major contributor for the NO overproduction in the hemodynamic derangements in portal hypertension (for review see Wiest and Groszmann[37]), and the role of eNOS is further supported in a recent study using gene-knockout mice[38]. Nevertheless, a persistent vascular hyporeactivity to vasoconstrictor stimulation in portal hypertensive human and animals was still observed in endothelium-free aortic ring or following pre-incubation with NOS inhibitor[13,39]. This observation was further supported in endothelial denuded hepatic artery from cirrhotic patients[22,40]. Therefore, in addition to nitric oxide, other factors are also involved in the pathogenesis of vascular hyporeactivity in portal hypertension. A number of studies have found an attenuated vascular reactivity to vasoconstrictors (such as angiotensin II, vasopressin, and α1-adrenoceptor agonists), but these receptors were not down regulated or the receptor numbers and affinities were unchanged[15-18,41]. These studies lead to a hypothesis of a defect in signal transduction cascade in portal hypertension. Accordingly, if the post-receptor defect does exist in portal hypertension, a generalized hyporesponsiveness to any receptor and G-protein linked vasoconstrictors should be observed in cirrhotic human and animals with portal hypertension.
In the current study, we found an expected decrease in contractile responses to PEP (a α1-adrenoceptor agonist) and AVP (a V1-AVP receptor agonist) in the endothelium-denuded aortic ring from cirrhotic rats compared to those from sham-operated rats. By contrast, we found a normal contractile response to U-46619 (a TXA2 receptor agonist) in the aortic rings from cirrhotic rats compared to those from the sham-operated rats. TXA2 receptor is a G-protein-coupled and PLC-linked transmembrane receptor as well as those of α1, V1-AVP or angiotensin II receptors. Van Obbergh et al, and Schepke et al, have demonstrated similar observations of a normal contractile response to U-46619 in cirrhotic rats and humans[22,23]. In addition, another study in cirrhotic patients also showed an increased vascular response following 5-hydroxytryptamine stimulation[24]. Taken together, it is conceivable that the vascular hyporeactivity to vasoconstrictors in portal hypertension is a selective agonist-mediated phenomenon. In other words, decreased vascular reactivity in portal hypertension may occur in some agonists but do not occur in others. Please note that the vascular reactivity in the current study was performed in endothelium-denuded aortic rings from cirrhotic rats. Similar to previous observations, we also observed a lack of the role of iNOS on the vascular hyporesponsiveness in portal hypertension evidenced by both ex vivo vascular tension study with L-NIL preincubation and aortic iNOS protein expression[35,36]. Therefore, the role of NO on vascular reactivity has been minimized to the least extent. Moreover, we found a normal contractile response by NaF/AlCl3 from the aortic ring of cirrhotic rats compared to those from sham-operated rats. A similar observation showing a normal contractile response to NaF/AlCl3 in the endothelium-denuded hepatic artery in cirrhotic patients was reported by Schepke et al[22]. Previously, an impairment of contractile response to NaF/AlCl3 had been reported in the intestinal microcirculation of portal hypertensive rats[42]. In that study, vascular contractility was evaluated by perfusion of mesenteric beds with intact endothelium. It is possible that the different vascular preparation and the presence of NO may contribute to such discrepancy. Because NaF/AlCl3 is a direct G protein activator, it produces smooth muscle cells’ contraction independent to the receptor and G-protein coupling[31,32]. Our results suggest that the contractile response of aortic smooth muscle cell downstream from G-protein in portal hypertensive humans or animals is similar to that in non-portal hypertensive ones. In other words, it is possible that the signal transduction cascade downstream from G-protein activation may be intact in cirrhotic rats with portal hypertension. However, the current study cannot exclude the minor G-protein-related RhoA/Rho kinase pathway. Further studies are needed to clarify this point.
Phosphoinositide metabolism is important in the signal transduction cascade for receptor-coupled vasoconstriction[31,43]. Its hydrolyzed products, IP3 and 1,2-diacylglycerol, are crucial second messengers for triggering and maintaining the contractile response of the vascular smooth muscle cells[31,44]. In the current study, we observed that the increased formation of IP3 after PEP and AVP stimulation in cirrhotic rats was significantly lower than that in sham-operated rats. In contrast, the increased formation of IP3 after U-46619 and NaF/AlCl3 stimulation was similar between the two experimental groups. The changes in IP3 formation following different agonists’ stimulation were parallel to the corresponding changes of vascular contractile response between cirrhotic and normal rats. In our previous study, we observed that, in portal hypertensive animals, the PEP-stimulated [3H] inositol phosphate formation in tail artery was attenuated, whereas the NaF/AlCl3-mediated [3H] inositol phosphate formation in portal vein was unaltered[21]. On the other hand, Hartleb et al[45], observed a lack of vascular hyporesponsiveness to L-type calcium channel activator in cirrhotic rats. Moreover, Castro et al[46], found that the intracellular calcium mobilization pathway is preserved in the vascular smooth muscle cells from cirrhotic rats. Together these results strengthened the presence of an intact signal transduction cascade in cirrhosis with portal hypertension that challenged the hypothesis of the post-receptor defect.
