

Abstract
Background and Objectives
Untreated phenylketonuria (PKU), a hereditary metabolic disorder caused by a genetic mutation in phenylalanine hydroxylase (PAH), is characterized by elevated blood phenylalanine (Phe) and severe neurologic disease. Sapropterin dihydrochloride, a synthetic preparation of naturally occurring PAH cofactor tetrahydrobiopterin (BH4), activates residual PAH in a subset of patients, resulting in decreased blood Phe and increased Phe tolerance. The objective of this study was to determine the appropriate dose of sapropterin in pediatric patients (0–6 years). The study design used D-optimization and was prospectively powered to achieve precise estimates of clearance and volume of distribution.
Methods
Oral sapropterin (5 or 20 mg/kg) was administered once daily. Sapropterin plasma concentrations were measured by a validated method. Population pharmacokinetic analysis was performed with NONMEM® version 7.2 on pooled data from 156 pediatric and adult PKU patients in two phase III clinical studies.
Results
The best pharmacokinetic model was a one-compartment model with an absorption lag, first-order input, and linear elimination, with a factor describing endogenous BH4 levels. Body weight was the only covariate significantly affecting sapropterin pharmacokinetics. Based on recommended dosing, exposure across age groups was comparable. The absorption rate and terminal half-life suggest flip-flop pharmacokinetic behavior where absorption is rate limiting.
Conclusion
The effect of weight on sapropterin pharmacokinetics was significant and exposure was comparable across age groups; thus, weight-based dosing is appropriate. The doses selected for pediatric patients provided similar exposure as in adults. Given the slow absorption and elimination half-life, once-daily dosing is justified.
Electronic supplementary material
The online version of this article (doi:10.1007/s40262-014-0196-4) contains supplementary material, which is available to authorized users.
Key Points
| Population pharmacokinetics of sapropterin dihydrochloride, a synthetic preparation of naturally occurring phenylalanine hydroxylase cofactor tetrahydrobiopterin (BH4), were evaluated in pediatric phenylketonuria patients. |
| The best pharmacokinetic model was a one-compartment model with an absorption lag, first-order input, and linear elimination, with a factor describing endogenous BH4 levels. Body weight was the only covariate significantly affecting sapropterin pharmacokinetics. |
| The doses selected for pediatric patients provided similar exposure as in adults. |
Introduction
Phenylketonuria (PKU) is a hereditary metabolic disorder caused by a genetic mutation and deficiency in phenylalanine hydroxylase (PAH), an enzyme required for the metabolism of phenylalanine (Phe). In PKU patients, PAH is mutated to varying degrees and if active PAH is not present in sufficient quantities, Phe accumulates to abnormally high levels in the blood and brain; this often results in mental retardation and brain damage, mental illness, seizures, tremors, and cognitive problems. Tetrahydrobiopterin (BH4), a cofactor for PAH, facilitates the hydroxylation of Phe to tyrosine, thereby maintaining appropriate levels of plasma Phe. Several studies have shown a reduction in plasma Phe levels in some PKU patients treated with a synthetic preparation of the dihydrochloride salt of naturally occurring BH4 (sapropterin dihydrochloride; sapropterin) [1–3].
A population pharmacokinetic model of data arising from study PKU-004, which assessed sapropterin levels in children (≥9 years of age) and adults, found a two-compartment model with endogenous BH4 provided the best description of the data [4]. The population pharmacokinetic model from this study was used to design an optimal pharmacokinetic sampling strategy for study PKU-015, a population pharmacokinetic study to characterize pharmacokinetic characteristics of BH4 in pediatric patients from 0 to 6 years of age, and provide dosing recommendations for this population. The study design used D-optimization [5–7] based on the previous pharmacokinetic model and was prospectively powered to achieve precise estimates of apparent total clearance of the drug from plasma after oral administration (CL/F) and apparent volume of distribution of the central compartment (Vc/F) in each age group.
Graphical examination of the concentration–time data arising from PKU-015 suggested that there were little data supporting a peripheral compartment, which was reflected in the poor precision of the estimates for the associated parameters in the previous evaluation (Electronic Supplementary Material Fig. 1S). Thus, pooled data from studies PKU-004 and PKU-015 were fit to ensure a pharmacokinetic model that was capable of describing data arising from all ages.
Methods
Study Design
Both studies (PKU-015 and PKU-004) were approved by institutional review boards or ethics committees at all centers and were performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. Informed written consent was obtained from all patients or their guardian, in the case of children, before inclusion in the study.
Study PKU-015
Study PKU-015 was an open-label, multicenter study conducted at 19 centers in the USA and Canada. The study was designed to evaluate the safety and population pharmacokinetics of sapropterin, and the effect of sapropterin on neurocognitive development and blood Phe levels in infants and young children with PKU. A total of 95 patients (0–6 years old) received oral sapropterin at a dose of 20 mg/kg once daily (at the same time each day) with food for 4 weeks. Subjects were eligible to participate in the population pharmacokinetic substudy and were enrolled by age at study entry. A total of 94 subjects were enrolled in the pharmacokinetic substudy and 80 of these subjects were evaluable. D-optimization was used to select the pharmacokinetic sample times and identify the number of patients needed to estimate the pharmacokinetic parameters. The study was prospectively powered to achieve precise estimates of CL/F and Vc/F in each age group.
