

Abstract
In higher eukaryotes the dynamics of replisome components during fork collapse and restart are poorly understood. Here, we reconstituted replication fork collapse and restart by inducing single-strand DNA (ssDNA) lesions that create a double-strand break (DSB) in one of the replicated sister chromatids after fork passage. We found that, upon fork collapse, the active CDC45–MCM–GINS (CMG) helicase complex loses its GINS subunit. A functional replisome is restored by the reloading of GINS and Pol epsilon onto DNA in a RAD51- and MRE11- dependent manner, but independently of replication origin assembly and firing. PCNA mutant alleles defective in break-induced replication (BIR) are unable to support restoration of replisome integrity. These results reveal that in higher eukaryotes replisomes are partially dismantled following fork collapse and fully re-established by a recombination-mediated process.
Introduction
The entire genomic DNA must be replicated prior cell division. However, DNA replication progression is frequently impaired by various factors such as protein-DNA complexes on the genome, secondary DNA structures formed in palindromic or repetitive sequences, covalent adducts and most importantly DNA lesions creating discontinuities in the template. Prokaryotic and eukaryotic organisms are both equipped with various systems that promote complete duplication of genomic DNA. In Escherichia coli (E. coli), where replication starts from a single origin, the progression of individual forks is assisted by restarting mechanisms such as repriming and homologous recombination (HR) that mitigate the risk of incomplete genome replication in the event of fork stalling1. Although these mechanisms promote efficient replication progression it is unclear whether they operate in eukaryotic organisms, where replication initiates from multiple origins. Many potential “dormant origins”, which are not utilized during unperturbed replication could be used to compensate for stalled forks caused by DNA damage or other factors2,3. However, recent fibre labelling techniques that allowed the visualization of fork restart occurring at individual stalled forks4 indicated that fork restart takes place in eukaryotic organisms.
Several pathways assist DNA replication in the event of DNA damage. When replication forks encounter DNA lesions present on the template, translesion synthesis (TLS) or template switching pathways enable lesion bypass5. TLS requires specialized polymerases such as zeta and eta, which are recruited onto chromatin depending on mono-ubiquitination of PCNA at Lys164 mediated by the RAD6–RAD18 complex6. However, the RAD6 pathway has recently been shown to be separable from chromosomal replication7,8, suggesting that TLS is dispensable for individual fork restart.
In E. coli recA recombinase plays a crucial role in fork restart1. Eukaryotic recA homolog RAD51 is required for fork restart at replication fork barriers (RFB) in fission yeast9 and for the restart of forks stalled by ssDNA gaps arising in nucleotide excision repair (NER) defective cells10 or by hydroxyurea11.
Other DNA repair factors such as the MRE11–RAD50–NBS1 (MRN) complex, which has nuclease and DNA tethering activities that could promote the repair of collapsed forks12 might also be involved in stalled or collapsed fork restart.
Genetic studies of break induced replication (BIR) in budding yeast Saccharomyces cerevisiae (S. cerevisiae) have given important clues to reveal the restart mechanism of collapsed forks13. Recently, it has been demonstrated that BIR requires all the essential replication fork factors14, indicating that the fork formed in BIR might work as a conventional replication fork, although there are important differences such as the requirement of Pol delta subunit Pol32 and the mutagenic behaviour13. A detailed biochemical analysis of replisome components during replication fork collapse that might lead to a better understanding of fork restart through BIR is currently missing. The components of replication machinery in budding yeast are well-characterized15. From late M to G1 phase, the ORC complex (consisting of ORC1–6), CDC6, CDT1, and the MCM complex (consisting of MCM2–7) are sequentially assembled onto replication origins to form the pre-replicative complex (pre-RC). At the onset of S phase, Cyclin dependent kinases (CDKs) and DBF dependent kinases (DDKs) trigger DNA replication initiation by attracting many initiation factors, which convert the pre-RC into an active replisome16. MCM is supposed to serve as a replicative helicase with help of CDC45 and GINS (consisting of SLD5–PSF1–2–3). The ternary complex of CDC45–MCM–GINS was first identified in Drosophila and called “CMG complex”17. After the initial synthesis of an RNA-DNA primer by Pol alpha, Pol delta and epsilon continue lagging and leading strand synthesis. Non-essential components such as TIPIN, TIM1 and CLASPIN are required for keeping the stalled forks stable and ready for restart18. In E. coli, such fork stabilization factors are absent. Therefore, replisome is easily dismantled upon fork stalling and must be re-loaded to resume DNA replication. It is unclear whether the same re-loading system exists in eukaryotes. Fork stabilization systems that prevent fork collapse might reduce the requirement for replisome reloading. However, these pathways cannot ensure fork progression on broken templates, which are likely to occur when forks encounter ssDNA lesions.
In this study, we set out to discover the molecular mechanisms underlying RAD51 and MRE11 dependent restart of forks collapsed by ssDNA lesions. We show that upon fork collapse Pol epsilon and GINS are uncoupled from the replisome and that RAD51 and MRE11 are required for their reloading onto DNA. Notably, PCNA mutant proteins defective in BIR do not support efficient replication and replisome integrity upon fork collapse. These results suggest in eukaryotes the replisome components lost during fork collapse are re-loaded by a recombination-mediated process.
Results
RAD51 mediated restart of forks collapsed by ssDNA lesions
RAD51 is required for replication restart in yeast and mammals9-11,19. However, the mechanism of replication fork reassembly after collapse remains obscure. Using Xenopus laevis (X. laevis) egg extract as model system we set out to uncover the mechanism underlying RAD51 mediated replication fork restart. We first tested which DNA lesions produce replication fork collapse that requires RAD51 to be restarted. To this end we analysed the effects of DNA damaging agents such as methyl methanesulfonate (MMS) and ultraviolet radiation (UV) on DNA replication in the absence of RAD51 bound to chromatin. RAD51 chromatin binding was inhibited by the BRC4 domain of BRCA2 protein fused to GST, as previously shown20. Replication products were resolved on neutral agarose gel21, where the major signals could be observed as two bands; the upper one contains branched DNA, whereas the lower one corresponds to branch-free DNA (Fig.1A). The signal present in the entire lane was quantified to measure DNA replication and reported in the accompanying graph. Consistent with previous results, although DNA damage decreased the number of active replicons due to physical blockage and activation of the S-phase checkpoint20 the absence of RAD51 bound to chromatin did not cause any further impairment of DNA replication (Fig.1A). As RAD51 is involved in HR dependent post-replication repair, which can be redundantly carried out by translesion polymerases20 we tested the contribution of translesion DNA synthesis to the DNA replication efficiency in the presence of UV and MMS- treated templates by using PCNA-K164R, which suppresses the chromatin loading of translesion polymerases20. The suppression of this pathway did not affect the efficiency of DNA replication of damaged templates under the conditions used in these experiments (Fig.1A). These observations suggest RAD51 and translesion synthesis play a minor role in replication restart during MMS- or UV-challenged replication in egg extract.
