

Abstract
Cardiomyopathies classification is based on morphological and functional phenotypes and subcategories of familial/genetic and non-familial/non-genetic disease. The non-compaction cardiomyopathy is a rare disorder which is considered to be an unclassified cardiomyopathy according to the ESC Working Group on Myocardial and Pericardial Diseases and the World Health Organization or a primary genetically-determined cardiomyopathy according to the American Heart Association. The diagnosis of non-compaction is challenging and its nosology is debated since this morphological trait can be shared by different cardiomyopathies and non-cardiomyopathy conditions. Myocardial structure has a spectrum from normal variants to the pathological phenotype of non-compaction cardiomyopathy, which reflects the embryonic structure of the human heart due to an arrest in the compaction process during the first trimester. However, when a definite diagnosis of non-compaction is made, the diagnostic process should orient towards a genetic disease with a relatively high probability of sarcomere mutations. Non-compaction cardiomyopathy is a diagnostically challenging entity. Nowadays there are some controversies associated with this cardiomyopathy, that it worth to be discussed.
Keywords: non-compaction, cardiomyopathy, genetic, phenotype
INTRODUCTION
A cardiomyopathy is defined as a myocardial disorder in which the heart muscle is structurally and functionally abnormal, in the absence of coronary artery disease, hypertension, valvular disease and congenital heart disease sufficient to cause the observed myocardial abnormality. Cardiomyopathies are grouped into specific morphological and functional phenotypes; each phenotype is then sub-classified into familial and non-familial forms. The spongy myocardium was first identified by Grant in 1926 (1). The isolated non-compaction cardiomyopathy (NCC) was reported for the first time in 1984 (2). But the first reported cases of non-compaction cardiomyopathy were described in association with congenital heart disease (obstructed outflow tract of the left and right ventricle, complex cyanotic congenital malformations and coronary anomalies) (3). In some patients, NNC is associated with left ventricular dilatation and systolic dysfunction, which can be transient in neonates. Non-compaction cardiomyopathy occurs in isolation and in association with congenital cardiac disorders such as Ebstein's anomaly or complex cyanotic heart disease and some neuromuscular diseases. However non-compaction diagnosis is challenging and its nosology is debated since this morphological trait can be shared by different cardiomyopathies and non-cardiomyopathy conditions. ❑
PREVALENCE OF NON-COMPACTION CARDIOMYOPATHY
A real population prevalence of isolated NNC is not known; it is reported in 0.014% of consecutive echocardiograms (4). In large pediatric series, NNC is reported to be the commonest cause of unclassified cardiomyopathies. However, this is a fact that is probably underestimated, since the image quality of this method has greatly improved in recent years.
Familial occurrence is frequent with autosomal dominant and X-linked transmissions. Different mutations in sarcomere protein genes were identified and there seems to be a shared molecular etiology of different cardiomyopathy phenotypes, including NCC, hypertrophic and dilated cardiomyopathies. More recent studies have determined that the prevalence of the disease was of around 18% to 50% among members of affected families (4). NNC is frequently familiar, with at least 25% of asymptomatic relatives having a range of echocardiographic abnormalities. Some cardiac abnormalities may be associated with NCC: the non-compacted myocardium with sinusoids and fistulas of the right coronary artery may present congenital abnormalities in the outflow tract of left and right ventricles (5); presence of Ebstein's anomaly, bicuspid aortic valve and transposition of great vessels (6-8). Patients with NCC may also have a ventricular septal defect (9). ❑
GENETICS OF NON-COMPACTION CARDIOMYOPATHY
The NCC can occur in metabolic diseases and genetic syndromes, including the Barth syndrome, the Charcot-Marie-Tooth disease and the Melnick- Needles syndrome (10). The NCC can be genetically sporadic or familial. Some affected individuals may be detected by tracking the asymptomatic relatives of affected patients. Genes in which causative mutations have been identified include: G 4.5 encoding taffazin (X-linked, responsible for the Barth syndrome) (4), alpha dystrobrevin (4) , ZASP (4), actin (4), lamin A/C (4); a locus on chromosome 11 p 15 (in a family with autosomal dominant penetrance) (11); the E101K mutation of the alpha-cardiac actin has been identified in families with NCC, septal defect and apical hypertrophic cardiomyopathy (12); a mutation (P121L) in the gene that codes the dystrobrevin alpha, cytoskeletal protein and transcription factor NKX2.5 was found in a family with NCC and congenital heart disease (13,14); a mutation of the gene for the cytoskeletal protein, CYPHER/ZASP, was found in one family and in three sporadic cases (15,16). Genetic heterogeneity, with an overlap of different phenotypes, and the variability of hereditary patterns, raise the questions whether there is a morphological trait from dilated/hypertrophic cardiomyopathy to NCC and what are the triggers and modifiers to develop either dilated, hypertrophic cardiomyopathy, or NCC in patients with the same mutation. ❑
NON-COMPACTION CARDIOMYOPATHY DIAGNOSIS
The non-compaction cardiomyopathy is a rare disorder which is considered to be an unclassified cardiomyopathy according to the ESC Working Group on Myocardial and Pericardial Diseases (like Barth syndrome) (4) and the World Health Organization or a primary genetically-determined cardiomyopathy according to the American Heart Association (17). It is characterized by the following aspects:
-
1.
