

Abstract
The Gram-positive anaerobic bacterium Clostridium difficile produces toxins A and B, which can cause a spectrum of diseases from pseudomembranous colitis to C. difficile-associated diarrhea. A limited number of C. difficile strains also produce a binary toxin that exhibits ADP ribosyltransferase activity. Here, the structure and the mechanism of action of these toxins as well as their role in disease are reviewed. Nosocomial C. difficile infection is often contracted in hospital when patients treated with antibiotics suffer a disturbance in normal gut microflora. C. difficile spores can persist on dry, inanimate surface for months. Metronidazole and oral vancomycin are clinically used for treatment of C. difficile infection but clinical failure and concern about promotion of resistance are motivating the search for novel non-antibiotic therapeutics. Methods for controlling both toxins and spores, replacing gut microflora by probiotics or fecal transplant, and killing bacteria in the anaerobic gut by photodynamic therapy are discussed.
Keywords: bile acids, binary toxin, Clostridium difficile, fecal transplant, monoclonal antibody, pathogenicity locus, photodynamic therapy, probiotics, spore germination, toxin A and B
Clostridium difficile is a Gram-positive, spore-forming and obligate anaerobe gastrointestinal (GI) bacterium that can cause a range of diseases like antibiotic-associated diarrhea (AAD), pseudomembranous colitis and toxic megacolon [1–4]. Human C. difficile-associated diarrhea (CDAD) is responsible for one-fourth of all AAD cases, with approximately three million cases per year [5–8]. More than 14,000 people in the USA alone are reported to have died of C. difficile in 2012 and treatment costs are estimated to be more than US$3 billion according to the Center for Disease Control and Prevention [401].
C. difficile infection (CDI) is an inflammatory condition of the large intestine characterized by diarrhea and the appearance of distinct plaques and neutrophil accumulation in the lumen of the intestinal lining [9,10]. The bacterium is transmitted by the fecal-oral route and can readily colonize people with suppressed microflora as a consequence of antibiotic treatment [11,12]. The pathogenesis of C. difficile is based upon the action of at least one of the two major toxins, A and B (encoded by TcdA and TcdB), that belong to the family of clostridial glucosylating toxins [13–17]. TcdA and TcdB are structurally large (270–308 kDa) single-subunit polypeptides, which can be divided into four main domains: N-terminal glucosyltransferase domain, cysteine protease domain, central translocation domain and C-terminal receptor-binding domain [17–20].
Anaerobic C. difficile vegetative cells are excreted by the host, but have to be in spore form to survive for long periods outside of the host environment [21]. Metabolically, the bacterial spore is dormant and highly resistant to many types of environmental insults, but when conditions become suitable, the spores germinate and grow out as vegetative cells and produce toxins. Spores are extremely resistant to disinfectants and can persist for more than 12 months in dry, inanimate environments with little loss of viability or pathogenicity [22]. Thus, the eradication of spores in the feces of infected patients is very difficult leading to infection or re-infection of cohabitating individuals through unintentional ingestion of infected materials [23,24]. Spores are also implicated in the 20–25% of CDI cases that relapse after antibiotic treatment [25].
Metronidazole and vancomycin are clinically used for treatment of infections by C. difficile. Metronidazole is inexpensive and is used for mild and moderate CDI but the number of treatment failures with this antibiotic is increasing. Vancomycin causes few side effects, but it is costly and there is concern about the increase of clinical failure caused by the appearance of resistant strains. Increasing failure and recurrence with these antibiotics has led researchers to investigate other antibiotics for treatment of CDI. Rifaximin, tigecycline and fidaxomicin are examples of these antibiotics; some of them, like fidaxomicin, are already US FDA approved for CDI, while other new antibiotics are now being studied for GI infections, especially C. difficile. Nosocomial infections with C. difficile often occur during antibiotic therapy that disrupts the normal microflora found in the colon [26]. When commonly used antibiotics such as clindamycin and fluoroquinolones are systemically administered, the increasing resistance of C. difficile to these antibiotics makes the colon vulnerable to colonization by opportunistic pathogens [27].
C. difficile toxins
Pathogenicity locus
The C. difficile pathogenicity locus (PaLoc) consists of TcdA and TcdB and three additional genes, negative (tcdC), positive (tcdD) regulators as well as a holin-like pore-forming protein (tcdE) as shown in Figure 1 [28–34].
Figure 1.
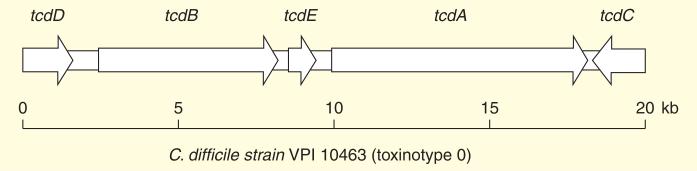
Schematic representation of Clostridium difficile PaLoc region coding for the TcdA and TcdB and three addition genes in reference strain VPI 10463 of toxinotype 0.
Strains of C. difficile are classified into two main groups: PCR ribotype and toxinotype. The first one is for typing the 16S-23S rRNA gene and the second one for identifying the restriction pattern of the toxin genes. PCR-RFLP (restriction fragment length polymorphism) analysis of the PaLoc has revealed that C. difficile strains consist of 31 toxinotypes (XXXI) [34,35]. Insertion, deletion and also mutations of each toxinotype have been characterized by comparison with the reference strain VPI 10463 (toxinotype 0).
Among all toxinotypes, only toxinotype X (strain 8864) and XI (strain SE923) showed significant changes in the PaLoc [35,36]. Strain 8864 has a 1.1 kb insertion and a 5.9 kb deletion when PaLoc boundaries are considered. Strain SE923 has a modification in the 5′-end extension of the toxinotype XI strain group which causes enteritis [30,36,37]. The deletion of large region of PaLoc at its 5′-end extension in toxinotype XI has been confirmed and is considered as a type of C. difficile reorganized genome within regions adjacent to the PaLoc [38].
A deletion in the tcdC locus of the virulent C. difficile strain NAP1/027 may cause high TcdA and TcdB production [11]. In fact C. difficile NAP1/BI/027 produces more toxin than reference strains and also produces a binary toxin (CDT) that triggers the formation of microtubule protrusions on the GI epithelial cells, leading to enhanced colonization of C. difficile (see section ‘Hypervirulent C. difficile’). CDT, which is an actin-specific ADP-ribosyltransferase, has been correlated with elevated resistance to fluoroquinolones due to mutations in DNA gyrase genes [39–47].
TcdA & TcdB
TcdA and TcdB, mapping at 19.6 kb of the PaLoc including strains of C. difficile that have deletions, insertions or polymorphic restriction sites in one or more of the genes on the PaLoc (31 different toxinotypes have been described up to now) [28,34,48]. These toxins that are known to exert their cytotoxic activity via a modification of cytoskeletal components are large multidomain proteins with high homology to each other [49,50]. Both are highly toxic when administered systemically to mice, and can cause disease in patients by disruption of the cytoskeleton leading to cytopathic effects in cultured cells within hours of intoxication [51–53]. Systemic toxemia may therefore contribute to extraintestinal disease complications sometimes associated with severe cases of CDI [54–57].
CDI can begin by the ingestion of vegetative organisms, or spores often in combination with antibiotic exposure [1,58,59]. The clinical manifestations are variable, ranging from asymptomatic carriage, to mild self-limiting diarrhea and severe pseudo-membranous colitis [60–64]. The repeating domains of toxins bind to the related carbohydrate moieties on glycoprotein receptor(s) that are present on the surface of mammalian cells. These cell surface interactions cause internalization and interruption of critical cell signaling events. One factor which is important in disease manifestation is that the surface layer proteins of C. difficile play an important role in bacterial colonization, and that antibodies raised against these proteins are partially protective [65,66].
Structure of TcdA & TcdB
Both large toxins A and B are glucosyltransferases and structurally consist of four polypeptide domains: the amino-terminal catalytic glucosyltransferase domain which possesses full biological activity; the autocatalytic cysteine protease domain that autoproteolytically regulates C. difficile glucosylating toxins by releasing a cytotoxic effector domain into target cells; the central translocation domain that is characterized by a small hydrophobic stretch and is thought to mediate membrane insertion during translocation processes; the carboxy-terminal host-cell-binding domain consisting of repetitive oligopeptides that are involved in receptor binding [17,30,67–74].
Not only a single polypeptide toxin, TcdB (~270 kDa; protein with 2366 amino acid residues), and TcdA genes, but also two regulators TcdC (negative regulator) and TcdR (activator factor) along with TcdE (a putative holin) all are part of toxin gene expression in the 19.6-kb chromosomal region PaLoc [30,75]. The ability of the toxin cell-wall-binding domains (CWB domains) to associate with cells has been implied from known functional aspects of related non-toxin protein domains derived from bacteria [76]. At least four functional domains contribute to cell entry in TcdB and glucosylation of small-GTPases within the cytosol of the cell [65]. Figure 2 illustrates the functional domain and the site action of each TcdA and TcdB.
Figure 2. Schematic structure of TcdA and TcdB with their functions and sites of action.

TcdA and TcdB consist of four main domains: GT (N-terminal glucosyltransferase domain); CPD (autocatalytic cysteine protease domain); TMD (central translocation domain) covering a hydrophobic region and RBD (highly repetitive C-terminal receptor binding domain).
Residues 365–516 belong to the substrate recognition sites in the GT domain. Residues 544–955 are the cysteine protease domain which is necessary for autoproteolytic activity and delivery of the enzymatic domain into the cytosol [66,77,78]. Residues between 956 and 1128 belong to a putative membrane-spanning domain and its effects on toxin activity are unknown. The carboxy-terminal region of the toxin is the fourth functional domain that is predicted to interact with receptors on target cells [19]. TcdB plays a prominent role in the pathogenesis of CDIs but the lack of suitable genetic tools to manipu late C. difficile and also the lack of isogenic mutants may hinder the elucidation of the exact mechanisms. Although the repeats in TcdA show a low level of sequence similarity, a number of fragments containing 5–15 repeats have been shown to form stable folded secondary structures [79].
The first 3D structural information of TcdA-f1 which is a fragment of C-terminal 127 residues of TcdA from C. difficile strain 48489 (toxinotype VI), indicated that this region may function as receptor binding. The structure analysis revealed the presence of four copies from a short repeat (SR) and one copy from a long repeat (LR). Each SR or LR contains a single β-hairpin consisting of a pair of five- to six-residue anti-parallel β-strands as illustrated in Figure 3.
Figure 3. Ribbon representation of the TcdA-f1 structure.

β-Hairpin, SRs, LRs, C-terminal and N-terminal have been shown. Data taken from [74].
The beginning and the end of the connecting loop preceding the following β-hairpin has been defined to coincide with the boundaries of each SR or LR. Except for the N- and C-terminal hairpins, each β-hairpin interacts with both the preceding one and the following one. The structure of each β-hairpin depends on the positions of residues at strand 1 and 2 which form a small hydrophobic cluster leading to consecutive pairs of β-hairpins to bind together. The second strand of each SR is followed by a loop of 7–10 residues that in the TcdA-f1, three instances of connecting loops found to play a neutral role in the overall arrangement of β-hairpins. The structural topographic analysis is valuable in dissecting the binding mechanism and explaining how large clostridial cytotoxins bind to cell surfaces. The mechanism elucidation will be helpful in developing novel CDAD treatments that may be based on blocking toxin binding to cell surfaces In the 1990s, toxin A-B+ strain type also were identified as causing severe cases of CDI, however, it accounts for a lesser proportion (2–11%) of patients affected by C. difficile in comparison with TcdA and TcdB. Molecular typing methods like restriction endonuclease analysis (REA), 16S-23S rRNA ribotyping, amplified fragment length polymorphism (AFLP), multilocus sequence typing (MLST) and toxinotyping (PCR-RFLP) have been applied for identification of A-B+ variants [80]. The study of eight A-B+ strains isolated from different sources showed that they belonged to the same sequence type and cluster in a very homogenous MLST phylogenetic lineage despite their origins from unrelated patients [81]. Three typing methods: serogrouping, 16S-23S rRNA PCR ribotyping and REA were used to show the relatedness of 23 A-B+ strains [82]. The results indicated that 21 of these 23 isolates had a 1.8 kb deletion of tcdA corresponding to toxinotype VIII. The two remaining A-B+ strains were strain 8864, which was unique by all three typing methods. Moreover, strain 8864 has been characterized as the first toxin variant strain [29,83].
The second category of A-B+ strain types which are located at the same position as strain 8864 and contain a 1.8 kb deletion in tcdA, is serogroup F strains (type strain 1470). Frequent isolation from asymptomatic infants and their lack of pathogenicity in animal models led this strain to be considered as non-pathogenic [84,85]. These organisms have been classified as toxinotype VIII which is the most clinically significant toxin variant strain type. It also contained a stop codon corresponding to amino acid position 47, leading to the truncation of TcdA-1470. Toxinotypes XVI and XVII are other newer A-B+ strains which contain the genes that code the CDT [86].
Mechanism of action of TcdA & TcdB
The crucial step for pathogenicity of the toxins TcdA and TcdB is the translocation of the catalytic domains that occur when the C-terminal region of toxins first interact with cell-surface carbohydrates (e.g., αGal(1→3)bGal(1→4)βGlcNac glycan in hamsters). Then both the C-terminal and central regions of the toxin facilitate entry into the cell through receptor-mediated endocytosis [71,87,88]. These TcdA and TcdB units catalyze the monoglucosylation of the threonine 35/37 residue of small GTP-binding proteins Rho, Rac and Cdc42 within target cells, and thus modulate several physiological cellular events resulting in cell death. In fact, glucosylation leading to the depolymerization of the actin cytoskeleton, followed by disruption of tight cellular junctions and ultimately leading to apoptosis in colonic epithelial cells (Figure 4) [89].
Figure 4. Mechanism of action of Clostridium difficile toxins.

Toxins bind to receptors of target cells and are endocytosed. Hydrophobic regions of the protein allow insertion into the membrane in acidification of the toxins in endosomes and the N-terminal catalytic domain is translocated into the cytosol. Toxin is a glucosyltransferase that transfers a glucose moiety from the donor substrate UDP-glucose to a threonine residue (Thr-37 in RhoA) and make it inactive.