Study PKU-004
Study PKU-004 was a multicenter, intra-patient, dose-escalation, open-label extension study conducted at 26 centers in North America (Canada and the USA) and Europe (France, Germany, Ireland, Italy, Poland, and the UK). The study was designed to evaluate the long-term safety and efficacy of various doses of sapropterin in patients ≥8 years old with PKU who had previously responded to sapropterin treatment. Study PKU-004 occurred in two parts. In part 1, patients received sapropterin in three consecutive 2-week courses of daily single oral doses of 5 mg/kg, followed by 20 mg/kg/day, and finally 10 mg/kg/day for 4 more weeks. Following completion of the 4-week 10 mg/kg/day period in part 1 of PKU-004, each patient was enrolled in part 2, a 16-week fixed-dose period during which the daily dose of sapropterin was fixed within the range of 5–20 mg/kg/day on the basis of the patient’s Phe level at the end of the 2-week 10 mg/kg treatment period. A total of 80 subjects were enrolled in study PKU-004. After completing the first 16 weeks of treatment, 78 subjects were enrolled and evaluable in the pharmacokinetic substudy.
Pharmacokinetic Sampling
In PKU-015, three plasma samples from each patient in the ≤1 year old age group and four plasma samples from each patient in the >1 year old age group were collected at the week 0 through week 4 visits according to a D-optimized design shown in Table 1.
Table 1.
D-optimal sampling design in study PKU-015 and study PKU-004
| Age group (years) | Dose (mg/kg/day) | Visit (week) | Optimal sampling time (h) | Post-dose sampling window | |
|---|---|---|---|---|---|
| Lower bound (h) | Upper bound (h) | ||||
| Study PKU-015 | |||||
| 0 to <1 | 20 | 1 | Pre-dose | – | – |
| 2 | Pre-dose | – | – | ||
| 1, 2, 3, or 4 | 4.9 | 4.5 | 5.5 | ||
| >1 to 6 | 20 | 0 | Pre-dose | ||
| 2, 3, and/or 4a | 0.22 | 0.12 | 1.2 | ||
| 2, 3, and/or 4a | 3.2 | 2.6 | 5.2 | ||
| 2, 3, and/or 4a | 7 | 6 | 8 | ||
| Study PKU-004 | |||||
| 9–50 | 5 | 16, 20, or 22 | Pre-dose | – | – |
| – | 0 | 0.1b | |||
| – | 1.2 | 3.7 | |||
| – | 5.6 | 8.0 | |||
| 9–50 | 20 | 16, 20, or 22 | – | 0 | 0.1 |
| – | 0.3 | 1.0 | |||
| – | 5 | 5.9 | |||
| – | 7.0 | 8.0 | |||
– not applicable
aSubjects could have all three samples drawn at one visit, at two of the three visits, or have one sample drawn at each of the three visits
bIt was recommended that one sample be taken before dosing and one within the first 10 min after dosing
In PKU-004, four plasma samples from each patient were collected from each patient at any point during the week 16, 20, or 22 visits according to a D-optimized design shown in Table 1. Patients receiving 10 mg/kg/day in the fixed-dose period could be assigned to Group 1 or Group 2, but were to follow the prescribed sample assessment schedule for that group once they had been assigned.
Tetrahydrobiopterin Assay
As BH4 is unstable in plasma, its concentration was measured indirectly by oxidizing BH4 to L-biopterin; measuring the concentration of L-biopterin, which has been shown to be stable [4, 8]; and correcting for the oxidative conversion of BH4 to L-biopterin. The collected plasma samples were shipped frozen on dry ice to a central laboratory for assay. Samples were oxidized and assayed for L-biopterin by a validated analytical liquid chromatography/tandem mass spectrometry method.
The plasma concentration of BH4 was calculated from the measured concentration of L-biopterin by correcting for the oxidation conversion ratio of BH4 to L-biopterin, which was determined in the assay validation as the molar ratio, according to equation (Eq. 1):
| 1 |
where MWBH4 is 241.2 g/mol and MWBiopterin is 237.2 g/mol. The nominal conversion ratio of BH4 to L-biopterin was determined to be 47.3 %. The conversion ratio was stable within at least 8 weeks of sample storage at −70 °C.
The lower limit of quantitation was 5.00 ng/mL for L-biopterin and 10.7 ng/mL for BH4.
Pharmacokinetic Modeling
The pharmacokinetic data used in the population analysis included all available concentration data collected from studies PKU-015 and PKU-004. Data from study PKU-015 were intended to be evaluated separately from study PKU-004, or if needed using an informative prior based on the previously developed population pharmacokinetic model [4]. However, initial graphical evaluation of data from study PKU-015, together with preliminary model-based evaluations, suggested that the data for both studies were adequately described using a one-compartment model with a parameter for endogenous BH4, rather than a two-compartment model as reported for PKU-004 [4]. Subsequently, the data from studies PKU-015 and PKU-004 were pooled for population pharmacokinetic model development.
Structural Model and Covariate Model
A series of different structural models were evaluated in the present analysis, including one- and two-compartment models with first-order input, with and without an absorption lag. The best pharmacokinetic model was a one-compartment model with an absorption lag, first-order input, and linear elimination, with a factor describing endogenous BH4 levels. Covariates evaluated in the model development are listed in Electronic Supplementary Material Table 1S and body weight was the only covariate significantly affecting sapropterin pharmacokinetics. The first-order conditional estimation (FOCE) method was used because the change in objective function value (∆OFV) has been found to more reliably follow a chi-square distribution.
Continuous covariates such as age or weight were modeled using a general power function, according to Eq. 2:
| 2 |
where the typical value of the parameter (TVP) represents the predicted pharmacokinetic parameter (CL/F, Vc/F) for the ‘reference’ individual with covariate value(s) covi, P pop represents the population central tendency for the pharmacokinetic parameter TVP, covi represents the individual value for that covariate normalized by the approximate median value for the patient population, and θ i represents a scale factor relating the covariate function to the pharmacokinetic parameter. A typical adult (e.g., 70 kg body weight) was chosen as the reference individual to allow comparison of the results in pediatrics with those in adults.
Categorical covariates (e.g., race) were modeled using the general Eq. 3:
| 3 |
where covi is either 0 for the standard or reference patient, or 1 for the comparative patient. P pop represents the value for the pharmacokinetic parameter when covi is 0 and θ i represents a scale factor for the influence of that covariate on the pharmacokinetic parameter.