Change in myocardial wall due to the prominence of its trabeculations with deep intertrabecular recesses, which may be secondary to the intrauterine arrest of myocardial compaction that occurs in the early stages of fetal development. The result is two layers of myocardium, a compacted one and non-compacted layer.
-
2.
Continuity between the ventricular cavity and the intertrabecular recesses, which are filled with blood from the ventricle and which have no communication with the epicardial coronary system.
-
3.
Decrease in the coronary flow reserve measured by PET-CT, observed in most segments that show ventricular wall motion abnormalities.
The diagnosis of NCC is often made by echocardiography. Characterization of the normal myocardial structure is the first step before definition of a pathological appearance of the myocardium. However, other imaging tests such as magnetic resonance imaging, which is the most chosen method, computed tomography and left ventriculography may diagnose or confirm the clinical suspicion. The main differential diagnoses of NCC include: dilated cardiomyopathy, hypertensive heart disease, apical hypertrophic cardiomyopathy, infiltrative cardiomyopathy and endomyocardial fibrosis.
The echocardiogram is used as the initial method in the diagnosis and monitoring. There are some echocardiographic criteria proposed for NCC diagnosis (18-21):
-
1.
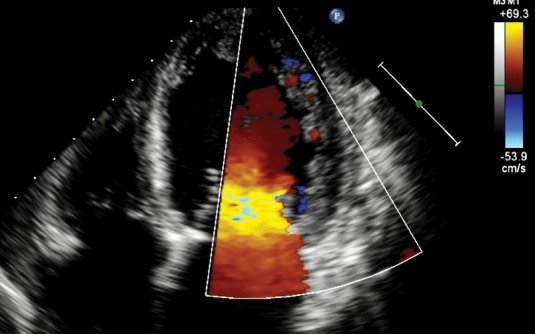
Absence of coexisting cardiac abnormalities; segmental thickening of myocardial wall of left ventricle with two layers: a thin epicardial layer and a thick endocardial layer with prominent trabeculations and deep recesses. The ratio of non-compacted myocardium to compact myocardium at the end of systole is > 2:1; the trabeculae are usually located on the apical/lateral, middle/bottom walls of the left ventricle; most non-compacted segments are hypokinetic; the flow between the intertrabecular recesses can be identified by using the color Doppler method (Figure 1).
-
2.
Presence of x/y < 0.5, where: X = distance from the epicardial surface to the trabecular recess; Y = distance from the epicardial surface to the peak of trabeculations. These criteria are applied to trabeculations of the left ventricular apex with subxiphoid or apical four-chamber views at the end of the diastole.
-
3.
Presence of more than three trabeculations in the left ventricular wall, with the papillary muscles located at the apex, visible in one image plane; intertrabecular spaces, perfused from the ventricular cavity, viewed by color Doppler imaging.
The most used in practice is the first criteria. The myocardial wall trabeculations are most commonly located at the apex and on lateral and bottom walls of the left ventricle (Figure 1a). Contrast echocardiogram or color Doppler (Figure 1b) is useful to better image intertrabecular spaces.
Figure 1A. Apical 4 chamber view in bidimensional echocardiography shown left ventricular non-compaction of apical and lateral wall.
