

Summary
During evolution of the vertebrate cardiovascular system, the vast endothelial surface area associated with branching vascular networks mandated the development of molecular processes to efficiently and specifically recruit circulating sentinel host defense cells and tissue repair cells at localized sites of inflammation/tissue injury. The forces engendered by high-velocity blood flow commensurately required the evolution of specialized cell surface molecules capable of mediating shear-resistant endothelial adhesive interactions, thus literally capturing relevant cells from the blood stream onto the target endothelial surface and permitting subsequent extravasation. The principal effectors of these shear-resistant binding interactions comprise a family of C-type lectins known as ‘selectins’ that bind discrete sialofucosylated glycans on their respective ligands. This review explains the ‘intelligent design’ of requisite reagents to convert native CD44 into the sialofucosylated glycoform known as hematopoietic cell E-/L-selectin ligand (HCELL), the most potent E-selectin counter-receptor expressed on human cells, and will describe how ex vivo glycan engineering of HCELL expression may open the ‘avenues’ for the efficient vascular delivery of cells for a variety of cell therapies.
Keywords: stem cell, mesenchymal stem cell, E-selectin, E-selectin ligand, HCELL, regenerative medicine
Introduction
The successful clinical implementation of adoptive cellular therapeutics critically depends on delivery of pertinent cells to the requisite tissue site(s) with maximum efficiency. To minimize morbidity, primary clinical principles dictate that transferred cells should be administered intravascularly, thereby exploiting the native processes governing cell migration. A thorough understanding of the molecular mechanisms that underlie cell trafficking in the vasculature is thus essential to the rational design and implementation of strategies to enable cell therapies such as in T/natural killer (NK) cell-based immunotherapy and stem cell-based regenerative medicine.
Cell migration requires the action of distinct classes of molecules that function in a sequential fashion. To exit the vasculature, circulating cells must first be capable of adhering to target vascular endothelium with sufficient strength to overcome the shear forces of blood flow. The most effective mediators of this initial decelerating/braking adhesive interaction are the selectins – E-selectin (CD62E, expressed on endothelium), P-selectin (CD62P, expressed on platelets and on endothelium), and L-selectin (CD62L, expressed on leukocytes and on hematopoietic stem cells). E- and P-selectins are typically referred to as the ‘vascular selectins’ because of their expression on endothelium, whereas L-selectin is called the ‘leukocyte selectin’. The selectins are integral membrane proteins comprising a family of C-type (i.e. requiring Ca2+ for activity) lectins that bind to sialofucosylated glycans displayed on their relevant ligands. These glycan structures can be displayed on protein scaffolds (i.e. glycoproteins) or on lipid scaffolds (i.e. glycolipids), and, in each case, the native display of these determinants is dependent on the expression and activity of specific Golgi glycosyltransferases that add the relevant sialic acid and fucose monosaccharides in the proper locations of the glycan polymer.
We have developed a platform technology called ‘glycosyltransferase-programmed stereosubstitution’ (GPS) to custom modify cell surface glycans without affecting cell viability or native phenotype. GPS has been used to convert membrane CD44 on live cells into the potent E-selectin ligand glycoform known as hematopoietic cell E-/L-selectin ligand (HCELL), thereby conferring tissue-specific migration of intravascularly administered cells to vascular beds expressing E-selectin in vivo. This article will explain the creation of GPS and will consider how enforced HCELL expression could be harnessed to optimize tissue delivery of infused cells in adoptive cellular therapeutics.
Regulation of cellular trafficking: the multi-step paradigm
The recruitment of discrete subsets of cells in blood flow into tissues is critical to a variety of physiologic and pathologic processes, including immunity, tissue repair, and cancer metastasis. Most commonly, extravasation occurs within post-capillary venules under fluid shear stress conditions of 1–4 dynes/cm2 (1). A variety of in vitro and in vivo studies have established that the migration of cells from vascular to extravascular compartments is governed by a cascade of molecular interactions, conventionally referred to as the ‘multi-step paradigm’ (2, 3). ‘Step 1’ in this process involves the capture of cells from the fluid stream onto the target tissue endothelium. This physiologic braking process, mediated principally by the selectins and their ligands, gives rise to initial tethering and then rolling interactions of cells directly on the endothelial surface, at velocities well below that of the prevailing fluid stream. Rolling interactions allow for intimate contact with the endothelium for distances ranging from a few micrometers to several millimeters; cells can thus be exposed to chemical signals (principally chemokines, but also cytokines and other inflammatory agents) present in the local milieu (Step 2), resulting in the activation-dependent upregulation of integrin adhesiveness leading to firm adhesion (Step 3), followed by endothelial transmigration (Step 4) (Fig. 1).
Fig. 1. The multistep model of transendothelial migration.
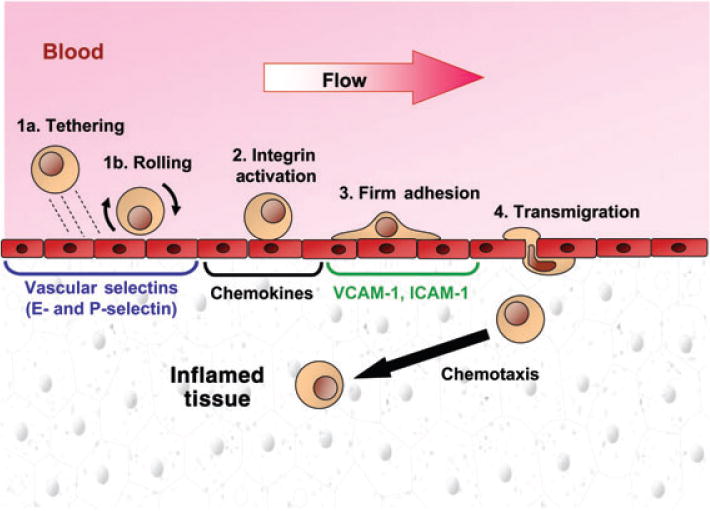
Schematic representation of the multiple steps involved in emigration of cells from the vascular compartment into tissue parenchyma. At all sites of tissue injury or inflammation, endothelial cells characteristically display vascular selectins, chemokines and integrin receptors as shown in the figure. Steps 1a and 1b together act to capture cells in hemodynamic flow onto the endothelial surface and are mediated principally by selectin receptor/ligand interactions. Chemokine-induced activation (Step 2) of integrin adhesiveness results in firm adhesion (Step 3) followed by transmigration (Step 4). See text for details.
Implicit in this model is the critical role of Step 2 in serving as a checkpoint for completion of the cascade. The major mediators of firm adherence are the integrins, very-late activation protein 4 (VLA-4) and lymphocyte function-associated antigen 1 (LFA-1), which are maintained in an inactive state in essentially all ‘resting’ circulating cells. The transition from Step 1 to Step 3 is thought to be orchestrated by the discrete combinatorial diversity of chemokine receptors and chemokines expressed on circulating cells and target endothelium, respectively. Chemokines comprise a superfamily of small proteins that function as potent chemotactic agents (reviewed in 4). Some chemokines are widely distributed, whereas some have a tissue-specific distribution and some are expressed in an inflammation-dependent manner. Additional complexity arises from the fact that the ligands for chemokines are G-protein-coupled receptors (GPCRs) that have a cell-specific distribution. Where pertinent, triggering of these GPCRs results in the activation of integrins LFA-1 and/or VLA-4, potentiating their binding to vascular endothelial counter-receptors, intercellular adhesion molecule 1 (ICAM-1) and vascular cell-adhesion molecule 1 (VCAM-1), respectively. Extravasation then ensues, typically driven by chemokine gradients present in the subendothelial compartment (5) (Fig. 1).
The conventional multi-step model holds that Step 1 interactions are reversible: if rolling cells possess the requisite receptors for those particular chemoattractants expressed in the perivascular microenvironment, integrin activation ensues, and they will then transition into firm adherence and undergo transmigration; in the absence of pertinent Step 2 receptors, rolling would be transient (lasting seconds to minutes), followed by detachment of cells and re-entry into the blood stream. To date, methods to augment cell migration have focused primarily on introducing chemokine receptors by genetic means (6–8) or upregulating their expression by manipulating cells in vitro (9–11). However, for every tissue target, the proximate hurdle to achieving cell recruitment is, literally, upstream of Step 2 events, as only those cells that can undergo efficient tethering/rolling interactions can become tissue residents. Thus, strategies to at least maintain or, as needed, enhance or create expression of Step 1 effectors on cell surfaces are essential to realize delivery of pertinent cells to sites where they are needed.
The ‘roll’ of selectin-dependent adhesive interactions
Although several molecules, including CD44 and some integrins [e.g. LPAM-1 (α4β7) (12)], are capable of mediating tethering and rolling adhesive interactions, the selectins are the most potent effectors of Step 1 and can maintain rolling at higher fluid shear levels than any other structures (reviewed in 1). The selectins are also unique in that they display optimal binding to their ligands under physiologic shear conditions, and, indeed, L-selectin will not adhere to any of its counter-receptors under static conditions (13–16). This unusual property of selectin receptor/ligand interactions is in part a consequence of fast on–off rates, typical of low affinity interactions between lectins and glycans. In the setting of fluid shear forces, these fast on–off binding interactions are translated into cellular torque, resulting in rolling interactions at velocities below the prevailing hemodynamic flow. However, more specifically, selectin receptor/ligand interactions have the conspicuous capacity to decrease their off rate within a certain force range (17–19), thereby increasing tether life times and commensurately increasing the number of bonds per tether with escalating wall shear stress. This process leads to stable, nearly constant selectin-dependent rolling velocities over a wide shear stress range (20), and to the paradoxical observation that selectin-dependent rolling velocity can actually decrease within a range of increasing application of shear stress (18, 21). This so-called ‘catch bond’ phenomenon yields to ‘slip bonds’ and to dissociation of the selectin receptor/ligand complex as higher mechanical tensile forces are applied; cells will thus detach from the endothelial surface if they encounter (higher) hemodynamic shear conditions whereby slip bonds dominate, but during stable cell rolling, this catch-slip interface occurs naturally as bonds initiated at the leading edge transition toward the trailing edge. The characteristic topographical distribution of L-selectin and of selectin ligands on the tips of microvilli of circulating cells may also be a contributing factor in this force–bond relationship, as the elastic spring constant of the microvillus can serve to further dampen bond dissociation rates (22). Membrane compliance is also contributory, as cell-substrate binding under shear stress causes cell deformation (flattening) that boosts the number of selectin receptor/ligand contacts (23).
The expression of selectins varies depending on the cell type, the anatomic location, the activation status of the cell(s), and the presence or absence of inflammation. As noted above, L-selectin is expressed on mature leukocytes and on hematopoietic stem cells, but its expression is conspicuously absent in intermediate stages of leukocyte development (24–27). Among lymphocytes, L-selectin is characteristically expressed on naive T cells and central memory cells, but its expression is generally low on effector T cells (reviewed in 28). L-selectin gene expression is thought to be constitutive on cells that express it, but transcriptional regulation induced by cytokines and post-transcriptional regulation of gene expression have each been recognized (29, 30). Among mature leukocytes, there is a rapid downregulation of surface L-selectin expression following cell activation, with shedding of the L-selectin ecto-domain (31); this membrane proximal proteolysis is mediated by the action of surface metalloproteases, at least one of which is a disintegrin and metalloprotease known as ADAM17 (a disintegrin and metalloprotease domain 17) or tumor necrosis factor (TNF)-α-converting enzyme (TACE) (32). This same enzyme mediates shedding of VCAM-1 of endothelial membranes (33). Although there is evidence of shedding of both P-selectin and E-selectin, the protease(s) mediating these processes is currently unknown. However, in contrast to L-selectin, the predominant mechanism regulating expression of both P-selectin and E-selectin is not via surface proteolysis, but by induction of surface expression. Notably, with the exception of the microvasculature of the skin and bone marrow (34–36), neither P-selectin nor E-selectin is constitutively expressed in any endothelial beds.
It is well recognized that the permanent expression of P- and E-selectin in bone and skin microvessels supports steady-state trafficking of circulating cells to these sites (reviewed in 37). However, in all microvessels (including those of the skin and marrow), upregulated expression of these vascular selectins is a critical feature of all inflammatory responses, and drives extravasation of cells bearing requisite counter-receptors. Importantly, inflammation induces distinctly different kinetics of expression for P- and E-selectin (38–40). Both of these structures are inducible membrane molecules whose transcription is markedly upregulated by inflammatory cytokines such as TNF-α and IL-1 (reviewed in 41). E-selectin is not stored in intracellular compartments, and its expression requires de novo synthesis over a period of hours (expression starts approximately 2–3 h after stimulus). However, in addition to cytokine-induced de novo synthesis, P-selectin is stored in the a-granules of platelets and in the Weibel–Palade bodies of endothelial cells and its surface expression can be rapidly upregulated via granular translocation (within seconds in platelets and minutes in endothelial cells) in response to inflammatory mediators like histamine and thrombin. Following surface expression on endothelium, both P-selectin and E-selectin are rapidly internalized by endocytosis, however, whereas E-selectin is degraded in lysosomes, P-selectin is recycled to the trans-Golgi network and is then returned to the Weibel–Palade bodies where it can then be remobilized (42).
Data obtained from murine studies indicate that the roles of P-selectin and E-selectin generally overlap in cellular recruitment. Mice genetically deficient in either E- or P-selectin appear healthy, whereas mice doubly deficient in P- and E-selectin display a marked increase in bacterial infections, especially in the skin and oral mucosa (43, 44). P-selectin-deficient mice show decreased neutrophil exudates at early time-points following chemical induction of peritonitis (43, 45–47), but E-selectin-deficient mice show no overt abnormalities in this model. However, depending on the inflammatory stimulus and the mouse strain utilized, E-selectin shows a dominant role over P-selectin in leukocyte recruitment (48–52).
Of the three selectins, the binding interactions mediated by E-selectin display the greatest resistance to detachment under shear (strongest adhesion) and the slowest rolling velocity (53–56). Importantly, in vivo studies in E-selectin-deficient mice have defined a unique role for this selectin in leukocyte recruitment during the inflammatory process. Intravital microscopy studies show that the E-selectin-deficient mice have markedly increased leukocyte transit times within vascular beds treated with TNF-α, and that E-selectin-mediated slow rolling velocity markedly reduces transit times, allowing for optimal responses to inflammatory agents/chemokines in the local milieu (57, 58). In those situations where chemokines and other chemoattractants are present in high concentration (e.g. as in experiments wherein they are superperfused in a localized site), the increased leukocyte transit times consequent to E-selectin deficiency is not a critical variable as reduced time of exposure to such agents does not affect the response to an overwhelming local concentration (57). However, under conditions where chemokines may be present in low concentration(s) and/or at a topographically limited site (such as during the initial stages of inflammation), it can be predicted that E-selectin receptor/ligand-mediated slow velocity rolling would provide essential contact time for Step 2 triggering of integrin adhesiveness and leukocyte emigration. Moreover, an important distinction between primates and mice bears mention regarding the biology of E-selectin and P-selectin, which may greatly impact extrapolation of results from mice to humans: the cytokine TNF-α induces both P- and E-selectin expression in murine endothelial cells, but only induces E-selectin on human and non-human primate endothelial cells (59–61). This variation in transcriptional control is secondary to species-specific differences in the P-selectin promoter (60). Thus, cell trafficking patterns under inflammatory conditions in humans may depend on the contribution of E-selectin to a much greater extent than that predicted on the basis of mouse studies. Collectively, these findings have profound implications for strategies to direct cell migration to sites of inflammation or tissue injury in human beings, suggesting that optimizing expression/activity of E-selectin ligands on requisite cells may be obligatory to most effectively enhance recruitment for clinical indications.
E-selectin counter-receptors on circulating cells
As stated above, all three selectins bind to sialofucosylated glycans bearing a specific terminal sialic acid (also known as neuraminic acid; ‘NeuAc’) in α(2, 3)-linkage to galactose (Gal) and a fucose (Fuc) in α(1, 3)-linkage to N-acetylglucosamine (GlcNAc), prototypically displayed as the tetrasaccharide structure known as ‘sialylated Lewis X’ (sLex; also known as CD15s): NeuAc α 2–3Gal β1–4[Fuc α 1–3]GlcNAc β1-R. The core lactosamine of sLex is known as a ‘Type 2’ unit, Galβ1–4GlcNAc β1-R. The isomeric ‘Type 1’ lactosamine unit, Galβ1–3GlcNAcβ1-R, can also be modified with α(2, 3)-linked-sialic acid on galactose, and when this structure contains a fucose substitution in α 1,4-linkage to N-acetylglucosamine, it is known as ‘sialylated Lewis a’ (sLea): NeuAc α 2–3Galβ1–3[Fuc α 1–4]GlcNAcβ1-R (Fig. 2).
Fig. 2. Structures of Type 1 and Type 2 lactosamines and of sLea and sLex.
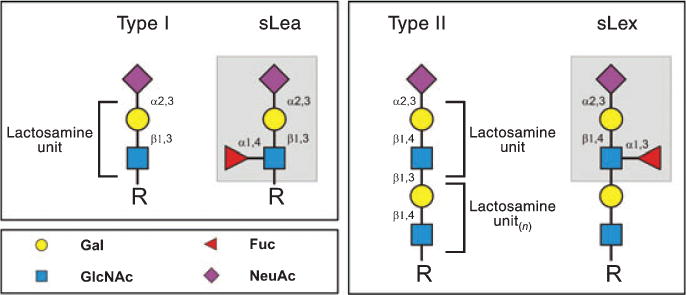
The subscript (n) shown on Type 2 lactosamine unit indicates that this disaccharide is often present as a chain of repeating units (polylactosamine). Color key figures correspond to respective monosaccharides. Pertinent linkages are shown on the structures.