Covariates were first examined for potential effect on structural parameters by graphical assessment in Xpose 4.3.5 running on R3.0.2, followed by a model-based analysis if any trend was observed. A standard approach was used in covariate evaluation. During individual covariate evaluations, a P value of 0.005 was used to select covariates; a full model was built with all identified covariates and a P value of 0.001 was used to reduce the model during backwards elimination. Covariate factors were considered clinically relevant and accepted in the model if the magnitude of the change of the parameter due to a covariate influence resulted in ≥20 % variation of the parameter. The precision of the estimated covariate effect was also considered; covariate factors were not accepted unless the parameter precision was <30 %.
Statistical Model
Inter-individual variability was described using the following exponential error model, according to Eq. 4:
| 4 |
where P j is the individual value for the pharmacokinetic parameter in the jth individual and η j is an independent random variable with a mean of zero and variance ω P 2. Between-occasion variability was also assessed.
The residual variability for the pharmacokinetic model was described using a constant coefficient of variation (CCV) model based on the log transform both sides (LTBS) approach according to Eq. 5:
| 5 |
where Ĉpij is the ith plasma concentration for the jth individual predicted by the model, Cpij is the measured concentration, and εij is the residual error normally distributed with mean zero and variance σ2.
Model Selection Criteria
Model building followed standard criteria [9, 10]. Decision making during model building was guided by evaluation of the ∆OFV, evaluation of the magnitude of inter-individual and residual variance, and examination of diagnostic residual plots. The chi-squared test (P < 0.005) for the log-likelihood ∆OFV between nested models with degrees of freedom equal to the difference in number of parameters between models was used to declare superiority of one model over another. This corresponds to a reduction in OFV of ≥7.88 (P < 0.005) for comparison of models that differ by one parameter. The superior model should also have an associated reduction in the magnitude of inter-individual and residual variance estimates, as well as improved diagnostic plots. The ∆OFV was not used to evaluate parameters describing variability. The covariance step was implemented for each model, and standard errors (SEs) for parameter estimates, as well as correlation between parameters, were evaluated. Models with parameter estimates with high associated SEs (>35 % of the parameter estimate), models with a high degree of correlation between parameters (>90 %), and models that included a covariate(s) whose effect on the estimated parameter value was negligible were carefully evaluated and re-parameterized, or possibly rejected.
Model Qualification
The model developed in this analysis was tested and qualified by evaluating precision of the final parameter estimates and the condition number, and determining both the symmetrical 95 % confidence intervals (CIs) from the asymptotic SEs of the parameter estimates, as well as non-parametrically bootstrapped 95 % CI. Standard diagnostic plots and visual and numerical predictive checks were also evaluated. Finally, the power to evaluate CL/F and Vc/F was evaluated using the final model and database to confirm the prospective evaluation.
In addition, a visual predictive check (VPC) [11] was conducted to compare the distribution of simulated observations from the final model to those obtained from the original data.
Systems
The concentration–time data collected in these studies were analyzed using mixed–effects modeling methods as implemented by the computer program NONMEM® (version 7, level 2; Icon Development Solutions, Ellicott City, MD, USA) [12, 13], compiled using Intel Fortran Parallel Studio 2011 (Intel Corporation, Santa Clara, CA, USA), and installed on a grid server system running Windows Server 2008 x64-bit. Diagnostic graphics, exploratory analyses, and post-processing of NONMEM® output was performed using the SPlus 6.2 Professional software [14] or R version 2.15.0 or later.
Results
Demographics
A total of 156 patients (80 females, 76 males) took part in this study. Age groups were <1 year (n = 10), 1 year to <2 years (n = 14), 2 years to <4 years (n = 28), 4 years to <7 years (n = 28), 7 years to <12 years (n = 10), and ≥12 years (n = 66). The overall summary statistics for baseline demographics are presented in Table 2.
Table 2.
Demographic characteristics of study patients
| Characteristic | Study PKU-015 (N = 80)a | Study PKU-004 (N = 78)a | Pooled studies PKU-015 and PKU-004 (N = 156)a |
|---|---|---|---|
| Age (years) | 3.28 (2.01) [0.107–6.98] | 21.1 (9.64) [9–50] | 12 (11.3) [0.107–50] |
| Age category [n (%)] | |||
| <1 year | 10 (12.5) | 0 | 10 (6.4) |
| 1 to <2 years | 14 (17.5) | 0 | 14 (9.0) |
| 2 to <4 years | 28 (35.0) | 0 | 28 (17.9) |
| 4 to <7 years | 28 (35.0) | 0 | 28 (17.9) |
| 7 to <12 years | 0 | 10 | 10 (6.4) |
| ≥12 years | 0 | 68 | 66 (42.3) |
| Sex [n (%)] | |||
| Females | 49 (61.3) | 33 (42.3) | 80 (51.3) |
| Males | 31 (38.7) | 45 (57.7) | 76 (48.7) |
| Ethnicity [n (%)] | |||
| Non-Hispanic | 78 (97.5) | 76 | 152 (97.4) |
| Hispanic | 2 (2.5) | 2 | 4 (2.6) |
| Body weight (kg) | 15.9 (6.36) [4.5–41.9] | 67.2 (21.8) [28.2–144] | 40.9 (30.3) [4.5–144] |
| Height (cm) | 95.6 (18.3) [56–128] | 165 (13.3) [126–191] | 129 (38.2) [56–191] |
| Body surface area (m2) | 0.635 (0.19) [0.259–1.18] | 1.72 (0.31) [1.05–2.65] | 1.17 (0.604) [0.259–2.65] |