Figure 1.

RAD51 is required for DNA replication in the presence of forks collapsed by a single strand break in the template. (a) The requirement of RAD51 and PCNA modification at Lys 164 for replication of undamaged sperm DNA (control) or MMS or UV treated sperm DNA was tested using GST-BRC4 (BRC4), which sequesters RAD51, and PCNA K164R mutant. Replication products (labelled with 32P-dATP) were resolved by neutral agarose gel electrophoresis and subjected to autoradiography (left). The quantification of the signal is shown in the graph as photon emission intensity (Intensity) expressed in arbitrary units (AU) (right). The data shown here and hereafter represent typical findings of 3 or more experiments. (b) A model for ssDNA specific endonuclease-dependent fork collapse and RAD51-dependent restart. (c) The requirement of RAD51 for DNA replication in the presence of S1 nuclease was tested using sperm nuclei (4000 nuclei per μl) incubated for 80 min in the presence or absence of 1 μg ml−1 aphidicolin (Low aph) and S1 nuclease (0, 2.92, 1.46, 0.73, 0.37 U μl−1). Replication products were detected by autoradiography with quantification shown on the right, as in 1a. Sybr Green staining shows total DNA (Sybr Green).
It is likely that these lesions do not require RAD51, as they do not break the template. A strand invasion step, which is a RAD51-depedent process, would instead be required to mediate replication fork restart following formation of a DSB in one of the replicated sister chromatids created by the fork passing across a single stranded DNA lesion in the template (one-sided DSB) (Fig.1B). To reproduce this condition in vitro we developed an assay based on the use of single strand specific endonucleases such as S1 and Mung bean, which are expected to cut unwound ssDNA regions generated at the passage of the fork and to induce structures that we refer as “collapsed forks” at a high rate. Extracts were also supplemented with low doses of aphidicolin, which slows down the rate of fork progression by inhibiting DNA polymerase alpha (Pol alpha)22, thereby increasing the amount of ssDNA available for endonuclease mediated cutting23. Using a physical method to detect DNA breaks such as the comet assay we showed that S1 nuclease is able to induce DNA breaks only in replicating nuclei, which contain regions of unwound ssDNA, and not in nuclei that were not incubated in egg extract or whose replication was inhibited by addition of recombinant geminin3 to egg extract (Supplementary Fig.1). This indicated that ssDNA endonuclease digestion of sperm chromatin targets replication structures. We then examined whether RAD51 is required for replication progression in the presence of different amounts of S1 nuclease. To this end we monitored DNA replication by measuring the incorporation of 32P-labelled nucleotide in genomic DNA separated on neutral agarose gels (Fig.1C) or acid-precipitated24,25 (Supplementary Fig.2). Egg extracts used in these experiments were highly efficient at replicating DNA as revealed by the amount of replicated DNA (13±0.6 ng DNA/μl extract) compared to the input (14 ng DNA/μl extract). High doses of S1 nuclease strongly inhibited DNA replication probably due to the high amount of DSBs induced in the template (Fig.1C). However, the absence of RAD51 bound to chromatin substantially suppressed DNA replication at lower S1 concentrations, which affected DNA replication only moderately in control samples (Fig.1C and Supplementary Fig.2). Staining of total genomic DNA also indicated that S1 nuclease did not affect the amount of template DNA. Depletion of RAD51 from egg extract also led to suppression of DNA replication in the presence of S1 nuclease. In these conditions DNA replication was restored by the addition of 100 nM recombinant RAD51 protein to egg extract (Supplementary Fig.2). These results confirmed the findings obtained with the interference of RAD51 binding to chromatin obtained using GST-BRC4. The effects of S1 nuclease on DNA replication in the absence of RAD51 bound to chromatin were enhanced by aphidicolin. In contrast, the low doses of aphidicolin used in these experiments did not substantially affect replication efficiency when added in the absence of S1 nuclease (Supplementary Fig.2). Importantly, when DSBs were induced by adding EcoRI to egg extract, DNA replication was inhibited irrespective of RAD51 status (Supplementary Fig.3). These results suggest that RAD51 is required for efficient DNA replication in the presence of forks collapsed by a DSB in the template.
RAD51 is required to maintain replisome integrity
We then examined the chromatin binding of replication fork proteins in the presence of S1 nuclease (Fig.2A). As expected, the induction of DNA breaks by S1 increased RAD51 binding to DNA, which was suppressed by GST-BRC4. Intriguingly, we found a substantial reduction of essential replication proteins such as SLD5 and PSF2, components of GINS complex, and polymerase epsilon (Pol epsilon) in the presence of GST-BRC4 and S1 nuclease, whereas MCM2, CDC45 and Pol alpha were not affected (Fig. 2A and 2B). In the presence of the replication origin assembly inhibitor geminin26, although RAD51 binding induced by S1 nuclease treatment was still observed the binding of all other fork proteins was inhibited. This indicated that RAD51 was able to de novo assemble onto chromatin independently of origin formation. Importantly, the fact that CDC45 binding was not affected also indicated that the inhibition of DNA replication was not due to origin firing suppression mediated by the activation of the DNA damage checkpoint, which would have inhibited CDC45 loading onto chromatin18,27,28. To establish whether the dissociation of replisome proteins occurs after origin firing S1 nuclease was added to mock- or RAD51-depleted extracts after DNA replication initiation (Fig.2C). Again, S1 nuclease reduced the binding of PSF2 in the absence of RAD51, whereas CDC45 was not affected indicating that the dissociation of GINS proteins and Pol epsilon occurs after DNA replication initiation. This conclusion is also supported by a time-course experiment, where PSF2 gradually dissociated from chromatin during the incubation with S1 nuclease in the absence of RAD51 (Fig.2D).
Figure 2.