All selectins can bind to both sLex and sLea (62–64), but not all cells express these structures, and some cells that express these structures do not bind to selectins. The minimum carbohydrate structure that serves as an E-selectin ligand is sLex and/or sLea, with E-selectin showing higher avidity binding to sLex (65). Human (and mouse) hematopoietic cells and all normal stem cells (both adult and embryonic) described to date characteristically express type 2 lactosamine units; thus, wherever found on such cells, the relevant selectin ligand(s) will display sLex-type sialofucosylated lactosamines. Notably, type 2 chains (but not type 1) are typically expressed as repeating units called ‘polylactosamines’ (also called ‘poly-N-acetyllactosamines’), and, therefore, the terminal sLex presented on such chains extends out significantly from the core scaffold (protein or lipid) and may be presented on multiple polylactosamine branches of a proximal lactosamine backbone (reviewed in 66). The sLea stereoisomer is not expressed on human leukocytes or hematopoietic stem cells, and is most typically found on glycans expressed on human epithelial cells and malignancies derived therefrom (67–70); indeed, the most extensively utilized marker for diagnosis of tumors of the gastrointestinal tract, ‘CA19-9’, is sLea (71). Expression of both sLea and sLex by human cancer cells has been correlated with hematogenous metastasis, etiologically related to the capacity of such cancer cells to bind vascular E-selectin under hemodynamic flow conditions (70, 72, 73).
Multivalent presentation of sLex and sLea enhances binding of all three selectins to these structures, and, in addition, specialized post-translational sulfate modifications on sLex itself (e.g. for L-selectin ligands expressed on lymph node high endothelial venules) or on tyrosines adjacent to the O-linked sLex structure of the protein scaffold [e.g. for P- and L-selectin binding to P-selectin glycoprotein ligand-1 (PSGL-1)] render higher affinity binding of L-selectin and of P- and L-selectin, respectively (reviewed in 74, 75). In contrast to P- and L-selectin, no additional structural modifications have been found to date that definitively enhance E-selectin binding to sLex or sLea (76), and high resolution nuclear magnetic resonance (NMR) spectroscopy studies have revealed that E-selectin has five fold and 10-fold higher affinity for sLex than do L-selectin and P-selectin, respectively (77). Related to this fact, there is a common misconception that E-selectin binds indiscriminately to the sLex/sLea tetrasaccharide structures if they are displayed on glycoproteins or glycolipids. This misperception traces its origins to the history of the cutaneous lymphocyte antigen (CLA), a structure originally identified by a rat immunoglobulin M (IgM) monoclonal antibody (mAb) known as HECA-452 (78). Earlier studies showed that the majority of lymphocytes in human skin, but only a minor population present in the circulation or in non-cutaneous sites, reacted with this mAb (79, 80); as such, skin-homing lymphocytes were operationally defined as expressing CLA, the epitope of HECA-452. Subsequent studies showed that CLA+ T cells bound E-selectin and HECA-452-immunoprecipitated protein from lymphocytes bound to E-selectin (81, 82), and that HECA-452 directly binds to both sLex and sLea (62). Thus, by inference, HECA-452 reactivity became (inappropriately) synonymous with E-selectin ligand activity. Subsequent biochemical studies showed that the CLA epitope is located on PSGL-1 (83, 84), and provided direct evidence that only CLA+PSGL-1 can function as an E-selectin ligand, whereas CLA− and CLA+ glycoforms of PSGL-1 equally bind P-selectin (84).
Although it is well-accepted that HECA-452 recognizes sLex/sLea structures, caution must be raised regarding the rather common practice of using this mAb as a reporter for E-selectin ligand expression, as human cell lines have been described that possess significant sialylation-dependent E-selectin ligand activity in shear-based assays but lack any detectable HECA-452 determinants (85, 86). Conspicuously, as shown by others (87) and repeatedly observed by us, this antibody (or other mAb recognizing sLex, for that matter) does not block E-selectin binding in any dynamic (flow-based) assay system. Thus, a clear distinction must be drawn between structures that possess features that can support E-selectin binding (e.g. recognizable by relevant mAb) and structures that do support E-selectin binding (some of which remain to be elucidated). A clear distinction must also be drawn between structure(s) that can support selectin binding in static assays versus dynamic assays, and between those structures that display ligand activity in vitro versus structures that (properly) support selectin binding in vivo. Apart from issues related to the presentation of adhesion molecules on native cell membranes (discussed below), many biochemical studies in vitro do not yield relevant insight on physiologic selectin ligands, in large part because the methods employed do not assess adhesion under appropriate shear conditions.
We have extensively investigated whether HECA-452 reactivity itself predicts E-selectin ligand activity. To this end, we have utilized a technique known as the ‘blot rolling assay’ (88), a methodology that we created expressly to identify a novel selectin ligand that we identified on human hematopoietic stem cells (89), which is now known as ‘HCELL’ (a potent ligand for E- and L-selectin; see below). The blot rolling assay is a powerful technique, capable of discriminating and elucidating those membrane molecules that support adhesive interactions under appropriate shear conditions. A great advantage of this method is that, within a complex mixture of membrane molecules, it allows identification of specific proteins that can mediate shear-resistant binding interactions under appropriate flow conditions without requiring prior enrichment or isolation of such proteins. In this assay, membranes are isolated from a cell type of interest, detergent solubilized, and the component proteins are resolved by gel electrophoresis and then transferred onto a support PVDF sheet (e.g. by western blot). The sheet is rendered translucent, then placed within a parallel plate flow chamber and mounted on the stage of an inverted microscope (Fig. 3). Particles or cells bearing known adhesion molecules of interest (e.g. Chinese hamster ovary cells stably transfected to express human E-selectin) are then introduced into the chamber under controlled flow conditions, and the presence of tethering and rolling interactions on discrete bands can be observed by video microscopy. Pertinent substrate molecules supporting tethering/rolling of the flowing cells can thus be identified by immunostaining (if protein is known and there are mAb that are reactive) or by extraction of the band for mass spectrometry analysis or protein microsequencing (i.e. for an unrecognized protein).
Fig. 3. Blot rolling assay apparatus.
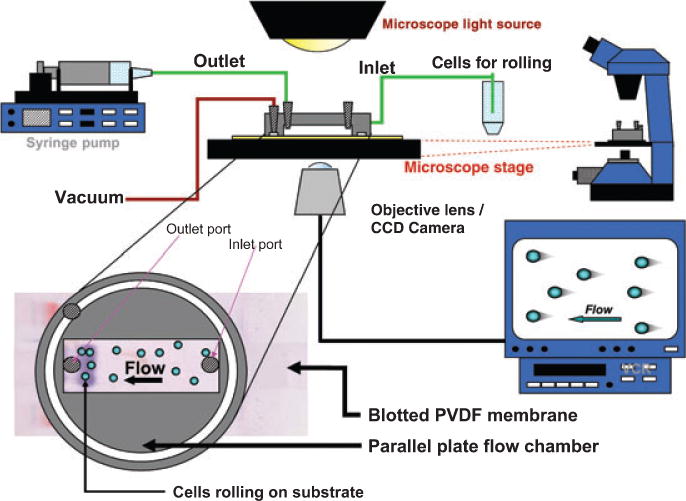
Components of the blot rolling assay are shown. Insert at left corner displays the relative placement of the parallel plate device over the PVDF membrane, in line with the relevant lane of SDS-PAGE. Cells (or particles) are brought into the parallel plate chamber under controlled flow conditions (at inlet port), and rolling interactions on pertinent substrate band(s) can be visualized directly.
Our numerous studies over the past decade using the blot rolling assay have clearly shown that HECA-452 reactivity does not directly correlate with E-selectin ligand activity, as several HECA452-reactive bands identified by western blot do not support E-selectin-dependent rolling interactions under the hemodynamic shear conditions employed (see 90). These findings underscore the dichotomy between antibody determinants and selectin binding determinants, and also highlight the difference between a static assay system (e.g. antibody staining) and shear-based adhesion assays. Insofar as selectins bind optimally under shear conditions, dynamic assay systems favor detection of functionally relevant selectin receptor-ligand interactions. At the same time, it is important to recognize that clustering of a given molecule within a defined region in space (i.e. an electrophoretic band) in either static or shear-based assays could itself induce binding properties that might not exist on the cell surface, as natively displayed structure(s) would certainly have different site density(ies) on a given scaffold (protein or lipid), copy number, orientation and topographic display. For instance, clustering of sLex or sLea alone on a microbead surface results in rolling on immobilized E-selectin under physiologic flow, varying with the relative site densities of the glycans on the beads and site densities of the substrate E-selectin (91, 92). Still, the blot rolling assay has been proven to be an extremely useful tool for identifying membrane-bound E-selectin ligands, and, in every case to date, a protein shown to support E-selectin binding by this technology has been found to support E-selectin binding as natively displayed on the intact cell membrane. Importantly, the E-selectin ligand HCELL, initially identified by blot rolling studies, is now known to mediate robust physiologic activity in vivo (93), indicating that this assay can reveal the identity of endogenous selectin ligands.
To date, three integral membrane glycoproteins have been identified as the principal E-selectin ligands on circulating human cells: CLA (PSGL-1), CD43, and HCELL (83, 90, 94, 95). Although a number of other membrane glycoproteins isolated from human neutrophils have been shown to express sLex and to support E-selectin binding in in vitro assays, including L-selectin (96, 97), b2-integrins LFA-1 and Mac-1 (98, 99), and CD66 (100, 101), the contribution of these structures to E-selectin binding on intact cells is, at best, modest. In addition to glycoproteins, it is well known that glycolipids can possess significant E-selectin ligand activity, especially those found on human myeloid cells (102–107). PSGL-1 is a disulfide-linked homodimeric mucin-like glycoprotein expressed on early hematopoietic cells and essentially in all leukocytes, and it is the prime leukocyte counterreceptor for P-selectin (reviewed in 108). As noted above, PSGL-1 is also an L-selectin ligand, and, as ‘CLA’, can also bind E-selectin; the CLA phenotype is conferred by extensive sLex modifications displayed on O-linked glycans (i.e. attached to threonine or serine residues) distributed throughout the stem region of the core protein (109, 110). All current studies indicate that sLex-bearing CD43 only binds E-selectin, and that it collaborates with CLA in promoting migration of Th1 cells to inflamed skin (111, 112). HCELL is a specialized glycoform of CD44 predominantly expressed on human hematopoietic stem cells, and is the most potent E-selectin and L-selectin ligand expressed on human cells (73, 90, 93, 113).
HCELL was initially identified by operational criteria as a distinct integral membrane glycoprotein capable of supporting L-selectin-dependent binding of lymphocytes to primary human hematopoietic progenitor cells and the human primitive hematopoietic cell line, KG1a, under shear-based (Stamper–Woodruff) assay conditions (114, 115). Similar to other selectin receptor/ligand interactions, this L-selectin binding activity was dependent on Ca2+, and was inhibited by sialidase treatment of ligand-bearing cells (i.e. sialylation was critical for ligand activity) (114). At the time of its discovery, no L-selectin ligand had yet been described on a non-endothelial cell type (114), and thus recognition of this structure opened our understanding of L-selectin function beyond its well-characterized role in mediating trafficking of lymphocytes to lymph node (reviewed in 1). Extensive biochemical studies performed over a period of several years showed that this novel L-selectin ligand was a sialofucosylated glycoprotein different from PSGL-1 (116), and was structurally unique from all other glycoprotein L-selectin ligands in that: (i) its binding activity was sulfation-independent and was resistant to digestion with O-sialoglycoprotease (i.e. sulfation-dependency and O-sialoglycoprotease sensitivity are properties of L-selectin ligands of lymph node high endothelial venules and of PSGL-1); (ii) it lacked reactivity with mAb MECA79 [which identifies L-selectin ligands displayed on lymph node high endothelial venules, reviewed in (1)]; and (iii) its L-selectin binding determinants were expressed on N-linked glycans (i.e. glycan attached to the peptide via asparagine residues) rather than the canonical O-linked glycans (as found in lymph node high endothelial venule ligands and PSGL-1) (116, 117). Despite abundant data on its distinctive biochemical features, the identification of this molecule was extremely elusive because it, like all other L-selectin ligands, required a threshold hemodynamic shear to engage L-selectin. Thus, immunoprecipitation of KG1a cells with an L-selectin-immunoglobulin (L-selectin–Ig) chimeric construct failed to isolate the molecule, and ligand blotting using L-selectin–Ig chimera did not reveal any immunostaining band(s). Immunoprecipitation of KG1a cell lysates with HECA-452 mAb was similarly uninformative, as a complex mixture of proteins was obtained, one of which was PSGL-1. However, our studies employing N-glycanase (to assess the contribution of N-glycans to ligand activity) provided a critical clue: for optimal enzyme digestion, it was necessary to subject membrane proteins to sodium dodecyl sulfate (SDS) and β-mercaptoethanol denaturing conditions. We observed that N-glycanase digestion abrogated L-selectin ligand activity, but control studies of buffer-treated, denatured membrane proteins showed that the L-selectin ligand activity was intact, similar to that of untreated membrane protein (116). With this finding, it was then realized that the ligand’s binding activity would likely withstand SDS-polyacrylamide gel electrophoresis (SDS-PAGE) conditions. Thus, the blot rolling assay was created, and this innovative technology led to the identification of the ligand as a 90–100 kDa band on SDS-PAGE gels; mass spectrometry revealed that the band consisted of a novel sialofucosylated glycoform of CD44 (90). Once isolated, further binding studies were undertaken to assess whether the molecule could bind E-selectin and/or P-selectin; these investigations revealed that this previously unrecognized structure was not a P-selectin ligand, but was the most potent E- and L-selectin ligand expressed natively on human cells (90, 113). Owing to its predominant expression on the earliest human hematopoietic progenitor cells (115), it was thus named ‘hematopoietic cell E-/L-selectin ligand’.
The glycans that mediate the E- and L-selectin ligand activity of HCELL are displayed on CD44, but it is inappropriate to simply call this structure ‘CD44’ or to state that ‘CD44 is a selectin ligand’. The working end of HCELL is not the CD44 peptide per se, it is the distinctive post-translational carbohydrate decorations that are presented upon the CD44 frame. In our early studies of HCELL based on Stamper–Woodruff assays, we consistently observed that L-selectin-mediated lymphocyte adherence was robust on KG1a cells that were treated by the crosslinking fixative glutaraldehyde, which markedly alters protein conformation but does not affect glycan structure (114). These results indicated that the protein was essentially inert with regard to ligand function(s). The central role of glycans in defining HCELL was further established by the finding that the expression of ligand activity was blocked by inhibitors of glycosylation, and that sialidase and/or fucosidase digestion alone abrogated ligand activity, thereby highlighting the key contribution of terminal α(2, 3)-linked sialic acid and α(1, 3)-linked fucose residues, likely presented as sLex, for ligand function (114, 116). Later studies showed that the E- and L-selectin ligand activity of CD44 was associated with HECA-452 reactivity (89, 90). Thus, just as ‘CLA’ defines the E-selectin binding glycoform of PSGL-1, ‘HCELL’ defines the specialized E-/L-selectin-binding glycoform of CD44. Besides ensuring consistency in such nomenclature, it is important to remember the logical precedent that HCELL (similar to CLA) was first described by unique operational characteristics, well before the scaffold protein was identified, and that the term HCELL accurately describes the function of this novel CD44 glycoform. This distinction is essential because CD44 is an extremely heterogenous and pleiotropic molecule, resulting from alternative splicing of nine encoding exons in humans (10 exons in mice) and a variety of post-translational modifications, including abundant N- and O-glycosylation, and even glycosaminoglycan substitutions (particularly chondroitin sulfate), of the core protein (reviewed in 118, 119). In fact, as a consequence of splice variations, there are presently over 20 well-characterized CD44 protein isoforms, each with its own name [e.g. ‘CD44v6’, containing variant exon 6 on the core (‘CD44H’) backbone]. Some of the common CD44 protein isoforms are routinely referred by titles (e.g. ‘epithelial’ CD44 is CD44v8–10, ‘standard’ CD44 is CD44H). This multistructural complexity, appropriately yielding to species-specific designations, underscores the need to clearly distinguish ‘HCELL’ from ‘CD44’.
The glycan substitutions that create HCELL confer a totally new biology on CD44. It is worth remembering that, prior to discovery of HCELL, it was thought that CD44 did not bind to any integral membrane protein(s). As an adhesion molecule, it was believed that CD44’s sole function was to bind extracellular matrix elements, principally hyaluronic acid, for which it is most commonly known as the ‘hyaluronic acid receptor’. Conspicuously, whereas sialylation of CD44 is critical to elaboration of HCELL, sialylation of CD44 inhibits its binding to hyaluronic acid (120–122). Moreover, CD44 binding to hyaluronic acid is not Ca2+-dependent, and, preceding the identification of HCELL, cation-dependency had not been described for any CD44 adhesive interactions. On the basis of this physical chemistry, it is understandable that no one considered the possibility that a glycoform of the hyaluronic acid receptor could be a selectin ligand. Indeed, in prior studies of E-selectin ligands in human cells (and that of other mammals), isolated CD44 was frequently used as the ‘negative control’, specifically because it was shown to be non-reactive with HECA-452 mAb and found to be devoid of selectin ligand activity, even in the presence of divalent cations (79, 82, 123, 124).
On human hematopoietic progenitor cells, HCELL is present predominantly on the ‘standard’ CD44 isoform (CD44H) that contains none of the alternatively spliced exon sequences (core mw 37 000) (89, 90). The HCELL glycoform migrates as a 90 000–100 000 mw band on SDS-PAGE gels, and is characterized by selectin ligand activity displayed exclusively on multi-antennary N-linked glycans (116). However, more recent studies have shown that human malignant cells, prominently colon cancer cells, can express an alternative HCELL structure characterized by display of sialofucosylated selectin binding determinants on O-linked glycans located on splice variant peptide sequences, yielding HCELL glycoforms of various molecular weights (most commonly ~150 000 mw) (72, 73, 125). This ‘colonic’ HCELL differs from ‘hematopoietic’ HCELL in CD44 splice isoforms and in glycan linkages, but is otherwise indistinguishable in functional characteristics, displaying essentially equivalent high potency in binding E- and L-selectin in shear-based assays (73). The fact that similar E- and L-selectin binding properties are found between hematopoietic and colonic HCELL, despite differences in regions of the scaffold CD44 protein that display the relevant binding determinants and differences in the core glycan linkage (i.e. N- versus O-) of these determinants, emphasizes the key role of terminal sialofucosylation (s) in licensing selectin ligand activity.