PKC plays an essential role in the regulation of vascular smooth muscle cell contraction[47]. In the current study, the PdBU-mediated contractile response was not different between the two experimental groups. The role of PKC in vascular contractility in portal hypertension is controversial. Previous studies have reported that the PdBU-mediated contractile response remains unchanged in aortic rings and mesenteric vascular bed of portal vein stenosed rats, which in line with the findings of the present studies in cirrhotic rats[21,47]. In contrast, other studies have demonstrated that the alteration of PKC plays a partial role for the decreased vascular reactivity in portal hypertension[42,48,49]. However, it has been suggested that overproduction of NO may contribute, in part, a role for the alteration of PKC activity in portal hypertension[50]. Therefore, in the current study, it is possible that a normal PdBU-induced contractile response was observed in endothelial-denuded aortic ring in cirrhotic rats, which suggested a normal PKC activity in cirrhotic rats. All together, the current study suggested that the G-protein and PLC pathway associated with the IP3 and 1,2-diacylglycerol actions were not impaired in cirrhotic rats with portal hypertension. The decreased vascular reactivity observed in the specific receptors may probably result from a defect in receptor-G-protein coupling. Further studies are needed to clarify this phenomenon.
In summary, this study demonstrated that, without the influence of NO, vascular hyporeactivity persisted in CBL rats with portal hypertension. However, the decreased vascular reactivity is an agonist-specific phenomenon. In addition, the current study is against the presence of a post-receptor defect in vascular hyporeactivity observed in portal hypertension.
Footnotes
Supported by the National Science Council of Taiwan, No. NSC91-2314-B-075-129 and the Taipei Veterans General Hospital of Taiwan, China, No. VGH91-28
Science Editor Guo SY Language Editor Elsevier HK
References
- 1.Albillos A, Colombato LA, Groszmann RJ. Vasodilatation and sodium retention in prehepatic portal hypertension. Gastroenterology. 1992;102:931–935. doi: 10.1016/0016-5085(92)90179-3. [DOI] [PubMed] [Google Scholar]
- 2.Schrier RW, Arroyo V, Bernardi M, Epstein M, Henriksen JH, Rodés J. Peripheral arterial vasodilation hypothesis: a proposal for the initiation of renal sodium and water retention in cirrhosis. Hepatology. 1988;8:1151–1157. doi: 10.1002/hep.1840080532. [DOI] [PubMed] [Google Scholar]
- 3.Vorobioff J, Bredfeldt JE, Groszmann RJ. Hyperdynamic circulation in portal-hypertensive rat model: a primary factor for maintenance of chronic portal hypertension. Am J Physiol. 1983;244:G52–G57. doi: 10.1152/ajpgi.1983.244.1.G52. [DOI] [PubMed] [Google Scholar]
- 4.Vorobioff J, Bredfeldt JE, Groszmann RJ. Increased blood flow through the portal system in cirrhotic rats. Gastroenterology. 1984;87:1120–1126. [PubMed] [Google Scholar]
- 5.Benoit JN, Barrowman JA, Harper SL, Kvietys PR, Granger DN. Role of humoral factors in the intestinal hyperemia associated with chronic portal hypertension. Am J Physiol. 1984;247:G486–G493. doi: 10.1152/ajpgi.1984.247.5.G486. [DOI] [PubMed] [Google Scholar]
- 6.Tahan V, Avsar E, Karaca C, Uslu E, Eren F, Aydin S, Uzun H, Hamzaoglu HO, Besisik F, Kalayci C, et al. Adrenomedullin in cirrhotic and non-cirrhotic portal hypertension. World J Gastroenterol. 2003;9:2325–2327. doi: 10.3748/wjg.v9.i10.2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pizcueta P, Piqué JM, Fernández M, Bosch J, Rodés J, Whittle BJ, Moncada S. Modulation of the hyperdynamic circulation of cirrhotic rats by nitric oxide inhibition. Gastroenterology. 1992;103:1909–1915. doi: 10.1016/0016-5085(92)91451-9. [DOI] [PubMed] [Google Scholar]