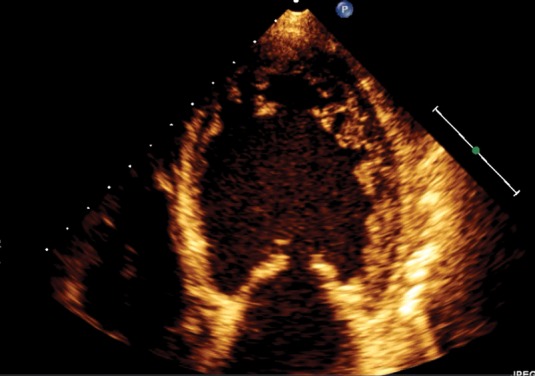
Figure 1B. Apical 4 chamber view in color Doppler echocardiography shown that blood is present between apical and lateral wall trabeculations.
New echocardiographic technique, such as tissue Doppler imaging, strain and strain rate, and speckle tracking, may help to evaluate the functional impact of an abnormal myocardial architecture and enable the clinician to distinguish between normally trabeculated myocardium from NCC (22-24). It seems that LV base and apex rotated in the same direction in all non-compaction patients (24).
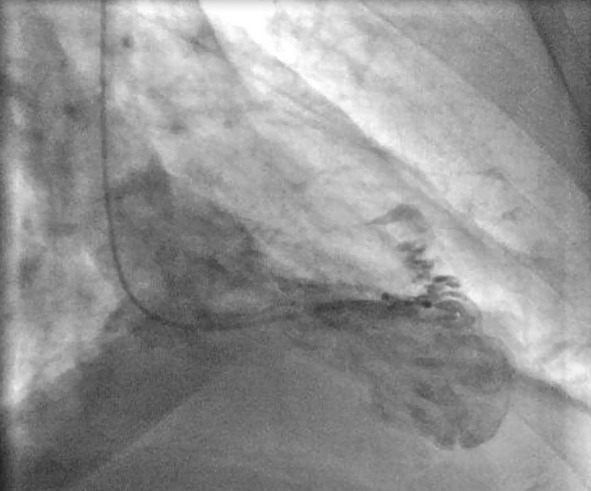
Because the apical region cannot be properly viewed by echocardiography, and this leads to underestimation of the degree of the left ventricular non-compaction, cardiac resonance has become the method of choice to confirm or rule out the diagnosis of NCC (25); it provides a more detailed description of the cardiac morphology in any image plane (Figure 2). A ratio of compacted myocardium to non-compacted myocardium > 2.3 produces the highest sensitivity (86%) and specificity (99%) in the diagnosis (25). Ventriculography can also be helpful (Figure 3).
Figure 2. Apical 4 chamber view in magnetic resonance image.
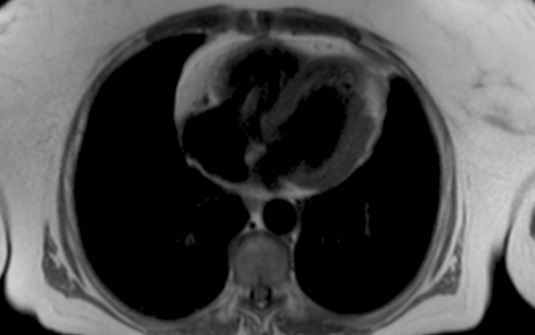
Figure 3. Ventriculography image that shown the presence of multiples trabeculations on apical segments.
Due to the high prevalence of neuromuscular disorders reported in patients with NCC (up to 82%) (21), neurological and musculoskeletal evaluations are also recommended, regardless of whether they have symptoms or not, because the neuromuscular disorder could not yet manifested it clinically.