RAD51 is required for stable chromatin association of fork proteins in the presence template breakage. In (a) fork proteins association to chromatin isolated from extracts treated with GST or GST-BRC4 and 0, 2.92, 0.97, and 0.32 U μl−1 S1 nuclease in the presence of 1μg ml−1 aphidicolin (Low aph) was analysed. In (b) we monitored the chromatin status of fork proteins, histone H2AX and PCNA in extracts treated with GST or GST-BRC4 2.92 (1/100) and 0.37 (1/800) U μl−1 S1 nuclease and aphidicolin. In (c) chromatin binding of PSF2 and CDC45 in the presence of 0.97 U μl−1 S1 nuclease was analysed in mock or RAD51 depleted extracts. In (d) chromatin binding of the indicated proteins over time in extracts treated with GST or GST-BRC4 and 1.46 U μl−1 of S1 nuclease and aphidicolin was monitored. In (e) nuclear Chk1 phosphorylation on Ser 345 was monitored in extracts treated with 1μg/ml aphidicolin alone or in combination with 1.46 U μl−1 of S1 nuclease. W.B in a–e were performed using antibodies against the indicated chromatin binding factors. Ext: 0.5 μl egg extract was loaded as control in (b) and (d). Chromatin and nuclear fractions were isolated 60 min after the addition of sperm DNA to egg extracts unless otherwise indicated.
Replication factors are loaded with different stoichiometry on chromatin during DNA replication. MCM is present in large excess at active and dormant origins whereas CDC45 and GINS are loaded in limiting amounts reflecting the actual number of active replication forks at any given time3,29-31. To verify whether the stoichiometry of DNA replication factors bound to chromatin affects their differential loss after template breakage we monitored their chromatin binding in the presence of limiting amount of MCM proteins loaded onto DNA3. To this end we added geminin after initiation of MCM loading to limit the amount of MCM complexes bound to DNA to a minimum still capable of supporting normal DNA replication efficiency as previously described3,32. Using this approach we showed that MCM loading and CDC45 were still unaffected by template breakage induced by S1 nuclease in the absence of RAD51 bound to chromatin whereas GINS components and Pol epsilon were gradually lost from DNA (Supplementary Fig.4).
To exclude the possibility that S1 nuclease dependent inhibition of DNA replication is due to the formation of more DSBs in the absence of RAD51, we also examined the phosphorylation status of histone H2AX33 (Fig.2B, 2D and 2E). At the concentration of S1 that inhibits PSF2 binding, H2AX phosphorylation levels, which correlate to the amounts of DSBs in the genome, were not affected by the presence or the absence of BRC4 throughout the time course (Fig.2D). This indicated that inhibition of RAD51 binding to chromatin did not induce more DSBs. We also monitored DNA checkpoint activation induced by S1 treatment by monitoring Chk1 phosphorylation33, which was phosphorylated in extracts treated with S1 nuclease (Fig.2E). However, Chk1 phosphorylation was not increased by BRC4 treatment (Fig.2E). This excluded the possibility of the DNA damage checkpoint being more active in the absence of RAD51. In addition, although the low levels of aphidicolin used were able to induce detectable Chk1 phosphorylation (Fig.2E) they did not compromise overall efficiency of DNA replication (Supplementary Fig.2), suggesting that the checkpoint signalling was not strong enough to inhibit DNA replication. Therefore, suppression of DNA replication in the absence of RAD51 could not be ascribed to differential checkpoint activation although we cannot exclude checkpoint contribution to the effects caused by template breakage on replisome components.
Origin firing independent GINS and Pol epsilon reloading
The requirement of RAD51 for DNA replication in the presence of S1 might be explained by the continuous need of HR to promote replication fork restart. To test this hypothesis we set up an assay based on chromatin transfer experiment (Fig.3). Replication forks were stalled by aphidicolin in the first extract, and chromatin was isolated and treated with Mung bean nuclease, another ssDNA specific endonuclease, which was able to collapse forks on isolated chromatin in vitro more efficiently than S1 nuclease (data not shown). Chromatin was then transferred to a second extract, which was mock- or RAD51- depleted. The replication products were then analyzed on alkaline or neutral agarose gels. As the second extract was also supplemented with geminin and roscovitine, which respectively suppress the assembly and the firing of new origins3,34,35, DNA replication could only take place following the restart of existing stalled and collapsed forks. Nuclei simply treated with aphidicolin did not require RAD51 to restart replication (Fig.3A). Inhibition of RAD51 function by BRC4 addition to the second extract, instead, markedly suppressed replication of Mung bean treated chromatin (Fig.3A). To confirm these data we used RAD51-depleted extracts supplemented with recombinant RAD51 protein in a similar assay. We found that replication fork restart was reduced in the RAD51-depleted extracts, and the replication restart was rescued by the addition of 100 nM RAD51 protein (Fig.3B). The addition of 100 nM recombinant RAD51 protein to egg extract was sufficient to restore endogenous levels of RAD51 bound to chromatin (Fig.3C). Furthermore, the extent of inhibition of DNA replication restart efficiency was greatly enhanced by addition of BRC4 to both the first and the second extract (Supplementary Fig.5). This was likely due to the removal of residual RAD51 bound to chromatin carried by DNA templates incubated in the first extract. These data suggest that restart of collapsed forks require de novo assembly of RAD51 onto chromatin.
Figure 3.

RAD51 is required for origin independent fork restart and reloading of replisome components after fork collapse. In (a) and (b) replication fork restart was monitored following incubation of sperm nuclei in the 1st extract for 60 min with or without 10 μg/ml aphidicolin and then transferring nuclear fractions that were untreated or briefly incubated with Mung bean nuclease to a 2nd extract containing 320 nM geminin, 1 mM roscovitine and GST or GST-BRC4 (a), or to mock or RAD51-depleted extracts containing 25, 50, 100 nM recombinant RAD51 (b). Replication products were monitored by incorporation of 32P-dATP added to the 2nd extract and resolved by alkaline (a) or neutral agarose gel (b) and subjected to autoradiography. Quantification of signals is shown at the bottom of the gel in (a) and in the graph (b). In (c) chromatin binding of RAD51 and CDC45 was monitored in egg extracts that were mock or RAD51 depleted and supplemented with the indicated amount of recombinant RAD51 (rRAD51). The status of replication fork proteins bound to chromatin isolated from extracts treated as in (a) is shown in (d).
We then examined the chromatin binding of replication fork proteins in this chromatin transfer experiment (Fig.3D). When RAD51 was available in the second extract (lanes 1 and 3), no differential binding of fork proteins was observed regardless of the presence or absence of RAD51 in the first extract. When RAD51 was available in the first extract but not in the second, some decrease of PSF2 binding was observed, consistent with the decreased replication activities in Fig.3A and B. When RAD51 binding was instead prevented both in the first and second extract, the binding of SLD5 and PSF2 was almost completely suppressed. These results suggest that RAD51 is required for the re-loading of GINS complex after the fork collapse, facilitating replication restart. A similar behaviour, although less pronounced, was observed for Pol epsilon but not for Pol alpha.