- 8.Silva G, Navasa M, Bosch J, Chesta J, Pilar Pizcueta M, Casamitjana R, Rivera F, Rodés J. Hemodynamic effects of glucagon in portal hypertension. Hepatology. 1990;11:668–673. doi: 10.1002/hep.1840110421. [DOI] [PubMed] [Google Scholar]
- 9.Sitzmann JV, Li SS, Lin PW. Prostacyclin mediates splanchnic vascular response to norepinephrine in portal hypertension. J Surg Res. 1989;47:208–211. doi: 10.1016/0022-4804(89)90109-1. [DOI] [PubMed] [Google Scholar]
- 10.Sogni P, Moreau R, Gadano A, Lebrec D. The role of nitric oxide in the hyperdynamic circulatory syndrome associated with portal hypertension. J Hepatol. 1995;23:218–224. doi: 10.1016/0168-8278(95)80339-4. [DOI] [PubMed] [Google Scholar]
- 11.Oberti F, Sogni P, Cailmail S, Moreau R, Pipy B, Lebrec D. Role of prostacyclin in hemodynamic alterations in conscious rats with extrahepatic or intrahepatic portal hypertension. Hepatology. 1993;18:621–627. [PubMed] [Google Scholar]
- 12.Braillon A, Cailmail S, Gaudin C, Lebrec D. Reduced splanchnic vasoconstriction to angiotensin II in conscious rats with biliary cirrhosis. J Hepatol. 1993;17:86–90. doi: 10.1016/s0168-8278(05)80526-4. [DOI] [PubMed] [Google Scholar]
- 13.Karatapanis S, McCormick PA, Kakad S, Chin JK, Islam M, Jeremy J, Harry D, McIntyre N, Burroughs AK, Jacobs M. Alteration in vascular reactivity in isolated aortic rings from portal vein-constricted rats. Hepatology. 1994;20:1516–1521. doi: 10.1002/hep.1840200622. [DOI] [PubMed] [Google Scholar]
- 14.Kiel JW, Pitts V, Benoit JN, Granger DN, Shepherd AP. Reduced vascular sensitivity to norepinephrine in portal-hypertensive rats. Am J Physiol. 1985;248:G192–G195. doi: 10.1152/ajpgi.1985.248.2.G192. [DOI] [PubMed] [Google Scholar]
- 15.Liao JF, Yu PC, Lin HC, Lee FY, Kuo JS, Yang MC. Study on the vascular reactivity and alpha 1-adrenoceptors of portal hypertensive rats. Br J Pharmacol. 1994;111:439–444. doi: 10.1111/j.1476-5381.1994.tb14755.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.MacGilchrist AJ, Sumner D, Reid JL. Impaired pressor reactivity in cirrhosis: evidence for a peripheral vascular defect. Hepatology. 1991;13:689–694. [PubMed] [Google Scholar]
- 17.Murray BM, Paller MS. Decreased pressor reactivity to angiotensin II in cirrhotic rats. Evidence for a post-receptor defect in angiotensin action. Circ Res. 1985;57:424–431. doi: 10.1161/01.res.57.3.424. [DOI] [PubMed] [Google Scholar]
- 18.Murray BM, Paller MS. Pressor resistance to vasopressin in sodium depletion, potassium depletion, and cirrhosis. Am J Physiol. 1986;251:R525–R530. doi: 10.1152/ajpregu.1986.251.3.R525. [DOI] [PubMed] [Google Scholar]
- 19.Hadoke PW, Hayes PC. In vitro evidence for vascular hyporesponsiveness in clinical and experimental cirrhosis. Pharmacol Ther. 1997;75:51–68. doi: 10.1016/s0163-7258(97)00022-3. [DOI] [PubMed] [Google Scholar]
- 20.Huang YT, Lin HC, Yu PC, Lee FY, Tsai YT, Lee SD, Yang MC. Decreased vascular reactivity of portal vein in rats with portal hypertension. J Hepatol. 1996;24:194–199. doi: 10.1016/s0168-8278(96)80029-8. [DOI] [PubMed] [Google Scholar]
- 21.Huang YT, Wang GF, Yang MC, Chang SP, Lin HC, Hong CY. Vascular hyporesponsiveness in aorta from portal hypertensive rats: possible sites of involvement. J Pharmacol Exp Ther. 1996;278:535–541. [PubMed] [Google Scholar]