Glycosyltransferases that regulate sLex expression
Post-translational glycosylation requires the action of a variety of glycosyltransferases, Golgi enzymes that function in a specific order to add monosaccharide units to the (growing) glycoconjugate acceptor (reviewed in 126). Within Golgi compartments, glycan synthesis is thought to proceed like an assembly line process, such that a glycan modified by a particular glycosyltransferase reaction becomes the acceptor for a subsequent glycosyltransferase. These enzymes are stereospecific, that is, they function to catalyze a glycosidic bond between a particular donor nucleotide-sugar (or, in some cases, lipid-phosphate-sugar) and the specific hydroxyl of a distinct acceptor structure, in precise anomeric linkage (α or β). Accordingly, the membrane glycan structures found on any given cell are a direct reflection of the expression of genes that encode the relevant glycosyltransferases that catalyze the requisite glycosidic linkages. While there can be redundancy in enzymes that perform a specific modification, more commonly the absence of a specific glycosyltransferase results in diminished expression of the associated determinant. However, absence of a particular determinant is not evidence that a relevant glycosyltransferase is not expressed. Importantly, expression of a known glycosyltransferase (or a set of glycosyltransferases) cannot predict, a priori, expression of a specified glycan determinant by a cell for a number of reasons, including: (i) absence of appropriate donor nucleotide-sugar(s); (ii) the (relative) proximal location of a given glycosyltransferase within the Golgi ‘assembly line’ yielding a glycan upstream that can not be an acceptor for other (downstream) glycosyltransferases; and (iii) concurrent competition between different Golgi glycosyltransferases that can each modify an identical acceptor (127). Moreover, recent studies from our laboratory (described below) indicate that postsynthetic glycosidase activity can affect expression of key cell surface glycans (128).
Over the past two decades, a multitude of studies have been undertaken to elucidate the regulation of expression of sLex. Soon after discovery of E-selectin, it was recognized that transfection of non-myeloid cells with α (1, 3)-fucosyltransferases (creating the Fuca1–3- linkage on GlcNAc within terminal type 2 lactosamines) resulted in de novo expression of sLex and E-selectin ligand activity (129, 130), indicating that sialylated type 2 acceptors are created in cells that do not express sLex and suggesting that expression of fucosyltransferase(s) could be limiting in synthesis of E-selectin ligand(s). Earlier studies also showed the importance of α(1, 3)-fucosyltransferase expression in elaborating P-selectin ligand activity by PSGL-1 (109). At present, there are six (validated) α(1, 3)-fu-cosyltransferases that can modify terminal lactosamines, each categorized by initials (FT) followed by a numeral (typically Roman, i.e. FTIII, FTIV, FTV, FTVI, FTVII, and FTIX) (reviewed in 131). Of these, FTIII, FTIV, FTV, and FTVI can modify both a sialylated Type 2 acceptor and an unsialylated (‘neutral’) Type 2 acceptor, yielding sLex or the trisaccharide Lex (also known as CD15), Gal β1–4[Fuc α 1–3]GlcNAc β1-R, respectively. In addition, FTIII and FTV can each modify Type 1 acceptors, yielding Lea or sLea (especially for FTIII). Discrete acceptor specificities are shown by FTVII and FTIX, in that FTVII is specific for sialylated Type 2 acceptors and FTIX has preference for unsialylated acceptors, producing sLex or Lex, respectively.
Of all the α(1, 3)-fucosyltransferases, only FTIV and FTVII are found on leukocytes. In transfected cell lines, FTIV shows a preference for glycolipid acceptors, whereas FTVII generates sLex predominantly on glycoproteins (132). Furthermore, FTIV tends to fucosylate GlcNAc residues in proximal lactosamine units of polylactosamine chains, whereas FTVII shows unique site-specificity in fucosylating only the distal GlcNAc and doing so only after sialylation of the Gal (i.e. the sole glycan acceptor for FTVII is the GlcNAc within a terminal sialylated type 2 lactosamine structure, NeuAcα2–3Gal β1–4GlcNAc β1-R) (133) (Fig. 4). Studies in genetically deficient mice have shown that though both of these enzymes are involved in E-selectin ligand expression in leukocytes, the predominant enzyme is FTVII: myeloid cell E-selectin (and P-selectin) ligand activity is markedly reduced in FTVII-null mice, but relatively intact in FTIV-null mice, whereas E-selectin (and P-selectin) ligands are completely absent in T-helper 1 (TH1) and T cytotoxic 1 (TC1) lymphocytes in FTVII-knock out animals and unaffected in FTIV-null mice (134, 135). Interestingly, in studies of human T lymphoblasts, E-selectin ligand activity requires higher levels of FTVII than that of P-selectin ligand activity, i.e. synthesis of P-selectin ligands occurs at lower levels of FucT-VII activity than that required for synthesis of E-selectin ligands (87). These same studies have shown that E-selectin, but not P-selectin, ligand activity correlates with surface sLex expression on T cells, with parallel increases in E-selectin ligand activity with increasing sLex expression. There is evidence that the contribution(s) of FTIV to selectin ligand synthesis in leukocytes may be lineage dependent, with FTIV contributing more in neutrophils than in lymphocytes, perhaps a function of the higher levels of FTIV enzyme activity attainable in myeloid cells (136, 137). Notably, a person carrying a homozygous missense mutation in the FTVII gene has been described, and neutrophils from this individual show a dramatic decrease in sLex expression and diminished staining with E-selectin–Ig chimera by flow cytometry, but normal staining with P-selectin–Ig chimera and normal engagement to recombinant P and E-selectin substrates in flow chamber studies (138). Quantitative reverse transcriptase-polymerase chain reaction (RT-PCR) analysis of FTIV and FTVII transcripts in neutrophils from this individual show a 100-fold increase in FTIV transcripts relative to those of unaffected persons, suggesting that upregulated expression of FTIV compensates for loss of functional FTVII in myeloid cells (138). However, there was no information provided on the level of FTIV transcripts, or the E- or P-selectin ligand activity, of T cells from this person. This individual does not suffer from recurrent infections or leukocytosis (139), in contrast to individuals possessing a global fucosylation defect known as leukocyte adhesion deficiency type II (LADII) that cannot create sLex and lack membrane selectin ligands; LADII is caused by a defect in the transporter that transfers the substrate GDP-fucose into the Golgi, but the deficit in selectin ligand expression can be overcome by oral administration of high doses of fucose (reviewed in 140).
Fig. 4. Endogenous synthesis of hematopoietic cell E-/L-selectin ligand (HCELL) versus enforced HCELL expression by glycosyltransferase-programmed stereosubstitution (GPS).
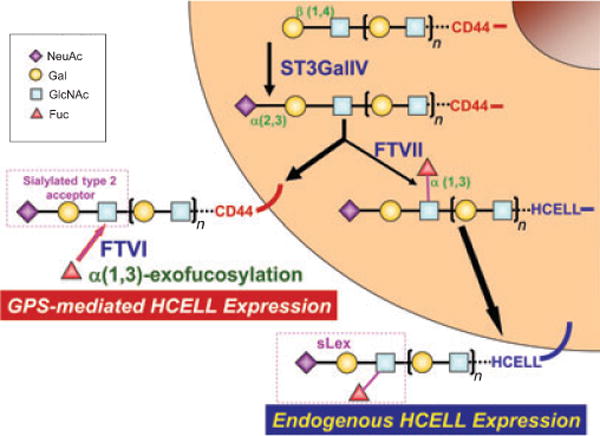
Relevant structures of the terminal sialylated Type 2 lactosamine acceptor and of sLex are shown for CD44 and HCELL, respectively. Conversion of native CD44 to the HCELL glycoform within the Golgi by sialofucosylation is shown in the figure, mediated by sequential action of the enzymes ST3GalIV and FTVII. In GPS, FTVI-mediated α(1, 3)-exofucosylation of the terminal sialylated Type 2 lactosamine acceptor of membrane-bound CD44 yields HCELL. The presence of polylactosamine structure(s) is depicted by brackets and subscript n; dashes represent the presence of additional (membrane-proximal) glycans linking to the core CD44 protein.
A family of six sialyltransferases (known as ST3GalI-VI) are capable of transferring sialic acid in α(2, 3)-linkage to terminal galactose, of which three, ST3GalIII, ST3GalIV and ST3GalVI, can sialylate type 2 lactosamines (reviewed in 141). Of these, ST3GalIII prefers type 1 lactosamines and is thus thought to be responsible for creation of sLea structures, and deficiency of this enzyme in the mouse is not associated with loss of E- or P-selectin ligand activity in neutrophils (142). However, deficiency in ST3GalIV results in reduced E-selectin and P-selectin ligand activity of neutrophils studied in vitro, whereas in vivo studies showed no effect on P-selectin-mediated rolling of neutrophils on inflamed endothelium, but E-selectin-mediated rolling velocity was increased (142). These results suggest that there is a residual α(2, 3)-sialylation of selectin ligands, likely secondary to ST3GalVI activity, and further studies employing mice deficient in ST3GalVI and/or deficient in both ST3GalIV and ST3GalVI are warranted.
In addition to fucosyltransferases and sialyltransferases, two other glycosyltransferases, core 2 β(1, 6)-N-acetylglucosaminyltransferase-I (C2GnT-I) and β-1,4-galactosyltransferase-I (β4GalT-1), have been studied for their role(s) in sLex expression and elaboration of selectin ligand activity in vivo. C2GnT-I is a member of a family of enzymes (including two others) that place GlcNAc on the 6th-position of the ‘core’ N-acetylgalactosamine linked to serine or threonine residues of the peptide chain, and thus initiates lactosamine unit synthesis on that ‘Core-2’ branch. Early in vitro studies showed that non-myeloid cells transfected with PSGL-1 and fucosyltransferase genes alone were unable to display sLex or selectin ligand activity (109, 143). These data, subsequently corroborated by others (144), showed that to display sLex on PSGL-1, cotransfection of a C2GnT is needed to extend lactosamine units from the O-glycan linkage. However, it must be emphasized that these studies have specifically revealed the critical role for Core 2 branch structures in elaborating sLex and selectin binding by PSGL-1 (and, by inference, any other ligands displaying sLex exclusively on Core 2 extensions). Importantly, leukocyte recruitment to inflamed peritoneum is significantly affected in mice lacking C2GnT-I, and, consistently, neutrophils and lymphocytes from these mice lack P-selectin ligand activity (145–147). However, neutrophils and lymphocytes from C2GnT-I-null mice retain significant E-selectin ligand activity, perhaps reflecting contribution(s) of non-PSGL-1-based E-selectin ligands and/or reflecting compensating expression of other C2GnT enzymes elaborating E-selectin ligands (e.g. C2GnT-III, see below) (146). Once initiated by linkage of GlcNAc onto either core N- or O-glycan structures, polylactosamine synthesis occurs by the alternate linkage of Gal and GlcNAc residues, respectively, generated by the sequential action of β-1,4-galactosyltransferase (β4GalT) and β-1,3-N-acteylglucosaminotransferase. Terminal Gal in polylactosamine chains can then be modified by α(2, 3)-linked sialic acid, followed by FTVII-mediated α(1, 3)-fucosylation of the ultimate GlcNAc, to yield sLex (Fig. 4). Although 6 β4GalTs (β4GalTI-VI) have been characterized that can catalyze lactosamine linkages (reviewed in 148), in vivo studies to date have been undertaken only in β4GalT-I-deficient mice (149). Both cell-mediated responses (e.g. contact hypersensitivity) and acute neutrophilic infiltrates were significantly suppressed in β4GalT-I-deficient mice. P-selectin ligand activity (as measured by flow cytometry using P-selectin–Ig chimera) was significantly decreased (with no difference in PSGL-1 protein levels), but the role of β4GalT-I in directing synthesis of lactosamines critical to E-selectin ligand expression/activity was not assessed (149).
A variety of cytokines mediating leukocyte development and differentiation affect glycosyltransferase gene expression, thus driving discrete post-translational modifications of cell surface proteins and lipids. Early studies revealed that CLA expression could be induced on naive human T cells in vitro by mitogen-activation in the presence of cytokines such as IL-2 and TGF-β (150). These results prompted further studies in mouse and human leukocytes to define how cytokines affect expression of fucosyltransferases, sialyltransferases, core 2 β(1,6)-N-acetylglucosaminyltransferases, and β-1,4-galac-tosyltransferases that control expression of sLex and of E-selectin and P-selectin ligands. Studies in vitro using human (151–153) and mouse lymphocytes (154, 155) have revealed a general pattern whereby mitogen- or TCR/antigen-driven activation of T cells in the presence of IL-12 (a TH1 polarizing cytokine) markedly enhances FTVII expression with commensurate induction of both E- and P-selectin ligands, whereas IL-4 (a TH2-polarizing cytokine) can dampen and/or oppose effects of IL-12 on FTVII expression and, in particular, on E-selectin ligand expression. In cultures of mitogen-activated mouse CD4 cells, IL-12 also stimulates expression of ST3GalIV (154), and IL-12 induces expression of ST3GalVI in CD4 cells obtained from ST3GalIV-null mice (156), indicating that IL-12-triggered induction of both of these sialyltransferases elicits heightened E-and P-selectin ligand activity. Studies in murine CD4+ T cells have shown that IL-12 induces C2GnT-1 expression (157), while studies in murine CD8+ cells have shown that C2GnT-1 expression is greatly stimulated by IL-2, and, to lesser extent, by IL-15 (158). In culture, IL-2 also induces expression of another core 2 β(1, 6)-N-acetylglucosaminyltransferase, C2GnT-III, which, in IL-2-treated mitogen-activated CD8+ cells from C2GnT-1-null mice, contributes to elaboration of P-selectin ligand activity (159). Compared with these other enzymes, very little is known at present regarding the effect(s) of cytokines on expression β4GalTs. One study of CD3-activated human T cells in culture showed a trend towards increasing β4GalT-I transcript levels with IL-12 incubation, and of suppression by IL-4, but neither of these changes were statistically significant (153).
In human myeloid cells, G-CSF (granulocyte-colony stimulating factor) induces a marked increase in E-selectin ligand activity, with accompanying markedly upregulated expression sLex (160). Biochemical studies of circulating myeloid cells from individuals receiving G-CSF and of human myeloid cells treated with G-CSF in vitro reveal marked increases in CLA and HCELL expression, together with induction of a currently unidentified 65 kDa E-selectin ligand. This profound increased E-selectin ligand expression is associated with significant increases in transcripts encoding ST3GalIV, FTIV, and FTVII (160). Besides increasing surface sLex expression, G-CSF administration is also associated with increased expression of Lex on myeloid cells (CD15) (128). The heightened surface Lex level is not associated with upregulated expression of FTIX (which fucosylates only neutral lactosamines, thus generating Lex) or with increased de novo synthesis of Lex, but results from the induction of a cell surface α(2, 3)-sialidase that cleaves off the terminal sialic acid of sLex, yielding Lex. Thus, in human myeloid cells, G-CSF-induced upregulation of glycosyltransferases creating sLex (in the Golgi) is accompanied by upregulation of a sLex-targeting membrane glycosidase (sialidase) such that cell surface expression of both sLex and Lex increases. These data unveil novel perspectives on the role(s) of post-Golgi glycosidases in the ‘biosynthesis’ of cell membrane glycans. Furthermore, the potential that selectin ligand activity may be controlled, postsynthetically, by expression of surface glycosidase(s) that remove key monosaccharides (e.g. sialic acid) warrants further investigation.
Exoglycosylation of cell surfaces: chronology and constraints
As described above, the formation of glycosidic linkages in vivo depends on the expression of pertinent glycosyltransferases within the Golgi compartment that catalyze reactions in a stereospecific fashion on discrete acceptors. In the late 1970s, the finding that the ordered biosynthetic pathways of the Golgi could be recapitulated in vitro using glycosyltransferases (isolated from natural sources) and relevant acceptor substrates (161), together with the increasing availability of specific glycosyltransferases, prompted investigators to use these enzymes as structural probes to characterize the oligosaccharides displayed on cell membranes (162). These studies generally consisted of treating target cells with a specific glycosidase (e.g. sialidase) to expose potential oligosaccharide acceptors, followed by reglycosylation using pertinent glycosyltransferase [e.g. α(2, 3)-sialyltransferase] together with radiolabelled donor sugar nucleotides (e.g. CMP-[3H]sialic acid), typically followed by SDS-PAGE and autoradiography to identify the exoglycosylated cell membrane proteins (reviewed in 163). In addition to information on the types of proteins bearing pertinent monosaccharide substitutions, these investigations also allowed for analysis of glycan structures and for assigning the specificity of carbohydrate-dependent binding interactions, such as between antibodies and target oligosaccharide antigens (162) or pathogen (virus/bacteria) and host cell target oligosaccharides (164, 165). Although these early investigations demonstrated that glycan modification of cell surface structures was feasible, these enzymatic reactions were optimized for intended biochemical analyses, not for preservation of cellular physiology. The effect(s) of cell manipulations and of exoglycosylation reaction(s) on cellular function/phenotype or viability were not considered (166, 167), and many such studies employed erythrocytes (reviewed 168). Even in the rare case where short-term viability of a nucleated cell was required to determine biological impact (e.g. viral production by infected cells), there was no formal assessment of cytotoxicity, of cell functional status, or of cell phenotype following glycosidase treatment/exoglycosylation (164, 169).