| ALT (IU/L) | 22.4 (8.15) [13–63] | 28.4 (18.3) [11–127] | 25.4 (14.5) [11–127] |
| AST (IU/L) | 38.1 (8.75) [23–63] | 25.7 (5.8) [14–43] | 32.2 (9.65) [14–63] |
| Bilirubin (mg/dL) | 7.51 (4.4) [1.71–34.2] | 0.55 (0.33) [0.1–1.9] | 4.12 (4.7) [0.1–34.2] |
| Albumin (g/dL) | 4.19 (0.274) [3.6–4.9] | 4.49 (0.23) [4–5] | 4.33 (0.29) [3.6–5] |
| CLCR (mL/min)b | 57.1 (22.2) [9.39–120] | 114 (26) [48–231] | 87.8 (45.1) [9.39–276] |
| Baseline phenylalanine (μmol/L) | 324 (140) [57.5–768] | 811 (393) [53–2190] | 562 (378) [53–2190] |
ALT alanine aminotransferase, AST aspartate aminotransferase, CL CR creatinine clearance
aValues are presented as mean (standard deviation) [range], unless specified otherwise
bCalculated from plasma creatinine
Final Model
The final dataset consisted of 475 observations from 156 patients from studies PKU-015 and PKU-004. The final pharmacokinetic model was a one-compartment model with first-order input and elimination. The model was parameterized in terms of CL/F, Vc/F, first-order absorption rate constant (k a), an absorption lag time (t lag), and endogenous level of BH4 (C0). The effects of weight on CL/F and Vc/F were described using a power function. No other covariates were identified as being predictive of pharmacokinetic variability. The residual error model was a CCV model (additive in the log domain) with separate terms for each study. Inter-individual variability was described for CL/F, Vc/F, and C0 with terms describing the correlation between CL/F and Vc/F. The equations for the final model parameters are provided in Eq. 6 and the parameter estimates for the final covariate model are presented in Table 3.
| 6 |
Table 3.
Estimated population pharmacokinetic parameters and bootstrap 95 % confidence interval for sapropterin dihydrochloride
| Parameter (units) | Parameter | Final model estimate (SE) | Bootstrap model estimate (95 % CI) |
|---|---|---|---|
| CL/F (L/h) | θ1 | 2,710 (9.8) | 2,708 (2,308–3,162) |
| Effect of weight on CL/F | θ6 | 0.864 (7.3) | 0.861 (0.751–0.980) |
| Vc/F (L) | θ2 | 3,010 (43.9) | 3,967 (1,509–8,596) |
| Effect of weight on Vc/F | θ7 | 0.644 (18.9) | 0.658 (0.399–0.827) |
| k a h-1 | θ3 | 0.235 (23.8) | 0.284 (0.158–0.524) |
| t lag (h) | θ4 | 0.321 (11.2) | 0.313 (0.246–0.400) |
| C0 (μg/L) | θ5 | 16.6 (4.1) | 16.39 (15.5–18.22) |
| Residual error study PKU-004 (% CV) | θ8 | 21.1 (9.2) | 20.47 (17.95–26.0) |
| Residual error study PKU-015 (% CV) | θ9 | 30.2 (12) | 30.312 (23.5–37.86) |
| IIVCL/F (% CV) | η1 | 45.61 (23.3) | 45.85 (35.05–56.07) |
| IIVVc/F (% CV) | η2 | 56.57 (39.4) | 54.67 (32.14–78.56) |
| IIVC0 (% CV) | η3 | 36.47 (30) | 36.16 (26.54–48.17) |
| Corr (CL/F, Vc/F) | 0.469 (NE) | 0.34 (0.20–0.50) |
C0 endogenous levels, CI confidence interval, CL/F apparent total clearance of the drug from plasma after oral administration, Corr correlation between parameters, CV coefficient of variation, IIV inter-individual variability, k a absorption rate constant, NE not estimated, SE standard error, t lag lag time, V c /F apparent volume of distribution of the central compartment
Parameter precision for the final model was generally good, with most parameters being estimated with relative SEs <25 %, with the exception of Vc/F and the associated inter-individual variability. For the addition of two covariate factors (weight on CL/F and weight on Vc/F), the objective function decreased 137.26 points. Inter-individual variability on CL/F decreased from 87 to 46 % and on Vc/F decreased from 78 to 57 %. The residual variability in the final model was acceptable, being 21 % for data arising from study PKU-004 and 30 % from data arising from study PKU-015; this variability was not affected by the addition of weight as a covariate to CL/F and Vc/F. Evaluation of the population mean value of half-life suggests a terminal half-life of 0.78 h, whereas the absorption half-life (calculated as 0.693/k a) is 2.95 h, suggesting flip-flop pharmacokinetic behavior [15], in which absorption becomes the rate-limiting metric of exposure. However, attempts to re-parameterize the model to force absorption to be faster than elimination were not successful. Given the absorption and elimination half-lives, once-daily dosing in pediatric patients is justified.
Final Model Stability and Predictability
The median values obtained from the non-parametric age-stratified bootstrap evaluation are in good agreement with the original estimated values (Table 3). The CIs for the parameters do not include the null, supporting the inclusion of weight as a covariate for CL/F and Vc/F. In general, the CIs are reassuringly narrow, although the intervals for Vc/F are wide, as would be inferred from the SEs of the parameter estimates. The condition number was low, suggesting minimal co-linearity; the shrinkage was low for CL/F, but high for Vc/F in the final model.
Predictive Performance of the Final Model
Goodness-of-fit plots are provided in Electronic Supplementary Material Fig. 2S and 3S. A plot of observed versus typical predicted BH4 concentration and the plot of observed versus individual predicted BH4 concentration show generally good agreement, with no apparent bias. Despite including weight in the model to describe CL/F and Vc/F, there was still significant unexplained variability in pharmacokinetic model parameters. The plots of conditional weighted residuals versus typical predicted concentrations and conditional weighted residuals versus time after dose show no evident trends or bias, and the range of conditional weighted residuals falls between ±3.