As the second extract contains replication origin assembly and firing inhibitors such as geminin and roscovitine, it is likely that the GINS complex re-assembles onto MCM-CDC45 complexes present on chromatin to re-establish active CMG complexes. These results indicate that RAD51 can promote GINS and Pol epsilon reloading at collapsed forks in the absence of replication origin firing.
MRE11 nuclease activity in replication fork restart
After the formation of one-sided DSBs during fork collapse, DSB end structures must be processed to produce ssDNA for RAD51-dependent strand invasion and/or annealing. In the case of HR mediated repair of DSBs, the MRE11 nuclease plays an important role in the initial end processing step12. MRE11 was also shown to be involved in replication restart in X. laevis and human cells36,37. To study the role of MRE11 nuclease in fork restart we used mirin, a MRE11 nuclease inhibitor (Fig.4). Mirin had little effect on DNA replication in the presence or absence of MMS induced DNA damage (Fig.4A), while it showed a substantial effect in the presence of S1 nuclease (Fig.4B and Supplementary Fig.2). We also examined the chromatin binding of fork proteins and found that SLD5, PSF2, and Pol epsilon were unable to stably associate to chromatin in the presence of S1 and mirin (Fig.4C). These results suggest that MRE11 nuclease activity is required for efficient replication restart. It is possible that MRE11 nuclease is involved in DSB resection and ssDNA generation required for the assembly of RAD51 nucleofilament. However, since mirin did not cause any clear difference in the total amount of RAD51 bound to chromatin (Fig.4C), we examined RAD51 protein levels present on individual forks. To this end we set up a chromatin immunoprecipitation (Ch-IP) assay using anti-CDC45 antibodies (Fig.4D) to isolate protein-DNA intermediates present at active replication forks. In this assay, chromatin fractions were isolated, proteins were cross-linked to DNA, which was subsequently fragmented and subjected to immunoprecipitation. The anti-CDC45 antibodies, but not control immunoglobulins, precipitated CDC45 as well as MCM7 and SLD5, consistent with previous results17. SLD5, instead, could not be detected in the presence of S1 nuclease and mirin. In addition, we found that RAD51 was also co-precipitated by anti CDC45 antibodies and that the amount bound to DNA was decreased by S1 and mirin. These data suggest that MRE11 nuclease activity promotes RAD51 and GINS association to restarting replication forks.
Figure 4.

MRE11 nuclease activity is required for DNA replication upon fork collapse. In (a) and (b) the effects of MRE11 nuclease inhibitor mirin on replication of sperm nuclei that were untreated or treated with MMS in the presence or absence of GST-BRC4 (a) or on sperm nuclei incubated in extracts treated with 0, 0.73, 0.37, 0.18 U μl−1 S1 nuclease and aphidicolin were monitored (b). Replication products were monitored by 32P-dATP incorporation and resolved by neutral agarose gels, which were subjected to autoradiography. Signal intensities were reported in the graphs. In (c) the effect of mirin on replication proteins bound to chromatin isolated after 50 min incubation in extracts treated with 0, 1.46, 0.73, 0.37 and 0.18 U μl−1 S1 nuclease was analysed. In (d) the binding of the indicated fork proteins to chromatin incubated for 45 min in egg extracts that were untreated or supplemented with 0.73 U μl−1 S1 nuclease and mirin was monitored following protein crosslinking, sonication induced DNA fragmentation and immunoprecipitation with control and anti-CDC45 serum. * non-specific band. Ext: 0.5 μl egg extract was loaded as a control in (c) and (d).
Fork restart requires PCNA dependent BIR
RAD51-dependent and RAD51-independent BIR has been hypothesized to be responsible for restoration of collapsed replication forks38-41. The fork structure produced by S1 or Mung bean nucleases (Fig.1B) might trigger BIR, which plays an important role in fork restart after DSB formation in S. cerevisiae42. Recently, PCNA alleles specifically defective in BIR pol30-89 (F248A F249A) and pol30-92 (R80A), which act as dominant negative inhibitors of BIR, have been described14. To verify whether BIR operates in higher eukaryotes and is responsible for fork restart we made the equivalent mutant (Y249A Y250A) of the PCNA allele that shows the most severe phenotype in yeast and tested its effect on DNA replication and chromatin association of replication proteins in the presence of S1 nuclease (Fig. 5). PCNA mutant proteins added in excess to egg extract equilibrate with endogenous PCNA forming mutant complexes that can be loaded onto chromatin20. Under these conditions similar replication activities were obtained in the presence of wild type PCNA or PCNA K164R and S1 nuclease (Fig 5A). However, DNA replication efficiency was substantially decreased in the presence of PCNA Y249A Y250A or PCNA K164R Y249A Y250A mutant proteins (Fig.5A), suggesting that S1 nuclease treatments require BIR to promote efficient DNA replication. Consistently, PCNA Y249A Y250A decreased the chromatin binding of PSF2 in the presence of S1 (Fig.5B). In addition, we found that Pol eta and RAD51 were also decreased by PCNA Y249A Y250A (Fig.5B), suggesting that the inability of this PCNA allele to support BIR is due to defective chromatin binding of Pol eta and RAD51. We then performed a pull-down assay to examine the physical interaction between PCNA and replication proteins in egg extracts (Fig.5C). Pol delta and Pol eta were efficiently pulled down by wild type PCNA and PCNA K164R, but not by PCNA Y249A Y250A and PCNA K164R Y249A Y250A, suggesting that physical interaction with PCNA is necessary to recruit polymerase eta (Pol eta). RAD51 was instead not pulled down by PCNA, suggesting that the effects of PCNA mutant alleles on RAD51 loading onto chromatin are not due to a direct interaction. Overall, these results indicate that the BIR-defective allele of PCNA is unable to support the proper loading of RAD51 and Pol eta onto chromatin to ensure efficient replication restart.
Figure 5.

The role of PCNA in DNA replication and chromatin association of replication proteins upon fork collapse. In (a) replication of sperm nuclei incubated in extracts for 80 min in the presence of 1 μg ml−1 aphidicolin and 0, 0.73, 0.37, 0.18 U μl−1 S1 nuclease and PCNA wild type (WT), PCNA K164R (KR), PCNA Y249A Y250A (YA) or PCNA K164R Y249A Y250A (KR YA) recombinant proteins. Replication products were resolved by neutral agarose gel and subjected to autoradiography (left). Signal intensities were quantified and reported in the graph (right). (b) Binding to chromatin of the indicated proteins was monitored by immunoblotting of chromatin treated with 200 J m−2 UV or incubated in extracts treated with 1 μg ml−1 aphidicolin, 0.97 U μl−1 S1 nuclease or 0.1 U μl−1 EcoR1 and recombinant PCNA wild type (WT), PCNA K164R (KR) or PCNA Y249A Y250A (YA) as indicated. 0.5 μl egg extract was loaded as a control (Ext). (c) The interaction of PCNA and replication proteins in egg extract was monitored by incubation of His-tagged wild type and mutant PCNA proteins followed by pull down with Ni-NTA sepharose. The interacting proteins were detected by immunoblotting as indicated.