Until the 1990s, enzymatic manipulation of glycans – whether present on the cell surface or in solution – was performed on a small scale owing to limitations of isolating sufficiently pure and active glycosyltransferases from natural sources (reviewed in 170). Glycan modification(s) could also be achieved by organic synthesis, but this approach is a laborious and low efficiency process, dominated by cumbersome protection–deprotection loops required to achieve stereoselectivity, and involving use of toxic reagents that are damaging to cells and can pose significant environmental hazards. To circumvent these issues, substantial efforts have been directed toward expanding the repertoire of available glycosyltransferases that can achieve highly regio- and stereospecific glycan alteration with high efficiency and with essentially no safety or environmental risk. Over the past two decades, the increasing ease of generating recombinant glycosyltransferases (from both eukaryotes and prokaryotes) has made large-scale glycan biocatalysis practical, creating an emerging technology platform to modify carbohydrates stereoselectively for clinical applications of pharmaceutically-relevant glycoconjugates. For example, in the production of recombinant glycoprotein therapeutic reagents such as humanized monoclonal antibodies, the capacity to amend glycans postproduction has enormous implications for protein stability and bioactivity (171). However, it is worth remembering that these pertinent enzymatic reagents and attendant reaction conditions have been formulated expressly for high-efficiency modification(s) of soluble molecules for medicinal chemistry, not for the preservation of cellular viability and function. As such, one cannot be confident that such reagents could be used ‘as is’ to modify cell surface glycans without causing cytotoxicity.
Our finding that HCELL is a specialized HECA-452-reactive glycoform of CD44 prompted us to determine whether enforced HCELL expression could be achieved by enzymatic exoglycosylation of native membrane CD44 to create sLex determinants. A critical consideration in ex vivo glycan engineering of cell surface structures is that several classes of glycosyltransferases require divalent cation, most effectively manganese, for elaboration of catalytic activity (172). Importantly, in the creation of the sLex determinant, the only glycosyltransferase-dependent linkage not requiring divalent cation activation is α(2, 3)-sialylation of Gal, i.e. divalent cations are required for enzymatic catalysis of α(1, 3)-fucosylation of N-GlcNAc, β(1, 4)-galactosylation of N-GlcNAc, and β(1, 3)-N-acetylglucosaminylation of Gal (173–178). Whereas input of divalent cation(s) is not an issue in glycosylation of soluble organic molecules, it poses a major constraint in the glycoengineering of surface molecules of living cells. Divalent cations, particularly manganese, are known to activate integrins (e.g. LFA-1 and VLA-4) at molar concentrations well below those necessary to activate glycosyltransferase reactions (179–182). In adoptive cell therapies, Mn2+-triggered integrin-dependent firm adhesion could cause irreversible arrest of cells on the endothelial surface, impeding cell emigration and profoundly impacting cell trafficking patterns. Moreover, exposure to manganese induces signal transduction (183, 184) and, most importantly, apoptotic cell death (184–187) at substantially lower concentrations than that needed to activate glycosyltransferases. Such deleterious effects are also seen with other divalent cations (e.g. cobalt) commonly used to catalyze glycosyltransferase reactions (again, at lower concentrations than used in enzymatic reactions) (188, 189).
In the 1990s, two groups performed studies of ex vivo α(1, 3)-fucosylation of cell surfaces using manganese-containing reaction buffers (190, 191), but neither group reported data regarding cellular toxicity. We thus initiated studies to directly determine whether enforced α(1, 3)-fucosylation of primary human cells isolated from marrow (including hematopoietic progenitor cells, mesenchymal stem cells and leukocytes) could affect cell viability. Consistent with results of others (191), enforced α(1, 3)-fucosylation with fucosyltransferase-VI (FTVI) yielded increased cell surface sLex expression (as determined by HECA-452 staining by flow cytometry). However, regardless of cell type, exofucosylation was accompanied by profound cell death (consistently <5% cell viability). We observed significant cytotoxicity from incubation of cells with the enzyme alone using the commercially available FTVI (at that time, from Calbiochem, San Diego, CA, USA) used by others (191). We found that glycerol, a common stabilizer used in the commercial enzyme preparation, itself induced cell death. More importantly, we observed marked cell death (occurring over a period of 24 h) following incubation of cells in enzyme buffer alone for as little as 15 min; further studies showed that this cytotoxicity was induced by manganese in the buffer (93). Thus, although α(1, 3)-fucosylation of cell membrane glycans could be readily achieved, we concluded that the observed attendant cytotoxicity would essentially obviate any desired biologic effect(s).
While studies on the cytotoxic effects of α(1, 3)-fucosylation were being undertaken in our laboratory, we and other investigators were examining if α(1, 3)-fucosylation improves homing of hematopoietic progenitor cells to bone marrow. As described above, marrow microvessels constitutively express E-selectin, which is co-localized uniquely with the chemokine CXCL12 (SDF-1) (192). Marrow-migrating hematopoietic progenitor cells express E-selectin ligands and the receptor for CXCL12, CXCR4 (reviewed in 37) (Fig. 5A). Thus, exofucosylation to enforce (higher) E-selectin ligand expression by hematopoietic progenitors would be expected to enhance Step 1 interactions and subsequent extravasation of these CXCR4-bearing cells into marrow parenchyma. However, the inherent cell toxicity of the commercial FTVI enzyme formulation and in the reaction buffer conditions was not considered by other investigators (193–195). Indeed, one report suggesting that α(1, 3)-fucosylation improved engraftment of human hematopoietic progenitor cells in immunocompromised mice compared outcomes only between fucosylated cells and buffer-treated cells, but did not analyze engraftment of untreated cells (194); the inclusion of the untreated control would have revealed the negative impact of the α(1, 3)-fucosylation reaction on cell function/viability. In our early studies, untreated (control) human hematopoietic progenitor cells showed better short-term homing to marrow of immunocompromised mice than that of α(1, 3)-fucosylated cells because of cell death in the latter group. Notably, these results were consistent with the published findings of others using intravital microscopy and short-term homing assays: α(1, 3)-fucosylated human hematopoietic progenitor cells showed increased rolling interactions on marrow microvessels, but without improved homing to the parenchyma (195). Therefore, though enhanced E-selectin ligand activity by enforced α(1, 3)-fucosylation promoted Step 1 interactions on marrow microvessels, hematopoietic progenitor cells were functionally incapable of transmigration because of cell death, perhaps coupled with augmented firm adhesion on the endothelial surface secondary to Mn2+-induced activation of integrins. We thus labored to overcome these critical constraints, developing the requisite FTVI enzyme formulation and novel reaction conditions necessary to preserve cell phenotype and viability, thereby accomplishing the intended biological effects of enforced HCELL expression: improved vascular delivery of functionally intact cells to tissues where they are needed. We call this technology ‘glycosyltransferase-programmed stereosubstitution’ (GPS).
Fig. 5. Molecular components of human hematopoietic stem cell (A) and mesenchymal stem cell (B) interaction with marrow microvascular endothelium.
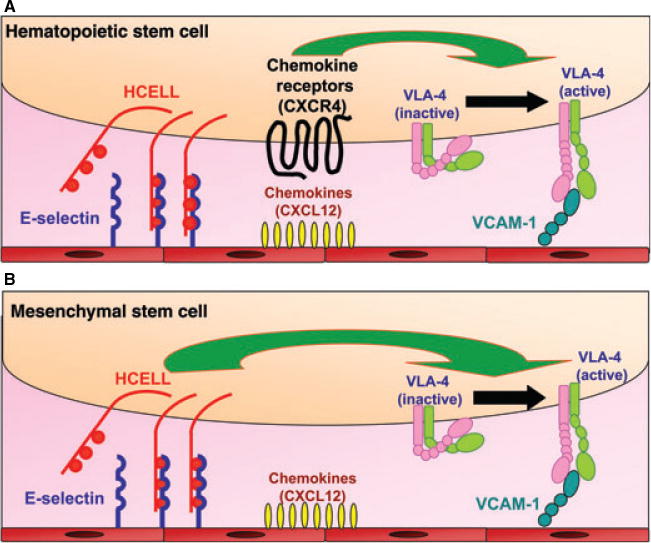
Relevant molecular effectors for cellular homing to marrow are shown. Schematic of mesenchymal stem cell lacking chemokine receptors depicts the proposed ‘Step 2-bypass pathway’. See text for details.
Development of GPS and evidence of its efficacy in navigating cellular trafficking
The description of reagents and methods created to enable GPS have been published recently (93), and thus only a brief overview will be presented here. The first objective in development of GPS was to produce high titer recombinant human FTVI suspended in a biologically compatible storage buffer, preferably lacking stabilizers. This was accomplished by using a Pichia pastoris expression system and final formulation of the recombinant FTVI in HBSS buffer alone (93). We then optimized enzymatic reaction conditions so as to avoid input divalent cations: multiple buffers were tested on multiple cell types, and high-efficiency α(1, 3)-fucosylation of cell surfaces on a variety of human cell types (hematopoietic and non-hematopoietic) was achieved using HBSS as the reaction buffer (93). Enforced fucosylation using these specifically formulated reagents and conditions had no effects on cell viability or function (except for rendering E-selectin ligand activity on cells bearing appropriate sialylated Type 2 lactosamines) (Fig. 4). Concurrent with the effort to reformulate FTVI and develop novel reaction conditions, we raised mAb against HCELL (isolated from KG1a cells) that could be used to probe the carbohydrate structure of CD44 on relevant target cells. A series of mAb were produced that possessed reactivity against glycosylated determinants of CD44, and all are mono-specific (i.e. recognize only CD44). One of these new anti-CD44 mAb, SACK-1, distinctly recognizes an α(2, 3)-sialylated glycoform of CD44 (93). This mAb has proved useful in predicting the generation of HCELL following α(1, 3)-fucosylation of CD44, and is now a key component of the GPS ‘tool kit’.
To test the efficacy of GPS in directing cell trafficking, we sought to analyze the effects of enforced HCELL expression on cell migration to a tissue that constitutively expresses E-selectin, such as the skin or the marrow, in the absence of injury. To this end, we employed a xenograft model, specifically to assess whether GPS would license migration of intravenously infused human mesenchymal stem cells (MSC) to mouse marrow under steady-state conditions. Our inspiration for this study derived from clinical trials in the late 1990s, showing that patients suffering from osteogenesis imperfecta could receive only transient therapeutic benefit from transplant of normal (allogeneic) human MSC (196, 197). Osteogenesis imperfecta is a genetic bone disorder, inherited in autosomal dominant fashion, characterized by extremely fragile bones owing to the production of abnormal collagen by osteoblasts (which are progeny of MSC), with resulting abnormal skeletal matrix. In every patient undergoing allogeneic MSC transplant for osteogenesis imperfecta, the fraction of donor-derived osteoblasts in the marrow was very low (never exceeding 2%), with fleeting benefits in osteoid development (196, 197).
Although multiple factors could be impeding osteogenesis following MSC transplant, the proximate obstacle is that MSCs home poorly to marrow (198, 199). We thus performed biochemical studies on human MSCs to understand the molecular basis of this homing defect. Our extensive analysis of the expression of adhesion molecules and chemokine receptors on human MSCs revealed that these cells do not express the principal molecular effectors of migration to marrow that are expressed on human hematopoietic stem cells, E-selectin ligands and CXCR4 (93) (Fig. 5A). Furthermore, we observed that human MSCs do not express PSGL-1, LFA-1 or LPAM-1. However, they do express CD44 and VLA-4; expression of VLA-4 is important because marrow vasculature also constitutively expresses its cognate ligand, VCAM-1 (34–36, 192) (Fig. 5).
Flow cytometry analysis of human MSCs using our panel of antibodies directed to CD44 glycoforms revealed these cells express SACK-1 determinants; we also observed that human MSCs lack reactivity with HECA-452 and with an sLex-specific mAb known as CSLEX-1. Collectively, these results indicated that absence of HCELL expression on human MSCs is due to lack of α(1, 3)-fucose modifications rendering sLex. Accordingly, we performed cell surface α(1, 3)-fucosylation and observed dramatic increases in HECA-452 and CSLEX-1 reactivity. Following enforced fucosylation, western blot showed that the only membrane glycoprotein staining with HECA-452 was CD44, indicating, specifically, that the native sialylated CD44 glycoform was converted into HCELL (93). Expression of HCELL was transient (stable for approximately 24 h), with loss secondary to cell surface protein turnover. Enforced HCELL expression conferred robust binding interactions with endothelial E-selectin under fluid shear conditions, displaying shear-resistant E-selectin binding at upwards of 30 dyn/cm2 (a shear stress level approximately 10-fold higher than that in postcapillary venules). As visualized by intravital microscopy, intravenously infused HCELL− human MSCs showed minimal interactions with marrow vessels, whereas infused HCELL+ MSCs displayed striking tethering and rolling interactions on marrow microvessels and transmigrated into the murine marrow parenchyma despite the absence of expression of CXCR4. Infiltrating human MSCs navigated to the endosteal surface, differentiatied into osteoblasts, and generated human osteoid matrix (Fig. 6). The observed osteotropism, achieved by selective expression of a single glycoprotein, HCELL, establishes this molecule as a ‘bone marrow homing receptor’. Furthermore, the finding that a discrete glycosylation can render a completely new biology on CD44 underscores the need to define this novel phenotype by its proper name, ‘HCELL’.
Fig. 6. Schematic model of marrow engraftment and differentiation of human mesenchymal stem cell (MSC) glycoengineered to express hematopoietic cell E-/L-selectin ligand (HCELL).
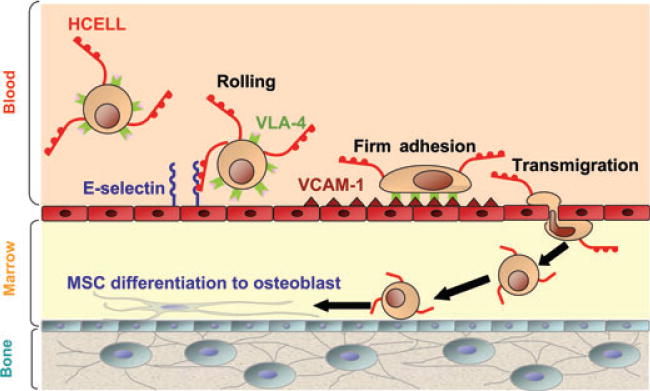
Interactions between HCELL and VLA-4 on the MSC surface with endothelial E-selectin and vascular cell-adhesion molecule 1 (VCAM-1), respectively, drive transmigration of MSC into marrow parenchyma. Due to the transient nature of enforced HCELL expression, reversion to native CD44 occurs as cells locomote to endosteal surface and subsequently differentiate to osteoblasts (loss of dots represents return to native CD44).
At present, α(1, 3)-fucosylation is the principal target of GPS. As stated above, the α(2, 3)-sialyltransferases do not require input divalent cation(s) for catalytic activity, and we have succeeded in converting undersialylated CD44 to HCELL on the cell surface by sequential treatment with α(2, 3)-N-sialyltransferase (from Roche Applied Science, Indianapolis, IN, USA) followed by FTVI. Additional studies are underway using specialized formulations of β4GalTs to create relevant Type 2 lactosamine backbone units (i.e. Gal β1–4GlcNAc β1-R) for appropriate sialofucosylation to create sLex. As mentioned above, a variety of cytokines affect the expression of glycosyltransferases that create the sLex determinant. Ongoing studies are thus also exploring how cytokine treatments in vitro may be combined with GPS to further augment HCELL expression, especially on lymphocytes.
Our development of GPS highlights several key guiding principles for future efforts in custom engineering of cell surface glycans: (i) identification of a relevant target glycoconjugate ‘acceptor’ on cells of interest (this can be accomplished using mAb, for example, SACK-1 mAb); (ii) use of pertinent enzymatic reagents and conditions to perform stereospecific carbohydrate substitution without affecting cell viability or generating unwanted phenotypic effects; and (iii) verification of target modification, as evidenced by appropriate biochemical and functional assays, including in vivo demonstration of the desired phenotypic effect (Table 1).
Table 1.
Guiding principles for glycosyltransferase-programmed stereosubstitution of cell surface gylcans
|
GPS: revising the multi-step paradigm and implications for cell therapeutics
The finding that enforced HCELL expression of human MSCs mediates E-selectin-dependent Step 1 interactions directing homing to bone marrow in the absence of CXCR4 engagement suggests that chemokine-triggered upregulation of integrin adhesiveness may not be compulsory for efficient transmigration. While the expression of chemokine receptors on human MSCs remains controversial (200–202), the expression of the Step 3 effector VLA-4 and its role in transendothelial migration of MSCs has been consistently observed (203–205). Our data provide direct evidence that the expression of a Step 1 effector (HCELL) on cells that express VLA-4 can license cell regulatory pathways driving transendothelial migration at sites where E-selectin and VCAM-1 are co-expressed. These findings are consistent with an emerging body of experimental evidence which indicates that the role(s) of Step 1 effectors in cell trafficking is much more complicated than previously perceived, certainly serving as more than just ‘molecular brakes’: Specifically, engagement of Step 1 molecules, alone (i.e. absent engagement of Step 2), may be sufficient to induce integrin adhesiveness, with accompanying firm adherence and transendothelial migration (reviewed in 1).