The clinical manifestations of NCC may vary widely between asymptomatic and symptoms of heart failure, arrhythmias (atrial fibrillation, ventricular tachycardia, branch blocks or bradycardia and Wolff Parkinson White Syndrome in pediatric population) or thromboembolism (26,27). The electrocardiographic findings are often abnormal in NCC, without any specific changes. It seems that a large portion of patients diagnosed with systolic heart failure met the criteria for NCC, at least when such criteria are applied a posteriori. Black people have a higher incidence of hypertrabeculation that meets current diagnostic criteria, regardless of the presence of left ventricular disease. Therefore the suspected diagnosis must be confirmed with the use of cardiac magnetic resonance imaging, so as to differentiate the trabeculae of aberrant bands, false tendons and abnormal insertion of papillary muscles. The differential diagnosis with thrombi, apical hypertrophic cardiomyopathy, fibroma, obliterative process, intramyocardial hematoma, cardiac metastases, intramyocardial abscesses and cardiac hemangiomas must be considered (28). In children, NCC must be differentiated from pulmonary valve atresia with intact interventricular septum and diseases that may lead to obstruction of the outflow of the left ventricle (29). However, the variety in clinical presentation, the genetic heterogeneity, and the phenotype of the first transgenetic animal model of an NCC-associated mutation question the hypothesis that NCC could be a distinct cardiomyopathy: it seems to be rather a distinct phenotype or phenotypic, morphological expression of different underlying diseases than a distinct cardiomyopathy (30). ❑
TREATMENT OF NON-COMPACTION CARDIOMYOPATHY
The main complications related to NCC are thromboembolism, arrhythmias and progressive heart failure. Chronic oral anticoagulation treatment is recommended in patients with decreased systolic function with ejection fraction below 40%, history of thromboembolism or atrial fibrillation. The use of aspirin is recommended for asymptomatic patients with normal systolic function. Extrapolating the indications of management in heart failure, betablockers and ACE inhibitors are also recommended in NCC. In the case of symptomatic ventricular arrhythmia and in the context of impaired systolic function, the prevention of a sustained event and potentially lethal arrhythmia is indicated by antiarrhythmic agents or implantable cardiac defibrillators. Patients with higher final diastolic diameter of left ventricle, low ejection fraction, functional class III-IV NYHA, persistent or permanent atrial fibrillation and bundle branch block are at high risk and they are, at an early stage, candidates for aggressive interventions, including the consideration of an implantable cardiac defibrillator, cardiac resynchronization therapy and evaluation for transplant (31). The prognosis of patients with NCC is determined by the degree and progression of heart failure, presence of thromboembolic events and arrhythmias. The occurrence of embolic events, ventricular arrhythmias and sudden death appears to be significantly lower in pediatric patients than in adults. ❑
CONTROVERSIES AND CHALENGES
Non-compaction cardiomyopathy is a diagnostically challenging entity. First of all, NCC is considered to be an unclassified cardiomyopathy according to the ESC Working Group on Myocardial and Pericardial Diseases and the World Health Organization and a primary genetically-determined cardiomyopathy according to the American Heart Association (4,17). Second, there is some controversy whether NCC is congenital due to an arrest of the normal compaction process of the developing myocardium or NCC is acquired. The ontogenetic development of the myocardium is critical to appreciate the morphological appearance of NCC. Emergence of trabeculations (at the end of the fourth week of gestation) and trabecular remodeling (after completion of ventricular septation at 8 weeks of gestation in human) are the key steps to understand NCC (32). The compaction process or trabecular remodeling gradually progresses from the epicardium to the endocardium, from the base to the apex and from the septum to the free wall in the LV, and is more pronounced in the left ventricle than in the right ventricle. The time of arrest of the normal embryonic myocardial maturation determines the severity and extension of NCC. Recent advances in genetics raise the question whether NCC can also develop postnatally. In addition, serial echocardiographic studies showed that non-compactation was not diagnosed on initial echocardiogram but was becoming evident in subsequent examinations. However it is not clear whether NCC is a separate cardiomyopathy, or merely a congenital or acquired morphological trait shared by many phenotypically distinct cardiomyopathies. There is the reality that a pathogenetic mutation in the same gene results in a different trabecular remodeling or maladaptive remodeling response of the embryonic myocardium, which can cause different phenotypes/cardiomyopathies.
This raises the question that at least for certain mutations, hypertrophic, restrictive, dilated cardiomyopathies, and non-compaction are not clearly distinct clinical and pathophysiological entities (33,34).