- 22.Schepke M, Heller J, Paschke S, Thomas J, Wolff M, Neef M, Malago M, Molderings GJ, Spengler U, Sauerbruch T. Contractile hyporesponsiveness of hepatic arteries in humans with cirrhosis: evidence for a receptor-specific mechanism. Hepatology. 2001;34:884–888. doi: 10.1053/jhep.2001.28794. [DOI] [PubMed] [Google Scholar]
- 23.Van Obbergh L, Leonard V, Chen H, Xu D, Blaise G. The endothelial and non-endothelial mechanism responsible for attenuated vasoconstriction in cirrhotic rats. Exp Physiol. 1995;80:609–617. doi: 10.1113/expphysiol.1995.sp003871. [DOI] [PubMed] [Google Scholar]
- 24.Islam MZ, Williams BC, Madhavan KK, Hayes PC, Hadoke PW. Selective alteration of agonist-mediated contraction in hepatic arteries isolated from patients with cirrhosis. Gastroenterology. 2000;118:765–771. doi: 10.1016/s0016-5085(00)70146-6. [DOI] [PubMed] [Google Scholar]
- 25.Kountouras J, Billing BH, Scheuer PJ. Prolonged bile duct obstruction: a new experimental model for cirrhosis in the rat. Br J Exp Pathol. 1984;65:305–311. [PMC free article] [PubMed] [Google Scholar]
- 26.Lee FY, Albillos A, Colombato LA, Groszmann RJ. The role of nitric oxide in the vascular hyporesponsiveness to methoxamine in portal hypertensive rats. Hepatology. 1992;16:1043–1048. doi: 10.1002/hep.1840160430. [DOI] [PubMed] [Google Scholar]
- 27.Lin HC, Yang MC, Hou MC, Li SM, Huang YT, Yu PC, Tsai YT, Lee SD. Effects of long-term administration of octreotide in portal vein-stenosed rats. Hepatology. 1996;23:537–543. doi: 10.1002/hep.510230319. [DOI] [PubMed] [Google Scholar]
- 28.Bredt DS, Mourey RJ, Snyder SH. A simple, sensitive, and specific radioreceptor assay for inositol 1,4,5-trisphosphate in biological tissues. Biochem Biophys Res Commun. 1989;159:976–982. doi: 10.1016/0006-291x(89)92204-3. [DOI] [PubMed] [Google Scholar]
- 29.Huang YT, Yu PC, Lee MF, Lin HC, Hong CY, Yang MC. Decreased vascular contractile and inositol phosphate responses in portal hypertensive rats. Can J Physiol Pharmacol. 1995;73:378–382. doi: 10.1139/y95-048. [DOI] [PubMed] [Google Scholar]
- 30.Challiss RA, Batty IH, Nahorski SR. Mass measurements of inositol(1,4,5)trisphosphate in rat cerebral cortex slices using a radioreceptor assay: effects of neurotransmitters and depolarization. Biochem Biophys Res Commun. 1988;157:684–691. doi: 10.1016/s0006-291x(88)80304-8. [DOI] [PubMed] [Google Scholar]
- 31.Hathaway DR, March KL, Lash JA, Adam LP, Wilensky RL. Vascular smooth muscle. A review of the molecular basis of contractility. Circulation. 1991;83:382–390. doi: 10.1161/01.cir.83.2.382. [DOI] [PubMed] [Google Scholar]
- 32.Somlyo AP, Somlyo AV. Signal transduction and regulation in smooth muscle. Nature. 1994;372:231–236. doi: 10.1038/372231a0. [DOI] [PubMed] [Google Scholar]
- 33.Sieber CC, Groszmann RJ. Nitric oxide mediates hyporeactivity to vasopressors in mesenteric vessels of portal hypertensive rats. Gastroenterology. 1992;103:235–239. doi: 10.1016/0016-5085(92)91118-n. [DOI] [PubMed] [Google Scholar]
- 34.Vallance P, Moncada S. Hyperdynamic circulation in cirrhosis: a role for nitric oxide? Lancet. 1991;337:776–778. doi: 10.1016/0140-6736(91)91384-7. [DOI] [PubMed] [Google Scholar]
- 35.Fernández M, García-Pagán JC, Casadevall M, Bernadich C, Piera C, Whittle BJ, Piqué JM, Bosch J, Rodés J. Evidence against a role for inducible nitric oxide synthase in the hyperdynamic circulation of portal-hypertensive rats. Gastroenterology. 1995;108:1487–1495. doi: 10.1016/0016-5085(95)90698-3. [DOI] [PubMed] [Google Scholar]
- 36.Niederberger M, Ginés P, Martin PY, Tsai P, Morris K, McMurtry I, Schrier RW. Comparison of vascular nitric oxide production and systemic hemodynamics in cirrhosis versus prehepatic portal hypertension in rats. Hepatology. 1996;24:947–951. doi: 10.1002/hep.510240432. [DOI] [PubMed] [Google Scholar]