Functioning as Step 2 effectors, chemokines contribute to transendothelial migration by binding to specific cell surface G-protein-coupled receptors (GPCRs) (206–208). Engagement of GPCRs results in the activation of G-proteins and triggers several downstream signaling molecules leading to ‘inside-out’ upregulation of integrin adhesiveness. Although CD44 was first identified by its role in binding to extracellular matrix structures such as hyaluronic acid, early studies showed that CD44 is a G-protein (209), suggesting that CD44-dependent G-protein-mediated signal transduction may activate integrins. Indeed, early studies pointed to a role for anti-CD44 mAb crosslinking in ‘outside-in’ triggering of LFA-1 adhesiveness in human T cells (210, 211). Recent studies have shown that CD44 crosslinking not only results in the activation of integrin adhesiveness, but also results in transendothelial migration in the absence of chemokines (212–214). Notably, a direct role for CD44 in VLA-4 adhesiveness was shown in murine T cells where CD44-dependent rolling interactions on plastic dishes coated with both hyaluronic acid and VCAM-1 directly primed VLA-4-dependent firm adhesion (214). These studies also provided compelling biochemical evidence that the cytoplasmic tail of CD44 promotes the assembly of a bimolecular complex with VLA-4, whereby engagement of CD44 results in synergistic upregulation of VLA-4 adhesiveness, leading to endothelial transmigration without chemokine engagement (214). Collectively, these findings add a new dimension of CD44-mediated mechanosignaling to the multi-step paradigm, defining, at a molecular level, the critical components of a ‘Step 2-bypass pathway’ (Fig. 5B).
The cytokines IL-1 and TNF-α each induce expression of E-selectin, VCAM-1 and ICAM-1, and, as such, the expression of these three adhesion molecules is the endothelial ‘address’ at essentially every site of inflammation or tissue injury (41, 215–219). As CD44 expression is, with few exceptions (e.g. 220), a characteristic feature of adult stem cells and embryonic stem cells, and of lymphocytes and NK cells, GPS-mediated enforced HCELL expression on such cells could license migration and infiltration of any of these types of cells for application(s) in a wide variety of physiologic and pathologic conditions, including regenerative medicine, infectious diseases, immune diseases, and cancer. The potential clinical application of GPS technology for regenerative therapeutics in generalized skeletal disease is already evident (Fig. 6), but another immediate application may be in promoting stem cell recruitment at sites of ischemic injury (221). Moreover, exploiting GPS to optimize vascular delivery of relevant progenitor/stem cells for tissue regeneration may be the only option for organs/tissues where local injection of such cells is impractical (e.g. lung). In infectious diseases, improved trafficking of in vitro-expanded antigen-specific lymphocytes could enhance the efficacy of adoptive immunotherapy (e.g. for cytomegalovirus infections following allogeneic hematopoietic stem cell transplantation, 222, 223). For immune diseases, the capacity to selectively promote migration of culture-expanded regulatory lymphocytes to sites of inflammation could heighten peripheral tolerance and re-establish immune ‘homeostasis’ (224, 225). Similarly, in cancer therapeutics, the potential of GPS to improve tumor infiltration of expanded tumor-antigen specific T cells could boost the effectiveness of this therapeutic approach (226). Indeed, a recent paper identifies an additional role of CD44 in regulating effector T cell locomotion within the tumor stroma: the intracellular domain of CD44 anchors cytoskeletal ERM (exrin, radixin, moesin) proteins, allowing the cells to stabilize their polarity, thereby enabling efficient scanning of tumor cells and more efficient tumoricidal effect (227). This function of CD44 would also likely enable infiltrated T cells to survey pathogens within pertinent sites of inflammation. Importantly, our studies show that enforced HCELL expression on the cell surface is transient, with reversion to endogenous CD44 phenotype within 48 h of exofucosylation (93). Thus, once cells have extravasated, it would be expected that the native biology of CD44 would be manifest (Fig. 6).
It is worth remembering that depending on the cell type, membrane structures other than CD44 (e.g. glycolipids and other proteins) may present relevant sialylated lactosamine glycans that could serve as acceptors for α(1, 3)-fucosylation. In our studies of human MSCs, CD44 was the only protein bearing the pertinent glycan acceptor, but we also found evidence of a minor contribution of glycolipids to E-selectin ligand activity following FTVI treatment (93). In prior studies of α(1, 3)-exofucosylation of cells, no biochemical data were presented regarding the actual glycoprotein target(s) of the reaction (190, 191, 193–195), but it was found that glycolipids can contribute to the observed increased E-selectin binding activity (193). In any cell type, a membrane molecule that contains E-selectin ligand activity would likely contribute to Step 1 interactions, and, therefore, should help in recruitment of cells at sites where endothelial E-selectin is expressed. However, the fact that CD44 can function in a Step 2-bypass pathway indicates that this structure, when modified to become HCELL, will serve to enhance transmigration in the absence (or presence) of chemokine input (i.e. Step 2) (Fig. 5B). Thus, the capacity to custom modify HCELL expression through surface glycan engineering of CD44 on cells co-expressing VLA-4 should enable vascular delivery of such cells to any site of tissue damage. However, in those cases where the site density of HCELL and/or VLA-4 on relevant cells is low, or, alternatively, where the site densities of endothelial E-selectin and/or VCAM-1, respectively, are low, the proposed Step 2-bypass pathway may not be sufficient to achieve endothelial transmigration. In such situations, chemokines and other Step 2 effectors would be obligatory to accomplish extravasation, but it would expected that HCELL/VLA-4 cross-talk would function to reduce the chemokine threshold needed to prime firm adherence and transmigration. In any case, HCELL adhesive interactions on endothelial E-selectin would still figure prominently, if not decisively, in the regulation of cell migration.
Conclusion
The capability to achieve appropriate patient- and disease-specific treatments through adoptive cellular therapies is critically dependent on getting the requisite cells to the sites where they are needed. Evolution has provided a complex tapestry of adhesion molecules and chemokines to provide fine regulatory control of cellular trafficking patterns. Characteristically, at all sites of tissue injury and/or inflammation, the affected endothelial bed(s) respond with upregulated expression of a handful of adhesion proteins, principally E-selectin, P-selectin, VCAM-1 and ICAM-1, which serve to literally capture cells in circulation that display relevant counter-receptors. This review has described the ‘intelligent design’ of the most potent E-selectin ligand expressed on human cells, HCELL, using a platform technology called GPS. For the first time, a cell type (human MSC) unable to achieve tissue-specific migration has been engineered to possess the capability of migrating to a predetermined anatomic site, and, most critically, retain viability and native phenotype. The methods and results presented here provide a striking example of how a distinct monosaccharide substitution in precise linkage of a single membrane glycoprotein (CD44) is sufficient to endow a totally new biology on the target protein and the target cell(s). A set of guiding principles is proposed here that should allow for application of GPS beyond glycan engineering of HCELL. By extension, through creation of requisite reagents and reaction conditions, the relevant sugars displayed by essentially any selected membrane structure, protein or lipid, may be altered so that it expresses a completely new function. Thus, beyond providing a roadmap for directing navigational patterns of cells in circulation, our increasing understanding of glycan structural biology and of glycosyltransferases will yield even greater opportunities for testing how programmed carbohydrate modification(s) of the cell surface can be harnessed to usher forth the paradigm-shifting therapeutics of the future.
Acknowledgments
This article reviews over 20 years of laboratory effort seeking to create strategies to deliver infused cells to a predetermined anatomic site. The inspiration for this work derives from my patients undergoing hematopoietic stem cell transplantation, where the problems of lack of hematopoietic engraftment, infectious diseases (e.g. cytomegalovirus disease) and recurrence of (primary) malignant disease posttransplant (e.g. lymphoma, leukemia) made evident the imperative to optimize vascular delivery of relevant cells (hematopoietic stem cells or immune effector cells) to requisite (affected) sites in order to realize long-term cure. Thus, I dedicate this review to my patients, alive and deceased, for their lessons in courage and in humanity. Moreover, over this period of time, many technicians, students, post-docs et al. have contributed significantly to this mission. Certainly, there are no words adequate to relay my gratitude for their devotion and commitment to this endeavor. In typical acknowledgements found in scientific publications, it is uncommon for one to thank the unsung heroes: teachers/mentors and family. Regrettably, several mentors have departed – Ramzi S. Cotran, Edgar Haber, Harvey R. Colten, Yee Hon Chin, J. Wayne Streilein, and Robert A. Good – without ever knowing my eternal gratitude. I would thus like to formally thank here Drs. Adel A. Yunis, Murray Epstein, Claude P. Lechene, Emil R. Unanue, Lawrence M. Fishman, Gerald J. Elfenbein, and E. Donnall Thomas, for their example, for sharing their infinite wisdom, and for their constant encouragement and support. Last, but certainly not least, I thank my parents, Harold and Rosalina Sackstein, my wife, Beth, and my children, David and Danielle, without whom none of this would be possible.
This work was supported in part by Veterans Career Development and Merit Awards, and by National Institutes of Health grants RO1 HL60528, RO1 CA84156, RO1 HL073714, and RO1 CA 121335.
References
- 1.Sackstein R. The lymphocyte homing receptors: gatekeepers of the multistep paradigm. Curr Opin Hematol. 2005;12:444–450. doi: 10.1097/01.moh.0000177827.78280.79. [DOI] [PubMed] [Google Scholar]
- 2.Butcher EC. Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell. 1991;67:1033–1036. doi: 10.1016/0092-8674(91)90279-8. [DOI] [PubMed] [Google Scholar]
- 3.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 4.Campbell DJ, Kim CH, Butcher EC. Chemokines in the systemic organization of immunity. Immunol Rev. 2003;195:58–71. doi: 10.1034/j.1600-065x.2003.00067.x. [DOI] [PubMed] [Google Scholar]
- 5.Schreiber TH, Shinder V, Cain DW, Alon R, Sackstein R. Shear flow-dependent integration of apical and subendothelial chemokines in T-cell transmigration: implications for locomotion and the multistep paradigm. Blood. 2007;109:1381–1386. doi: 10.1182/blood-2006-07-032995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kahn J, et al. Overexpression of CXCR4 on human CD34+ progenitors increases their proliferation, migration, and NOD/SCID repopulation. Blood. 2004;103:2942–2949. doi: 10.1182/blood-2003-07-2607. [DOI] [PubMed] [Google Scholar]
- 7.Cheng Z, et al. Targeted migration of mesenchymal stem cells modified with CXCR4 gene to infarcted myocardium improves cardiac performance. Mol Ther. 2008;16:571–579. doi: 10.1038/sj.mt.6300374. [DOI] [PubMed] [Google Scholar]
- 8.Zhang D, et al. Over-expression of CXCR4 on mesenchymal stem cells augments myoangiogenesis in the infarcted myocardium. J Mol Cell Cardiol. 2008;44:281–292. doi: 10.1016/j.yjmcc.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peled A, et al. Dependence of human stem cell engraftment and repopulation of NOD/SCID mice on CXCR4. Science. 1999;283:845–848. doi: 10.1126/science.283.5403.845. [DOI] [PubMed] [Google Scholar]
- 10.Kollet O, et al. Human CD34(+)CXCR4(−) sorted cells harbor intracellular CXCR4, which can be functionally expressed and provide NOD/SCID repopulation. Blood. 2002;100:2778–2786. doi: 10.1182/blood-2002-02-0564. [DOI] [PubMed] [Google Scholar]
- 11.Forster R, et al. Intracellular and surface expression of the HIV-1 coreceptor CXCR4/fusin on various leukocyte subsets: rapid internalization and recycling upon activation. J Immunol. 1998;160:1522–1531. [PubMed] [Google Scholar]
- 12.Berlin C, et al. alpha 4 integrins mediate lymphocyte attachment and rolling under physiologic flow. Cell. 1995;80:413–422. doi: 10.1016/0092-8674(95)90491-3. [DOI] [PubMed] [Google Scholar]
- 13.Lawrence MB, Kansas GS, Kunkel EJ, Ley K. Threshold levels of fluid shear promote leukocyte adhesion through selectins (CD62L,P,E) J Cell Biol. 1997;136:717–727. doi: 10.1083/jcb.136.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Finger EB, Puri KD, Alon R, Lawrence MB, von Andrian UH, Springer TA. Adhesion through L-selectin requires a threshold hydrodynamic shear. Nature. 1996;379:266–269. doi: 10.1038/379266a0. [DOI] [PubMed] [Google Scholar]
- 15.Alon R, Chen S, Puri KD, Finger EB, Springer TA. The kinetics of L-selectin tethers and the mechanics of selectin-mediated rolling. J Cell Biol. 1997;138:1169–1180. doi: 10.1083/jcb.138.5.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Resto VA, Burdick MM, Dagia NM, McCammon SD, Fennewald SM, Sackstein R. L-selectin-mediated lymphocyte-cancer cell interactions under low fluid shear conditions. J Biol Chem. 2008;283:15816–15824. doi: 10.1074/jbc.M708899200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alon R, Hammer DA, Springer TA. Lifetime of the P-selectin-carbohydrate bond and its response to tensile force in hydrodynamic flow. Nature. 1995;374:539–542. doi: 10.1038/374539a0. [DOI] [PubMed] [Google Scholar]
- 18.Yago T, Wu J, Wey CD, Klopocki AG, Zhu C, McEver RP. Catch bonds govern adhesion through L-selectin at threshold shear. J Cell Biol. 2004;166:913–923. doi: 10.1083/jcb.200403144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beste MT, Hammer DA. Selectin catch-slip kinetics encode shear threshold adhesive behavior of rolling leukocytes. Proc Natl Acad Sci USA. 2008;105:20716–20721. doi: 10.1073/pnas.0808213105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen S, Springer TA. An automatic braking system that stabilizes leukocyte rolling by an increase in selectin bond number with shear. J Cell Biol. 1999;144:185–200. doi: 10.1083/jcb.144.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marshall BT, Long M, Piper JW, Yago T, McEver RP, Zhu C. Direct observation of catch bonds involving cell-adhesion molecules. Nature. 2003;423:190–193. doi: 10.1038/nature01605. [DOI] [PubMed] [Google Scholar]
- 22.Park EY, et al. Comparison of PSGL-1 microbead and neutrophil rolling: microvillus elongation stabilizes P-selectin bond clusters. Biophys J. 2002;82:1835–1847. doi: 10.1016/S0006-3495(02)75534-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lei X, Lawrence MB, Dong C. Influence of cell deformation on leukocyte rolling adhesion in shear flow. J Biomech Eng. 1999;121:636–643. doi: 10.1115/1.2800866. [DOI] [PubMed] [Google Scholar]
- 24.Kansas GS, Wood GS, Fishwild DM, Engleman EG. Functional characterization of human T lymphocyte subsets distinguished by monoclonal anti-leu-8. J Immunol. 1985;134:2995–3002. [PubMed] [Google Scholar]
- 25.Kansas GS, Dailey MO. Expression of adhesion structures during B cell development in man. J Immunol. 1989;142:3058–3062. [PubMed] [Google Scholar]
- 26.Picker LJ, Treer JR, Ferguson-Darnell B, Collins PA, Buck D, Terstappen LW. Control of lymphocyte recirculation in man. I. Differential regulation of the peripheral lymph node homing receptor L-selectin on T cells during the virgin to memory cell transition. J Immunol. 1993;150:1105–1121. [PubMed] [Google Scholar]