Frequency histogram plots of normalized prediction distribution errors, quantile–quantile plots, and plots of observed concentrations overlaid with typical predicted concentrations versus time after dose suggested that the final model described the observed data properly.
Visual Predictive Check
To visualize the predictive performance of the final model, several VPC plots were generated. In a plot showing the 2.5th, 50th, and 97.5th prediction intervals for BH4 concentrations overlaid with the observed data (Fig. 1), most of the observed concentrations fall within the prediction intervals, and there are no obvious patterns for observations that fall outside the prediction intervals, which indicates that the model can capture the variability in the observed data. The percentiles of the observed data are consistent with the prediction intervals of the simulated data. The VPC plots stratified by age or weight suggest that the observed and simulated percentiles are in reasonable agreement, even for the lowest age and weight groups in which the amount of data is sparse. A VPC plot stratified by study suggested that, despite simplification of the model originally used to describe data arising from PKU-004, the data are adequately reproduced by the new model, and confirmed that there are no inherent differences between the two study populations.
Fig. 1.
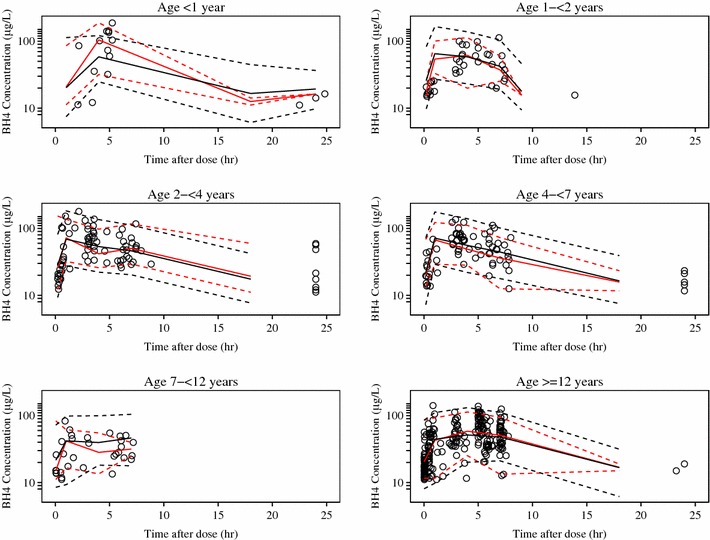
Visual predictive check by age group. Open blue circles are observed data, the solid red line is the median observed concentration, dashed red lines are the upper and lower 95 % observed intervals, the solid black line is the median of the simulated data, and dashed black lines are the upper and lower 95th percentiles of the simulated data. BH4 tetrahydrobiopterin
Confirmation of Study Power
The desired precision was to target the 95 % CI of the CL/F and Vc/F to be within the intervals of 60 and 166 % of the point estimate for the geometric mean for each designated age group. A prospective evaluation of study power indicated that PKU-015 had sufficient power to meet these criteria. The power was re-evaluated and the results demonstrate that the study had sufficient power to meet the pre-specified criteria for each age group (Fig. 2).
Fig. 2.
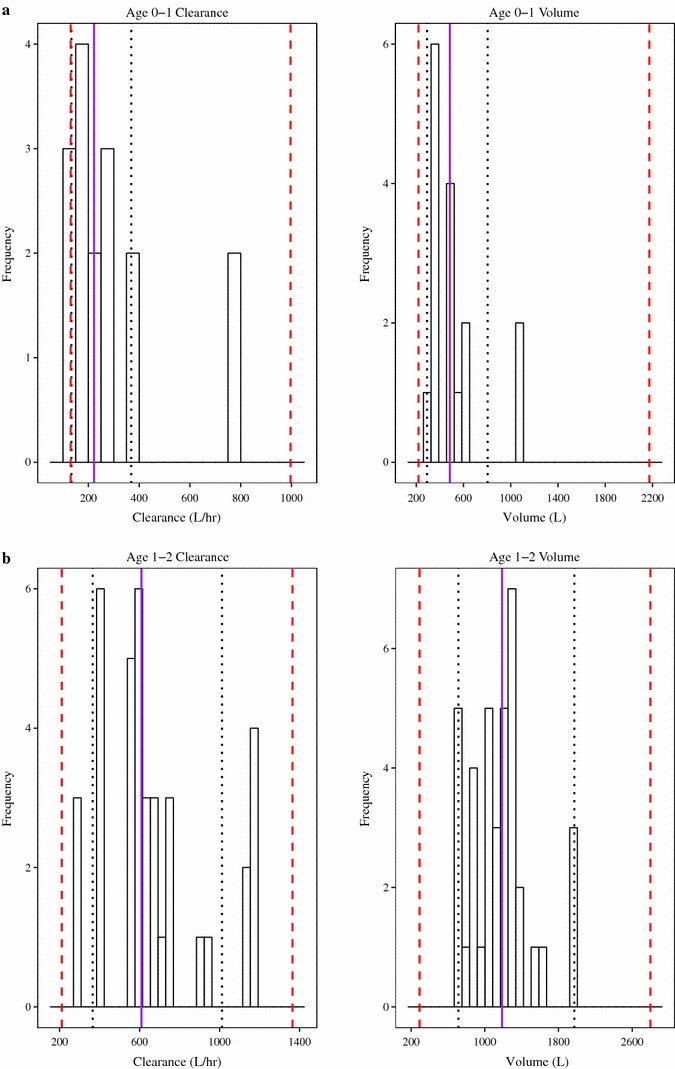
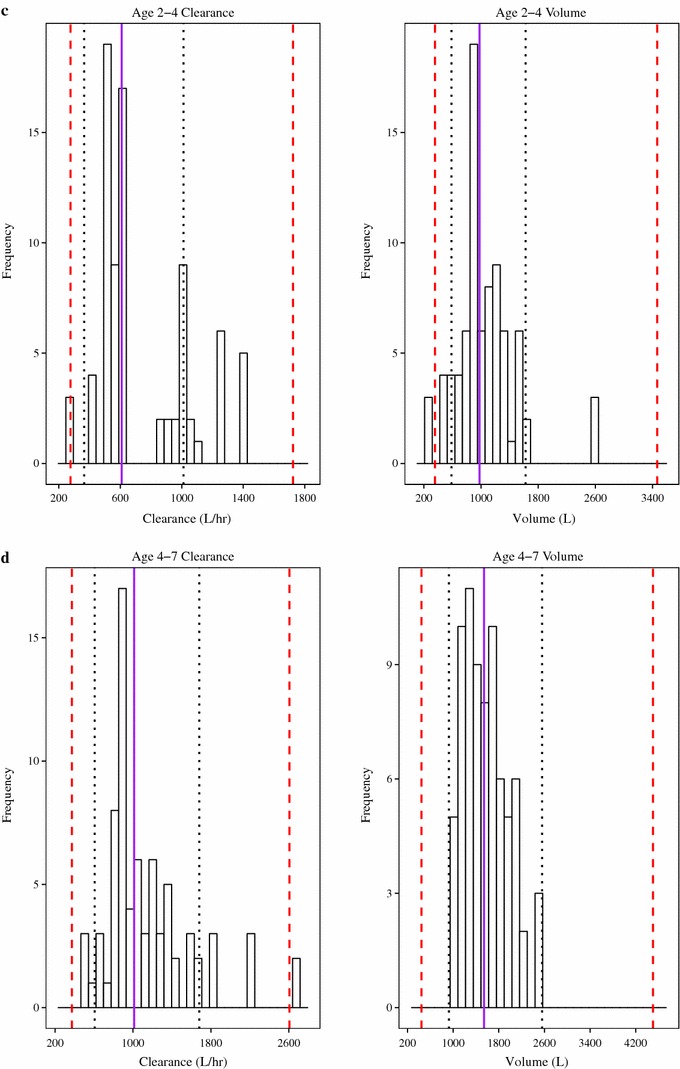
Frequency histograms of individual parameter estimates. Confirmation of power for apparent total clearance of the drug from plasma after oral administration and apparent volume of distribution of the central compartment in the age group 0–1 years (a), 1–2 years (b), 2–4 years (c), and 4–7 years (d). The solid purple line is the median value, dotted black lines are the 95 % confidence interval, and the red dashed lines are 60–166 % of point estimate of the mean