Two main mechanisms have been known for action of TcdA and TcdB:
They are glucosyltransferases that irreversibly inactivate small Rho GTPases, leading to disruption of cytoskeleton and tight junctions and subsequent cell rounding, detachment and cell death.
The toxins themselves or acting synergistically with other mediators, induce intestinal injury and inflammation via disruption of the intestinal epithelial barrier, induction of proinflammatory mediators and cytokines, causing cell apoptosis or necrosis in epithelial and immune cells and contributing to mucosal damage.
The role of Ca2+ in the mechanism of action of toxins
It has been assumed that metal ion binding is important for both intracellular and extracellular actions of the toxins, as metal ions are involved in carbohydrate or phospholipid proteins binding to their respective ligands [90–92]. Therefore, domains have been tested for their ability to coordinate to metal ions such as Ca2+ or Mg2+ and for the potential requirement of these metal ions for CWB-domain association with mammalian cells [79]. The results indicated that the recognition of mammalian cell surface of the C. difficile CWB domains is Ca2+ ion dependent. Although intracellular Ca2+ levels did not show any effect on toxin activity, extracellular calcium modulated protein (calmodulin) has been shown to have an effect on toxin internalization due to cytoskeletal changes [92,93].
Intracellular levels of Ca2+ were found to be rapidly elevated to a high steady state concentration in the presence of toxin B [94]. Ca2+ was associated with the CWB domains of toxin A and on the surface of Chinese hamster ovary (CHO) cells. Therefore, inhibition of toxin endocytosis leads to accumulation of toxin on the cell surface. It is also interesting to note that CWB domains of toxin A could also be detected on the surface of CHO cells under attenuated Ca2+ conditions by elevating the protein concentration. The effect of toxin A is highly dependent on a complex set of environmental factors, including extracellular Ca2+ and upstream Rho-inactivation. Collectively, Ca2+ plays a role as a toxin sensitizer. The choline moiety is completely surrounded by aromatic residues of the β-solenoid fold, and moreover choline binding has been reported to stabilize the structures of the CWB domains similar to the toxin A CWB-construct [79]. However, choline binding did not induce changes in the circular dichroism spectra of the toxin A and B CWB domains.
The role of pH in the mechanism of action of toxins
The autoprotease domain is known to undergo structural rearrangement in the presence of inositol hexakisphosphate (InsP6) which causes the release of the glucosyltransferase domain into the cell [66,95]. In eukaryotic cells, InsP6 binds to the domain adjacent to the monoglucosyltransferase domain and activates an intramolecular cleavage reaction [66,77]. Thus, a multistep mechanism of receptor-mediated endocytosis, membrane translocation, autoproteolytic processing and monoglucosylation are important to the action of TcdA and TcdB on mammalian target cells. A 3D structure of TcdA after autoprocessing and after exposure to acidic pH has been carried out in order to study the toxin's action at neutral pH. At neutral pH, glucosyltransferase domain is in contact with the binding domain and causes heterogeneity of cleaved TcdA by elimination of this domain.
Significant changes in the pincer-like head of the delivery domain that result in its extension away from the binding domain happen at low pH. This change may occur through the decoupling of the two lobes of the head, effectively opening the pincer. It has been suggested that at low pH, the conformational change which is required for translocation of the glucosyltransferase domain into the host cytosol will be significant [96]. In fact, at the low pH environment of the endosomal compartment, toxins undergo a conformational change that cause membrane insertion and channel formation [97–99]. InsP6 acts as a cofactor to trigger CPD-mediated autocatalytic cleavage of the toxins, and the subsequent release of the N-terminus GT domain into the cytosol [77].
Autocleavage mechanism
The intracellular vesicular H+-ATPase induces an increase in the acidification of early endosomes and a subsequent increase in hydrophobicity. Surface hydrophobicity enables the corresponding part of the toxin to insert into the membrane and the catalytic domain can translocate into the cytosol. This mechanism is called the ‘short trip model’ of bacterial exotoxin uptake [99–102]. Due to the separation of the first 543 amino acids, a short trip model may be accomplished by the intrinsic properties of the toxin itself. Triggering of autoproteolysis by dithiothreitol (DTT) and/or InsP6 have been identified by two independent studies:
Proteolytic activity by a putative aspartate protease domain located in the C-terminal part of the translocation domain.
Function of an intrinsic CPD located adjacent to the autocleavage site in the N-terminal part of the translocation domain [66,77]. The onset of autoproteolysis under the influence of DTT points toward regulation of the CPD. This is because of inhibition by N-ethylmaleimide (NEM, a common inhibitor of cysteine proteases).
InsP6 may also use a similar mechanism; proteolysis is faster compared with DTT. It is interesting to remark that InsP6 can have a synergistic effect on the proteolysis, in combination with low DTT concentrations. In summary, translocation domain of clostridial glucosylating toxins comprises a CPD which is responsible for the autocatalytic processing of the toxins. The proteolysis is activated by reducing conditions and/or InsP6 which is essential for cytotoxic activity. Finding an intrinsic proteolytic activity of the toxins is the major step of toxin mechanism. This autoproteolytic activity is induced by InsP6 and/or DTT which is responsible for the separation of the catalytic domain from the holotoxin [66,77].
Apoptosis-inducing mechanism
In many human cells, C. difficile toxins have also been shown to induce apoptosis. The role of cysteine-aspartic protease (caspase) activation in TcdA-induced cell death has been examined by using two cell-culture models: human colonic carcinoma and ovarian carcinoma cell lines. A novel mechanism has been demonstrated in which TcdA-induced cell death involves a mitochondrial-dependent, caspase- and death receptor-independent pathway [103]. In apoptosis induction whether it is initiated by death receptor activation or by a mitochondrial-dependent pathway, effector caspases especially caspase 3, 6 and 7 are normally activated. Activation of these caspases causes cleavage of cellular macromolecules and subsequent changes in morphology.
In the mitochondrial-dependent pathway, activation of effector caspases cause an increase in pro-apoptosis protein family members Bax and Bak, the formation of channels in the outer membrane of mitochondria, producing release of cytochrome c and formation of the apoptosome. However, anti-apoptotic protein family members, such as Bcl-2 and Bcl-XL oppose the insertion of Bax and Bak into the mitochondrial membrane by binding and neutralizing them [104–106]. There are several mechanisms of pathogen-induced cell death based on apoptosis. The bacteria may eliminate host immune cells that are primed to undergo apoptosis, thereby decreasing the immune response, and prevent the host inflammatory response. Reduction of anti-apoptotic proteins of the Bcl-2 family can regulate the activation of the mitochondria independently from the death-receptor signaling cascade and form caspase activation, leading to C. difficile TcdA-induced cell death in epithelial ovarian and colonic cancer cells [103].
In the death receptor pathway, activation of caspase 8 is produced by binding of death ligands such as tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) to the death receptor. Activated TRAIL receptor recruits the fas-associated death domain, resulting in death-inducing signaling complex, activation of procaspase 8, cleavage of downstream effector caspases and subsequently apoptosis [107–109]. It has been demonstrated that the death receptor pathway is not involved in TcdA-induced cell death. This is because TRAIL-induced cell death is completely inhibited by the caspase 8 inhibitor while the inhibitor did not block TcdA-induced cell death.
Hypervirulent C. difficile
About 10% of C. difficile strains (including the epidemic NAP1/027 strain) belong to variant toxinotypes III, IV, V, VII, IX and XIII and produce binary toxin (CDT) that causes the production of tcdB and/or tcdA (different from TcdA and TcdB produced by historical strains) [110,111]. This binary toxin that belongs to the family of clostridial iota-like toxins has two components: ADP ribosyltransferase encoded by the genes cdtA (enzymatic component) and cdtB (binding component). CDT induces the formation of microtubule-based cell protrusions and increases the adherence of bacteria to intestinal epithelium [41]. Toxin B from hypervirulent C. difficile strain (tcdB) exhibits broader tropism and cytotoxicity in vivo in comparison with toxin B (TcdB) from historical strains, undergoes hydrophobic conformational changes at a higher pH which cause more rapid cell entry and more severe illness than TcdB (from reference strains of this organism) [78]. Some hypotheses have been proposed to explain these differences:
Rapid cell entry of cdtB could lead to more efficient cell killing by providing an endocytic environment for the toxin rather than possible destruction of cells by lysosomal proteases.
The data from the lysosomotropic inhibitor assays support the idea that tcdB does not reside within the endosome as long as TcdB.
Different sensitivity to the levels of InsP6 that trigger autoproteolytic processing associated with translocation.
Difference in the sequence of the tcdB hydrophobic region that has been proposed to mediate membrane insertion.
These mechanisms allow tcdB to insert into the membrane at an earlier stage of cell entry. This is similar to the mechanism identified for the cell-binding component of the anthrax toxin which is called protective antigen (PA). Therefore, there should be a difference in the pH-induced transition of these two forms of the toxin with the hydrophobic regions of tcdB, which becomes exposed at a pH higher (less acidic) than the pH that was necessary for triggering this transition in TcdB. In fact, the transition of TcdB occurs gradually, while tcdB demonstrates a sudden shift upon lowering the pH. The tcdB is able to translocate at an earlier point in endocytosis and this contributes, at least in part, to a more efficient toxin action. The expanded tropism, along with more efficient cell entry could combine to enhance the in vivo toxicity of tcdB. This toxin targets a broader array of cells in vivo, and patients infected with this strain are reported to have lower clinical cure rates and higher rates of CDI recurrence than patients infected with TcdB.
C. difficile spore formation
As the environment of the large bowel is anaerobic, the only way oxygen-sensitive vegetative cells of C. difficile could survive out of this environment would be in the spore form [112]. As Figure 5 shows, spores are coated with a peptidoglycan cortex and several layers of protein which allow them to survive in many harsh environmental conditions and for a long time in the presence of air [113].
Figure 5. Clostridium difficile spore structure.

Exosporium, coat, cortex, membrane, ribosomes and core have been indicated.
Spores are metabolically dormant after release from the mother cell, and they can be excreted by the host to survive for a long time in the outside until being reconsumed by another host where they can germinate and grow out as vegetative cells and produce toxins. Spores from C. difficile-infected patients excreted into the environment can survive against commonly employed disinfectants and can lead to an increase in the percentage of patients colonized by C. difficile. Figure 6 shows how spore layers are formed.
Figure 6. Clostridium difficile spore formation process.

Spores that form in response to nutrient limitation are composed of three major components: the coat, the cortex and the core. The coat layer protects the spore against environment insults. DNA is in the core and protected by bound with some protein.
There are two common reasons leading to development of CDAD: disruption of the microflora in the host intestine by antibiotic treatment regimens and ingestion of C. difficile spores [114,115]. The M68 strain for instance is a representative ribotype of C. difficile which can cause human disease [116]. By studying a murine model which was a carrier of M68 it has been found that antibiotic treatment could trigger sporulation, followed by spore excretion causing host-to-host transmission of C. difficile.
The stable intestinal microbiota community which can protect the host by preventing colonization by pathogens can be disrupted by antibiotics; these also lead C. difficile to sporulate and become infectious to other patients [117,118]. Intestinal damage and inflammation in naturally infected immunocompetent mice that have received antibiotics can be resolved after stopping antibiotic use and allowing the recovery of intestinal microbiota. The same results are expected for infection by C. difficile in humans [119].
Mechanism of spore germination
It is believed that the existence of normal microflora allows ingested C. difficile spores to remain quiescent in the absence of antibiotics [120,121]. However, antibiotic therapy causes transmission of the spores between infected mice [122]. Antimicrobial treatment causes disruption of gut microflora leading to germination of C. difficile in the intestines, outgrowth of vegetative cells, multiplying and filling the niches in the gut, and eventually producing toxins [123]. The germination process from the phase bright spore to a phase dark vegetative cell comprises the following steps: interaction of germinant to germination receptor; release of Ca2+-DPA (dipicolinic acid) by some enzymatic reactions; core rehydration; cortex degradation and the outgrowth of a vegetative cell [124,125].
Bile is a substance which helps in digestion by emulsifying fat and cholesterol and allowing their absorption in the small intestine, and is produced in the liver and stored in the gall bladder. Normal bile consists mainly of a mixture of cholate and chenodeoxycholate conjugated with taurine or glycine which can be recovered from the distal ileum to the liver Figure 7.
Figure 7. Chemical structure of initially bile acid components.

The bile acids consist of 30–40% of cholic acid, 30–40% of chenodeoxycholic acid, 20–25% of deoxycholic acid and 1–2% of lithocholic acid.
The colonization of C. difficile in spore-fed mice (both antibiotic treated and un-treated) has been examined in cecal and intestinal segments with regard to cholate metabolism. Results showed that the cholate was conjugated with glycine in the bile and formed glycocholate. This conjugated compound would then be deconjugated by normal flora during the passage through the small bowel to glycine and cholate component that are suitable germinants to cause germination and outgrowth of C. difficile spores [126].
In a healthy human when spores are ingested they are passed through the duodenum, to reach the jejeunum where they germinate because of the high concentration of the bile. The germinated spores pass through the ileum and reach the aerobic environment of the cecum. Here, metabolization of cholate derivatives (primary bile salt) happens by normal microflora to convert it to deoxycholate (secondary bile salt) which prevents vegetative growth of spores. A little germination caused by deoxycholate is of no consequence in the aerobic environment of the cecum [127–129].
But when antibiotics have been employed the normal microbial flora in the cecum is perturbed causing a relative increase in the concentration of primary bile salts (more cholate and less deoxycholate) and more germination of spores leading to colonization. In summary, normal microbial flora play an inhibitory role in C. difficile colonization by metabolizing cholate to deoxycholate. This mechanism could be an idea for inhibition of CDI, for example, by adding some deoxycholate to antibiotic regimens of infected patients.
Bile salts as spore germinators & inhibitors
Bile salts which have been reported to act as in vivo germination factors during the CDI treatments are divided in two groups regarding their physical properties: the primary bile salts like cholate and taurocholate which cause the stimulation of germination and the secondary bile salts like deoxycholate and chenodeoxycholate that inhibit the germination and growth of vegetative cells.
Taurocholate acts as a germinant for C. difficile spores and enhances colony formation with lysozyme and thioglycholate [130–132]. Studies showed that taurocholate and glycine are enough for germination of C. difficile spores since they act as cogerminants. Although taurocholate is capable of germinating the C. difficile spores rapidly, other cholates like glycocholate and deoxycholate just induce colony formation.