- 27.Lund-Johansen F, Terstappen LW. Differential surface expression of cell adhesion molecules during granulocyte maturation. J Leukoc Biol. 1993;54:47–55. doi: 10.1002/jlb.54.1.47. [DOI] [PubMed] [Google Scholar]
- 28.Sackstein R. A revision of Billingham’s tenets: the central role of lymphocyte migration in acute graft-versus-host disease. Biol Blood Marrow Transplant. 2006;12:2–8. doi: 10.1016/j.bbmt.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 29.Evans SS, Collea RP, Appenheimer MM, Gollnick SO. Interferon-alpha induces the expression of the L-selectin homing receptor in human B lymphoid cells. J Cell Biol. 1993;123:1889–1898. doi: 10.1083/jcb.123.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sackstein R, Meng L, Xu XM, Chin YH. Evidence of post-transcriptional regulation of L-selectin gene expression in rat lymphoid cells. Immunology. 1995;85:198–204. [PMC free article] [PubMed] [Google Scholar]
- 31.Kahn J, Ingraham RH, Shirley F, Migaki GI, Kishimoto TK. Membrane proximal cleavage of L-selectin: identification of the cleavage site and a 6-kD transmembrane peptide fragment of L-selectin. J Cell Biol. 1994;125:461–470. doi: 10.1083/jcb.125.2.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y, Brazzell J, Herrera A, Walcheck B. ADAM17 deficiency by mature neutrophils has differential effects on L-selectin shedding. Blood. 2006;108:2275–2279. doi: 10.1182/blood-2006-02-005827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garton KJ, et al. Stimulated shedding of vascular cell adhesion molecule 1 (VCAM-1) is mediated by tumor necrosis factor-alpha-converting enzyme (ADAM 17) J Biol Chem. 2003;278:37459–37464. doi: 10.1074/jbc.M305877200. [DOI] [PubMed] [Google Scholar]
- 34.Schweitzer KM, et al. Constitutive expression of E-selectin and vascular cell adhesion molecule-1 on endothelial cells of hematopoietic tissues. Am J Pathol. 1996;148:165–175. [PMC free article] [PubMed] [Google Scholar]
- 35.Frenette PS, Subbarao S, Mazo IB, von Andrian UH, Wagner DD. Endothelial selectins and vascular cell adhesion molecule-1 promote hematopoietic progenitor homing to bone marrow. Proc Natl Acad Sci USA. 1998;95:14423–14428. doi: 10.1073/pnas.95.24.14423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weninger W, et al. Specialized contributions by alpha(1,3)-fucosyltransferase-IV and FucT-VII during leukocyte rolling in dermal microvessels. Immunity. 2000;12:665–676. doi: 10.1016/s1074-7613(00)80217-4. [DOI] [PubMed] [Google Scholar]
- 37.Sackstein R. The bone marrow is akin to skin: HCELL and the biology of hematopoietic stem cell homing. J Invest Dermatol. 2004;122:1061–1069. doi: 10.1111/j.0022-202X.2004.09301.x. [DOI] [PubMed] [Google Scholar]
- 38.Eppihimer MJ, Wolitzky B, Anderson DC, Labow MA, Granger DN. Heterogeneity of expression of E- and P-selectins in vivo. Circ Res. 1996;79:560–569. doi: 10.1161/01.res.79.3.560. [DOI] [PubMed] [Google Scholar]
- 39.Asako H, et al. Role of H1 receptors and P-selectin in histamine-induced leukocyte rolling and adhesion in postcapillary venules. J Clin Invest. 1994;93:1508–1515. doi: 10.1172/JCI117129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kubes P, Kanwar S. Histamine induces leukocyte rolling in post-capillary venules. A P-selectin-mediated event. J Immunol. 1994;152:3570–3577. [PubMed] [Google Scholar]
- 41.Vestweber D, Blanks JE. Mechanisms that regulate the function of the selectins and their ligands. Physiol Rev. 1999;79:181–213. doi: 10.1152/physrev.1999.79.1.181. [DOI] [PubMed] [Google Scholar]
- 42.Subramaniam M, Koedam JA, Wagner DD. Divergent fates of P- and E-selectins after their expression on the plasma membrane. Mol Biol Cell. 1993;4:791–801. doi: 10.1091/mbc.4.8.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bullard DC, et al. Infectious susceptibility and severe deficiency of leukocyte rolling and recruitment in E-selectin and P-selectin double mutant mice. J Exp Med. 1996;183:2329–2336. doi: 10.1084/jem.183.5.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frenette PS, Mayadas TN, Rayburn H, Hynes RO, Wagner DD. Susceptibility to infection and altered hematopoiesis in mice deficient in both P- and E-selectins. Cell. 1996;84:563–574. doi: 10.1016/s0092-8674(00)81032-6. [DOI] [PubMed] [Google Scholar]
- 45.Bullard DC, et al. P-selectin/ICAM-1 double mutant mice: acute emigration of neutrophils into the peritoneum is completely absent but is normal into pulmonary alveoli. J Clin Invest. 1995;95:1782–1788. doi: 10.1172/JCI117856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Labow MA, et al. Characterization of E-selectin-deficient mice: demonstration of overlapping function of the endothelial selectins. Immunity. 1994;1:709–720. doi: 10.1016/1074-7613(94)90041-8. [DOI] [PubMed] [Google Scholar]
- 47.Mayadas TN, Johnson RC, Rayburn H, Hynes RO, Wagner DD. Leukocyte rolling and extravasation are severely compromised in P selectin-deficient mice. Cell. 1993;74:541–554. doi: 10.1016/0092-8674(93)80055-j. [DOI] [PubMed] [Google Scholar]
- 48.Ramos CL, et al. Differential effect of E-selectin antibodies on neutrophil rolling and recruitment to inflammatory sites. Blood. 1997;89:3009–3018. [PubMed] [Google Scholar]
- 49.Tamaru M, Tomura K, Sakamoto S, Tezuka K, Tamatani T, Narumi S. Interleukin-1beta induces tissue- and cell type-specific expression of adhesion molecules in vivo. Arterioscler Thromb Vasc Biol. 1998;18:1292–1303. doi: 10.1161/01.atv.18.8.1292. [DOI] [PubMed] [Google Scholar]
- 50.Hickey MJ, Kanwar S, McCafferty DM, Granger DN, Eppihimer MJ, Kubes P. Varying roles of E-selectin and P-selectin in different microvascular beds in response to antigen. J Immunol. 1999;162:1137–1143. [PubMed] [Google Scholar]
- 51.Kulidjian AA, Issekutz AC, Issekutz TB. Differential role of E-selectin and P-selectin in T lymphocyte migration to cutaneous inflammatory reactions induced by cytokines. Int Immunol. 2002;14:751–760. doi: 10.1093/intimm/dxf045. [DOI] [PubMed] [Google Scholar]
- 52.Reinhardt RL, Bullard DC, Weaver CT, Jenkins MK. Preferential accumulation of antigen-specific effector CD4 T cells at an antigen injection site involves CD62E-dependent migration but not local proliferation. J Exp Med. 2003;197:751–762. doi: 10.1084/jem.20021690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lawrence MB, Springer TA. Neutrophils roll on E-selectin. J Immunol. 1993;151:6338–6346. [PubMed] [Google Scholar]
- 54.Lawrence MB, Berg EL, Butcher EC, Springer TA. Rolling of lymphocytes and neutrophils on peripheral node addressin and subsequent arrest on ICAM-1 in shear flow. Eur J Immunol. 1995;25:1025–1031. doi: 10.1002/eji.1830250425. [DOI] [PubMed] [Google Scholar]
- 55.Jung U, Bullard DC, Tedder TF, Ley K. Velocity differences between L- and P-selectin-dependent neutrophil rolling in venules of mouse cremaster muscle in vivo. Am J Physiol. 1996;271:H2740–H2747. doi: 10.1152/ajpheart.1996.271.6.H2740. [DOI] [PubMed] [Google Scholar]
- 56.Jung U, Ley K. Mice lacking two or all three selectins demonstrate overlapping and distinct functions for each selectin. J Immunol. 1999;162:6755–6762. [PubMed] [Google Scholar]
- 57.Ley K, Allietta M, Bullard DC, Morgan S. Importance of E-selectin for firm leukocyte adhesion in vivo. Circ Res. 1998;83:287–294. doi: 10.1161/01.res.83.3.287. [DOI] [PubMed] [Google Scholar]
- 58.Jung U, Norman KE, Scharffetter-Kochanek K, Beaudet AL, Ley K. Transit time of leukocytes rolling through venules controls cytokine-induced inflammatory cell recruitment in vivo. J Clin Invest. 1998;102:1526–1533. doi: 10.1172/JCI119893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Burns SA, DeGuzman BJ, Newburger JW, Mayer JE, Jr, Neufeld EJ, Briscoe DM. P-selectin expression in myocardium of children undergoing cardiopulmonary bypass. J Thorac Cardiovasc Surg. 1995;110:924–933. doi: 10.1016/s0022-5223(05)80159-x. [DOI] [PubMed] [Google Scholar]
- 60.Pan J, Xia L, McEver RP. Comparison of promoters for the murine and human P-selectin genes suggests species-specific and conserved mechanisms for transcriptional regulation in endothelial cells. J Biol Chem. 1998;273:10058–10067. doi: 10.1074/jbc.273.16.10058. [DOI] [PubMed] [Google Scholar]
- 61.Yao L, Setiadi H, Xia L, Laszik Z, Taylor FB, McEver RP. Divergent inducible expression of P-selectin and E-selectin in mice and primates. Blood. 1999;94:3820–3828. [PubMed] [Google Scholar]
- 62.Berg EL, Robinson MK, Mansson O, Butcher EC, Magnani JL. A carbohydrate domain common to both sialyl Le(a) and sialyl Le(X) is recognized by the endothelial cell leukocyte adhesion molecule ELAM-1. J Biol Chem. 1991;266:14869–14872. [PubMed] [Google Scholar]
- 63.Berg EL, Magnani J, Warnock RA, Robinson MK, Butcher EC. Comparison of L-selectin and E-selectin ligand specificities: the L-selectin can bind the E-selectin ligands sialyl Le(x) and sialyl Le(a) Biochem Biophys Res Commun. 1992;184:1048–1055. doi: 10.1016/0006-291x(92)90697-j. [DOI] [PubMed] [Google Scholar]
- 64.Handa K, Nudelman ED, Stroud MR, Shiozawa T, Hakomori S. Selectin GMP-140 (CD62; PADGEM) binds to sialosyl-Le(a) and sialosyl-Le(x), and sulfated glycans modulate this binding. Biochem Biophys Res Commun. 1991;181:1223–1230. doi: 10.1016/0006-291x(91)92069-v. [DOI] [PubMed] [Google Scholar]
- 65.Tyrrell D, et al. Structural requirements for the carbohydrate ligand of E-selectin. Proc Natl Acad Sci USA. 1991;88:10372–10376. doi: 10.1073/pnas.88.22.10372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Renkonen O. Enzymatic in vitro synthesis of I-branches of mammalian polylactosamines: generation of scaffolds for multiple selectin-binding saccharide determinants. Cell Mol Life Sci. 2000;57:1423–1439. doi: 10.1007/PL00000627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kasai K, Kameya T, Okuda T, Terasaki PI, Iwaki Y. Immunohistochemical examination of lung cancers using monoclonal antibodies reacting with sialosylated Lewisx and sialosylated Lewisa. Virchows Arch A Pathol Anat Histopathol. 1986;410:253–261. doi: 10.1007/BF00710832. [DOI] [PubMed] [Google Scholar]
- 68.Zhang K, Baeckstrom D, Brevinge H, Hansson GC. Secreted MUC1 mucins lacking their cytoplasmic part and carrying sialyl-Lewis a and x epitopes from a tumor cell line and sera of colon carcinoma patients can inhibit HL-60 leukocyte adhesion to E-selectin-expressing endothelial cells. J Cell Biochem. 1996;60:538–549. doi: 10.1002/(SICI)1097-4644(19960315)60:4%3C538::AID-JCB10%3E3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 69.Hey NA, Aplin JD. Sialyl-Lewis x and Sialyl-Lewis a are associated with MUC1 in human endometrium. Glycoconj J. 1996;13:769–779. doi: 10.1007/BF00702341. [DOI] [PubMed] [Google Scholar]
- 70.Kannagi R. Carbohydrate-mediated cell adhesion involved in hematogenous metastasis of cancer. Glycoconj J. 1997;14:577–584. doi: 10.1023/a:1018532409041. [DOI] [PubMed] [Google Scholar]
- 71.Magnani JL. The discovery, biology, and drug development of sialyl Lea and sialyl Lex. Arch Biochem Biophys. 2004;426:122–131. doi: 10.1016/j.abb.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 72.Hanley WD, Burdick MM, Konstantopoulos K, Sackstein R. CD44 on LS174T colon carcinoma cells possesses E-selectin ligand activity. Cancer Res. 2005;65:5812–5817. doi: 10.1158/0008-5472.CAN-04-4557. [DOI] [PubMed] [Google Scholar]
- 73.Burdick MM, Chu JT, Godar S, Sackstein R. HCELL is the major E- and L-selectin ligand expressed on LS174T colon carcinoma cells. J Biol Chem. 2006;281:13899–13905. doi: 10.1074/jbc.M513617200. [DOI] [PubMed] [Google Scholar]
- 74.Rosen SD. Ligands for L-selectin: homing, inflammation, and beyond. Annu Rev Immunol. 2004;22:129–156. doi: 10.1146/annurev.immunol.21.090501.080131. [DOI] [PubMed] [Google Scholar]
- 75.McEver RP. Selectins: lectins that initiate cell adhesion under flow. Curr Opin Cell Biol. 2002;14:581–586. doi: 10.1016/s0955-0674(02)00367-8. [DOI] [PubMed] [Google Scholar]
- 76.Kanamori A, et al. Distinct sulfation requirements of selectins disclosed using cells that support rolling mediated by all three selectins under shear flow. L-selectin prefers carbohydrate 6-sulfation totyrosine sulfation, whereas p-selectin does not. J Biol Chem. 2002;277:32578–32586. doi: 10.1074/jbc.M204400200. [DOI] [PubMed] [Google Scholar]
- 77.Poppe L, Brown GS, Philo JS, Nikrad PV, Shah BH. Conformation of sLex tetrasaccaride, free and bound to E-, P-, and L-selectin. J Am Chem Soc. 1997;1736:1727–1736. [Google Scholar]
- 78.Duijvestijn AM, et al. High endothelial differentiation in human lymphoid and inflammatory tissues defined by monoclonal antibody HECA-452. Am J Pathol. 1988;130:147–155. [PMC free article] [PubMed] [Google Scholar]
- 79.Picker LJ, Michie SA, Rott LS, Butcher EC. A unique phenotype of skin-associated lymphocytes in humans. Preferential expression of the HECA-452 epitope by benign and malignant T cells at cutaneous sites. Am J Pathol. 1990;136:1053–1068. [PMC free article] [PubMed] [Google Scholar]
- 80.Picker LJ, Terstappen LW, Rott LS, Streeter PR, Stein H, Butcher EC. Differential expression of homing-associated adhesion molecules by T cell subsets in man. J Immunol. 1990;145:3247–3255. [PubMed] [Google Scholar]
- 81.Picker LJ, Kishimoto TK, Smith CW, Warnock RA, Butcher EC. ELAM-1 is an adhesion molecule for skin-homing T cells. Nature. 1991;349:796–799. doi: 10.1038/349796a0. [DOI] [PubMed] [Google Scholar]
- 82.Berg EL, et al. The cutaneous lymphocyte antigen is a skin lymphocyte homing receptor for the vascular lectin endothelial cell-leukocyte adhesion molecule 1. J Exp Med. 1991;174:1461–1466. doi: 10.1084/jem.174.6.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fuhlbrigge RC, Kieffer JD, Armerding D, Kupper TS. Cutaneous lymphocyte antigen is a specialized form of PSGL-1 expressed on skin-homing T cells. Nature. 1997;389:978–981. doi: 10.1038/40166. [DOI] [PubMed] [Google Scholar]
- 84.Fuhlbrigge RC, King SL, Dimitroff CJ, Kupper TS, Sackstein R. Direct real-time observation of E- and P-selectin-mediated rolling on cutaneous lymphocyte-associated antigen immobilized on Western blots. J Immunol. 2002;168:5645–5651. doi: 10.4049/jimmunol.168.11.5645. [DOI] [PubMed] [Google Scholar]
- 85.Wagers AJ, Lowe JB, Kansas GS. An important role for the alpha 1,3 fucosyltransferase, FucT-VII, in leukocyte adhesion to E-selectin. Blood. 1996;88:2125–2132. [PubMed] [Google Scholar]
- 86.Wagers AJ, Stoolman LM, Craig R, Knibbs RN, Kansas GS. An sLex-deficient variant of HL60 cells exhibits high levels of adhesion to vascular selectins: further evidence that HECA-452 and CSLEX1 monoclonal antibody epitopes are not essential for high avidity binding to vascular selectins. J Immunol. 1998;160:5122–5129. [PubMed] [Google Scholar]
- 87.Knibbs RN, et al. Alpha(1,3)-fucosyltrans-ferase VII-dependent synthesis of P- and E-selectin ligands on cultured T lymphoblasts. J Immunol. 1998;161:6305–6315. [PubMed] [Google Scholar]
- 88.Sackstein R, Fuhlbrigge R. The blot rolling assay: a method for identifying adhesion molecules mediating binding under shear conditions. Methods Mol Biol. 2006;341:217–226. doi: 10.1385/1-59745-113-4:217. [DOI] [PubMed] [Google Scholar]
- 89.Dimitroff CJ, Lee JY, Fuhlbrigge RC, Sackstein R. A distinct glycoform of CD44 is an L-selectin ligand on human hematopoietic cells. Proc Natl Acad Sci USA. 2000;97:13841–13846. doi: 10.1073/pnas.250484797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dimitroff CJ, Lee JY, Rafii S, Fuhlbrigge RC, Sackstein R. CD44 is a major E-selectin ligand on human hematopoietic progenitor cells. J Cell Biol. 2001;153:1277–1286. doi: 10.1083/jcb.153.6.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brunk DK, Goetz DJ, Hammer DA. Sialyl Lewis(x)/E-selectin-mediated rolling in a cell-free system. Biophys J. 1996;71:2902–2907. doi: 10.1016/S0006-3495(96)79487-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Brunk DK, Hammer DA. Quantifying rolling adhesion with a cell-free assay: E-selectin and its carbohydrate ligands. Biophys J. 1997;72:2820–2833. doi: 10.1016/S0006-3495(97)78924-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sackstein R, et al. Ex vivo glycan engineering of CD44 programs human multipotent mesenchymal stromal cell trafficking to bone. Nat Med. 2008;14:181–187. doi: 10.1038/nm1703. [DOI] [PubMed] [Google Scholar]