Clinical Significance of Identified Covariates
To assess the clinical significance of the effect of weight, the final population model was used to calculate pharmacokinetic parameter values for patients that represented the extremes of covariate influences within this study population. The effect of weight on CL/F and Vc/F is presented in Electronic Supplementary Material Table 2S. At the extremes of weight, a 5 kg patient had a CL/F and Vc/F value approximately 10 and 18 % of the reference 70 kg patient, respectively. Conversely, a patient weighing 145 kg had CL/F and Vc/F that was 190 and 160 % of the reference patient, respectively. Although the addition of weight to the model significantly decreased the inter-individual variability in CL/F and Vc/F, significant between-patient variability remained.
Both CL/F (Fig. 3a) and Vc/F (Fig. 3b) increase in a non-linear manner with increasing weight, although individual predictions still vary around the typical individual predictions. The intervals for Vc/F are broad, reflecting the precision with which Vc/F was estimated. Although the majority of individual estimated values for Vc/F fall at or below the median, there are patients (particularly patients with low body weight) whose estimated Vc/F values fill the predicted 95 % CI for this parameter.
Fig. 3.
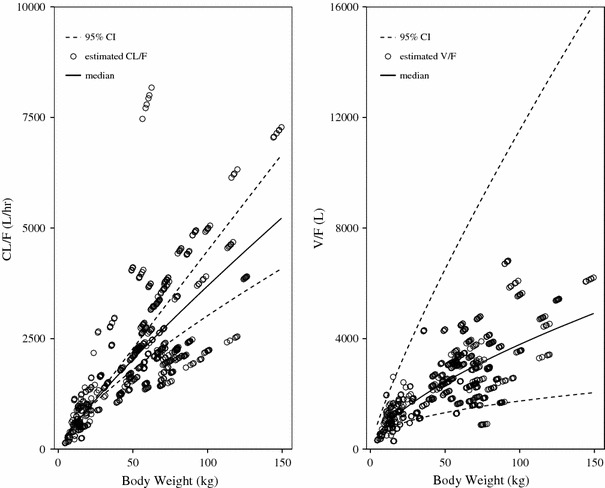
Relationship between weight and clearance (a) and weight and volume of distribution (b). Dotted lines are the upper and lower 95 % confidence intervals of median expected parameter value, the solid line is the median expected parameter value, and open circles are the individual estimated parameter values from final model. CI confidence interval, CL/F clearance, V/F volume of distribution
Exposure
In simulated concentration–time profiles following 20 mg/kg dosing for a range of weights, concentrations remain above the endogenous level for the dose interval (Electronic Supplementary Material Fig. 4S). Overall, BH4 exposure (area under the concentration–time curve at steady state; AUCss) based on administered dose and individual clearance estimates was comparable across all age groups, although there is a slight visual trend towards higher exposure (AUCss) as the age decreases (Fig. 4). However, the number of patients in the youngest age group is small, so this trend should be considered with caution. Maximum (peak) drug concentration (C max) could not be determined as the shrinkage on Vc/F was high and there were no terms for inter-individual variability on k a, making parameters used to calculate C max unlikely to reflect individual values. No other covariates were identified.
Fig. 4.