The left ventricular non-compaction definitions are variable, and it is difficult to know which echocardiographic (California (18), Zurich (19), Vienna (21), Milwaukee (35)) or MRI (25, 36) criteria are "best" for making a valid diagnosis of NCC. However the echocardiographic suspected diagnosis must be confirmed with the use of cardiac magnetic resonance imaging. It seems that delineation, hence measurements, of the compacted and non-compacted layers of the myocardium are more precise at end-diastole than at end-systole (35). This approach is consistent with the American Society of Echocardiography's convention of chamber and wall thickness measurements, which are performed at end-diastole.
Universally accepted definition of NCC is still lacking both on echocardiography and on MRI (37,38). Differential diagnoses are challenging because there is no diagnostic "gold standard" and also NCC shares many features with hypertrophic and dilated cardiomyopathies (39). ❑
CONCLUSION
Non-compaction cardiomyopathy is a disease that has been increasingly recognized in clinical practice during the last 25 years. Its clinical presentation is highly variable; it may be asymptomatic in many cases, or it may lead to severe heart failure and sudden death in other cases. The high incidence of non-compaction cardiomyopathy in recent years, mainly due to improvements in echocardiographic techniques and use of cardiac resonance, suggests that hypertrabeculation also occurs in other comorbidities and that it may be falsely diagnosed as ventricular non-compaction. Non-compaction cardiomyopathy diagnosis should be carefully evaluated by imaging methods to avoid inappropriate and exaggerated diagnoses. The variety in clinical presentation, the genetic heterogeneity and varies phenotypes raises the question if non-compaction cardiomyopathy can be a distinct cardiomyopathy. This implies that patients with non-compaction cardiomyopathy and their first degree relatives undergo a comprehensive diagnostic assessment by a multidisciplinary team, including cardiologists and geneticists.
CONFLICT OF INTEREST
none declared.
FINANCIAL SUPPORT
none declared.
References
- 1.Grant RT. An unusual anomaly of the coronary vessels in the malformed heart of a child. Heart. 1926;13:273–283. [Google Scholar]
- 2.Engberding R, Bender F. Identification of a rare congenital anomaly of the myocardium by two-dimensional echocardiography: persistence of isolated myocardial sinusoids. Am J Cardiol. 1984;53:1733–1734. doi: 10.1016/0002-9149(84)90618-0. [DOI] [PubMed] [Google Scholar]
- 3.Bellet S, Gouley BA. Congenital heart disease with multiple cardiac anomalies: report of a case showing aortic atresia, fibrous scar in myocardium and embryonic sinusoidal remains. Am J Med Sci. 1932;183:458–65. [Google Scholar]
- 4.Elliott P, Andersson B, Arbustini E, et al. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008;29:270–276. doi: 10.1093/eurheartj/ehm342. [DOI] [PubMed] [Google Scholar]
- 5.Lauer RM, Fink HP, Petry EL, et al. Angiographic demonstration of intramyocardial sinusoids in pulmonary-valve atresia with intact ventricular septum and hypoplastic right ventricle. N Engl J Med. 1964;271:68–72. doi: 10.1056/NEJM196407092710203. [DOI] [PubMed] [Google Scholar]
- 6.Attenhofer Jost CH, Connolly HM, O'Leary PW, et al. Left heart lesions in patients with Ebstein anomaly. Mayo Clin Proc. 2005;80:361–8. doi: 10.4065/80.3.361. [DOI] [PubMed] [Google Scholar]
- 7.Friedberg MK, Ursell PC, Silverman NH. Isomerism of the left atrial appendage associated with ventricular noncompaction. Am J Cardiol. 2005;96:985–90. doi: 10.1016/j.amjcard.2005.05.063. [DOI] [PubMed] [Google Scholar]
- 8.Spoladore R, Gianni U, Castella A. Multimodality imaging of mid ventricular obstruction in left ventricular noncompaction. Eur Heart J Cardiovasc Imaging. doi: 10.1093/ehjci/jet283. [DOI] [PubMed] [Google Scholar]