We also noticed that an abnormal upper band of PCNA, whose mobility was different from ubiquitinated PCNA and appeared only in the presence of PCNA Y249A Y250A (Fig.5B). This band turned out to be sumoylated PCNA as it could be removed by Ulp1 desumoylating enzyme43 (Supplementary Fig.6A). Mutation analysis (data not shown) led to identification of Lys254 as a novel PCNA sumoylation site, which was modified only in PCNA Y249A Y250A mutant. Mutation of Lys254 (K254R) in the context of PCNA Y249A Y250A affected neither chromatin loading of Pol eta and RAD51 or DNA replication (Supplementary Fig.6B, 6C and 6D). In addition, desumoylation of PCNA Y249A Y250A by Ulp1 did not rescue inhibition of DNA replication induced by S1 nuclease (Supplementary Fig.2). These results indicate that the effect of PCNA Y249A Y250A on DNA replication fork restart did not depend upon this extra modification, the significance of which requires further investigation.
Discussion
When a replication fork encounters a nick in the template or is subjected to nuclease attack, the newly synthesized strand and the parental nicked template form a DSB. We established an in vitro system that recapitulates occurrence of a DSB in one of the replicated sister chromatid after fork passage.
As a discontinuity in the template would likely affect the progression of the putative replicative helicase, we investigated the behavior of the CMG complex subunits MCM2-7, CDC45 and GINS. We observed the specific loss of the GINS subunit accompanied by the detachment of Pol epsilon upon induction of ssDNA lesions in the template and their reloading following RAD51 dependent fork restart. The binding to chromatin of CDC45 and the MCM complex was instead unaffected. The uncoupling of GINS from the CMG complex was unexpected, considering that CDC45 and GINS are recruited onto replication forks interdependently during the initiation of DNA replication44,45.
The release of GINS at the passage of the fork across a discontinuous template might be due to the structural configuration that the GINS subunit adopts within the CMG complex.
The CMG complex architecture was recently revealed46. The GINS complex and CDC45 were shown to associate with the exterior of MCM 2, 3 and 5 proteins, closing a gap at the interface between MCM2 and MCM5. ATP binding to the complex was shown to generate two channels, one through the MCM ring and the other one on its outer perimeter, each one probably engaged with a DNA strand46. This arrangement could make the CMG complex resistant to ssDNA lesions, as the breakage of one DNA strand would not induce complete unloading of the complex from the DNA (Fig.6), facilitating fork restart without the need to reload the helicase.
Figure 6.
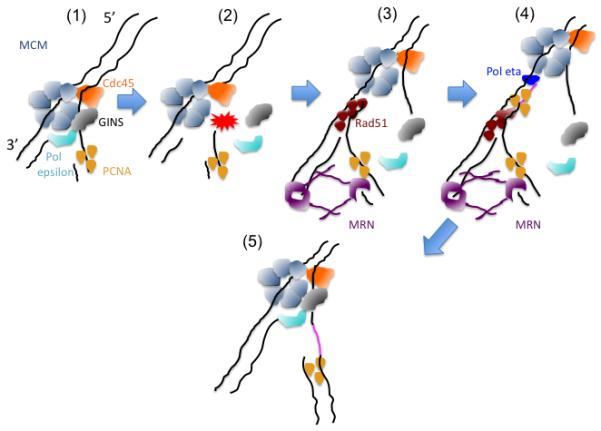
A model of replication fork collapse and restart: The presence of a ssDNA lesion in the template creates one-sided DSB at the passage of the replisome (1) leading to the dissociation of the GINS and Pol epsilon from the fork, whereas MCM and CDC45 remain stably bound to collapsed forks (2). The one-sided DSB undergoes MRE11 mediated nuclease resection and RAD51 dependent strand annealing/invasion of the intact template. The MRE11 complex might also tether the broken DNA strand to the intact one (3). This process requires BIR proficient PCNA, which promotes Pol eta dependent strand extension (4). Reloading of the GINS and Pol epsilon in an origin independent fashion promotes reassembly of functional replisome (5).
In this structure the GINS complex and CDC45 appear to be positioned asymmetrically with respect to MCM pore46. This could explain the selective loss of the GINS complex from the CMG complex, especially if the GINS complex is more loosely attached to DNA than CDC45 and therefore more prone to detachment following template breakage. Phosphorylation of GINS components such as PSF2 by checkpoint kinases33 might also actively promote GINS detachment following template breakage. Intriguingly, persistence of CDC45 bound to the CMG complex in the absence of the GINS complex implies that although the GINS complex and CDC45 depend on each other to load onto chromatin at origins their functions become independent after replication starts. GINS modular binding to MCM and CDC45, which takes place at origins by displacing Sld3 from the MCM-CDC4547, might be consistent with this behaviour.
A consequence of GINS detachment would be the slowing of helicase progression due to the loss of a major activator of the complex. This would also limit the extent of ssDNA accumulation potentially arising from DNA unwinding in the absence of DNA synthesis. The reloading of GINS onto MCM-CDC45 complex during fork restart could then reactivate the stalled helicase.
The detachment of Pol epsilon from DNA, which is consistent with previous reports36,48-50, could be due to its preferential association to the GINS complex51. Other replication factors such as PCNA have been shown to play a fundamental role in fork restart promoting BIR14. We showed that PCNA alleles that do not support BIR fail to promote fork restart preventing the binding of RAD51 and Pol eta to restarting forks. Therefore, it is likely that replication efficiency in the presence of ssDNA endonuclease is maintained through BIR and relies upon RAD51 and Pol eta mediated restarting events (Fig.6). Pol eta was shown to promote DNA synthesis after strand invasion mediated by RAD51 in addition to its well-known TLS activity52,53. The defective loading of Pol eta can be explained by the fact that Pol eta cannot associate with BIR defective PCNA mutant proteins. The suppression of RAD51 chromatin binding by BIR defective PCNA alleles indicates that PCNA is also involved in loading RAD51 onto restarting forks.