There are two mechanisms for germination of C. difficile by taurocholate. The first one is by triggering germination without permeabilization of the spore coat, and the second mechanism involves taurocholate being recognized by some receptors which are activated by small molecules [133,134]. A kinetic method was used to find which of the two mechanisms (germination receptor or spore membrane disruption) was produced by taurocholate in C. difficile strains 630 and VPI 10463 [135]. The kinetic results revealed that taurocholate germination was consistent with a germination receptor [135].
Glycine as an amino acid has no spore membrane disruption effect on its own, but in the presence of taurocholate acts as co-germinator acting synergistically such that one compound binding increases the affinity of another compound binding in a separate binding site [135]. Studies found that in the presence of high concentrations of glycine the germination speed was increased while there was no change in the germination rate when varying the taurocholate concentration [135]. In the lumen, the taurocholate binds to spores and later glycine could bind to this pair to make a taurocholate/glycine-bound spore better able to carry out the germination process [135].
The high concentration of chenodeoxycholate in the healthy human cecum indicates that this type of bile salt can act as inhibitor against C. difficile germination which is produced by deconjugation of two inactive bile salts, glycochenodeoxycholate and taurochenodeoxycholate. The dormancy of C. difficile spores in the human lower intestine does not depend on the concentration of glycine [135]. By increasing the concentration of taurocholate, the concentration of inhibitory chenodeoxycholate will decrease. As there are limitations for solubility of chenodeoxycholate in water, it cannot induce germination of C. difficile spore.
The only difference between cholate and chenodeoxycholate is in the 12α-hydroxy group [136,137]. In the presence of flora, taurocholate could be converted to deoxycholate with 7α-dehydroxylation and/or chenodeoxycholate with 12α-dehydroxylation both of which compounds can inhibit spore germination [138]. Furthermore, a human study confirmed this when in the stool of antibiotic-treated patients the level of primary bile salt was increased and the amount of 7α-dehydroxylation decreased, while in healthy human stool samples the amount of chenodeoxycholate was sufficient to cause inhibition of germination [120,139].
Treatment
Antibiotics
Metronidazole is used as the standard therapy for treatment of moderate CDI as it is less expensive and has the same efficacy as vancomycin. However, for pregnant women and children metronidazole is considered inadvisable. For patients with severe or complicated CDI, oral vancomycin is recommended with the dose of 4 × 125 mg/day for 10–15 days duration. Although vancomycin is US FDA approved, it is not in the first-line CDI therapy because of its price and also concern of promotion of resistance like vancomycin-resistant enterococci (VRE).
Both vancomycin and metronidazole can reduce the number of viable C. difficile cells but are unable to inhibit toxin and spore formation since vancomycin causes inhibition of cell wall synthesis and metronidazole causes DNA damage mediated by a radical pathway. An increase in the number of clinical therapeutic failures, and the recurrences rate of CDI after both vancomycin and metronidazole treatments, emphasis are on the need of new antibiotic options.
The macrocyclic and FDA-approved antibiotic, fidaxomicin, showed higher activity than vancomycin against C. difficile as well as reduced recurrences up to 45%. In addition, fidaxomicin is a more selective therapy for CDI, producing a higher fecal concentration and having less effect on resident microflora in comparison with vancomycin. Nevertheless, with the price being 120-fold more expensive than metronidazole and about three-times higher than oral vancomycin, these differences cause metronidazole to still be considered the first-line therapy for CDI [140].
Tigecycline, a derivative of minocycline, is another FDA-approved antibiotic that has been shown to have activity against severe CDIs. Similar to metronidazole, tigecycline is used intravenously which is favorable in comparison with oral administration of vancomycin, and studies indicated that tigecycline is more active than metronidazole and produces higher fecal concentrations. Case reports have suggested use of tigecycline would be a feasi ble alternative to other antibiotics for treatment of severe CDI. However, clinical failure in a patient with severe CDI treated with tigecycline for 2 weeks duration has been reported [141].
Rifaximin also can be used for treatment of GI infection because of its excellent safety profile. The mechanism of action of rifaximin is similar to fidaxomicin but it has higher fecal concentration and minimal alteration of microflora. Bacterial resistance and being more costly in comparison with clinically used antibiotics, however, could be a limitation of rifaximin used for CDI treatment [142].
Ramoplanin, rifalazil and nitazoxanide are other antibiotics with broad-spectrum antimicrobial activity against Gram-positive and Gram-negative bacteria. Although ramoplanin showed similar activity in comparison with vancomycin for CDI treatment, there was a suppression of spore formation. In Phase I and Phase II clinical trials, ramoplanin was found to control CDAD as well as vancomycin [143,144]. The most attractive property of rifalazil is its in vivo long half-life that makes it more effective against CDI. Nitazoxanide showed similar activity in comparison with metronidazole and vancomycin but the higher cost was the limitation for this antibiotic to be a first-line therapy for CDI. It is interesting to note that during antibiotic treatment with all of the above antibiotics, only the total of C. difficile vegetative cell counts reduced and there was no change in the number of spores [145,146].
Probiotics
The density of microorganisms living in the GI tract is the highest known. In humans, the gut is the residence of up to 10(14) bacteria belonging to hundreds of species [147]. Sterile at birth, the intestine is colonized immediately after delivery by a few pioneer species. Initial colonization by facultative anaerobes lowers the oxygen tension of the intestinal lumen, and allows subsequent colonization by anaerobic species. The complexity of the flora increases with age and the change of diet during the first year of life. Anaerobic bacteria belonging to the genera Bacteroides, Bifidobacterium, Eubacterium, Clostridium, Peptococcus, Peptostreptococcus and Ruminococcus are predominant, whereas aerobes (Escherichia, Enterobacter, Enterococcus, Klebsiella, Lactobacillus and Proteus) are less abundant. The composition of the gut microflora is difficult to permanently change because a system of immune tolerance to resident microbes is established that prevents new colonization taking place. When probiotic preparations are taken orally, the composition of the gut bacteria is only changed temporarily and reverts back to steady state when the probiotics are stopped [148].
Nevertheless, probiotics have been used as therapeutics for antibiotic-mediated CDI and a Cochrane systematic review [149] concluded “moderate quality evidence suggests that probiotics are both safe and effective for preventing C. difficile-associated diarrhea”. Bacteria such as Bifidobacterium and Lactobacillus spp. can inhibit the adhesion of C. difficile toxins to the host colon epithelial cells, and can act as stimulators of the host immune response. In addition, bacteria like Lactobacillus can have a direct antimicrobial activity by secretion of bacteriocins and other antimicrobial peptides [150]. Investigations by McFarland showed there were 26% fewer patients with vancomycin-associated CDI in comparison with placebo group [151]. But there was no distinction between CDI patients and C. difficile colonized patients in this study. In another study, patients who received high doses of vancomycin plus probiotics showed significantly lower CDI recurrence rate in comparison with subjects receiving high doses of vancomycin plus placebo. They postulated that probiotics could prevent C. difficile pathogenesis due to toxin neutralization. Only a few studies so far have been reported concerning the use of probiotics for antibiotic-associated CDI, and therefore well-designed clinical trials are needed for further investigations [152].
Fecal transplantation
Fecal transplantation can be considered the ‘hot topic’ in CDI therapy in the 21st century although it was actually first described in 1958 [153]. Considering that most CDI infections are due to depletion of healthy intestinal microflora after antibiotic use, it became logical to attempt to replace these lost ‘good bugs’ with bacteria obtained from healthy individuals. In fecal bacteriotherapy as a matter of fact, stool from healthy people was transferred to the large intestine of C. difficile-infected patients via enemas, hence that is called stool or fecal transplantation. A landmark paper in New England Journal of Medicine [154] reported 80% success rate for treatment of recurrent CDI by duodenal infusion of donor feces in comparison with vancomycin therapy.
This novel method which is effective for recurrent CDI patients who were already treated with oral vancomycin is also named fecal microbiome therapy (FMT). FMT can be carried out via the lower proximal or upper entrances to the GI tract. The stool delivery may be carried out with colonoscopy, enema or rectal tube for introduction into the lower GI tract, and via nasogastric tube or endoscopy/gastroscopy for upper GI tract passage into the duodenum [155]. Although the screened stool that was delivered via the upper GI tract showed a better treatment success (92%) in comparison with enema (79%), the delivery route and equipment can be selected based on the clinician's evaluations, apparatus availability and the patient's circumstances and preferences.
The mechanism of action of stool transplantation has not yet been completely understood, however, studies have proposed the replacement of healthy microbiome by the donor material in the gut of CDI patients which had been previously depleted with antibiotics. Up to now, FMT has been shown to be more effective for recurrent CDI patients [156]. Accordingly, FMT should be safer overall as it avoids use of further antibiotics leading to lower occurrence of allergies and adverse reactions that might be caused in antibiotic-treated patients. In addition, this therapy seems highly effective and will probably be inexpensive in comparison with newly developed antibiotics.
Although fecal transplantation looks promising for treatment of gut infections by C. difficile [157] and there have been predictions that it will become the treatment of choice [158], there are some challenges that need to be addressed in further studies. One challenge is the donor's fecal sample has to be screened before transplantation and there is always the risk of new infections with undetected pathogenic bacteria in the stool of the donor patient. While the screening procedure is time consuming, on the other hand the stool needs to be used as fresh during the transplantation procedure [159]. There is also no investigation and not even a suggestion to use FMT against C. difficile spores.
Antibodies
Differences in the restriction patterns of C. difficile strains have been observed by toxinotyping methods that would be valuable for antibody treatments. For instance, a 13% difference in toxinotype 0 and III in C-terminal region of toxin B is important for antibody binding [160,161]. The theoretical justification for antibody treatment of CDAD is based on this observation that a person with only a mild case of CDAD possesses high levels of anti-toxin A IgG serum titers, while patients with severe case of CDAD have shown low levels of anti-TcdA Ig titers, specifically IgM, IgG2 and IgG3 isotypes [161–163].
Theoretically, there are two mechanisms that explain the neutralizing of TcdA and TcdB by anti-toxin antibodies which have been discovered by measuring the increased level of IgG in the stool [161]. First, anti-toxin antibodies migrate to the GI tract through a leaky mucosal barrier which allows easy access of antibodies to the lumen [164,165]. In the second mechanism, the IgGs may be actively transported to the lumen via the neonatal IgG Fc receptor (FcRn) [165,166].
An effective antibody is one that interacts with an epitope on the outer part of a toxin molecule [301]. Protein epitopes are recognized by antibodies and although large proteins have a huge number of sequential amino acid possibilities, just a few epitopes of a protein are able to generate antibodies [301]. C. difficile has six surface proteins, SlpA (surface protein which often refer to P36 and P47), FliC (the main flagellum component), FilD (the flagellum tip or cap protein), Cwp84 and Cwp66 (two cell wall proteins) which just one of each SlpA (P36 and P47) are suitable for generation of antibodies [301].
Polyclonal antibodies
A composition of polyclonal ovine antibody has been suggested for treatment of CDI by binding to toxin and neutralizing its biological effects while having a low immunogenic effect on the patient [160]. This antibody can interact with an epitope of each N-terminal (1-957), mid-region (958-1831) or C-terminal domain (1832-2710) [160]. The raising of an ovine antibody in a sheep involves some steps including: administering an immunogen comprising a C. difficile toxin; allowing the generation of antibody; obtaining sufficient antibody from the sheep serum.
The immunogen preparation could be obtained either from a chemical treatment or via a recombinant method. In the first method, the immunogen is derived from treatment of toxin by some chemical like formaldehyde, glutaraldehyde or peroxides. An antibody which is obtained from recombinant method can selectively inactivate the active site of toxin by mutation or deletion like modification of aspartate-any residue-aspartate (DXD) motif in N-terminal domain [160]. The interval and dosage range of the immunogen depends on parameters such as nature of immunogen, the nature of formulation, the route of administration and the judgment of the attending person.
Differential centrifugation, polyethylene glycol precipitation of protein and salt fraction with ammonium sulfate are examples of some methods for preparation of antibody from egg yolk by separating proteinaceous material from natural sources. The reasonable pure antibodies can be obtained from egg yolk by known procedure like gel filtration, ion-exchange chromatography, isoelectric focusing and affinity chromatography for isolation of Igs [302]. Thus, first the whole egg yolk is converted to powder by spray-drying or freeze-drying then the lipids are removed [302]. Separation of lipids by solvent extraction including consequence sedimentation of lipids with water at temperature between about 0 and 6°C then sanitization by filtration and finally lyophilization. The lipid separation process could be monitored by HPLC, UV absorption, chemical colorimetric assay and/or ELISA.
Monoclonal antibodies
It has been suggested that the symptoms of CDAD-infected patients could be treated by active and passive immunization by infusion of intravenous immune globulin containing antitoxin A and B [167–169]. It is thought that this protection against CDAD is because of elevated concentration of serum anti-toxin A Ig [170]. From two points active and passive immunization against C. difficile toxins are important: preventing initial episodes and reducing risk of recurrent [171,172]. Investigations by Babcock et al. indicated that antibodies may block the C-terminal cell-receptor binding domain region of TcdA, or prevent the internalization of the toxin [173].
Humanized monoclonal antibodies (HuMAbs) were characterized against both toxin A (HuMAb CDA1) and toxin B (MDX-1388). The safety and pharmacokinetic studies of HuMAb CDA1 revealed that this antibody is safe and well tolerated and can use in dose between 0.3 and 20 mg/kg [174]. HuMAb CDA1 not only could protect hamsters from mortality when used alone, but also significantly more protection was observed when a combination therapy was administered [175].
Some symptomless carriers have been reported that had an increased efficacy of their serum IgG concentrations against toxin A [162]. It has been found that concentration of IgG in patients who had recurrent C. difficile was higher than patients with non-recurrent diarrhea [176]. The host immune response against C. difficile has an essential role in determining the clinical outcome of infection [162]. Intravenous immunization with IgG against C. difficile has shown success in treating children infected with recurrent C. difficile colitis [167]. By utilizing ELISA to measure the concentration levels of IgA, IgG and IgM, it was found that patients who only had a single bout of CDI and recovered had higher serum antibody responses than those who developed recurrent infection and those who died [163].