- 37.Wiest R, Groszmann RJ. The paradox of nitric oxide in cirrhosis and portal hypertension: too much, not enough. Hepatology. 2002;35:478–491. doi: 10.1053/jhep.2002.31432. [DOI] [PubMed] [Google Scholar]
- 38.Theodorakis NG, Wang YN, Skill NJ, Metz MA, Cahill PA, Redmond EM, Sitzmann JV. The role of nitric oxide synthase isoforms in extrahepatic portal hypertension: studies in gene-knockout mice. Gastroenterology. 2003;124:1500–1508. doi: 10.1016/s0016-5085(03)00280-4. [DOI] [PubMed] [Google Scholar]
- 39.Michielsen PP, Boeckxstaens GE, Sys SU, Herman AG, Pelckmans PA. The role of increased nitric oxide in the vascular hyporeactivity to noradrenaline in long-term portal vein ligated rats. J Hepatol. 1995;23:341–347. [PubMed] [Google Scholar]
- 40.Heller J, Schepke M, Gehnen N, Molderings GJ, Müller A, Erhard J, Spengler U, Sauerbruch T. Altered adrenergic responsiveness of endothelium-denuded hepatic arteries and portal veins in patients with cirrhosis. Gastroenterology. 1999;116:387–393. doi: 10.1016/s0016-5085(99)70136-8. [DOI] [PubMed] [Google Scholar]
- 41.MacGilchrist AJ, Deighton NM, Hamilton CA, Reid JL. Binding studies of platelet alpha 2- and lymphocyte beta 2-adrenoceptors in patients with cirrhosis. Br J Clin Pharmacol. 1990;30:644–647. doi: 10.1111/j.1365-2125.1990.tb03828.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu ZY, Benoit JN. Nonreceptor-mediated intestinal vasoconstriction in portal hypertensive rats. Am J Physiol. 1994;267:H370–H375. doi: 10.1152/ajpheart.1994.267.1.H370. [DOI] [PubMed] [Google Scholar]
- 43.Heagerty AM, Ollerenshaw JD. The phosphoinositide signaling system and the pathogenesis of hypertension. In Hypertension: Pathophysiology, diagnosis, and management. Edited by Laragh JH and Brenner BM. Raven Press New York; 1999. pp. 1601–1615. [Google Scholar]
- 44.Rasmussen H, Takuwa Y, Park S. Protein kinase C in the regulation of smooth muscle contraction. FASEB J. 1987;1:177–185. [PubMed] [Google Scholar]
- 45.Hartleb M, Moreau R, Gaudin C, Lebrec D. Lack of vascular hyporesponsiveness to the L-type calcium channel activator, Bay K 8644, in rats with cirrhosis. J Hepatol. 1995;22:202–207. doi: 10.1016/0168-8278(95)80430-7. [DOI] [PubMed] [Google Scholar]
- 46.Castro A, Ros J, Jiménez W, Clària J, Llibre J, Leivas A, Arroyo V, Rivera F, Rodés J. Intracellular calcium concentration in vascular smooth muscle cells of rats with cirrhosis. J Hepatol. 1994;21:521–526. doi: 10.1016/s0168-8278(94)80096-0. [DOI] [PubMed] [Google Scholar]
- 47.Atucha NM, Ortíz MC, Martínez C, Quesada T, García-Estañ J. Role of protein kinase C in mesenteric pressor responses of rats with portal hypertension. Br J Pharmacol. 1996;118:277–282. doi: 10.1111/j.1476-5381.1996.tb15399.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tazi KA, Moreau R, Heller J, Poirel O, Lebrec D. Changes in protein kinase C isoforms in association with vascular hyporeactivity in cirrhotic rat aortas. Gastroenterology. 2000;119:201–210. doi: 10.1053/gast.2000.8522. [DOI] [PubMed] [Google Scholar]
- 49.Trombino C, Tazi KA, Gadano A, Moreau R, Lebrec D. Protein kinase C alterations in aortic vascular smooth muscle cells from rats with cirrhosis. J Hepatol. 1998;28:670–676. doi: 10.1016/s0168-8278(98)80292-4. [DOI] [PubMed] [Google Scholar]
- 50.Chagneau C, Tazi KA, Heller J, Sogni P, Poirel O, Moreau R, Lebrec D. The role of nitric oxide in the reduction of protein kinase C-induced contractile response in aortae from rats with portal hypertension. J Hepatol. 2000;33:26–32. doi: 10.1016/s0168-8278(00)80155-5. [DOI] [PubMed] [Google Scholar]