BIR might have a central role in vertebrate cells where it could be facilitated by the presence of repetitive sequence that could allow homologous pairing of the one sided-DSB with DNA segments downstream or upstream the lesion. Although this type of repair could lead to loss or duplication of the intervening DNA sequence as shown for tumor cells54 it might be essential for cell survival in the presence of collapsed forks. The MRE11 nuclease appears to play a key role in this process. MRE11 nuclease dead cells were shown to be sensitive to replication fork stalling agents indicating that MRE11 is involved in the repair of these structures55. We showed that inhibition of MRE11 activity impairs replication fork restart and replisome integrity after fork collapse. Together with previous observations showing that MRE11 nuclease activity is required with RAD51 for the processing of replication intermediates arising during unchallenged DNA replication20 our findings suggest that MRE11 and RAD51 functions are coordinated to ensure efficient DNA replication under stressful conditions. Therefore, MRE11 and RAD51 dependent fork repair leading to reloading of the GINS onto the MCM-CDC45 complex still engaged with the DNA could be sufficient to restore a functional CMG helicase complex and promote replication fork restart following template breakage in higher eukaryotes (Fig.6).
Methods
Recombinant proteins and antibodies
Recombinant GST, GST-BRC4, RAD51, his-tagged wild type PCNA, his-tagged PCNA-K164R, and his-tagged geminin were prepared and used in egg extract as described20. GST and GST-BRC4 were used at 0.5 mg ml−1 in egg extracts. Site directed mutations were introduced into the pET28-based expression vectors of wild type and mutant (K164R) X. laevis PCNA (gifts from Dr H. Ulrich, Cancer Research UK), and his-tagged recombinant proteins of PCNA Y249A Y250A, PCNA K164A Y249A Y250A, PCNA K254R, PCNA K164R K254R, PCNA Y249A Y250A K254R and PCNA K164R K249A K250A K254R were prepared in the same way as wild type PCNA and PCNA K164R. Recombinant PCNA proteins were used at 0.2 mg ml−1 in egg extracts. Recombinant his-tagged Ulp1 was provided by Dr H. Ulrich (Cancer Research UK) and used at 60 ng μl−1 in egg extracts. Antibodies against RAD51, Pol alpha, MCM7, PCNA, gamma-H2AX were previously described 20. Anti-human Chk1 phospho-Ser345 antibody that reacts with phospho-Ser344 of X. laevis Chk1 was purchased from Cell Signaling Technology. Antibodies against MCM2, SLD5, PSF2, and Pol epsilon p60 subunit were provided by Dr H. Takisawa (Osaka University), Pol delta p125 and p66 subunits were by Dr S. Waga (Japan Women’s University), and Pol eta and kappa were by Dr M. Akiyama (Nara Institute of Technology). Anti MRE11, ORC2 and CDC45 antibodies were previously described 20,36.
X. laevis egg extracts and replication assay
Egg extracts and chromatin were prepared as described 20. To isolate nuclear fractions 4,000 sperm nuclei per μl were incubated in extract, diluted with 10 volumes of EB buffer (100 mM KCl, 50 mM Hepes-KOH pH 7.5, 2.5 mM MgCl2) and layered onto 200 μl EB-buffer + 30% (w/v) sucrose cushion. The nuclei were spun at 10,000 × g for 2 min at 4°C, washed with 300 μl of EB-buffer and spun again at 10,000 × g for 2 min at 4°C and analysed by immunoblotting. Replication assay with neutral and alkaline agarose gel was previously described20. Signal intensity was measured with a phosphoimager (Molecular Dynamics). S1 nuclease was purchased from Sigma. Roscovitine was purchased from Calbiochem. Mirin was provided by Dr J. Gautier (Columbia University) and used at 100 μM in egg extract to inhibit MRE11 nuclease activity.
Comet assay
4,000 sperm nuclei per μl were incubated in egg extract in the presence or in the absence of geminin or with EB-buffer for 80 min. Reactions were supplemented with 0.55 U μl−1 S1 nuclease. Nuclei were isolated and alkaline comet assay was performed as described21 and quantified using Comet IV software (Perceptive instruments).
RAD51 depletion
For 100 μl depleted extracts 20 μl glutathione Sepharose beads (GE healthcare) were incubated with 150 μg of GST or GST-BRC4 proteins for 60 min at 4°C, washed with EB three times and equally divided. 10 μl beads were then incubated with 100 μl egg extract for 30 min at 4°C twice. The flow through fraction was used as RAD51-depleted extract.
Replication restart assay
Sperm nuclei were incubated in the first extracts for the indicated times, diluted with 20 volume of EB-buffer containing 0.002% Triton X-100 and layered onto 30% Sucrose cushion made of the same buffer. The nuclear fraction was spun at 10.000 × g for 3 min at 4°C, and suspended with 20 μl Mung bean reaction buffer (1 × NEBuffer 1, 1 mM ZnSO4, 0.002% Triton X-100, 10 U Mung bean nuclease, New England Biolab). After incubation for 20 min at 23°C, 10 volumes of EB-buffer were added, the nuclear fraction was spun at 10.000 × g for 1 min at 4°C and re-suspended with the second extract for replication restart.
Chromatin immunoprecipitation
Sperm nuclei were incubated for 45 min in 100 μl egg extracts, and mixed with 1 ml EB buffer supplemented with 0.2% (v/v) Triton and 1% (v/v) formaldehyde, and incubated for 10 min on ice. Then, chromatin fractions were prepared and resuspended with 600 μl EB buffer supplemented 0.2% Triton (v/v), and sonicated until average DNA size was 500 bp. After the mixture was subjected to centrifugation at 2400 × g for 5 min, the supernatant was collected and mixed with 3 μl protein-A-sepharose cross linked to 1.5 μl control or anti-CDC45 serum. After incubating for 1 hr at 4°C, the sepharose was washed with EB supplemented with 0.2 % Triton (v/v) and analysed by immunoblotting.
Pull down assay
30 μl egg extract with or without recombinant his-tagged PCNA proteins (0.2 mg ml−1) were diluted with 120 μl EB-buffer supplemented with 40 mM imidazole, incubated for 25 min at R.T., and supplemented with 5 μl Ni-NTA agarose (QIAGEN). After incubating for 30 min at 4°C, the agarose beads were washed three times with EB-buffer containing 0.05 % tween (v/v) and 40 mM imidazole and one time with 50 mM Tris-HCl (pH 7.5), resuspended with SDS-PAGE sample buffer and analysed by immunoblotting.