Active or passive immunization by IgA should be protective against toxins exerting cytotoxic effects [177]. Passive immunizations have been shown to protect axenic mice (mice with only one species of gut bacteria) from pseudomembranous colitis by mAb against toxin A. Although an inadequate antibody cannot completely protect against severe disease, passive immunotherapy with human Ig can benefit infected patients even in severe cases [167–169,175]. Immunized cattle providing IgG and immunized hens providing IgY antibodies have both shown the ability to inhibit TcdA-induced cytotoxicity [178]. However, administration of IgY against recombinant TcdA and TcdB caused the complete inhibition of CDAD relapse meaning that neutralization of TcdB is also important in treatment of CDAD [179].
Recombinant antibody (rAb) fragments can be designed with greater efficacy by binding the epitopes that are not accessible with conventional antibodies. One study reported the binding of a rAb with TcdB in single-chain variable fragment (scFv) format. Single domain antibodies (sdAbs), fragment antigen binding (Fab) and scFv are some common rAb techniques [180,181]. High tissue penetration properties, high chemical and thermal stability and amenability to in vitro production are some properties of sdAbs [182–185]. The best feature of this rAb is its small size which consists of a heavy chain variable (VH) domain and light chain variable (VL) domain of Igs that makes it able to access immuno-silent cavities in enzyme, receptor and infectious agents [186–189]. These properties make the sdAbs a promising C. difficile toxin-neutralizing agent that may show a greater efficacy in the GI tract [190].
In summary, both traditional antibodies like IgY, IgG, and also recombinant antibodies like Fab, scFv have been used for CDAD immunotherapy. Single-domain rAb, VHH, have shown a potential activity for binding to toxins.
Photodynamic therapy
Photodynamic therapy (PDT) is a promising alternative methodology for inactivating both cancer and bacterial cells [191,192]. The antimicrobial PDT (aPDT) approach is based on the PDT concept, which involves the administration of a photoactive dye called a photosensitizer (PS) targeting microbial cells, that is able to produce reactive oxygen species (ROS), consisting of singlet oxygen and/or free radicals, upon irradiation with light to eradicate bacteria as well as fungi, viruses and protozoa [174,193,194].
Two oxidative mechanisms of photoinactivation (PI) are implicated in the inactivation of the target cells. The type I pathway involves electron/hydrogen atom-transfer reactions from the PS excited state, with the participation of a substrate to produce radical ions, while the type II pathway involves energy transfer from the long-lived triplet state to molecular oxygen to produce singlet oxygen (1O2). Both processes lead to highly toxic ROS such as 1O2 and free radicals which can irreversibly alter vital components of cells resulting in oxidative and lethal damage [195–197].
Non-thermal red light with a specific wavelength is produced by the desired light source which is chosen based on its power density, penetration and wavelength. Power density determines the time needed for delivery of the desired dose, penetration depends on the depth of the infected target tissue and wavelength is determined by the absorption spectra of the employed photosensitizer [198].
Several PSs have been examined for their potential use in aPDT including; for example, rose bengal and the phenothiazinium salts methylene blue and toluidine blue Figure 8 [196,199–201].
Figure 8.

Chemical structures of some representative photosensitizers that have been investigated for use in antimicrobial PDT.
It is known that cationic photosensitizers are highly effective against pathogenic drug-resistant bacteria such as methicillin-resistant Staphylococcus aureus and vancomycin-resistant Enterococcus faecalis [202,203]. Molecules with a permanent positive charge such as phenothiazinium salts are able to effectively target both Gram-positive and Gram-negative bacteria. The outer-membrane permeability barrier typical of Gram-negative bacteria is disrupted by cationic molecules, while Gram-positive species have negatively charged but permeable cell walls [204]. Because of the structural differences in the cell walls, Gram-positive bacteria like C. difficile are generally more susceptible to PDT as compared with Gram-negative species [205].
Wainwright et al., however, discussed a couple of challenges that will need to be overcome before PDT treatment for CDIs can be carried out in the colon [206]. Lack of oxygen or low levels of oxygen partial pressure in the cecum and nearby sections of the gut is the first highlighted problem. According to the accepted mechanism of PDT, oxygen is necessary for the type II pathway to produce singlet oxygen, but to what extent oxygen is required for the type I pathway is still uncertain. Methylene blue, for instance, produces singlet oxygen and free radicals through both type I and type II pathways. In the anaerobic environment of the colon, could free radical formation by photoexcited methylene blue be sufficient for eradication of CDI?
Another serious problem is the oral delivery of PS to the large intestine. Cassidy et al. [207] reported a new strategy for delivery of PS to the colon which was called a ‘smart vehicle’. According to this strategy, the PS (MB or a cationic porphyrin) was encapsulated in a Eudragit® 25-based drug delivery system, via hot melt extrusion. This material was stable in the acidic environment of the stomach but the PS was released in the higher pH environment of the colon. There was also addition of tetrachlorodecaoxide (TCDO) that released oxygen in the colon. Although this strategy looks promising in vitro, it is far from being clinically used and further in vivo examination in animal models would be needed [207].
In another novel method being developed by Rineh et al., a Nile blue derivative with a benzophenothiazinium dye structure known as EtNBS, was used to produce free radicals upon photoexcitation via type I mechanism Figure 9. EtNBS has previously been studied for aPDT [208] and anti-cancer PDT at low oxygen concentrations [209]. The preliminary in vitro results indicated that EtNBS was able to eradicate the C. difficile bacteria in an anaerobic chamber in the absence of oxygen while methylene blue was only active in the presence of oxygen Figure 10.
Figure 9.
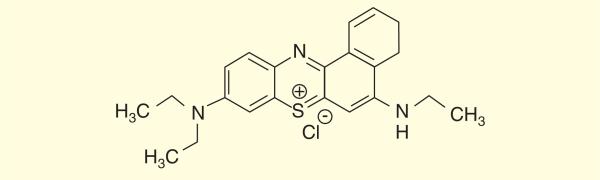
Chemical structure of a small cationic, methylene blue-like molecule EtNBS (5-ethylamino-9-diethyl-amino-benzo[a]phenothiazinium chloride).
Figure 10. EtNBS-PDT in vitro results.

EtNBS was utilized as photosensitizer in PDT against Clostridium difficile and compares with methylene blue. The experiment was carried out in anaerobic chamber. The result revealed that the C. difficile bacteria population was reduced in zero oxygen by 6 log scales value when EtNBS was administrated as photosensitizer.
Utilizing EtNBS as a PS in the colon may allow oxygen-independent PDT to destroy the C. difficile bacteria without damaging the host intestine. The ongoing in vivo investigations demonstrated the PS could be delivered via the anus to its target site in the large intestine area. A diffusing optical fiber was introduced for illumination of EtNBS with red light in the infected area. Preliminary results testing this method revealed that treatment of mice with CDI with EtNBS-PDT did not cause the mice to lose any body weight during the 2-week treatment in comparison with vancomycin-treated mice (unpublished data).
Conclusion & perspective for novel non-antibiotic drug development
Although vancomycin and metronidazole have been shown to have inhibitory effects on AAD, but as they cause perturbation of microbial flora they may not be ideal drugs. The recent emergence of antibiotic-resistant C. difficile strains and hypervirulent C. difficile that cause increased morbidity and mortality has required the development of novel non-antibiotic-based treatment regimens.
The C-terminal region of TcdA and TcdB causes these toxins bind to the surface of epithelial cells with interactions with putative cell-surface carbohydrate receptors. Structural studies of this cell receptor-binding domain from TcdA and TcdB revealed a β-solenoid fold. Protein–protein or protein–carbohydrate interactions have been suggested to play a role in solenoid-like structure of TcdA.
TcdA and TcdB use different types of receptors; the receptor for TcdB appears to be in basolateral cell surface, while in TcdA the receptor is on the apical cell surface. Based on these data, several novel approaches for treating CDIs have been proposed to competitive inhibition of toxin binding to host epithelial cells. In one study, a pentasaccharide containing the LeA-LacNAc and a related tetrasaccharide have been synthesized to bind to TcdA by applying orthogonally protected donors and acceptors and armed-disarmed principle [210]. As the pathogenicity of C. difficile depends on its glucosylating protein toxins and its severity seems to correlate with the amount of toxin, development of glycan-mimicking compounds should be one of the potential approaches to block the receptor-binding domain [211,212].
Polyclonal antitoxin A serum dose not directly or indirectly cross-react with toxin B but it does react with glucan-binding protein (GBP). Polyclonal anti-GBP sera, however, cross-react with toxin A because maybe the toxin A repeats are more similar to the GBP repeats than to the toxin B repeats. Passive immunotherapy induced by polyclonal and monoclonal antibodies have been shown to neutralize C. difficile toxins. Recent studies have investigated using rAb fragments especially single-domain antibody fragments (VHHs) are more effective for neutralizing C. difficile toxins and CDAD. Research indicated that C. difficile remains in the gut in the form of spores and then can be germinated due to the lack of normal microbial flora after antibiotic treatment [146,213]. Germination of the spores and how this germination is controlled in the intestinal environment are two fundamental aspects of C. difficile infections. Germination will happen when one or more small molecules bind to the spore and stimulate it with the help of primary bile salts like cholate, taurocholate and glycocholate.
The identification of effectors that can cause C. difficile spore germination gives a clue to how inhibition of germination may be achieved. A general principle of action of cholate as a germinant for C. difficile spores is the importance of the 12α-hydroxy group which chenodeoxycholate lacks in this position. Then the 12α position can help researchers identify inhibitors of C. difficile germination. So one idea for inhibition of C. difficile spores would be introducing deoxycholate in the patient's normal diet during antibiotic treatment or using a different bacterial strain to convert the primary cholate to a secondary derivative by reducing the 7α-hydroxyl group.
Expert commentary
CDI is rapidly becoming a major health problem. Widespread use of antibiotics in hospitalized patients leads to depletion of normal gut microflora and hardy C. difficile spores are lying in wait to colonize or infect these patients as disinfectants fail to destroy them. C. difficile binary toxins are more virulent than the normal toxins and can cause severe GI infections. Although investigations into toxin structures and their mechanism of actions may lead to development of some promising monoclonal antibodies, another major challenge is C. difficile produce spores that can be germinated when the gut conditions become favorable. Antibiotic therapy is not the best strategy for controlling CDI because of failure and recurrence, high costs, development of resistance and depletion of gut microflora. In these difficult cases, non-antibiotic treatments such as antibodies, administration of appropriate bile acids, probiotics, fecal transplants and PDT could be promising. There are only a few studies on the use aPDT for eradication of infections by anaerobic bacteria. Recent studies show that PDT mediated by Nile blue derivative EtNBS, could produce ROS in low oxygen environments and may eradicate C. difficile both in vitro and in vivo.
Five-year view
New macrocyclic antibiotics, fidaxomicin and tigecycline were approved or are on course to gain regulatory approval, but although these antibiotics show considerably better activity than vancomycin or metronidazole against CDI, it is still likely that antibiotic therapy of C. difficile infection is not the best choice due to the inevitable disruption of the normal microflora. Therefore, other non-antibiotic strategies should be investigated. Probiotics and the newly introduced fecal transplant procedure are likely to grow in effectiveness as they become better understood. Advances in antibody production technology mean that a greater range of antibody-based therapeutics will be studied. Molecular drug discovery efforts may provide pharmaceuticals that can neutralize toxins and inhibit spore germination. PDT may be used for eradication of anaerobic C. difficile infections if PS that follow the type I pathway can be developed.
Key issues.
The numbers of deaths and the costs associated with Gram-positive Clostridium difficile infections (CDIs) are increasing.
Metronidazole and vancomycin are the first-line therapy for CDIs but treatment failures and recurrences are increasing.
Although new macrocyclic antibiotics are under investigation for CDI therapy, the disruption of microflora is a major issue after antibiotic treatments.
Monoclonal antibodies have been shown to be an effective non-antibiotic approach against CDIs.
Vegetative cells produce spores that can stay for a long time in hospital environments and germination in patients is controlled by bile salts.
Fecal transplant is a new alternative therapy that may gain traction in the near future.
In the future, photodynamic therapy may be effective in the anaerobic environment of the colon since photosensitizer such as EtNBS may act in an oxygen-independent manner.
Acknowledgments
Research in the Hamblin laboratory is supported by US NIH grant R01AI050875.