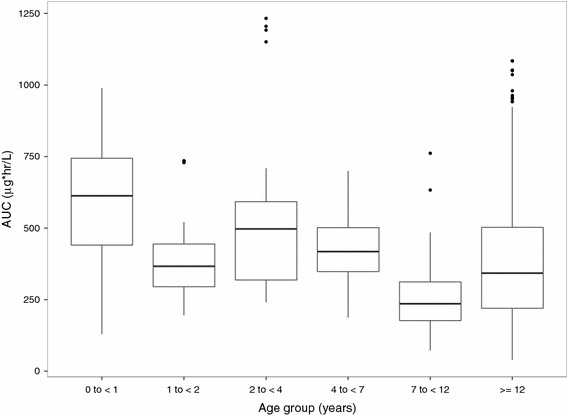
Comparison of tetrahydrobiopterin exposure by age groups. Whiskers represent 10th and 90th percentile, boxes represent 25th and 75th percentile, center lines represent median, and solid dots represent outliers. AUC area under the concentration–time curve
Comparison of Previous and Current Pharmacokinetic Models
In order to evaluate the differences in exposure expected from the original two-compartment model and the current one-compartment model, simulated concentration–time profiles for the reference patient were generated. Differences between the expected concentration times arising from these two models is minimal (Fig. 5). The simplification from a two-compartment structural model to a one-compartment structural model used in the current evaluation is justified based further on the poor precision with which the inter-compartmental clearance term (Q/F) and the apparent volume of distribution of the peripheral compartment (Vp/F) were identified in the original model. The inability to utilize the previous model may be partly attributed to the change in NONMEM® versions (6.1–7.2) that involved underlying changes to the FOCE estimation method and changes in the compilers used for NONMEM®.
Fig. 5.
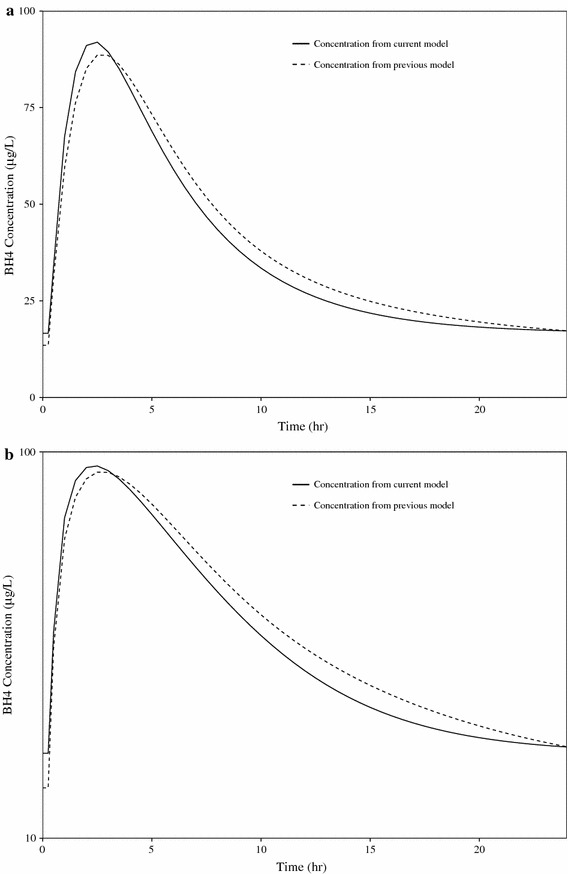
Linear (a) and semi-log (b) comparison of simulated concentration–time profiles for a 20 mg/kg dose for the reference patient. BH4 tetrahydrobiopterin
Discussion
This analysis suggests that the pharmacokinetics of BH4 can be described by a one-compartment model first-order input following a t lag and first-order elimination. A term describing the endogenous concentration is also included in the model. Inter-individual variability was included on CL/F, Vc/F, and C0, with correlation described between CL/F and Vc/F. The model was fit using the FOCE method and the LTBS approach was used. A proportional residual error model (additive in the log domain) was incorporated with separate residual error terms estimated for each study.
A D-optimal, sparsely sampled population pharmacokinetic approach was used in this study for several reasons: this approach suggests windows of time where sampling will be most informative relative to a proposed model, without undue penalty against the identification of alternative models; allows patients to have fewer blood samples drawn than with traditional pharmacokinetic sample designs, which often require eight or more pharmacokinetic samples per patient; and weighs various sample schemes based on the efficiency of a proposed design, the expected bias and precision of estimated parameters, and practical considerations. The fact that patients in this study received different dosing regimens of sapropterin further improved the information content of the data obtained. The final model was evaluated using multiple methods including non-parametric bootstrapping and VPCs. All evaluations suggested the model adequately described the data and was acceptable.
Limitations of this study include variability in Phe levels and a small sample size. Phe levels are impacted by multiple factors and can change rapidly in patients with PKU. The timing of Phe determinations relative to pharmacokinetic sampling was not known. Although study PKU-015 was adequately powered and enrollment was consistent with other pediatric studies, a relatively small number of pediatric subjects (n = 80) are included in this analysis.
Total body weight was the only significant covariate and improved the model fit to the data when included on both CL/F and Vc/F. The effect of body weight on CL/F and Vc/F was substantial and supports a dose adjustment strategy based on weight in the pediatric population. While the addition of weight to the model significantly decreased the inter-individual variability in CL/F and Vc/F, there was still significant between-patient variability remaining. Evaluation of the population mean value of half-life suggests a terminal half-life of 0.78 h, whereas the absorption half-life (calculated as 0.693/k a) is 2.95 h, suggesting flip-flop pharmacokinetic behavior where absorption becomes the rate-limiting metric of exposure. The estimated elimination half-life from the current model is different from the previous model, which could be due to the use of endogenous concentrations to estimate half-life in the previous model.
Based on recommended dosing, exposure across age groups was comparable. Given the absorption half-life and elimination half-life, once-daily dosing is justified in pediatric patients.