- 9.Lilje C, Razek V, Joyce JJ, et al. Complications of non-compaction of the left ventricular myocardium in a paediatric population: a prospective study. Eur Heart J. 2006;27:1855–60. doi: 10.1093/eurheartj/ehl112. [DOI] [PubMed] [Google Scholar]
- 10.Zaragoza MV, Arbustini E, Narula J. Noncompaction of the left ventricle: primary cardiomyopathy with an elusive genetic etiology. Curr Opin Pediatr. 2007;19:619–27. doi: 10.1097/MOP.0b013e3282f1ecbc. [DOI] [PubMed] [Google Scholar]
- 11.Monserrat L, Hermida-Prieto M, Fernandez X, et al. Mutation in the alpha-cardiac actin gene associated with apical hypertrophic cardiomyopathy, left ventricular non-compaction, and septal defects. Eur Heart J. 2007;28:1953–61. doi: 10.1093/eurheartj/ehm239. [DOI] [PubMed] [Google Scholar]
- 12.Conces DJ Jr, Ryan T, Tarver RD. Non-compaction of ventricular myocardium: CT appearance. Am J Roentgenol. 1991;156:717–8. doi: 10.2214/ajr.156.4.2003432. [DOI] [PubMed] [Google Scholar]
- 13.Ichida F, Tsubata S, Bowles KR, et al. Novel gene mutations in patients with left ventricular noncompaction or Barth syndrome. Circulation. 2001;103:1256–63. doi: 10.1161/01.cir.103.9.1256. [DOI] [PubMed] [Google Scholar]
- 14.Xing Y, Ichida F, Matsuoka T, et al. Genetic analysis in patients with left ventricular noncompaction and evidence for genetic heterogeneity. Mol Genet Metab. 2006;88:71–7. doi: 10.1016/j.ymgme.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 15.Vatta M, Mohapatra B, Jimenez S, et al. Mutations in cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non-compaction. J Am Coll Cardiol. 2003;42:2014–27. doi: 10.1016/j.jacc.2003.10.021. [DOI] [PubMed] [Google Scholar]
- 16.Sasse-Klaassen S, Probst S, Gerull B, et al. Novel gene locus for autosomal dominant left ventricular noncompaction maps to chromosome 11p15. Circulation. 2004;109:2720–3. doi: 10.1161/01.CIR.0000131865.21260.56. [DOI] [PubMed] [Google Scholar]
- 17.Maron BJ, Towbin JA, Thiene G, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee;Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups;and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–16. doi: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- 18.Chin TK, Perloff JK, Williams RG, et al. Isolated non-compaction of left ventricular myocardium: a study of eight cases. Circulation. 1990;82:507–13. doi: 10.1161/01.cir.82.2.507. [DOI] [PubMed] [Google Scholar]
- 19.Jenni R, Oechslin E, Schneider J, et al. Echocardiographic and pathoanatomical characteristics of isolated left ventricular non-compaction: a step towards classification as a distinct cardiomyopathy. Heart. 2001;86:666–71. doi: 10.1136/heart.86.6.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frischknecht BS, Jost CH, Oechslin EN, et al. Validation of noncompaction criteria in dilated cardiomyopathy, and valvular and hypertensive heart disease. J Am Soc Echocardiogr. 2005;18:865–72. doi: 10.1016/j.echo.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 21.Stollberger C, Finsterer J, Blazek G. Left ventricular hypertrabeculation/ noncompaction and association with additional cardiac abnormalities and neuromuscular disorders. Am J Cardiol. 2002;90:899–902. doi: 10.1016/s0002-9149(02)02723-6. [DOI] [PubMed] [Google Scholar]
- 22.McMahon CJ, Pignatelli RH, Nagueh SF, et al. Left ventricular non-compaction cardiomyopathy in children: characterization of clinical status using tissue Doppler-derived indices of left ventricular diastolic relaxation. Heart. 2007;93:676–681. doi: 10.1136/hrt.2006.093880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eidem BW. Noninvasive evaluation of left ventricular noncompaction: what's new in 2009? Pediatr Cardiol. 2009;30:682–689. doi: 10.1007/s00246-008-9372-3. [DOI] [PubMed] [Google Scholar]