Footnotes
Financial & competing interests disclosure
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as
• of interest
•• of considerable interest
- 1.Bartlett JG. Narrative review: the new epidemic of Clostridium difficile-associated enteric disease. Ann. Intern. Med. 2006;145:758–764. doi: 10.7326/0003-4819-145-10-200611210-00008. [DOI] [PubMed] [Google Scholar]
- 2.Brazier JS. Clostridium difficile: from obscurity to superbug. Br. J. Biomed. Sci. 2008;65:39–44. doi: 10.1080/09674845.2008.11732796. [DOI] [PubMed] [Google Scholar]
- 3.Ozaki E, Kato H, Kita H, et al. Clostridium difficile colonization in healthy adults: transient colonization and correlation with enterococcal colonization. J. Med. Microbiol. 2004;53:167–172. doi: 10.1099/jmm.0.05376-0. [DOI] [PubMed] [Google Scholar]
- 4•.Hurley BW, Nguyen CC. The spectrum of pseudomembranous enterocolitis and antibiotic-associated diarrhea. Arch. Intern. Med. 2002;162:2177–2184. doi: 10.1001/archinte.162.19.2177. [Covers the types of disease associated with Clostridium difficile infection (CDI).] [DOI] [PubMed] [Google Scholar]
- 5.Bartlett JG. Antibiotic-associated diarrhea. Clin. Infect. Dis. 1992;15:573–581. doi: 10.1093/clind/15.4.573. [DOI] [PubMed] [Google Scholar]
- 6.Bean NH, Griffin PM, Goulding JS, Ivey CB. Foodborne disease outbreaks, 5-year summary, 1983–1987. MMWR CDC Surveill. Summ. 1990;39:15–57. [PubMed] [Google Scholar]
- 7.Johnson S, Clabots CR, Linn FV, Olson MM, Peterson LR, Gerding DN. Nosocomial Clostridium difficile colonisation and disease. Lancet. 1990;336:97–100. doi: 10.1016/0140-6736(90)91605-a. [DOI] [PubMed] [Google Scholar]
- 8.Mcfarland LV, Mulligan ME, Kwok RY, Stamm WE. Nosocomial acquisition of Clostridium difficile infection. N. Engl. J. Med. 1989;320:204–210. doi: 10.1056/NEJM198901263200402. [DOI] [PubMed] [Google Scholar]
- 9.Adams SD, Mercer DW. Fulminant Clostridium difficile colitis. Curr. Opin. Crit. Care. 2007;13:450–455. doi: 10.1097/MCC.0b013e3282638879. [DOI] [PubMed] [Google Scholar]
- 10.Bartlett JG, Chang TW, Gurwith M, Gorbach SL, Onderdonk AB. Antibiotic-associated pseudomembranous colitis due to toxin-producing clostridia. N. Engl. J. Med. 1978;298:531–534. doi: 10.1056/NEJM197803092981003. [DOI] [PubMed] [Google Scholar]
- 11••.Mcdonald LC, Killgore GE, Thompson A, et al. An epidemic, toxin gene-variant strain of Clostridium difficile. N. Engl. J. Med. 2005;353:2433–2441. doi: 10.1056/NEJMoa051590. [Description of the recently discovered epidemic toxin gene-variant strain of C. difficile isolated from patients in the USA.] [DOI] [PubMed] [Google Scholar]
- 12.Warny M, Pepin J, Fang A, et al. Toxin production by an emerging strain of Clostridium difficile associated with outbreaks of severe disease in North America and Europe. Lancet. 2005;366:1079–1084. doi: 10.1016/S0140-6736(05)67420-X. [DOI] [PubMed] [Google Scholar]
- 13.Sullivan NM, Pellett S, Wilkins TD. Purification and characterization of toxins A and B of Clostridium difficile. Infect. Immun. 1982;35:1032–1040. doi: 10.1128/iai.35.3.1032-1040.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rifkin GD, Fekety FR, Silva J., Jr Antibiotic-induced colitis implication of a toxin neutralised by Clostridium sordellii antitoxin. Lancet. 1977;2:1103–1106. doi: 10.1016/s0140-6736(77)90547-5. [DOI] [PubMed] [Google Scholar]
- 15.Bartlett JG, Onderdonk AB, Cisneros RL, Kasper DL. Clindamycin-associated colitis due to a toxin-producing species of Clostridium in hamsters. J. Infect. Dis. 1977;136:701–705. doi: 10.1093/infdis/136.5.701. [DOI] [PubMed] [Google Scholar]
- 16.Voth DE, Ballard JD. Clostridium difficile toxins: mechanism of action and role in disease. Clin. Microbiol. Rev. 2005;18:247–263. doi: 10.1128/CMR.18.2.247-263.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17•.Von E-SC, Boquet P, Sauerborn M, Thelestam M. Large clostridial cytotoxins–a family of glycosyltransferases modifying small GTP-binding proteins. Trends Microbiol. 1996;4:375–382. doi: 10.1016/0966-842X(96)10061-5. [Review covering the biochemistry and cell biology of C. difficile toxins.] [DOI] [PubMed] [Google Scholar]
- 18.Dove CH, Wang SZ, Price SB, et al. Molecular characterization of the Clostridium difficile toxin A gene. Infect. Immun. 1990;58:480–488. doi: 10.1128/iai.58.2.480-488.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Von E-SC, Laufenberg-Feldmann R, Sartingen S, Schulze J, Sauerborn M. Comparative sequence analysis of the Clostridium difficile toxins A and B. Mol. Gen. Genet. 1992;233:260–268. doi: 10.1007/BF00587587. [DOI] [PubMed] [Google Scholar]
- 20.Triadafilopoulos G, Pothoulakis C, O'brien MJ, Lamont JT. Differential effects of Clostridium difficile toxins A and B on rabbit ileum. Gastroenterology. 1987;93:273–279. doi: 10.1016/0016-5085(87)91014-6. [DOI] [PubMed] [Google Scholar]
- 21.Jump RLP, Pultz MJ, Donskey CJ. Vegetative Clostridium difficile survives in room air on moist surfaces and in gastric contents with reduced acidity: a potential mechanism to explain the association between proton pump inhibitors and C. difficile-associated diarrhea? Antimicrob. Agents Chemother. 2007;51:2883–2887. doi: 10.1128/AAC.01443-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilcox MH. Gastrointestinal disorders and the critically ill. Clostridium difficile infection and pseudomembranous colitis. Best Pract. Res. Clin. Gastroenterol. 2003;17:475–493. doi: 10.1016/s1521-6918(03)00017-9. [DOI] [PubMed] [Google Scholar]
- 23.Gerding DN, Muto CA, Owens RC., Jr Measures to control and prevent Clostridium difficile infection. Clin. Infect. Dis. 2008;46(Suppl. 1):S43–S49. doi: 10.1086/521861. [DOI] [PubMed] [Google Scholar]
- 24.Riggs MM, Sethi AK, Zabarsky TF, Eckstein EC, Jump RLP, Donskey CJ. Asymptomatic carriers are a potential source for transmission of epidemic and nonepidemic Clostridium difficile strains among long-term care facility residents. Clin. Infect. Dis. 2007;45:992–998. doi: 10.1086/521854. [DOI] [PubMed] [Google Scholar]
- 25.Bartlett JG. Clostridium difficile: old and new observations. J. Clin. Gastroenterol. 2007;41(Suppl. 1):S24–S29. [Google Scholar]
- 26.Bartlett JG. Clinical practice. Antibiotic-associated diarrhea. N. Engl. J. Med. 2002;346:334–339. doi: 10.1056/NEJMcp011603. [DOI] [PubMed] [Google Scholar]
- 27••.Gorbach SL. Antibiotics and Clostridium difficile. N. Engl. J. Med. 1999;341:1690–1691. doi: 10.1056/NEJM199911253412211. [Points out the dangers of overusing antibiotics and the rleationship with nocosomial CDI.] [DOI] [PubMed] [Google Scholar]
- 28.Rupnik M, Braun V, Soehn F, et al. Characterization of polymorphisms in the toxin A and B genes of Clostridium difficile. FEMS Microbiol. Lett. 1997;148:197–202. doi: 10.1111/j.1574-6968.1997.tb10288.x. [DOI] [PubMed] [Google Scholar]
- 29.Torres JF. Purification and characterisation of toxin B from a strain of Clostridium difficile that does not produce toxin A. J. Med. Microbiol. 1991;35:40–44. doi: 10.1099/00222615-35-1-40. [DOI] [PubMed] [Google Scholar]
- 30.Hammond GA, Johnson JL. The toxigenic element of Clostridium difficile strain VPI 10463. Microb. Pathol. 1995;19:203–213. doi: 10.1016/s0882-4010(95)90263-5. [DOI] [PubMed] [Google Scholar]
- 31.Hundsberger T, Braun V, Weidmann M, Leukel P, Sauerborn M, Von E-SC. Transcription analysis of the genes tcdA-E of the pathogenicity locus of Clostridium difficile. Eur. J. Biochem. 1997;244:735–742. doi: 10.1111/j.1432-1033.1997.t01-1-00735.x. [DOI] [PubMed] [Google Scholar]
- 32.Mani N, Dupuy B. Regulation of toxin synthesis in Clostridium difficile by an alternative RNA polymerase sigma factor. Proc. Natl Acad. Sci. USA. 2001;98:5844–5849. doi: 10.1073/pnas.101126598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Braun V, Hundsberger T, Leukel P, Sauerborn M, Von E-SC. Definition of the single integration site of the pathogenicity locus in Clostridium difficile. Gene. 1996;181:29–38. doi: 10.1016/s0378-1119(96)00398-8. [DOI] [PubMed] [Google Scholar]
- 34.Rupnik M, Avesani V, Janc M, Von E-SC, Delmee M. A novel toxinotyping scheme and correlation of toxinotypes with serogroups of Clostridium difficile isolates. J. Clin. Microbiol. 1998;36:2240–2247. doi: 10.1128/jcm.36.8.2240-2247.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rupnik M. Heterogeneity of large clostridial toxins: importance of Clostridium difficile toxinotypes. FEMS Microbiol. Rev. 2008;32:541–555. doi: 10.1111/j.1574-6976.2008.00110.x. [DOI] [PubMed] [Google Scholar]
- 36.Geric B, Carman RJ, Rupnik M, et al. Binary toxin-producing, large clostridial toxin-negative Clostridium difficile strains are enterotoxic but do not cause disease in hamsters. J. Infect. Dis. 2006;193:1143–1150. doi: 10.1086/501368. [DOI] [PubMed] [Google Scholar]
- 37.Avbersek J, Janezic S, Pate M, et al. Diversity of Clostridium difficile in pigs and other animals in Slovenia. Anaerobe. 2009;15:252–255. doi: 10.1016/j.anaerobe.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 38.Geric SB, Rupnik M. Clostridium difficile toxinotype XI (A-B-) exhibits unique arrangement of PaLoc and its upstream region. Anaerobe. 2010;16:393–395. doi: 10.1016/j.anaerobe.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 39.Akerlund T, Persson I, Unemo M, et al. Increased sporulation rate of epidemic Clostridium difficile Type 027/NAP1. J. Clin. Microbiol. 2008;46:1530–1533. doi: 10.1128/JCM.01964-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maccannell DR, Louie TJ, Gregson DB, et al. Molecular analysis of Clostridium difficile PCR ribotype 027 isolates from Eastern and Western Canada. J. Clin. Microbiol. 2006;44:2147–2152. doi: 10.1128/JCM.02563-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schwan C, Stecher B, Tzivelekidis T, et al. Clostridium difficile toxin CDT induces formation of microtubule-based protrusions and increases adherence of bacteria. PLoS Pathol. 2009;5(10):e1000626. doi: 10.1371/journal.ppat.1000626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bourgault A-M, Lamothe F, Loo VG, Poirier L. In vitro susceptibility of Clostridium difficile clinical isolates from a multi-institutional outbreak in southern Quebec, Canada. Antimicrob. Agents Chemother. 2006;50:3473–3475. doi: 10.1128/AAC.00479-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Drudy D, Quinn T, O'mahony R, Kyne L, O'gaora P, Fanning S. High-level resistance to moxifloxacin and gatifloxacin associated with a novel mutation in gyrB in toxin-A-negative, toxin-B-positive Clostridium difficile. J. Antimicrob. Chemother. 2006;58:1264–1267. doi: 10.1093/jac/dkl398. [DOI] [PubMed] [Google Scholar]
- 44.Drudy D, Kyne L, O'mahony R, Fanning S. gyrA mutations in fluoroquinolone-resistant Clostridium difficile PCR-027. Emerging Infect. Dis. 2007;13:504–505. doi: 10.3201/eid1303.060771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carter GP, Lyras D, Allen DL, et al. Binary toxin production in Clostridium difficile is regulated by CdtR, a LytTR family response regulator. J. Bacteriol. 2007;189:7290–7301. doi: 10.1128/JB.00731-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mcmaster-Baxter NL, Musher DM. Clostridium difficile: recent epidemiologic findings and advances in therapy. Pharmacotherapy. 2007;27:1029–1039. doi: 10.1592/phco.27.7.1029. [DOI] [PubMed] [Google Scholar]
- 47.Blossom DB, Mcdonald LC. The challenges posed by reemerging Clostridium difficile infection. Clin. Infec. Dis. 2007;45:222–227. doi: 10.1086/518874. [DOI] [PubMed] [Google Scholar]
- 48•.Cohen SH, Tang YJ, Silva J., Jr Analysis of the pathogenicity locus in Clostridium difficile strains. J. Infect. Dis. 2000;181:659–663. doi: 10.1086/315248. [Looks at the differences in pathogenicity locus between different C. difficile strains.] [DOI] [PubMed] [Google Scholar]
- 49.Donelli G, Fiorentini C. Bacterial protein toxins acting on the cell cytoskeleton. New Microbiol. 1994;17(4):345–362. [PubMed] [Google Scholar]
- 50.Just I, Hofmann F, Genth H, Gerhard R. Bacterial protein toxins inhibiting low-molecular-mass GTP-binding proteins. Int. J. Med. Microbiol. 2001;291(4):243–250. doi: 10.1078/1438-4221-00127. [DOI] [PubMed] [Google Scholar]
- 51.Halabi-Cabezon I, Huelsenbeck J, May M, et al. Prevention of the cytopathic effect induced by Clostridium difficile Toxin B by active Rac1. FEBS Lett. 2008;582(27):3751–3756. doi: 10.1016/j.febslet.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 52.Hamm EE, Voth DE, Ballard JD. Identification of Clostridium difficile toxin B cardiotoxicity using a zebrafish embryo model of intoxication. Proc. Natl Acad. Sci. USA. 2006;103(38):14176–14181. doi: 10.1073/pnas.0604725103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pavliakova D, Moncrief JS, Lyerly DM, et al. Clostridium difficile recombinant toxin A repeating units as a carrier protein for conjugate vaccines: Studies of pneumococcal type 14, Escherichia coli K1, and Shigella flexneri type 2a polysaccharides in mice. Infect. Immun. 2000;68(4):2161–2166. doi: 10.1128/iai.68.4.2161-2166.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54•.Lyras D, O'connor JR, Howarth PM, et al. Toxin B is essential for virulence of Clostridium difficile. Nature. 2009;458(7242):1176–1179. doi: 10.1038/nature07822. [Points out the importance of toxin B in the pathogenicity of C. difficile.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shin BM, Kuak EY, Yoo SJ, Shin WC, Yoo HM. Emerging toxin A-B+ variant strain of Clostridium difficile responsible for pseudomembranous colitis at a tertiary care hospital in Korea. Diagn. Microbiol. Infect. Dis. 2008;60(4):333–337. doi: 10.1016/j.diagmicrobio.2007.10.022. [DOI] [PubMed] [Google Scholar]