- 94.Matsumoto M, et al. CD43 functions as a ligand for E-Selectin on activated T cells. J Immunol. 2005;175:8042–8050. doi: 10.4049/jimmunol.175.12.8042. [DOI] [PubMed] [Google Scholar]
- 95.Fuhlbrigge RC, King SL, Sackstein R, Kupper TS. CD43 is a ligand for E-selectin on CLA+ human T cells. Blood. 2006;107:1421–1426. doi: 10.1182/blood-2005-05-2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Picker LJ, Warnock RA, Burns AR, Doerschuk CM, Berg EL, Butcher EC. The neutrophil selectin LECAM-1 presents carbohydrate ligands to the vascular selectins ELAM-1 and GMP-140. Cell. 1991;66:921–933. doi: 10.1016/0092-8674(91)90438-5. [DOI] [PubMed] [Google Scholar]
- 97.Zollner O, et al. L-selectin from human, but not from mouse neutrophils binds directly to E-selectin. J Cell Biol. 1997;136:707–716. doi: 10.1083/jcb.136.3.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kotovuori P, et al. The vascular E-selectin binds to the leukocyte integrins CD11/CD18. Glycobiology. 1993;3:131–136. doi: 10.1093/glycob/3.2.131. [DOI] [PubMed] [Google Scholar]
- 99.Zen K, Cui LB, Zhang CY, Liu Y. Critical role of mac-1 sialyl lewis x moieties in regulating neutrophil degranulation and transmigration. J Mol Biol. 2007;374:54–63. doi: 10.1016/j.jmb.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 100.Kuijpers TW, et al. CD66 nonspecific cross-reacting antigens are involved in neutrophil adherence to cytokine-activated endothelial cells. J Cell Biol. 1992;118:457–466. doi: 10.1083/jcb.118.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Stocks SC, Kerr MA. Neutrophil NCA-160 (CD66) is the major protein carrier of selectin binding carbohydrate groups LewisX and sialyl lewisX. Biochem Biophys Res Commun. 1993;195:478–483. doi: 10.1006/bbrc.1993.2068. [DOI] [PubMed] [Google Scholar]
- 102.Tiemeyer M, et al. Carbohydrate ligands for endothelial-leukocyte adhesion molecule 1. Proc Natl Acad Sci USA. 1991;88:1138–1142. doi: 10.1073/pnas.88.4.1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Alon R, Feizi T, Yuen CT, Fuhlbrigge RC, Springer TA. Glycolipid ligands for selectins support leukocyte tethering and rolling under physiologic flow conditions. J Immunol. 1995;154:5356–5366. [PubMed] [Google Scholar]
- 104.Stroud MR, et al. Monosialogangliosides of human myelogenous leukemia HL60 cells and normal human leukocytes. 2. Characterization of E-selectin binding fractions, and structural requirements for physiological binding to E-selectin. Biochemistry. 1996;35:770–778. doi: 10.1021/bi952461g. [DOI] [PubMed] [Google Scholar]
- 105.Burdick MM, Bochner BS, Collins BE, Schnaar RL, Konstantopoulos K. Glycolipids support E-selectin-specific strong cell tethering under flow. Biochem Biophys Res Commun. 2001;284:42–49. doi: 10.1006/bbrc.2001.4899. [DOI] [PubMed] [Google Scholar]
- 106.Schnaar RL. Glycolipid-mediated cell-cell recognition in inflammation and nerve regeneration. Arch Biochem Biophys. 2004;426:163–172. doi: 10.1016/j.abb.2004.02.019. [DOI] [PubMed] [Google Scholar]
- 107.Nimrichter L, et al. E-selectin receptors on human leukocytes. Blood. 2008;112:3744–3752. doi: 10.1182/blood-2008-04-149641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yang J, Furie BC, Furie B. The biology of P-selectin glycoprotein ligand-1: its role as a selectin counterreceptor in leukocyte-endothelial and leukocyte-platelet interaction. Thromb Haemost. 1999;81:1–7. [PubMed] [Google Scholar]
- 109.Li F, Wilkins PP, Crawley S, Weinstein J, Cummings RD, McEver RP. Post-translational modifications of recombinant P-selectin glycoprotein ligand-1 required for binding to P- and E-selectin. J Biol Chem. 1996;271:3255–3264. [PubMed] [Google Scholar]
- 110.Tauxe C, Xie X, Joffraud M, Martinez M, Schapira M, Spertini O. P-selectin glycoprotein ligand-1 decameric repeats regulate selectin-dependent rolling under flow conditions. J Biol Chem. 2008;283:28536–28545. doi: 10.1074/jbc.M802865200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Matsumoto M, Shigeta A, Furukawa Y, Tanaka T, Miyasaka M, Hirata T. CD43 collaborates with P-selectin glycoprotein ligand-1 to mediate E-selectin-dependent T cell migration into inflamed skin. J Immunol. 2007;178:2499–2506. doi: 10.4049/jimmunol.178.4.2499. [DOI] [PubMed] [Google Scholar]
- 112.Alcaide P, King SL, Dimitroff CJ, Lim YC, Fuhlbrigge RC, Luscinskas FW. The 130-kDa glycoform of CD43 functions as an E-selectin ligand for activated Th1 cells in vitro and in delayed-type hypersensitivity reactions in vivo. J Invest Dermatol. 2007;127:1964–1972. doi: 10.1038/sj.jid.5700805. [DOI] [PubMed] [Google Scholar]
- 113.Dimitroff CJ, Lee JY, Schor KS, Sandmaier BM, Sackstein R. differential L-selectin binding activities of human hematopoietic cell L-selectin ligands, HCELL and PSGL-1. J Biol Chem. 2001;276:47623–47631. doi: 10.1074/jbc.M105997200. [DOI] [PubMed] [Google Scholar]
- 114.Oxley SM, Sackstein R. Detection of an L-selectin ligand on a hematopoietic progenitor cell line. Blood. 1994;84:3299–3306. [PubMed] [Google Scholar]
- 115.Sackstein R. Expression of an L-selectin ligand on hematopoietic progenitor cells. Acta Haematol. 1997;97:22–28. doi: 10.1159/000203656. [DOI] [PubMed] [Google Scholar]
- 116.Sackstein R, Dimitroff CJ. A hematopoietic cell L-selectin ligand that is distinct from PSGL-1 and displays N-glycan-dependent binding activity. Blood. 2000;96:2765–2774. [PubMed] [Google Scholar]
- 117.Sackstein R, Fu L, Allen KL. A hematopoietic cell L-selectin ligand exhibits sulfate-independent binding activity. Blood. 1997;89:2773–2781. [PubMed] [Google Scholar]
- 118.Deguchi T, Komada Y. Homing-associated cell adhesion molecule (H-CAM/CD44) on human CD34+ hematopoietic progenitor cells. Leuk Lymphoma. 2000;40:25–37. doi: 10.3109/10428190009054878. [DOI] [PubMed] [Google Scholar]
- 119.Naor D, Nedvetzki S, Golan I, Melnik L, Faitelson Y. CD44 in cancer. Crit Rev Clin Lab Sci. 2002;39:527–579. doi: 10.1080/10408360290795574. [DOI] [PubMed] [Google Scholar]
- 120.Katoh S, Zheng Z, Oritani K, Shimozato T, Kincade PW. Glycosylation of CD44 negatively regulates its recognition of hyaluronan. J Exp Med. 1995;182:419–429. doi: 10.1084/jem.182.2.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Skelton TP, Zeng C, Nocks A, Stamenkovic I. Glycosylation provides both stimulatory and inhibitory effects on cell surface and soluble CD44 binding to hyaluronan. J Cell Biol. 1998;140:431–446. doi: 10.1083/jcb.140.2.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Katoh S, et al. Cutting edge: an inducible sialidase regulates the hyaluronic acid binding ability of CD44-bearing human monocytes. J Immunol. 1999;162:5058–5061. [PubMed] [Google Scholar]
- 123.Walcheck B, Watts G, Jutila MA. Bovine gamma/delta T cells bind E-selectin via a novel glycoprotein receptor: first characterization of a lymphocyte/E-selectin interaction in an animal model. J Exp Med. 1993;178:853–863. doi: 10.1084/jem.178.3.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jutila MA, et al. Cell surface P- and E-selectin support shear-dependent rolling of bovine gamma/delta T cells. J Immunol. 1994;153:3917–3928. [PubMed] [Google Scholar]
- 125.Hanley WD, Napier SL, Burdick MM, Schnaar RL, Sackstein R, Konstantopoulos K. Variant isoforms of CD44 are P- and L-selectin ligands on colon carcinoma cells. FASEB J. 2006;20:337–339. doi: 10.1096/fj.05-4574fje. [DOI] [PubMed] [Google Scholar]
- 126.Czlapinski JL, Bertozzi CR. Synthetic glycobiology: Exploits in the Golgi compartment. Curr Opin Chem Biol. 2006;10:645–651. doi: 10.1016/j.cbpa.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 127.Lowe JB. Glycosylation in the control of selectin counter-receptor structure and function. Immunol Rev. 2002;186:19–36. doi: 10.1034/j.1600-065x.2002.18603.x. [DOI] [PubMed] [Google Scholar]
- 128.Gadhoum SZ, Sackstein R. CD15 expression in human myeloid cell differentiation is regulated by sialidase activity. Nat Chem Biol. 2008;4:751–757. doi: 10.1038/nchembio.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lowe JB, Stoolman LM, Nair RP, Larsen RD, Berhend TL, Marks RM. ELAM-1-dependent cell adhesion to vascular endothelium determined by a transfected human fucosyltransferase cDNA. Cell. 1990;63:475–484. doi: 10.1016/0092-8674(90)90444-j. [DOI] [PubMed] [Google Scholar]
- 130.Goelz SE, et al. ELFT: a gene that directs the expression of an ELAM-1 ligand. Cell. 1990;63:1349–1356. doi: 10.1016/0092-8674(90)90430-m. [DOI] [PubMed] [Google Scholar]
- 131.Ma B, Simala-Grant JL, Taylor DE. Fucosylation in prokaryotes and eukaryotes. Glycobiology. 2006;16:158R–184R. doi: 10.1093/glycob/cwl040. [DOI] [PubMed] [Google Scholar]
- 132.Huang MC, Laskowska A, Vestweber D, Wild MK. The alpha (1,3)-fucosyltransferase Fuc-TIV, but not Fuc-TVII, generates sialyl Lewis X-like epitopes preferentially on glycolipids. J Biol Chem. 2002;277:47786–47795. doi: 10.1074/jbc.M208283200. [DOI] [PubMed] [Google Scholar]
- 133.Niemela R, et al. Complementary acceptor and site specificities of Fuc-TIV and Fuc-TVII allow effective biosynthesis of sialyl-TriLex and related polylactosamines present on glycoprotein counterreceptors of selectins. J Biol Chem. 1998;273:4021–4026. doi: 10.1074/jbc.273.7.4021. [DOI] [PubMed] [Google Scholar]
- 134.Homeister JW, et al. The alpha(1,3) fucosyltransferases FucT-IV and FucT-VII exert collaborative control over selectin-dependent leukocyte recruitment and lymphocyte homing. Immunity. 2001;15:115–126. doi: 10.1016/s1074-7613(01)00166-2. [DOI] [PubMed] [Google Scholar]
- 135.Smithson G, et al. Fuc-TVII is required for T helper 1 and T cytotoxic 1 lymphocyte selectin ligand expression and recruitment in inflammation, and together with Fuc-TIV regulates naive T cell trafficking to lymph nodes. J Exp Med. 2001;194:601–614. doi: 10.1084/jem.194.5.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Knibbs RN, et al. The fucosyltransferase FucT-VII regulates E-selectin ligand synthesis in human T cells. J Cell Biol. 1996;133:911–920. doi: 10.1083/jcb.133.4.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wagers AJ, Stoolman LM, Kannagi R, Craig R, Kansas GS. Expression of leukocyte fucosyltransferases regulates binding to E-selectin: relationship to previously implicated carbohydrate epitopes. J Immunol. 1997;159:1917–1929. [PubMed] [Google Scholar]
- 138.Bengtson PLA, Larson G, Påhlsson P. Polymorphonuclear leukocytes from individuals carrying the G329A mutation in the alpha 1,3-fucosyltransferase VII gene (FUT7) roll on E- and P-selectins. J Immunol. 2002;69:3940–3946. doi: 10.4049/jimmunol.169.7.3940. [DOI] [PubMed] [Google Scholar]
- 139.Bengtson PLC, Lundblad A, Larson G, Påhlsson P. Identification of a missense mutation (G329A;Arg(110)–> GLN) in the human FUT7 gene. J Biol Chem. 2001;276:31575–31582. doi: 10.1074/jbc.M104165200. [DOI] [PubMed] [Google Scholar]
- 140.Yakubenia S, Wild MK. Leukocyte adhesion deficiency II. Advances and open questions. FEBS J. 2006;273:4390–4398. doi: 10.1111/j.1742-4658.2006.05438.x. [DOI] [PubMed] [Google Scholar]
- 141.Harduin-Lepers A, Vallejo-Ruiz V, Krzewinski-Recchi MA, Samyn-Petit B, Julien S, Delannoy P. The human sialyltransferase family. Biochimie. 2001;83:727–737. doi: 10.1016/s0300-9084(01)01301-3. [DOI] [PubMed] [Google Scholar]
- 142.Ellies LG, et al. Sialyltransferase specificity in selectin ligand formation. Blood. 2002;100:3618–3625. doi: 10.1182/blood-2002-04-1007. [DOI] [PubMed] [Google Scholar]
- 143.Kumar R, Camphausen RT, Sullivan FX, Cumming DA. Core2 beta-1, 6-N-acetylglucosaminyltransferase enzyme activity is critical for P-selectin glycoprotein ligand-1 binding to P-selectin. Blood. 1996;88:3872–3879. [PubMed] [Google Scholar]
- 144.Martinez M, et al. Regulation of PSGL-1 interactions with L-selectin, P-selectin, and E-selectin: role of human fucosyltransferase-IV and -VII. J Biol Chem. 2005;280:5378–5390. doi: 10.1074/jbc.M410899200. [DOI] [PubMed] [Google Scholar]
- 145.Ellies LG, Tsuboi S, Petryniak B, Lowe JB, Fukuda M, Marth JD. Core 2 oligosaccharide biosynthesis distinguishes between selectin ligands essential for leukocyte homing and inflammation. Immunity. 1998;9:881–890. doi: 10.1016/s1074-7613(00)80653-6. [DOI] [PubMed] [Google Scholar]
- 146.Snapp KR, Heitzig CE, Ellies LG, Marth JD, Kansas GS. Differential requirements for the O-linked branching enzyme core 2 beta1-6-N-glucosaminyltransferase in biosynthesis of ligands for E-selectin and P-selectin. Blood. 2001;97:3806–3811. doi: 10.1182/blood.v97.12.3806. [DOI] [PubMed] [Google Scholar]
- 147.Sperandio M, Thatte A, Foy D, Ellies LG, Marth JD, Ley K. Severe impairment of leukocyte rolling in venules of core 2 glucos-aminyltransferase-deficient mice. Blood. 2001;97:3812–3819. doi: 10.1182/blood.v97.12.3812. [DOI] [PubMed] [Google Scholar]
- 148.Qasba PK, Ramakrishnan B, Boeggeman E. Structure and function of beta -1,4-galactosyltransferase. Curr Drug. 2008;9:292–309. doi: 10.2174/138945008783954943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Asano M, et al. Impaired selectin-ligand biosynthesis and reduced inflammatory responses in beta-1,4-galactosyltransferase-I-deficient mice. Blood. 2003;102:1678–1685. doi: 10.1182/blood-2003-03-0836. [DOI] [PubMed] [Google Scholar]
- 150.Picker LJ, Treer JR, Ferguson-Darnell B, Collins PA, Bergstresser PR, Terstappen LW. Control of lymphocyte recirculation in man. II. Differential regulation of the cutaneous lymphocyte-associated antigen, a tissue-selective homing receptor for skin-homing T cells. J Immunol. 1993;150:1122–1136. [PubMed] [Google Scholar]
- 151.Leung DY, Gately M, Trumble A, Ferguson-Darnell B, Schlievert PM, Picker LJ. Bacterial superantigens induce T cell expression of the skin-selective homing receptor, the cutaneous lymphocyte-associated antigen, via stimulation of interleukin 12 production. J Exp Med. 1995;181:747–753. doi: 10.1084/jem.181.2.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wagers AJ, Waters CM, Stoolman LM, Kansas GS. Interleukin 12 and interleukin 4 control T cell adhesion to endothelial selectins through opposite effects on alpha1,3-fucosyltransferase VII gene expression. J Exp Med. 1998;188:2225–2231. doi: 10.1084/jem.188.12.2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Nakayama F, et al. Expression of cutaneous lymphocyte-associated antigen regulated by a set of glycosyltransferases in human T cells: involvement of alpha1, 3-fucosyltransferase VII and beta1,4-galactosyltransferase I. J Invest Dermatol. 2000;115:299–306. doi: 10.1046/j.1523-1747.2000.00032.x. [DOI] [PubMed] [Google Scholar]
- 154.Blander JM, Visintin I, Janeway CA, Jr, Medzhitov R. Alpha(1,3)-fucosyltransferase VII and alpha(2,3)-sialyltransferase IV are up-regulated in activated CD4 T cells and maintained after their differentiation into Th1 and migration into inflammatory sites. J Immunol. 1999;163:3746–3752. [PubMed] [Google Scholar]
- 155.Lim YC, Henault L, Wagers AJ, Kansas GS, Luscinskas FW, Lichtman AH. Expression of functional selectin ligands on Th cells is differentially regulated by IL-12 and IL-4. J Immunol. 1999;162:3193–3201. [PubMed] [Google Scholar]
- 156.Underhill GH, Zisoulis DG, Kolli KP, Ellies LG, Marth JD, Kansas GS. A crucial role for T-bet in selectin ligand expression in T helper 1 (Th1) cells. Blood. 2005;106:3867–3873. doi: 10.1182/blood-2005-03-0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lim YC, et al. IL-12, STAT4-dependent up-regulation of CD4 (+) T cell core 2 beta-1,6-n-acetylglucosaminyltransferase, an enzyme essential for biosynthesis of P-selectin ligands. J Immunol. 2001;167:4476–4484. doi: 10.4049/jimmunol.167.8.4476. [DOI] [PubMed] [Google Scholar]
- 158.Carlow DA, Corbel SY, Williams MJ, Ziltener HJ. IL-2, -4, and -15 differentially regulate O-glycan branching and P-selectin ligand formation in activated CD8 T cells. J Immunol. 2001;167:6841–6848. doi: 10.4049/jimmunol.167.12.6841. [DOI] [PubMed] [Google Scholar]