DNA replication assay by TCA precipitation
DNA replication was quantified as described24. Briefly sperm nuclei (4000 nuclei per μl) were incubated in 20 μl egg extracts supplemented with α32P-dATP. Extracts were treated as described in Supplementary Fig.2 legend and incubated at 23°C for 120 min. Replication reactions were terminated by the addition of 200 μl Stop buffer (20 mM Tris HCl, pH 7.5, 5 mM EDTA, 0.5% SDS) with 0.6 mg ml−1 proteinase K, incubated at 42 °C for 1 hour, spotted on a GF/C glass filter (Whatman) and precipitated in 5% cold TCA and 2% pyrophosphate solution (20 ml per filter) for 20 minutes. Filters were allowed to dry after being washed 4 times with 5% TCA and once with ethanol. Radioactivity was counted with Cerenkov counter and plotted on graph. The amount of replicated template was calculated assuming extract dATP concentration is 50 μM (usually 60 pmoles μl−1 extract) using the following formula: pg DNA synthesized= (% dATP incorporated) × (pmoles dATP in the assay) × 4 × 330. 4000 nuclei per μl extract were used for each sample. The amount of replicated DNA was calculated considering 1 nucleus = 3.5 pg.
Supplementary Material
Acknowledgements
We are grateful to J. Haber and J. Lydeard for sharing unpublished results on PCNA BIR defective mutants. We thank members of the genome stability lab for their insightful comments. We thank H. Mahbubani and J. Gannon for technical support with X. laevis. This work was funded by Cancer Research UK. VC is also supported by the European Research Council (ERC) start up grant (206281), the Lister Institute of Preventive Medicine and the EMBO Young Investigator Program (YIP).
References
- 1.Michel B, Grompone G, Flores MJ, Bidnenko V. Multiple pathways process stalled replication forks. Proc Natl Acad Sci U S A. 2004;101:12783–8. doi: 10.1073/pnas.0401586101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Santocanale C, Sharma K, Diffley JF. Activation of dormant origins of DNA replication in budding yeast. Genes Dev. 1999;13:2360–4. doi: 10.1101/gad.13.18.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Woodward AM, et al. Excess Mcm2-7 license dormant origins of replication that can be used under conditions of replicative stress. J Cell Biol. 2006;173:673–83. doi: 10.1083/jcb.200602108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Petermann E, Helleday T. Pathways of mammalian replication fork restart. Nat Rev Mol Cell Biol. 2010;11:683–7. doi: 10.1038/nrm2974. [DOI] [PubMed] [Google Scholar]
- 5.Chang DJ, Cimprich KA. DNA damage tolerance: when it’s OK to make mistakes. Nat Chem Biol. 2009;5:82–90. doi: 10.1038/nchembio.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andersen PL, Xu F, Xiao W. Eukaryotic DNA damage tolerance and translesion synthesis through covalent modifications of PCNA. Cell Res. 2008;18:162–73. doi: 10.1038/cr.2007.114. [DOI] [PubMed] [Google Scholar]
- 7.Daigaku Y, Davies AA, Ulrich HD. Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature. 2010;465:951–5. doi: 10.1038/nature09097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karras GI, Jentsch S. The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase. Cell. 2010;141:255–67. doi: 10.1016/j.cell.2010.02.028. [DOI] [PubMed] [Google Scholar]
- 9.Lambert S, et al. Homologous recombination restarts blocked replication forks at the expense of genome rearrangements by template exchange. Mol Cell. 2010;39:346–59. doi: 10.1016/j.molcel.2010.07.015. [DOI] [PubMed] [Google Scholar]
- 10.Moriel-Carretero M, Aguilera A. A postincision-deficient TFIIH causes replication fork breakage and uncovers alternative Rad51- or Pol32-mediated restart mechanisms. Mol Cell. 2010;37:690–701. doi: 10.1016/j.molcel.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 11.Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell. 2010;37:492–502. doi: 10.1016/j.molcel.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huertas P. DNA resection in eukaryotes: deciding how to fix the break. Nat Struct Mol Biol. 2010;17:11–6. doi: 10.1038/nsmb.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Llorente B, Smith CE, Symington LS. Break-induced replication: what is it and what is it for? Cell Cycle. 2008;7:859–64. doi: 10.4161/cc.7.7.5613. [DOI] [PubMed] [Google Scholar]
- 14.Lydeard JR, et al. Break-induced replication requires all essential DNA replication factors except those specific for pre-RC assembly. Genes Dev. 2010;24:1133–44. doi: 10.1101/gad.1922610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gambus A, et al. GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks. Nat Cell Biol. 2006;8:358–66. doi: 10.1038/ncb1382. [DOI] [PubMed] [Google Scholar]
- 16.Tanaka S, Araki H. Regulation of the initiation step of DNA replication by cyclin-dependent kinases. Chromosoma. 2010;119:565–74. doi: 10.1007/s00412-010-0291-8. [DOI] [PubMed] [Google Scholar]
- 17.Moyer SE, Lewis PW, Botchan MR. Isolation of the Cdc45/Mcm2-7/GINS (CMG) complex, a candidate for the eukaryotic DNA replication fork helicase. Proc Natl Acad Sci U S A. 2006;103:10236–41. doi: 10.1073/pnas.0602400103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Errico A, Costanzo V. Differences in the DNA replication of unicellular eukaryotes and metazoans: known unknowns. EMBO Rep. 2010;11:270–8. doi: 10.1038/embor.2010.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Haro LP, et al. Metnase promotes restart and repair of stalled and collapsed replication forks. Nucleic Acids Res. 2010;38:5681–91. doi: 10.1093/nar/gkq339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hashimoto Y, Chaudhuri AR, Lopes M, Costanzo V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol. 2010;17:1305–11. doi: 10.1038/nsmb.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trenz K, Errico A, Costanzo V. Plx1 is required for chromosomal DNA replication under stressful conditions. EMBO J. 2008;27:876–85. doi: 10.1038/emboj.2008.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krokan H, Wist E, Krokan RH. Aphidicolin inhibits DNA synthesis by DNA polymerase alpha and isolated nuclei by a similar mechanism. Nucleic Acids Res. 1981;9:4709–19. doi: 10.1093/nar/9.18.4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sutherland GR. The role of nucleotides in human fragile site expression. Mutat Res. 1988;200:207–13. doi: 10.1016/0027-5107(88)90084-x. [DOI] [PubMed] [Google Scholar]
- 24.Balestrini A, Cosentino C, Errico A, Garner E, Costanzo V. GEMC1 is a TopBP1-interacting protein required for chromosomal DNA replication. Nat Cell Biol. 12:484–91. doi: 10.1038/ncb2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chong JP, Mahbubani HM, Khoo CY, Blow JJ. Purification of an MCM-containing complex as a component of the DNA replication licensing system. Nature. 1995;375:418–21. doi: 10.1038/375418a0. [DOI] [PubMed] [Google Scholar]