Conclusion
Overall, the pharmacokinetic model developed in the present evaluation provides reasonable estimates that are consistent with the previous evaluation [4], despite the simplification of the model. The effect of body weight on CL/F and Vc/F was substantial and supports a dose adjustment strategy based on weight in the pediatric population. The doses selected for pediatric patients provided similar exposure. Overall, the exposure across all age groups was comparable and given the absorption and elimination half-lives, once-daily dosing is justified for pediatric patients.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
Analysis of plasma samples for pharmacokinetic analysis was performed by QPS, LLC. (Newark, DE, USA). Database assembly and population pharmacokinetic analysis were conducted by Projections Research Inc. (Phoenixville, PA, USA). Medical writing and editorial assistance were provided by Gary Witherell, PhD (InClin Inc., San Mateo, CA, USA). The authors would like to thank the 19 investigators in study PKU-015 and the additional 24 investigators in study PKU-004 involved in patient recruitment and retention.
Conflicts of interest
DRM has provided paid consultancy support to BioMarin Pharmaceutical Inc. YQ, HZ, MM, and DGM are employees of BioMarin Pharmaceutical Inc. and own stock and stock options in BioMarin Pharmaceutical Inc. Projections Research Inc. and InClin, Inc. (San Mateo, CA, USA) were compensated for providing assistance with the study, data analysis, and/or manuscript preparation.
Author contributions
YQ, DRM, HZ, MM, and DGM contributed towards the writing and interpretation of the data. The final manuscript was approved by all authors. DRM completed the population pharmacokinetic modeling.
Role of the funding source
This study was sponsored by BioMarin Pharmaceutical Inc., which had a significant role in the study design; the collection, analysis and interpretation of data; and the writing of the report. The study protocol was drafted and developed by the study sponsor. Representatives or employees of the sponsor were responsible for the administration and monitoring of the study.
References
- 1.Lambruschini N, Pérez-Dueñas B, Vilaseca MA, Mas A, Artuch R, Gassió R, Gómez L, Gutiérrez A, Campistol J. Clinical and nutritional evaluation of phenylketonuric patients on tetrahydrobiopterin monotherapy. Mol Genet Metab. 2005;86(Suppl 1):S54–S60. doi: 10.1016/j.ymgme.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 2.Hennermann JB, Bührer C, Blau N, Vetter B, Mönch E. Long-term treatment with tetrahydrobiopterin increases phenylalanine tolerance in children with severe phenotype of phenylketonuria. Mol Genet Metab. 2005;86(Suppl 1):S86–S90. doi: 10.1016/j.ymgme.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 3.Trefz FK, Scheible D, Frauendienst-Egger G, Korall H, Blau N. Long-term treatment of patients with mild and classical phenylketonuria by tetrahydrobiopterin. Mol Genet Metab. 2005;86(Suppl 1):S75–S80. doi: 10.1016/j.ymgme.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 4.Feillet F, Clarke L, Meli C, Baker J, Lipson M, Bergoffen J, et al.; Sapropterin Research Group. Pharmacokinetics of sapropterin dihydrochloride in patients with phenylketonuria. Clin Pharmacokinet. 2008;47(12):817–25. [DOI] [PubMed]
- 5.Retout S, Duffull S, Mentre F. Development and implementation of the population Fisher information matrix for the evaluation of population pharmacokinetic designs. Comput Methods Programs Biomed. 2001;65(2):141–151. doi: 10.1016/S0169-2607(00)00117-6. [DOI] [PubMed] [Google Scholar]
- 6.Duffull SB, Retout S, Mentre F. The use of simulated annealing for finding optimal population designs. Comput Methods Programs Biomed. 2002;69(1):25–35. doi: 10.1016/S0169-2607(01)00178-X. [DOI] [PubMed] [Google Scholar]
- 7.Mentre F, Mallet A, Baccar D. Optimal design in random-effects regression models. Biometrika. 1997;84(2):429–442. doi: 10.1093/biomet/84.2.429. [DOI] [Google Scholar]
- 8.Mandema JW, Verotta D, Sheiner LB. Building population pharmacokinetic-pharmacodynamic models. J Pharmacokinet Biopharm. 1992;20:511–528. doi: 10.1007/BF01061469. [DOI] [PubMed] [Google Scholar]
- 9.Guidance for Industry: Population Pharmacokinetics. US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER), February 1999. http://www.fda.gov/downloads/Drugs/Guidances/UCM072137.pdf. Accessed 6 Oct 2014.
- 10.European Medicines Agency. Guideline on reporting the results of population pharmacokinetic analyses. CHMP June 2007. CHMP/EWP/185990/06. http://www.emea.europa.eu/pdfs/human/ewp/18599006enfin.pdf. Accessed 13 Oct 2014.
- 11.Yano Y, Beal SL, Sheiner LB. Evaluating pharmacokinetic/pharmacodynamic models using the predictive check. J Pharmacokinet Pharmacodyn. 2001;28(2):171–192. doi: 10.1023/A:1011555016423. [DOI] [PubMed] [Google Scholar]
- 12.Beal SL, Sheiner LB. The NONMEM system. Am Stat. 1980;34:118. doi: 10.2307/2684123. [DOI] [Google Scholar]
- 13.Boeckmann AJ, Beal SL, Sheiner LB. NONMEM users guide—part III, NONMEM installation guide. San Francisco: NONMEM Project Group, University of California; 1998 Mar.
- 14.SPlus version 6.2 Professional. Copyright © 1988–1999. Seattle: Data Analysis Products Division, MathSoft: 1988–1999
- 15.Yáñez JA, Remsberg CM, Sayre CL, Forrest ML, Davies NM. Flip-flop pharmacokinetics-delivering a reversal of disposition: challenges and opportunities during drug development. Ther Deliv. 2011;2(5):643–672. doi: 10.4155/tde.11.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.