- 159.Merzaban JS, Zuccolo J, Corbel SY, Williams MJ, Ziltener HJ. An alternate core 2 beta1,6-N-acetylglucosaminyltransferase selectively contributes to P-selectin ligand formation in activated CD8 T cells. J Immunol. 2005;174:4051–4059. doi: 10.4049/jimmunol.174.7.4051. [DOI] [PubMed] [Google Scholar]
- 160.Dagia NM, et al. G-CSF induces E-selectin ligand expression on human myeloid cells. Nat Med. 2006;12:1185–1190. doi: 10.1038/nm1470. [DOI] [PubMed] [Google Scholar]
- 161.Beyer TA, Rearick JI, Paulson JC, Prieels JP, Sadler JE, Hill RL. Biosynthesis of mammalian glycoproteins. Glycosylation pathways in the synthesis of the nonreducing terminal sequences. J Biol Chem. 1979;254:12531–12534. [PubMed] [Google Scholar]
- 162.Sadler JE, Paulson JC, Hill RL. The role of sialic acid in the expression of human MN blood group antigens. J Biol Chem. 1979;254:2112–2119. [PubMed] [Google Scholar]
- 163.Whiteheart SW, Passaniti A, Reichner JS, Holt GD, Haltiwanger RS, Hart GW. Glycosyltransferase probes. Methods Enzymol. 1989;179:82–95. doi: 10.1016/0076-6879(89)79116-3. [DOI] [PubMed] [Google Scholar]
- 164.Markwell MA, Paulson JC. Sendai virus utilizes specific sialyloligosaccharides as host cell receptor determinants. Proc Natl Acad Sci USA. 1980;77:5693–5697. doi: 10.1073/pnas.77.10.5693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Liukkonen J, Haataja S, Tikkanen K, Kelm S, Finne J. Identification of N-acetylneuraminyl alpha 2 → 3 poly-N-acetyllactosamine glycans as the receptors of sialic acid-binding Streptococcus suis strains. J Biol Chem. 1992;267:21105–21111. [PubMed] [Google Scholar]
- 166.Viitala J, Finne J. Specific cell-surface labeling of polyglycosyl chains in human erythrocytes and HL-60 cells using endo-beta-galactosidase and galactosyltransferase. Eur J Biochem. 1984;138:393–397. doi: 10.1111/j.1432-1033.1984.tb07928.x. [DOI] [PubMed] [Google Scholar]
- 167.Whiteheart SW, Hart GW. Sialyltransferases as specific cell surface probes of terminal and penultimate saccharide structures on living cells. Anal Biochem. 1987;163:123–135. doi: 10.1016/0003-2697(87)90102-3. [DOI] [PubMed] [Google Scholar]
- 168.Paulson JC, Rogers GN. Resialylated erythrocytes for assessment of the specificity of sialyloligosaccharide binding proteins. Methods Enzymol. 1987;138:162–168. doi: 10.1016/0076-6879(87)38013-9. [DOI] [PubMed] [Google Scholar]
- 169.Carroll SM, Paulson JC. Differential infection of receptor-modified host cells by receptor-specific influenza viruses. Virus Res. 1985;3:165–179. doi: 10.1016/0168-1702(85)90006-1. [DOI] [PubMed] [Google Scholar]
- 170.Palcic MM. Glycosyltransferases in glycobiology. Methods Enzymol. 1994;230:300–316. doi: 10.1016/0076-6879(94)30020-8. [DOI] [PubMed] [Google Scholar]
- 171.Warnock D, et al. In vitro galactosylation of human IgG at 1 kg scale using recombinant galactosyltransferase. Biotechnol Bioeng. 2005;92:831–842. doi: 10.1002/bit.20658. [DOI] [PubMed] [Google Scholar]
- 172.Lairson LL, Henrissat B, Davies GJ, Withers SG. Glycosyltransferases: structures, functions, and mechanisms. Annu Rev Biochem. 2008;77:521–555. doi: 10.1146/annurev.biochem.76.061005.092322. [DOI] [PubMed] [Google Scholar]
- 173.Witsell DL, Casey CE, Neville MC. Divalent cation activation of galactosyltransferase in native mammary Golgi vesicles. J Biol Chem. 1990;265:15731–15737. [PubMed] [Google Scholar]
- 174.Stults CL, Macher BA. Beta 1-3-N-acetylglucosaminyltransferase in human leukocytes: properties and role in regulating neolacto glycosphingolipid biosynthesis. Arch Biochem Biophys. 1993;303:125–133. doi: 10.1006/abbi.1993.1263. [DOI] [PubMed] [Google Scholar]
- 175.Murray BW, Takayama S, Schultz J, Wong CH. Mechanism and specificity of human alpha-1,3-fucosyltransferase V. Biochemistry. 1996;35:11183–11195. doi: 10.1021/bi961065a. [DOI] [PubMed] [Google Scholar]
- 176.Shinoda K, Tanahashi E, Fukunaga K, Ishida H, Kiso M. Detailed acceptor specificities of human alpha1,3-fucosyltransferases, Fuc-TVII and Fuc-TVI. Glycoconj J. 1998;15:969–974. doi: 10.1023/a:1006933808303. [DOI] [PubMed] [Google Scholar]
- 177.Palma AS, Morais VA, Coelho AV, Costa J. Effect of the manganese ion on human alpha3/4 fucosyltransferase III activity. Biometals. 2004;17:35–43. doi: 10.1023/a:1024485718843. [DOI] [PubMed] [Google Scholar]
- 178.Ramakrishnan B, Boeggeman E, Ramasamy V, Qasba PK. Structure and catalytic cycle of beta-1,4-galactosyltransferase. Curr Opin Struct Biol. 2004;14:593–600. doi: 10.1016/j.sbi.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 179.Altieri DC. Occupancy of CD11b/CD18 (Mac-1) divalent ion binding site(s) induces leukocyte adhesion. J Immunol. 1991;147:1891–1898. [PubMed] [Google Scholar]
- 180.Alon R, Kassner PD, Carr MW, Finger EB, Hemler ME, Springer TA. The integrin VLA-4 supports tethering and rolling in flow on VCAM-1. J Cell Biol. 1995;128:1243–1253. doi: 10.1083/jcb.128.6.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Takamatsu Y, Simmons PJ, Levesque JP. Dual control by divalent cations and mitogenic cytokines of alpha 4 beta 1 and alpha 5 beta 1 integrin avidity expressed by human hemopoietic cells. Cell Adhes Commun. 1998;5:349–366. doi: 10.3109/15419069809010781. [DOI] [PubMed] [Google Scholar]
- 182.Chigaev A, et al. Real time analysis of the affinity regulation of alpha 4-integrin. The physiologically activated receptor is intermediate in affinity between resting and Mn (2+) or antibody activation. J Biol Chem. 2001;276:48670–48678. doi: 10.1074/jbc.M103194200. [DOI] [PubMed] [Google Scholar]
- 183.de Bruyn KM, Rangarajan S, Reedquist KA, Figdor CG, Bos JL. The small GTPase Rap1 is required for Mn (2+)- and antibody-induced LFA-1- and VLA-4-mediated cell adhesion. J Biol Chem. 2002;277:29468–29476. doi: 10.1074/jbc.M204990200. [DOI] [PubMed] [Google Scholar]
- 184.Suzuki T, Tsukamoto I. Manganese-induced apoptosis in hepatocytes after partial hepatectomy. Eur J Pharmacol. 2005;525:48–53. doi: 10.1016/j.ejphar.2005.09.061. [DOI] [PubMed] [Google Scholar]
- 185.Desole MS, Sciola L, Delogu MR, Sircana S, Migheli R. Manganese and 1-methyl-4-(2′-ethylpheny1)-1,2,3,6-tetrahydropyridine induce apoptosis in PC12 cells. Neurosci Lett. 1996;209:193–196. doi: 10.1016/0304-3940(96)12645-8. [DOI] [PubMed] [Google Scholar]
- 186.Schrantz N, Blanchard DA, Mitenne F, Auffredou MT, Vazquez A, Leca G. Manganese induces apoptosis of human B cells: caspase-dependent cell death blocked by bcl-2. Cell Death Differ. 1999;6:445–453. doi: 10.1038/sj.cdd.4400508. [DOI] [PubMed] [Google Scholar]
- 187.Yang H, Sun Y, Zheng X. Manganese-induced apoptosis in rat myocytes. J Biochem Mol Toxicol. 2007;21:94–100. doi: 10.1002/jbt.20172. [DOI] [PubMed] [Google Scholar]
- 188.Sultana C, Shen Y, Johnson C, Kalra VK. Cobalt chloride-induced signaling in endothelium leading to the augmented adherence of sickle red blood cells and transendothelial migration of monocyte-like HL-60 cells is blocked by PAF-receptor antagonist. J Cell Physiol. 1999;179:67–78. doi: 10.1002/(SICI)1097-4652(199904)179:1<67::AID-JCP9>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 189.Manome H, Aiba S, Tagami H. Simple chemicals can induce maturation and apoptosis of dendritic cells. Immunology. 1999;98:481–490. doi: 10.1046/j.1365-2567.1999.00916.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Srivastava G, Kaur KJ, Hindsgaul O, Palcic MM. Enzymatic transfer of a preassembled trisaccharide antigen to cell surfaces using a fucosyltransferase. J Biol Chem. 1992;267:22356–22361. [PubMed] [Google Scholar]
- 191.Ramachandran V, et al. Dimerization of a selectin and its ligand stabilizes cell rolling and enhances tether strength in shear flow. Proc Natl Acad Sci USA. 2001;98:10166–10171. doi: 10.1073/pnas.171248098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Sipkins DA, et al. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature. 2005;435:969–973. doi: 10.1038/nature03703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Kobzdej MM, Leppanen A, Ramachandran V, Cummings RD, McEver RP. Discordant expression of selectin ligands and sialyl Lewis x-related epitopes on murine myeloid cells. Blood. 2002;100:4485–4494. doi: 10.1182/blood-2002-06-1799. [DOI] [PubMed] [Google Scholar]
- 194.Xia L, McDaniel JM, Yago T, Doeden A, McEver RP. Surface fucosylation of human cord blood cells augments binding to P-selectin and E-selectin and enhances engraftment in bone marrow. Blood. 2004;104:3091–3096. doi: 10.1182/blood-2004-02-0650. [DOI] [PubMed] [Google Scholar]
- 195.Hidalgo A, Frenette PS. Enforced fucosylation of neonatal CD34+ cells generates selectin ligands that enhance the initial interactions with microvessels but not homing to bone marrow. Blood. 2005;105:567–575. doi: 10.1182/blood-2004-03-1026. [DOI] [PubMed] [Google Scholar]
- 196.Horwitz EM, et al. Transplantability and therapeutic effects of bone marrow-derived mesenchymal cells in children with osteogenesis imperfecta. Nat Med. 1999;5:309–313. doi: 10.1038/6529. [DOI] [PubMed] [Google Scholar]
- 197.Horwitz EM, et al. Isolated allogeneic bone marrow-derived mesenchymal cells engraft and stimulate growth in children with osteogenesis imperfecta: Implications for cell therapy of bone. Proc Natl Acad Sci USA. 2002;99:8932–8937. doi: 10.1073/pnas.132252399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Rombouts WJ, Ploemacher RE. Primary murine MSC show highly efficient homing to the bone marrow but lose homing ability following culture. Leukemia. 2003;17:160–170. doi: 10.1038/sj.leu.2402763. [DOI] [PubMed] [Google Scholar]
- 199.Devine SM, et al. Mesenchymal stem cells are capable of homing to the bone marrow of non-human primates following systemic infusion. Exp Hematol. 2001;29:244–255. doi: 10.1016/s0301-472x(00)00635-4. [DOI] [PubMed] [Google Scholar]
- 200.Wynn RF, et al. A small proportion of mesenchymal stem cells strongly expresses functionally active CXCR4 receptor capable of promoting migration to bone marrow. Blood. 2004;104:2643–2645. doi: 10.1182/blood-2004-02-0526. [DOI] [PubMed] [Google Scholar]
- 201.Bhakta S, Hong P, Koc O. The surface adhesion molecule CXCR4 stimulates mesenchymal stem cell migration to stromal cell-derived factor-1 in vitro but does not decrease apoptosis under serum deprivation. Cardiovasc Revasc Med. 2006;7:19–24. doi: 10.1016/j.carrev.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 202.Shi M, et al. Regulation of CXCR4 expression in human mesenchymal stem cells by cytokine treatment: role in homing efficiency in NOD/SCID mice. Haematologica. 2007;92:897–904. doi: 10.3324/haematol.10669. [DOI] [PubMed] [Google Scholar]
- 203.Chen CP, et al. Trafficking of multipotent mesenchymal stromal cells from maternal circulation through the placenta involves vascular endothelial growth factor receptor-1 and integrins. Stem Cells. 2008;26:550–561. doi: 10.1634/stemcells.2007-0406. [DOI] [PubMed] [Google Scholar]
- 204.Kumar S, Ponnazhagan S. Bone homing of mesenchymal stem cells by ectopic alpha 4 integrin expression. FASEB J. 2007;21:3917–3927. doi: 10.1096/fj.07-8275com. [DOI] [PubMed] [Google Scholar]
- 205.Steingen C, Brenig F, Baumgartner L, Schmidt J, Schmidt A, Bloch W. Characterization of key mechanisms in transmigration and invasion of mesenchymal stem cells. J Mol Cell Cardiol. 2008;44:1072–1084. doi: 10.1016/j.yjmcc.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 206.Bokoch GM. Chemoattractant signaling and leukocyte activation. Blood. 1995;86:1649–1660. [PubMed] [Google Scholar]
- 207.Ganju RK, et al. The alpha-chemokine, stromal cell-derived factor-1alpha, binds to the transmembrane G-protein-coupled CXCR-4 receptor and activates multiple signal transduction pathways. J Biol Chem. 1998;273:23169–23175. doi: 10.1074/jbc.273.36.23169. [DOI] [PubMed] [Google Scholar]
- 208.Rossi D, Zlotnik A. The biology of chemokines and their receptors. Annu Rev Immunol. 2000;18:217–242. doi: 10.1146/annurev.immunol.18.1.217. [DOI] [PubMed] [Google Scholar]
- 209.Lokeshwar VB, Bourguignon LY. The lymphoma transmembrane glycoprotein GP85 (CD44) is a novel guanine nucleotide-binding protein which regulates GP85 (CD44)-ankyrin interaction. J Biol Chem. 1992;267:22073–22078. [PubMed] [Google Scholar]
- 210.Shimizu Y, Van Seventer GA, Siraganian R, Wahl L, Shaw S. Dual role of the CD44 molecule in T cell adhesion and activation. J Immunol. 1989;143:2457–2463. [PubMed] [Google Scholar]
- 211.Koopman G, van Kooyk Y, de Graaff M, Meyer CJ, Figdor CG, Pals ST. Triggering of the CD44 antigen on T lymphocytes promotes T cell adhesion through the LFA-1 pathway. J Immunol. 1990;145:3589–3593. [PubMed] [Google Scholar]
- 212.Katakai T, et al. Chemokine-independent preference for T-helper-1 cells in transendothelial migration. J Biol Chem. 2002;277:50948–50958. doi: 10.1074/jbc.M204133200. [DOI] [PubMed] [Google Scholar]
- 213.Wang HS, et al. CD44 cross-linking induces integrin-mediated adhesion and transendothelial migration in breast cancer cell line by up-regulation of LFA-1 (alpha L beta2) and VLA-4 (alpha4beta1) Exp Cell Res. 2005;304:116–126. doi: 10.1016/j.yexcr.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 214.Nandi A, Estess P, Siegelman M. Bimolecular complex between rolling and firm adhesion receptors required for cell arrest; CD44 association with VLA-4 in T cell extravasation. Immunity. 2004;20:455–465. doi: 10.1016/s1074-7613(04)00077-9. [DOI] [PubMed] [Google Scholar]
- 215.Pober JS, Bevilacqua MP, Mendrick DL, Lapierre LA, Fiers W, Gimbrone MA., Jr Two distinct monokines, interleukin 1 and tumor necrosis factor, each independently induce biosynthesis and transient expression of the same antigen on the surface of cultured human vascular endothelial cells. J Immunol. 1986;136:1680–1687. [PubMed] [Google Scholar]
- 216.Pober JS. Effects of tumour necrosis factor and related cytokines on vascular endothelial cells. Ciba Found Symp. 1987;131:170–184. doi: 10.1002/9780470513521.ch12. [DOI] [PubMed] [Google Scholar]
- 217.Bevilacqua MP, Pober JS, Mendrick DL, Cotran RS, Gimbrone MA., Jr Identification of an inducible endothelial-leukocyte adhesion molecule. Proc Natl Acad Sci USA. 1987;84:9238–9242. doi: 10.1073/pnas.84.24.9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Osborn L, et al. Direct expression cloning of vascular cell adhesion molecule 1, a cytokine-induced endothelial protein that binds to lymphocytes. Cell. 1989;59:1203–1211. doi: 10.1016/0092-8674(89)90775-7. [DOI] [PubMed] [Google Scholar]
- 219.Carlos TM, Harlan JM. Leukocyte-endothelial adhesion molecules. Blood. 1994;84:2068–2101. [PubMed] [Google Scholar]
- 220.Reyes M, Lund T, Lenvik T, Aguiar D, Koodie L, Verfaillie CM. Purification and ex vivo expansion of postnatal human marrow mesodermal progenitor cells. Blood. 2001;98:2615–2625. doi: 10.1182/blood.v98.9.2615. [DOI] [PubMed] [Google Scholar]
- 221.Mocco J, et al. HuEP5C7 as a humanized monoclonal anti-E/P-selectin neurovascular protective strategy in a blinded placebo-controlled trial of nonhuman primate stroke. Circ Res. 2002;91:907–914. doi: 10.1161/01.res.0000042063.15901.20. [DOI] [PubMed] [Google Scholar]
- 222.Einsele H, et al. Infusion of cytomegalovirus (CMV)-specific T cells for the treatment of CMV infection not responding to antiviral chemotherapy. Blood. 2002;99:3916–3922. doi: 10.1182/blood.v99.11.3916. [DOI] [PubMed] [Google Scholar]
- 223.Peggs KS, et al. Adoptive cellular therapy for early cytomegalovirus infection after allogeneic stem-cell transplantation with virus-specific T-cell lines. Lancet. 2003;362:1375–1377. doi: 10.1016/S0140-6736(03)14634-X. [DOI] [PubMed] [Google Scholar]
- 224.Huehn J, et al. Developmental stage, phenotype, and migration distinguish naive- and effector/memory-like CD4+ regulatory T cells. J Exp Med. 2004;199:303–313. doi: 10.1084/jem.20031562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Sather BD, et al. Altering the distribution of Foxp3(+) regulatory T cells results in tissue-specific inflammatory disease. J Exp Med. 2007;204:1335–1347. doi: 10.1084/jem.20070081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Mrass P, Kinjyo I, Ng LG, Reiner SL, Pure E, Weninger W. CD44 mediates successful interstitial navigation by killer T cells and enables efficient antitumor immunity. Immunity. 2008;29:971–985. doi: 10.1016/j.immuni.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]