- 26.McGarry TJ, Kirschner MW. Geminin, an inhibitor of DNA replication, is degraded during mitosis. Cell. 1998;93:1043–53. doi: 10.1016/s0092-8674(00)81209-x. [DOI] [PubMed] [Google Scholar]
- 27.Costanzo V, et al. Reconstitution of an ATM-dependent checkpoint that inhibits chromosomal DNA replication following DNA damage. Mol Cell. 2000;6:649–59. doi: 10.1016/s1097-2765(00)00063-0. [DOI] [PubMed] [Google Scholar]
- 28.Falck J, Petrini JH, Williams BR, Lukas J, Bartek J. The DNA damage-dependent intra-S phase checkpoint is regulated by parallel pathways. Nat Genet. 2002;30:290–4. doi: 10.1038/ng845. [DOI] [PubMed] [Google Scholar]
- 29.Edwards MC, et al. MCM2-7 complexes bind chromatin in a distributed pattern surrounding the origin recognition complex in Xenopus egg extracts. J Biol Chem. 2002;277:33049–57. doi: 10.1074/jbc.M204438200. [DOI] [PubMed] [Google Scholar]
- 30.Pacek M, Tutter AV, Kubota Y, Takisawa H, Walter JC. Localization of MCM2-7, Cdc45, and GINS to the site of DNA unwinding during eukaryotic DNA replication. Mol Cell. 2006;21:581–7. doi: 10.1016/j.molcel.2006.01.030. [DOI] [PubMed] [Google Scholar]
- 31.Mimura S, Takisawa H. Xenopus Cdc45-dependent loading of DNA polymerase alpha onto chromatin under the control of S-phase Cdk. EMBO J. 1998;17:5699–707. doi: 10.1093/emboj/17.19.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Errico A, et al. Tipin/Tim1/And1 protein complex promotes Pol alpha chromatin binding and sister chromatid cohesion. EMBO J. 2009;28:3681–92. doi: 10.1038/emboj.2009.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsuoka S, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–6. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 34.Sclafani RA, Holzen TM. Cell cycle regulation of DNA replication. Annu Rev Genet. 2007;41:237–80. doi: 10.1146/annurev.genet.41.110306.130308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meijer L, et al. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur J Biochem. 1997;243:527–36. doi: 10.1111/j.1432-1033.1997.t01-2-00527.x. [DOI] [PubMed] [Google Scholar]
- 36.Trenz K, Smith E, Smith S, Costanzo V. ATM and ATR promote Mre11 dependent restart of collapsed replication forks and prevent accumulation of DNA breaks. EMBO J. 2006;25:1764–74. doi: 10.1038/sj.emboj.7601045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bryant HE, et al. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 2009;28:2601–15. doi: 10.1038/emboj.2009.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ira G, Haber JE. Characterization of RAD51-independent break-induced replication that acts preferentially with short homologous sequences. Mol Cell Biol. 2002;22:6384–92. doi: 10.1128/MCB.22.18.6384-6392.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davis AP, Symington LS. RAD51-dependent break-induced replication in yeast. Mol Cell Biol. 2004;24:2344–51. doi: 10.1128/MCB.24.6.2344-2351.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Malkova A, et al. RAD51-independent break-induced replication to repair a broken chromosome depends on a distant enhancer site. Genes Dev. 2001;15:1055–60. doi: 10.1101/gad.875901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Signon L, Malkova A, Naylor ML, Klein H, Haber JE. Genetic requirements for RAD51- and RAD54-independent break-induced replication repair of a chromosomal double-strand break. Mol Cell Biol. 2001;21:2048–56. doi: 10.1128/MCB.21.6.2048-2056.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kraus E, Leung WY, Haber JE. Break-induced replication: a review and an example in budding yeast. Proc Natl Acad Sci U S A. 2001;98:8255–62. doi: 10.1073/pnas.151008198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li SJ, Hochstrasser M. The Ulp1 SUMO isopeptidase: distinct domains required for viability, nuclear envelope localization, and substrate specificity. J Cell Biol. 2003;160:1069–81. doi: 10.1083/jcb.200212052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kubota Y, et al. A novel ring-like complex of Xenopus proteins essential for the initiation of DNA replication. Genes Dev. 2003;17:1141–52. doi: 10.1101/gad.1070003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takayama Y, et al. GINS, a novel multiprotein complex required for chromosomal DNA replication in budding yeast. Genes Dev. 2003;17:1153–65. doi: 10.1101/gad.1065903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Costa A, et al. The structural basis for MCM2-7 helicase activation by GINS and Cdc45. Nat Struct Mol Biol. 2011;18:471–7. doi: 10.1038/nsmb.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bruck I, Kaplan DL. GINS and Sld3 Compete with One Another for Mcm2-7 and Cdc45 Binding. J Biol Chem. 2011;286:14157–67. doi: 10.1074/jbc.M111.218305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bjergbaek L, Cobb JA, Tsai-Pflugfelder M, Gasser SM. Mechanistically distinct roles for Sgs1p in checkpoint activation and replication fork maintenance. EMBO J. 2005;24:405–17. doi: 10.1038/sj.emboj.7600511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cobb JA, et al. Replisome instability, fork collapse, and gross chromosomal rearrangements arise synergistically from Mec1 kinase and RecQ helicase mutations. Genes Dev. 2005;19:3055–69. doi: 10.1101/gad.361805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tittel-Elmer M, Alabert C, Pasero P, Cobb JA. The MRX complex stabilizes the replisome independently of the S phase checkpoint during replication stress. EMBO J. 2009;28:1142–56. doi: 10.1038/emboj.2009.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Muramatsu S, Hirai K, Tak YS, Kamimura Y, Araki H. CDK-dependent complex formation between replication proteins Dpb11, Sld2, Pol (epsilon}, and GINS in budding yeast. Genes Dev. 2010;24:602–12. doi: 10.1101/gad.1883410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kawamoto T, et al. Dual roles for DNA polymerase eta in homologous DNA recombination and translesion DNA synthesis. Mol Cell. 2005;20:793–9. doi: 10.1016/j.molcel.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 53.McIlwraith MJ, et al. Human DNA polymerase eta promotes DNA synthesis from strand invasion intermediates of homologous recombination. Mol Cell. 2005;20:783–92. doi: 10.1016/j.molcel.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 54.Stephens PJ, et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature. 2009;462:1005–10. doi: 10.1038/nature08645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Buis J, et al. Mre11 nuclease activity has essential roles in DNA repair and genomic stability distinct from ATM activation. Cell. 2008;135:85–96. doi: 10.1016/j.cell.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.