- 24.van Dalen BM, Caliskan K, Soliman OI, et al. Left ventricular solid body rotation in non-compaction cardiomyopathy: a potential new objective and quantitative functional diagnostic criterion? Eur J Heart Fail. 2008;10:1088–1093. doi: 10.1016/j.ejheart.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 25.Petersen SE, Selvanayagam JB, Wiesmann F, et al. Left ventricular non-compaction: insights from cardiovascular magnetic resonance imaging. J Am Coll Cardiol. 2005;46:101–5. doi: 10.1016/j.jacc.2005.03.045. [DOI] [PubMed] [Google Scholar]
- 26.Ichida F, Hanamichi Y, Miyawaki T, et al. Clinical features of isolated noncompaction of the ventricular myocardium: long-term clinical course, hemodynamic properties, and genetic background. J Am Coll Cardiol. 1999;34:233–40. doi: 10.1016/s0735-1097(99)00170-9. [DOI] [PubMed] [Google Scholar]
- 27.Salerno JC, Chun TU, Rutledge JC. Sinus bradycardia, Wolff Parkinson White, and left ventricular noncompaction: an embrylogic connection? Pediatr Cardiol. 2008;29:679–82. doi: 10.1007/s00246-007-9043-9. [DOI] [PubMed] [Google Scholar]
- 28.Stollberger C, Finsterer J. Pitfalls in the diagnosis of left ventricular hypertrabeculation/non-compaction. Postgrad Med J. 2006;82:679–83. doi: 10.1136/pgmj.2006.046169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alhabshan F, Smallhorn JF, Golding F, et al. Extent of myocardial non-compaction: comparison between MRI and echocardiographic evaluation. Pediatr Radiol. 2005;35:1147–51. doi: 10.1007/s00247-005-1551-2. [DOI] [PubMed] [Google Scholar]
- 30.Oechslin E, Jenni R. Left ventricular non-compaction revisited: a distinct phenotype with genetic heterogeneity? Eur Heart J. 2011;32:1446–56. doi: 10.1093/eurheartj/ehq508. [DOI] [PubMed] [Google Scholar]
- 31.Rosa LV, Salemi VM, Alexandre LM, et al. Noncompaction cardiomyopathy: a current view. Arq Bras Cardiol. 2011;97:13–9. doi: 10.1590/s0066-782x2011000900021. [DOI] [PubMed] [Google Scholar]
- 32.Sedmera D, Pexieder T, Vuillemin M, et al. Developmental patterning of the myocardium. Anat Rec. 2000;258:319–337. doi: 10.1002/(SICI)1097-0185(20000401)258:4<319::AID-AR1>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 33.Sen-Chowdhry S, McKenna WJ. Left ventricular noncompaction and cardiomyopathy: cause, contributor, or epiphenomenon? Curr Opin Cardiol. 2008;23:171–175. doi: 10.1097/HCO.0b013e3282fdc939. [DOI] [PubMed] [Google Scholar]
- 34.Xu Q, Dewey S, Nguyen S, et al. Malignant and benign mutations in familial cardiomyopathies: insights into mutations linked to complex cardiovascular phenotypes. J Mol Cell Cardiol. 2010;48:899–909. doi: 10.1016/j.yjmcc.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 35.Paterick TE, Tajik JA. Left Ventricular Noncompaction – A Diagnostically Challenging Cardiomyopathy. Circ J. 2012;76:1556–1562. doi: 10.1253/circj.cj-12-0666. [DOI] [PubMed] [Google Scholar]
- 36.Jacquier A, Thuny F, Jop B, et al. Measurement of trabeculated left ventricular mass using cardiac magnetic resonance imaging in the diagnosis of left ventricular non-compaction. Eur Heart J. 2010;31:1098–1104. doi: 10.1093/eurheartj/ehp595. [DOI] [PubMed] [Google Scholar]
- 37.Rapezzi C, Arbustin Ai, Caforio ALP, et al. Diagnostic work-up in cardiomyopathies: bridging the gap between clinical phenotypes and final diagnosis. A position statement from the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2013;34:1448–58. doi: 10.1093/eurheartj/ehs397. [DOI] [PubMed] [Google Scholar]
- 38.Corrado G. Left ventricular non-compaction: troubles and traps of current imaging techniques. Eur Heart J Cardiovasc Imaging. 2013;14:930–931. doi: 10.1093/ehjci/jet090. [DOI] [PubMed] [Google Scholar]
- 39.Vieira da Rosa L, Cury Salemi VM, Machado Alexandre L, et al. Non-compaction Cardiomyopathy - a Current View. Arq Bras Cardiol. 2011;97:13–19. doi: 10.1590/s0066-782x2011000900021. [DOI] [PubMed] [Google Scholar]