- 56.He X, Wang J, Steele J, et al. An ultrasensitive rapid immunocytotoxicity assay for detecting Clostridium difficile toxins. J. Microbiol. Methods. 2009;78(1):97–100. doi: 10.1016/j.mimet.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Steele J, Feng H, Parry N, Tzipori S. Piglet models of acute or chronic Clostridium difficile illness. J. Infect. Dis. 2010;201(3):428–434. doi: 10.1086/649799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58•.Dubberke ER, Reske KA, Noble-Wang J, et al. Prevalence of Clostridium difficile environmental contamination and strain variability in multiple health care facilities. Am. J. Infec. Control. 2007;35(5):315–318. doi: 10.1016/j.ajic.2006.12.006. [Underlines the dangers posed by C. difficile spore contamination in healthcare facilities.] [DOI] [PubMed] [Google Scholar]
- 59.Roberts K, Smith CF, Snelling AM, et al. Aerial dissemination of Clostridium difficile spores. BMC Infect. Dis. 2008;8:7. doi: 10.1186/1471-2334-8-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Johnson S, Kent SA, O’leary KJ, et al. Fatal pseudomembranous colitis associated with a variant Clostridium difficile strain not detected by toxin A immunoassay. Ann. Intern. Med. 2001;135(6):434–438. doi: 10.7326/0003-4819-135-6-200109180-00012. [DOI] [PubMed] [Google Scholar]
- 61.Jacob SS, Sebastian JC, Hiorns D, Jacob S, Mukerjee PK. Clostridium difficile and acute respiratory distress syndrome. Heart Lung. 2004;33(4):265–268. doi: 10.1016/j.hrtlng.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 62.Dobson G, Hickey C, Trinder J. Clostridium difficile colitis causing toxic megacolon, severe sepsis and multiple organ dysfunction syndrome. Intensive Care Med. 2003;29(6):1030. doi: 10.1007/s00134-003-1754-7. [DOI] [PubMed] [Google Scholar]
- 63.Cunney RJ, Magee C, Mcnamara E, Smyth EG, Walshe J. Clostridium difficile colitis associated with chronic renal failure. Nephrol. Dial. Transplant. 1998;13(11):2842–2846. doi: 10.1093/ndt/13.11.2842. [DOI] [PubMed] [Google Scholar]
- 64.Sakurai T, Hajiro K, Takakuwa H, Nishio A, Aihara M, Chiba T. Liver abscess caused by Clostridium difficile. Scand. J. Infect. Dis. 2001;33(1):69–70. doi: 10.1080/003655401750064112. [DOI] [PubMed] [Google Scholar]
- 65.Just I, Selzer J, Wilm M, Mann M, Aktories K. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature. 1995;375(6531):500–503. doi: 10.1038/375500a0. [DOI] [PubMed] [Google Scholar]
- 66.Egerer M, Giesemann T, Jank T, Fullner Satchell KJ, Aktories K. Auto-catalytic cleavage of Clostridium difficile toxins A and B depends on cysteine protease activity. J. Biol. Chem. 2007;282(35):25314–25321. doi: 10.1074/jbc.M703062200. [DOI] [PubMed] [Google Scholar]
- 67.Pfeifer G, Schirmer J, Leemhuis J, et al. Cellular uptake of Clostridium difficile toxin B. Translocation of the N-terminal catalytic domain into the cytosol of eukaryotic cells. J. Biol. Chem. 2003;278(45):44535–44541. doi: 10.1074/jbc.M307540200. [DOI] [PubMed] [Google Scholar]
- 68.Jank T, Giesemann T, Aktories K. Rho-glucosylating Clostridium difficile toxins A and B: new insights into structure and function. Glycobiology. 2007;17(4):15R–19R. doi: 10.1093/glycob/cwm004. [DOI] [PubMed] [Google Scholar]
- 69.Hofmann F, Busch C, Prepens U, Just I, Aktories K. Localization of the glucosyltransferase activity of Clostridium difficile toxin B to the N-terminal part of the holotoxin. J. Biol. Chem. 1997;272(17):11074–11078. doi: 10.1074/jbc.272.17.11074. [DOI] [PubMed] [Google Scholar]
- 70.Faust C, Ye B, Song KP. The enzymatic domain of Clostridium difficile toxin A is located within its N-terminal region. Biochem. Biophys. Res. Commun. 1998;251(1):100–105. doi: 10.1006/bbrc.1998.9383. [DOI] [PubMed] [Google Scholar]
- 71.Tucker KD, Wilkins TD. Toxin A of Clostridium difficile binds to the human carbohydrate antigens I, X, and Y. Infect. Immun. 1991;59(1):73–78. doi: 10.1128/iai.59.1.73-78.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wren BW. A family of clostridial and streptococcal ligand-binding proteins with conserved C-terminal repeat sequences. Mol. Microbiol. 1991;5(4):797–803. doi: 10.1111/j.1365-2958.1991.tb00752.x. [DOI] [PubMed] [Google Scholar]
- 73.Frisch C, Gerhard R, Aktories K, Hofmann F, Just I. The complete receptor-binding domain of Clostridium difficile toxin A is required for endocytosis. Biochem. Biophys. Res. Commun. 2003;300(3):706–711. doi: 10.1016/s0006-291x(02)02919-4. [DOI] [PubMed] [Google Scholar]
- 74.Ho JGS, Greco A, Rupnik M, Ng KKS. Crystal structure of receptor-binding C-terminal repeats from Clostridium difficile toxin A. Proc. Natl Acad Sci USA. 2005;102(51):18373–18378. doi: 10.1073/pnas.0506391102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kai Soo T, Boon Yu W, Keang Peng S. Evidence for holin function of tcdE gene in the pathogenicity of Clostridium difficile. J. Med. Microbiol. 2001;50(7):613–619. doi: 10.1099/0022-1317-50-7-613. [DOI] [PubMed] [Google Scholar]
- 76.Ferretti JJ, Gilpin ML, Russell RRB. Nucleotide sequence of a glucosyltransferase gene from Streptococcus sobrinus MFe28. J. Bacteriol. 1987;169(9):4271–4278. doi: 10.1128/jb.169.9.4271-4278.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Reineke J, Tenzer S, Rupnik M, et al. Autocatalytic cleavage of Clostridium difficile toxin B. Nature. 2007;446(7134):415–419. doi: 10.1038/nature05622. [DOI] [PubMed] [Google Scholar]
- 78••.Lanis JM, Barua S, Ballard JD. Variations in Tcdb activity and the hypervirulence of emerging strains of Clostridium difficile. PLoS Pathogens. 2010;6(8):65–66. doi: 10.1371/journal.ppat.1001061. [Important study on the biochemical basis for hypervirulent C. difficile.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Demarest SJ, Salbato J, Elia M, et al. Structural characterization of the cell wall binding domains of Clostridium difficile toxins A and B; evidence that Ca2+ plays a role in toxin A cell surface association. J. Mol. Biol. 2005;346(5):1197–1206. doi: 10.1016/j.jmb.2004.12.059. [DOI] [PubMed] [Google Scholar]
- 80.Elliott B, Squire MM, Thean S, et al. New types of toxin A-negative, toxin B-positive strains among clinical isolates of Clostridium difficile in Australia. J. Med. Microbiol. 2011;60(Pt 8):1108–1111. doi: 10.1099/jmm.0.031062-0. [DOI] [PubMed] [Google Scholar]
- 81.Lemee L, Dhalluin A, Pestel-Caron M, Lemeland JF, Pons JL. Multilocus sequence typing analysis of human and animal Clostridium difficile isolates of various toxigenic types. J. Clin. Microbiol. 2004;42(6):2609–2617. doi: 10.1128/JCM.42.6.2609-2617.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Johnson S, Sambol SP, Brazier JS, et al. International typing study of toxin A-negative, toxin B-positive Clostridium difficile variants. J. Clin. Microbiol. 2003;41(4):1543–1547. doi: 10.1128/JCM.41.4.1543-1547.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Limaye AP, Turgeon DK, Cookson BT, Fritsche TR. Pseudomembranous colitis caused by a toxin A−B+ strain of Clostridium difficile. J. Clin. Microbiol. 2000;38(4):1696–1697. doi: 10.1128/jcm.38.4.1696-1697.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Depitre C, Delmee M, Avesani V, et al. Serogroup F strains of Clostridium difficile produce toxin B but not toxin A. J. Med. Microbiol. 1993;38(6):434–441. doi: 10.1099/00222615-38-6-434. [DOI] [PubMed] [Google Scholar]
- 85.Delmee M, Avesani V. Virulence of ten serogroups of Clostridium difficile in hamsters. J. Med. Microbiol. 1990;33(2):85–90. doi: 10.1099/00222615-33-2-85. [DOI] [PubMed] [Google Scholar]
- 86.Rupnik M, Kato N, Grabnar M, Kato H. New types of toxin A-negative, toxin B-positive strains among Clostridium difficile isolates from Asia. J. Clin. Microbiol. 2003;41(3):1118–1125. doi: 10.1128/JCM.41.3.1118-1125.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pothoulakis C, Gilbert RJ, Cladaras C, et al. Rabbit sucrase-isomaltase contains a functional intestinal receptor for Clostridium difficile toxin A. J. Clin. Invest. 1996;98(3):641–649. doi: 10.1172/JCI118835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pothoulakis C, Galili U, Castagliuolo I, et al. A human antibody binds to a-galactose receptors and mimics the effects of Clostridium difficile toxin A in rat colon. Gastroenterology. 1996;110(6):1704–1712. doi: 10.1053/gast.1996.v110.pm8964394. [DOI] [PubMed] [Google Scholar]
- 89.Aktories K, Barbieri JT. Bacterial cytotoxins: targeting eukaryotic switches. Nat. Rev. Microbiol. 2005;3(5):397–410. doi: 10.1038/nrmicro1150. [DOI] [PubMed] [Google Scholar]
- 90.Cho W. Membrane Targeting by C1 and C2 Domains. J. Biol. Chem. 2001;276(35):32407–32410. doi: 10.1074/jbc.R100007200. [DOI] [PubMed] [Google Scholar]
- 91.Dodd RB, Drickamer K. Lectin-like proteins in model organisms: Implications for evolution of carbohydrate-binding activity. Glycobiology. 2001;11(5):71R–79R. doi: 10.1093/glycob/11.5.71r. [DOI] [PubMed] [Google Scholar]
- 92.Ciesla WP, Bobak DA. Clostridium difficile toxins A and B are cation-dependent UDP-glucose hydrolases with differing catalytic activities. J. Biol. Chem. 1998;273(26):16021–16026. doi: 10.1074/jbc.273.26.16021. [DOI] [PubMed] [Google Scholar]
- 93.Fiorentini C, Donelli G, Nicotera P, Thelestam M. Clostridium difficile toxin A elicits Ca2+-independent cytotoxic effects in cultured normal rat intestinal crypt cells. Infect. Immun. 1993;61(9):3988–3993. doi: 10.1128/iai.61.9.3988-3993.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gilbert RJ, Pothoulakis C, Lamont JT, Yakubovich M. Clostridium difficile toxin B activates calcium influx required for actin disassembly during cytotoxicity. Am. J. Physiol. 1995;268(3 Pt 1):G487–G495. doi: 10.1152/ajpgi.1995.268.3.G487. [DOI] [PubMed] [Google Scholar]
- 95.Pruitt RN, Chagot B, Cover M, Chazin WJ, Spiller B, Lacy DB. Structure-function analysis of inositol hexakisphosphate-induced autoprocessing in Clostridium difficile toxin A. J. Biol. Chem. 2009;284(33):21934–21940. doi: 10.1074/jbc.M109.018929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pruitt RN, Chambers MG, Ng KKS, Ohi MD, Lacy DB. Structural organization of the functional domains of Clostridium difficile toxins A and B. Proc. Natl Acad. Sci. USA. 2010;107(30):13467–13472. doi: 10.1073/pnas.1002199107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Florin I, Thelestam M. Lysosomal involvement in cellular intoxication with Clostridium difficile toxin B. Microb. Pathog. 1986;1(4):373–385. doi: 10.1016/0882-4010(86)90069-0. [DOI] [PubMed] [Google Scholar]
- 98.Giesemann T, Jank T, Gerhard R, et al. Cholesterol-dependent pore formation of Clostridium difficile toxin A. J. Biol. Chem. 2006;281(16):10808–10815. doi: 10.1074/jbc.M512720200. [DOI] [PubMed] [Google Scholar]
- 99.Qa'dan M, Spyres LM, Ballard JD. pH-induced conformational changes in Clostridium difficile toxin B. Infect. Immun. 2000;68(5):2470–2474. doi: 10.1128/iai.68.5.2470-2474.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Barth H, Pfeifer G, Hofmann F, Maier E, Benz R, Aktories K. Low pH-induced formation of ion channels by Clostridium difficile toxin B in target cells. J. Biol. Chem. 2001;276(14):10670–10676. doi: 10.1074/jbc.M009445200. [DOI] [PubMed] [Google Scholar]
- 101.Sandvig K, Spilsberg B, Lauvrak SU, Torgersen ML, Iversen TG, Van Deurs B. Pathways followed by protein toxins into cells. Int. J. Med. Microbiol. 2004;293(7–8):483–490. doi: 10.1078/1438-4221-00294. [DOI] [PubMed] [Google Scholar]
- 102.Florin I, Thelestam M. Internalization of Clostridium difficile cytotoxin into cultured human lung fibroblasts. Biochim. Biophys. Acta. 1983;763(4):383–392. doi: 10.1016/0167-4889(83)90100-3. [DOI] [PubMed] [Google Scholar]
- 103.Matte I, Lane D, Côté É , et al. Antiapoptotic proteins Bcl-2 and Bcl-XL inhibit Clostridium difficile toxin A-induced cell death in human epithelial cells. Infect. Immun. 2009;77(12):5400–5410. doi: 10.1128/IAI.00485-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Reed JC. Bcl-2 family proteins. Oncogene. 1998;17(25):3225–3236. doi: 10.1038/sj.onc.1202591. [DOI] [PubMed] [Google Scholar]
- 105.Tait SWG, Green DR. Caspase-independent cell death: Leaving the set without the final cut. Oncogene. 2008;27(50):6452–6461. doi: 10.1038/onc.2008.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008;9(3):231–241. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- 107.Bodmer JL, Holler N, Reynard S, et al. TRAIL receptor-2 signals apoptosis through FADD and caspase-8. Nature Cell Biol. 2000;2(4):241–243. doi: 10.1038/35008667. [DOI] [PubMed] [Google Scholar]
- 108.Kischkel FC, Hellbardt S, Behrmann I, et al. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO. 1995;14(22):5579–5588. doi: 10.1002/j.1460-2075.1995.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Muzio M, Stockwell BR, Stennicke HR, Salvesen GS, Dixit VM. An induced proximity model for caspase-8 activation. J. Biol. Chem. 1998;273(5):2926–2930. doi: 10.1074/jbc.273.5.2926. [DOI] [PubMed] [Google Scholar]
- 110.Gonçalves C, Decré D, Barbut F, Burghoffer B, Petit JC. Prevalence and Characterization of a Binary Toxin (Actin-Specific ADP-Ribosyltransferase) from Clostridium difficile. J. Clin. Microbiol. 2004;42(5):1933–1939. doi: 10.1128/JCM.42.5.1933-1939.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Geric B, Johnson S, Gerding DN, Grabnar M, Rupnik M. Frequency of binary toxin genes among Clostridium difficile strains that do not produce large clostridial toxins. J. Clin. Microbiol. 2003;41(11):5227–5232. doi: 10.1128/JCM.41.11.5227-5232.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112•.Sorg JA, Sonenshein AL. Bile salts and glycine as cogerminants for Clostridium difficile spores. J. Bacteriol. 2008;190(7):2505–2512. doi: 10.1128/JB.01765-07. [Points out the role of bile acids and glycine in germination of C. difficile spores.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Henriques AO, Moran CP., Jr Structure, assembly, and function of the spore surface layers. Annu. Rev. Microbiol. 2007;61:555–588. doi: 10.1146/annurev.micro.61.080706.093224. [DOI] [PubMed] [Google Scholar]
- 114.Jarvis WR, Schlosser J, Jarvis AA, Chinn RY. National point prevalence of Clostridium difficile in US health care facility inpatients, 2008. Am. J. Infect. Control. 2009;37(4):263–270. doi: 10.1016/j.ajic.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 115.Vollaard EJ. Clasener HaL. Colonization resistance. Antimicrob. Agents Chemother. 1994;38(3):409–414. doi: 10.1128/aac.38.3.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Drudy D, Harnedy N, Fanning S, O'mahony R, Kyne L. Isolation and characterisation of toxin A-negative, toxin B-positive Clostridium difficile in Dublin, Ireland. Clin. Microbiol. Infec. 2007;13(3):298–304. doi: 10.1111/j.1469-0691.2006.01634.x. [DOI] [PubMed] [Google Scholar]
- 117.Stecher B, Hardt WD. The role of microbiota in infectious disease. Trends Microbiol. 2008;16(3):107–114. doi: 10.1016/j.tim.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 118.Stecher B, Robbiani R, Walker AW, et al. Salmonella enterica serovar typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol. 2007;5(10):2177–2189. doi: 10.1371/journal.pbio.0050244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kuipers EJ, Surawicz CM. Clostridium difficile infection. Lancet. 2008;371(9623):1486–1488. doi: 10.1016/S0140-6736(08)60635-2. [DOI] [PubMed] [Google Scholar]
- 120.Powell RC, Nunes WT, Harding RS, Vacca JB. The influence of nonabsorbable antibiotics on serum lipids and the excretion of neutral sterols and bile acids. Am. J. Clin. Nutr. 1962;11:156–168. doi: 10.1093/ajcn/11.2.156. [DOI] [PubMed] [Google Scholar]
- 121.Rolfe RD, Helebian S, Finegold SM. Bacterial interference between Clostridium difficile and normal fecal flora. J. Infect. Dis. 1981;143(3):470–475. doi: 10.1093/infdis/143.3.470. [DOI] [PubMed] [Google Scholar]
- 122.Giel JL, Sorg JA, Sonenshein AL, Zhu J. Metabolism of bile salts in mice influences spore germination in Clostridium difficile. PLoS ONE. 2010;5(1) doi: 10.1371/journal.pone.0008740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cloud J, Kelly CP. Update on Clostridium difficile associated disease. Curr. Opp. Gastroenterol. 2007;23(1):4–9. doi: 10.1097/MOG.0b013e32801184ac. [DOI] [PubMed] [Google Scholar]
- 124.Setlow P. Spore germination. Curr. Opp. Microbiol. 2003;6(6):550–556. doi: 10.1016/j.mib.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 125.Wax R, Freese E, Cashel M. Separation of two functional roles of L-alanine in the initiation of Bacillus subtilis spore germination. J. Bacteriol. 1967;94(3):522–529. doi: 10.1128/jb.94.3.522-529.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Corzo G, Gilliland SE. Bile salt hydrolase activity of three strains of Lactobacillus acidophilus. J. Dairy Sci. 1999;82(3):472–480. doi: 10.3168/jds.S0022-0302(99)75256-2. [DOI] [PubMed] [Google Scholar]
- 127.Carey JB, Jr, Watson CJ. Isolation of deoxycholic acid from normal human feces. J. Biol. Chem. 1955;216(2):847–850. [PubMed] [Google Scholar]
- 128.Makita M, Wells WW. Quantitative analysis of fecal bile acids by gas-liquid chromatography. Anal. Biochem. 1963;5(6):523–530. doi: 10.1016/0003-2697(63)90072-1. [DOI] [PubMed] [Google Scholar]
- 129.Thomas LA, Veysey MJ, French G, Hylemon PB, Murphy GM, Dowling RH. Bile acid metabolism by fresh human colonic contents: a comparison of caecal versus faecal samples. Gut. 2001;49(6):835–842. doi: 10.1136/gut.49.6.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kamiya S, Yamakawa K, Ogura H, Nakamura S. Recovery of spores of Clostridium difficile altered by heat or alkali. J. Med. Microbiol. 1989;28(3):217–221. doi: 10.1099/00222615-28-3-217. [DOI] [PubMed] [Google Scholar]
- 131.Weese JS, Staempfli HR, Prescott JF. Isolation of environmental Clostridium difficile from a veterinary teaching hospital. J. Vet. Diagnost. Investig. 2000;12(5):449–452. doi: 10.1177/104063870001200510. [DOI] [PubMed] [Google Scholar]
- 132.Wilson KH, Kennedy MJ, Fekety FR. Use of sodium taurocholate to enhance spore recovery on a medium selective for Clostridium difficile. J. Clin. Microbiol. 1982;15(3):443–446. doi: 10.1128/jcm.15.3.443-446.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Fisher N, Hanna P. Characterization of Bacillus anthracis germinant receptors in vitro. J. Bacteriol. 2005;187(23):8055–8062. doi: 10.1128/JB.187.23.8055-8062.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hornstra LM, De Vries YP, Wells-Bennik MHJ, De Vos WM, Abee T. Characterization of germination receptors of Bacillus cereus ATCC 14579. Appl. Environmen. Microbiol. 2006;72(1):44–53. doi: 10.1128/AEM.72.1.44-53.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ramirez N, Liggins M, Abel-Santos E. Kinetic evidence for the presence of putative germination receptors in Clostridium difficile spores. J. Bacteriol. 2010;192(16):4215–4222. doi: 10.1128/JB.00488-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Freeman J, Wilcox MH. Antibiotics and Clostridium difficile. Microb. Infect. 1999;1(5):377–384. doi: 10.1016/s1286-4579(99)80054-9. [DOI] [PubMed] [Google Scholar]
- 137•.Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006;47(2):241–259. doi: 10.1194/jlr.R500013-JLR200. [Shows how bile salts are trasnformed by normal human gut microbial flora.] [DOI] [PubMed] [Google Scholar]
- 138.Edenharder R. Dehydroxylation of cholic acid at C12 and epimerization at C5 and C7 by Bacteroides species. J. Steroid Biochem. 1984;21(4):413–420. doi: 10.1016/0022-4731(84)90304-2. [DOI] [PubMed] [Google Scholar]
- 139.Hashimoto S, Igimi H, Uchida K, Satoh T, Benno Y, Takeuchi N. Effects of β-lactam antibiotics on intestinal microflora and bile acid metabolism in rats. Lipids. 1996;31(6):601–609. doi: 10.1007/BF02523830. [DOI] [PubMed] [Google Scholar]
- 140•.Lancaster JW, Matthews SJ. Fidaxomicin: the newest addition to the armamentarium against Clostridium difficile infections. Clin. Ther. 2012;34:1–13. doi: 10.1016/j.clinthera.2011.12.003. [Discusses fidaxomicin that has been proposed and approved as a new drug against CDI.] [DOI] [PubMed] [Google Scholar]
- 141.Kopterides P, Papageorgiou C, Antoniadou A, et al. Failure of tigecycline to treat severe Clostridium difficile infection. Anaesth. Intensive Care. 2010;38(4):755–758. doi: 10.1177/0310057X1003800339. [DOI] [PubMed] [Google Scholar]
- 142.Johnson S, Schriever C, Galang M, Kelly CP, Gerding DN. Interruption of recurrent Clostridium difficile-associated diarrhea episodes by serial therapy with vancomycin and rifaximin. Clin. Infect. Dis. 2007;44:846–848. doi: 10.1086/511870. [DOI] [PubMed] [Google Scholar]
- 143.Pelaez T, Alcala L, Alonso R, et al. In vitro activity of ramoplanin against Clostridium difficile, including strains with reduced susceptibility to vancomycin or with resistance to metronidazole. Antimicrob. Agents Chemother. 2005;49:1157–1159. doi: 10.1128/AAC.49.3.1157-1159.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Freeman J, Baines SD, Jabes D, Wilcox MH. Comparison of the efficacy of ramoplanin and vancomycin in both in vitro and in vivo models of clindamycin-induced Clostridium difficile infection. J. Antimicrob. Chemother. 2005;56:717–725. doi: 10.1093/jac/dki321. [DOI] [PubMed] [Google Scholar]
- 145.Chen YX, Cabana B, Kivel N, Michaelis A. Effect of food on the pharmacokinetics of rifalazil, a novel antibacterial, in healthy male volunteers. J. Clin. Pharmacol. 2007;47:841–849. doi: 10.1177/0091270007300745. [DOI] [PubMed] [Google Scholar]
- 146.Anton PM, O'brien M, Kokkotou E, et al. Rifalazil treats and prevents relapse of Clostridium difficile-associated diarrhea in hamsters. Antimicrob. Agents Chemother. 2004;48:3975–3979. doi: 10.1128/AAC.48.10.3975-3979.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zoetendal EG, Vaughan EE, De Vos WM. A microbial world within us. Mol. Microbiol. 2006;59(6):1639–1650. doi: 10.1111/j.1365-2958.2006.05056.x. [DOI] [PubMed] [Google Scholar]
- 148.Mercenier A, Muller-Alouf H, Grangette C. Lactic acid bacteria as live vaccines. Curr. Issues Mol. Biol. 2000;2(1):17–25. [PubMed] [Google Scholar]
- 149.Goldenberg JZ, Ma SS, Saxton JD, et al. Probiotics for the prevention of Clostridium difficile-associated diarrhea in adults and children. Cochrane Database Syst. Rev. 2013;5:CD006095. doi: 10.1002/14651858.CD006095.pub3. [DOI] [PubMed] [Google Scholar]
- 150.Jena PK, Trivedi D, Chaudhary H, Sahoo TK, Seshadri S. Bacteriocin PJ4 active against enteric pathogen produced by Lactobacillus helveticus PJ4 isolated from gut microflora of wistar rat (Rattus norvegicus): partial purification and characterization of bacteriocin. Appl. Biochem. Biotechnol. 2013;169(7):2088–2100. doi: 10.1007/s12010-012-0044-7. [DOI] [PubMed] [Google Scholar]
- 151••.Mcfarland LV, Surawicz CM, Greenberg RN, et al. A randomized placebo-controlled trial of Saccharomyces boulardii in combination with standard antibiotics for Clostridium difficile disease. JAMA. 1994;271:1913–1918. [Important clinical trial of probiotics for CDI.] [PubMed] [Google Scholar]
- 152.Surawicz CM, Mcfarland LV, Greenberg RN, et al. The search for a better treatment for recurrent Clostridium difficile disease: Use of high-dose vancomycin combined with Saccharomyces boulardii. Clin. Infect. Dis. 2000;31:1012–1017. doi: 10.1086/318130. [DOI] [PubMed] [Google Scholar]
- 153.Eiseman B, Silen W, Bascom GS, Kauvar AJ. Fecal enema as an adjunct in the treatment of pseudomembranous enterocolitis. Surgery. 1958;44(5):854–859. [PubMed] [Google Scholar]
- 154••.Van NE, Vrieze A, Nieuwdorp M, et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N. Engl. J. Med. 2013;368:407–415. doi: 10.1056/NEJMoa1205037. [Important clinical trial of fecal transplants for CDI.] [DOI] [PubMed] [Google Scholar]
- 155.Rohlke F, Stollman N. Fecal microbiota transplantation in relapsing Clostridium difficile infection. Therap. Adv. Gastroenterol. 2012;5:403–420. doi: 10.1177/1756283X12453637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Chang JY, Antonopoulos DA, Kalra A, et al. Decreased diversity of the fecal Microbiome in recurrent Clostridium difficile-associated diarrhea. J. Infect. Dis. 2008;197:435–438. doi: 10.1086/525047. [DOI] [PubMed] [Google Scholar]
- 157.Kelly CP. Fecal microbiota transplantation -an old therapy comes of age. N. Engl. J. Med. 2013;368(5):474–475. doi: 10.1056/NEJMe1214816. [DOI] [PubMed] [Google Scholar]
- 158.Vyas D, L'esperance H E, Vyas A. Stool therapy may become a preferred treatment of recurrent Clostridium difficile? World J. Gastroenterol. 2013;19(29):4635–4637. doi: 10.3748/wjg.v19.i29.4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Allen-Vercoe E, Petrof EO. Artificial stool transplantation: progress towards a safer, more effective and acceptable alternative. Expert Rev. Gastroenterol. Hepatol. 2013;7:291–293. doi: 10.1586/egh.13.16. [DOI] [PubMed] [Google Scholar]
- 160.Shone C, Landon J. Antibodies to Clostridium difficile toxins. Antibodies to Clostridium difficile toxins (WO2010094970 A1) 2010:53. [Google Scholar]
- 161.Warny M, Vaerman JP, Avesani V, Delmee M. Human antibody response to Clostridium difficile toxin A in relation to clinical course of infection. Infect. Immun. 1994;62(2):384–389. doi: 10.1128/iai.62.2.384-389.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kyne L, Warny M, Qamar A, Kelly CP. Association between antibody response to toxin A and protection against recurrent Clostridium difficile diarrhoea. Lancet. 2001;357(9251):189–193. doi: 10.1016/S0140-6736(00)03592-3. [DOI] [PubMed] [Google Scholar]
- 163.Phillips C. Serum antibody responses to Clostridium difficile toxin A: predictive and protective? Gut. 2001;49(2):167–168. doi: 10.1136/gut.49.2.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Giannasca PJ, Warny M. Active and passive immunization against Clostridium difficile diarrhea and colitis. Vaccine. 2004;22(7):848–856. doi: 10.1016/j.vaccine.2003.11.030. [DOI] [PubMed] [Google Scholar]
- 165••.Kyne L. Clostridium difficile - Beyond antibiotics. N. Engl. J. Med. 2010;362(3):264–265. doi: 10.1056/NEJMe0910055. [Covers new experimental therapies against CDI.] [DOI] [PubMed] [Google Scholar]
- 166.Yoshida M, Kobayashi K, Kuo TT, et al. Neonatal Fc receptor for IgG regulates mucosal immune responses to luminal bacteria. J. Clin. Invest. 2006;116(8):2142–2151. doi: 10.1172/JCI27821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Leung DYM, Kelly CP, Boguniewicz M, Pothoulakis C, Lamont JT, Flores A. Treatment with intravenously administered gamma globulin of chronic relapsing colitis induced by Clostridium difficile toxin. J. Pediatrics. 1991;118(4 I):633–637. doi: 10.1016/s0022-3476(05)83393-1. [DOI] [PubMed] [Google Scholar]
- 168.Salcedo J, Keates S, Pothoulakis C, et al. Intravenous immunoglobulin therapy for severe Clostridium difficile colitis. Gut. 1997;41(3):366–370. doi: 10.1136/gut.41.3.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Wilcox MH. Descriptive study of intravenous immunoglobulin for the treatment of recurrent Clostridium difficile diarrhoea. J. Antimicrob. Chemother. 2004;53(5):882–884. doi: 10.1093/jac/dkh176. [DOI] [PubMed] [Google Scholar]
- 170.Kyne L, Warny M, Qamar A, Kelly CP. Asymptomatic carriage of Clostridium difficile and serum levels of IgG antibody against toxin A. N. Engl. J. Med. 2000;342(6):390–397. doi: 10.1056/NEJM200002103420604. [DOI] [PubMed] [Google Scholar]
- 171.Kelly CP. Immune response to Clostridium difficile infection. Eur. J. Gastroenterol. Hepatol. 1996;8(11):1048–1053. doi: 10.1097/00042737-199611000-00004. [DOI] [PubMed] [Google Scholar]
- 172.Kyne L, Kelly CP. Prospects for a vaccine for Clostridium difficile. BioDrugs. 1998;10:173–181. doi: 10.2165/00063030-199810030-00001. [DOI] [PubMed] [Google Scholar]
- 173.Lyerly DM, Phelps CJ, Toth J, Wilkins TD. Characterization of toxins A and B of Clostridium difficile with monoclonal antibodies. Infect. Immun. 1986;54(1):70–76. doi: 10.1128/iai.54.1.70-76.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Jori G, Fabris C, Soncin M, et al. Photodynamic therapy in the treatment of microbial infections: basic principles and perspective applications. Lasers Surg. Med. 2006;38:468–481. doi: 10.1002/lsm.20361. [DOI] [PubMed] [Google Scholar]
- 175.Babcock GJ, Broering TJ, Hernandez HJ, et al. Human monoclonal antibodies directed against toxins A and B prevent Clostridium difficile-induced mortality in hamsters. Infect. Immun. 2006;74(11):6339–6347. doi: 10.1128/IAI.00982-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Johnson S, Gerding DN, Janoff EN. Systemic and mucosal antibody responses to toxin A in patients infected with Clostridium difficile. J. Infect. Dis. 1992;166(6):1287–1294. doi: 10.1093/infdis/166.6.1287. [DOI] [PubMed] [Google Scholar]
- 177.Fekety R. Guidelines for the diagnosis and management of Clostridium difficile-associated diarrhea and colitis. Am. J. Gastroenterol. 1997;92(5):739–750. [PubMed] [Google Scholar]
- 178.Kelly CP, Pothoulakis C, Vavva F, et al. Anti-Clostridium difficile bovine immunoglobulin concentrate inhibits cytotoxicity and enterotoxicity of C. difficile toxins. Antimicrob. Agents Chemother. 1996;40(2):373–379. doi: 10.1128/aac.40.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Giannasca PJ, Zhang ZX, Lei WD, et al. Serum antitoxin antibodies mediate systemic and mucosal protection from Clostridium difficile disease in hamsters. Infect. Immun. 1999;67(2):527–538. doi: 10.1128/iai.67.2.527-538.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Powers DB, Amersdorfer P, Poul MA, et al. Expression of single-chain Fv-Fc fusions in Pichia pastoris. J. Immunol. Methods. 2001;251(1–2):123–135. doi: 10.1016/s0022-1759(00)00290-8. [DOI] [PubMed] [Google Scholar]
- 181.Zhang J, Mackenzie R, Durocher Y. Production of chimeric heavy-chain antibodies. Methods Mol. Biol. 2009;525:323–336. doi: 10.1007/978-1-59745-554-1_17. [DOI] [PubMed] [Google Scholar]
- 182.Harmsen MM, De Haard HJ. Properties, production, and applications of camelid single-domain antibody fragments. Appl. Microbiol. Biotechnol. 2007;77(1):13–22. doi: 10.1007/s00253-007-1142-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Harmsen MM, Van Solt CB, Van Zijderveld-Van Bemmel AM, Niewold TA, Van Zijderveld FG. Selection and optimization of proteolytically stable llama single-domain antibody fragments for oral immunotherapy. Appl. Microbiol. Biotechnol. 2006;72(3):544–551. doi: 10.1007/s00253-005-0300-7. [DOI] [PubMed] [Google Scholar]
- 184.Famm K, Hansen L, Christ D, Winter G. Thermodynamically stable aggregation-resistant antibody domains throughdirected evolution. J. Mol. Biol. 2008;376(4):926–931. doi: 10.1016/j.jmb.2007.10.075. [DOI] [PubMed] [Google Scholar]
- 185.Jespers L, Schon O, Famm K, Winter G. Aggregation-resistant domain antibodies selected on phage by heat denaturation. Nature Biotechnol. 2004;22(9):1161–1165. doi: 10.1038/nbt1000. [DOI] [PubMed] [Google Scholar]
- 186.Stijlemans B, Conrath K, Cortez-Retamozo V, et al. Efficient targeting of conserved cryptic epitopes of infectious agents by single domain antibodies: African trypanosomes as paradigm. J. Biol. Chem. 2004;279(2):1256–1261. doi: 10.1074/jbc.M307341200. [DOI] [PubMed] [Google Scholar]
- 187.De Genst E, Silence K, Decanniere K, et al. Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc. Natl Acad. Sci. USA. 2006;103(12):4586–4591. doi: 10.1073/pnas.0505379103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Stanfield RL, Dooley H, Flajnik MF, Wilson IA. Crystal structure of a shark single-domain antibody V region in complex with lysozyme. Science. 2004;305(5691):1770–1773. doi: 10.1126/science.1101148. [DOI] [PubMed] [Google Scholar]
- 189.Desmyter A, Transue TR, Ghahroudi MA, et al. Crystal structure of a camel single-domain V(H) antibody fragment in complex with lysozyme. Nat. Structural. Biol. 1996;3(9):803–811. doi: 10.1038/nsb0996-803. [DOI] [PubMed] [Google Scholar]
- 190•.Hussack G, Tanha J. Toxin-specific antibodies for the treatment of Clostridium difficile: current status and future perspectives. Toxins. 2010;2:998–1018. doi: 10.3390/toxins2050998. [Can antibodies against C. difficile toxins be used as a therapy?.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Jori G, Brown SB. Photosensitized inactivation of microorganisms. Photochem. Photobiol. Sci. 2004;3:403–405. doi: 10.1039/b311904c. [DOI] [PubMed] [Google Scholar]
- 192.Bonnett R. Chemical aspects of photodynamic therapy. Vol. 451. Gordon & Breach; UK: 2000. [Google Scholar]
- 193.Wainwright M. Photodynamic antimicrobial chemotherapy (PACT). J. Antimicrob. Chemother. 1998;42:13–28. doi: 10.1093/jac/42.1.13. [DOI] [PubMed] [Google Scholar]
- 194.Calin MA, Parasca SV. Light sources for photodynamic inactivation of bacteria. Lasers Med. Sci. 2009;24:453–460. doi: 10.1007/s10103-008-0588-5. [DOI] [PubMed] [Google Scholar]
- 195•.Hamblin MR, Hasan T. Photodynamic therapy: a new antimicrobial approach to infectious disease? Photochem. Photobiol. Sci. 2004;3:436–450. doi: 10.1039/b311900a. [Important review covering antimicrobial photodynamic therapy (PDT).] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Harris F, Chatfield LK, Phoenix DA. Phenothiazinium based photosensitisers -photodynamic agents with a multiplicity of cellular targets and clinical applications. Curr. Drug Targets. 2005;6:615–627. doi: 10.2174/1389450054545962. [DOI] [PubMed] [Google Scholar]
- 197.Ergaieg K, Chevanne M, Cillard J, Seux R. Involvement of both Type I and Type II mechanisms in Gram-positive and Gram-negative bacteria photosensitization by a meso-substituted cationic porphyrin. Sol. Energy. 2008;82:1107–1117. [Google Scholar]
- 198.Van DBH, Mizeret J, Theumann JF, et al. Light distributors for photodynamic therapy. Proc. SPIE Int. Soc. Opt. Eng. 1995;2631:173–198. [Google Scholar]
- 199.Tegos GP, Demidova TN, Arcila-Lopez D, et al. Cationic fullerenes are effective and selective antimicrobial photosensitizers. Chem. Biol. 2005;12:1127–1135. doi: 10.1016/j.chembiol.2005.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Huang L, Huang Y-Y, Mroz P, et al. Stable synthetic cationic bacteriochlorins as selective antimicrobial photosensitizers. Antimicrob. Agents Chemother. 2010;54:3834–3841. doi: 10.1128/AAC.00125-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Castano AP, Demidova TN, Hamblin MR. Mechanisms in photodynamic therapy: part one-photosensitizers, photochemistry and cellular localization. Photodiagn. Photodyn. Ther. 2005;1:279–293. doi: 10.1016/S1572-1000(05)00007-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Wainwright M, Phoenix DA, Laycock SL, Wareing DRA, Wright PA. Photobactericidal activity of phenothiazinium dyes against methicillin-resistant strains of Staphylococcus aureus. FEMS Microbiol. Lett. 1998;160:177–181. doi: 10.1111/j.1574-6968.1998.tb12908.x. [DOI] [PubMed] [Google Scholar]
- 203.Wainwright M, Phoenix DA, Gaskell M, Marshall B. Photobactericidal activity of methylene blue derivatives against vancomycin-resistant Enterococcus spp. J. Antimicrob. Chemother. 1999;44:823–825. doi: 10.1093/jac/44.6.823. [DOI] [PubMed] [Google Scholar]
- 204.Demidova TN, Gad F, Zahra T, Francis KP, Hamblin MR. Monitoring photodynamic therapy of localized infections by bioluminescence imaging of genetically engineered bacteria. J. Photochem. Photobiol. B. 2005;81:15–25. doi: 10.1016/j.jphotobiol.2005.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Caminos DA, Spesia MB, Durantini EN. Photodynamic inactivation of Escherichia coli by novel meso-substituted porphyrins by 4-(3-N,N,N-trimethylammoniumpropoxy)phenyl and 4-(trifluoromethyl)phenyl groups. Photochem. Photobiol. Sci. 2006;5(1):56–65. doi: 10.1039/b513511g. [DOI] [PubMed] [Google Scholar]
- 206.Wainwright M, Dai T, Hamblin MR. Antimicrobial photodynamic therapy in the colon: delivering a light punch to the guts? Photochem. Photobiol. 2011;87:754–756. doi: 10.1111/j.1751-1097.2011.00925.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207••.Cassidy CM, Tunney MM, Caldwell DL, Andrews GP, Donnelly RF. Development of novel oral formulations prepared via hot melt extrusion for targeted delivery of photosensitizer to the colon. Photochem. Photobiol. 2011;87:867–876. doi: 10.1111/j.1751-1097.2011.00915.x. [First paper to seriously propose that PDT could actually be uased to treat C. difficileinfection in the anerobic environment of the colon.] [DOI] [PubMed] [Google Scholar]
- 208.Foley JW, Song X, Demidova TN, Jilal F, Hamblin MR. Synthesis and properties of benzo[a]phenoxazinium chalcogen analogues as novel broad-spectrum antimicrobial photosensitizers. J. Med. Chem. 2006;49:5291–5299. doi: 10.1021/jm060153i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209•.Evans CL, Abu-Yousif AO, Park YJ, et al. Killing hypoxic cell populations in a 3D tumor model with EtNBS-PDT. PLoS ONE. 2011;6(8):e23434. doi: 10.1371/journal.pone.0023434. [Shows that the benzophenthiazinium dye EtNBS is efficient at mediating PDT at very low oxygen concentrations.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Zhang P, Ng K, Ling CC. Total synthesis of LeA-LacNAc pentasaccharide as a ligand for Clostridium difficile toxin A. Org. Biomol. Chem. 2010;8(1):128–136. doi: 10.1039/b914193f. [DOI] [PubMed] [Google Scholar]
- 211.Bartlett JG, Perl TM. The new Clostridium difficile - What does it mean? N. Engl. J. Med. 2005;353(23):2503–2505. doi: 10.1056/NEJMe058221. [DOI] [PubMed] [Google Scholar]
- 212.Reinert DJ, Jank T, Aktories K, Schulz GE. Structural basis for the function of Clostridium difficile toxin B. J. Mol. Biol. 2005;351(5):973–981. doi: 10.1016/j.jmb.2005.06.071. [DOI] [PubMed] [Google Scholar]
- 213.Pear SM, Williamson TH, Bettin KM, Gerding DN, Galgiani JN. Decrease in nosocomial Clostridium difficile-associated diarrhea by restricting clindamycin use. Ann. Intern. Med. 1994;120(4):272–277. doi: 10.7326/0003-4819-120-4-199402150-00003. [DOI] [PubMed] [Google Scholar]
- 301.Fang L, Marquardt RR, Sellen RT. 2008 US20110020356.
- 302.Songer JG, Cama VA, Sterling CR. 1999 WO1999002188.
- 401.Healthcare-associated infections. Clostridium difficile Infection. www.cdc.gov/HAI/organisms/cdiff/Cdiff_infect.html.