

Abstract
Neuromyelitis optica (NMO) is an inflammatory demyelinating disease of the central nervous system in which anti-aquaporin-4 (AQP4) autoantibodies (AQP4-IgG) cause damage to astrocytes by complement-dependent cytotoxicity (CDC). Various approaches have been attempted to produce NMO lesions in rodents, some involving genetically modified mice with altered immune cell function. Here, we found that mouse serum strongly inhibits complement from multiple species, preventing AQP4-IgG-dependent CDC. Effects of mouse serum on complement activation were tested in CDC assays in which AQP4-expressing cells were incubated with AQP4-IgG and complement from different species. Biochemical assays and mass spectrometry were used to characterize complement inhibitor(s) in mouse serum. Sera from different strains of mice produced almost no AQP4-IgG-dependent CDC compared with human, rat and guinea pig sera. Remarkably, addition of mouse serum prevented AQP4-IgG-dependent CDC caused by human, rat or guinea pig serum, with 50% inhibition at <5% mouse serum. Hemolysis assays indicated that the inhibitor(s) in mouse serum target the classical and not the alternative complement pathway. We found that the complement inhibitor(s) in mouse serum were contained in a serum fraction purified with protein-A resin; however, the inhibitor was not IgG as determined using serum from IgG-deficient mice. Mass spectrometry on the protein A-purified fraction produced several inhibitor candidates. The low intrinsic complement activity of mouse serum and the presence of complement inhibitor(s) limit the utility of mouse models to study disorders, such as NMO, involving the classical complement pathway.
Keywords: NMO, Aquaporin-4, Complement-dependent cytotoxicity, AQP4-IgG, Mouse models
1. Introduction
Neuromyelitis optica (NMO) is an autoimmune disease of the central nervous system (CNS) associated with inflammatory demyelinating lesions in spinal cord, optic nerve and, to a lesser extent, brain (de Seze et al., 2002; Jacob et al., 2013; Wingerchuk et al., 2007). The major clinical manifestations of NMO include paralysis and visual impairment. Most NMO patients have in their serum IgG1-type autoantibodies against astrocyte water channel protein aquaporin-4 (AQP4), called AQP4-IgG (Lennon et al., 2005). It is believed that NMO pathogenesis involves AQP4-IgG binding to AQP4, which initiates complement-dependent cytotoxicity (CDC) and astrocyte damage, producing an inflammatory reaction with granulocyte and macrophage infiltration, and disruption of the blood–brain barrier (Fujihara, 2011; Jarius and Wildemann, 2010; Papadopoulos, 2012). Secondary damage to oligodendrocytes and neurons cause demyelination and neurological deficit. Antibody-dependent cell-mediated cytotoxicity (ADCC) may also contribute to astrocyte damage in NMO (Bennett et al., 2009; Ratelade et al., 2013; Vincent et al., 2008), and AQP4-sensitized T-cells may be involved in blood–brain barrier disruption (Pohl et al., 2011).
Development of animal models of NMO is a high priority in the NMO field, both for elucidation of pathogenesis mechanisms and for testing of potential therapeutics. Rodent models have supported the pathogenicity of AQP4-IgG in NMO. Peripheral administration of AQP4-IgG worsened inflammatory CNS lesions in rats with pre-existing experimental autoimmune encephalomyelitis (EAE) (Bennett et al., 2009; Bradl et al., 2009; Kinoshita et al., 2009) or in rats pre-treated with complete Freund’s adjuvant (Kimura et al., 2010). However, the background hyper-inflammatory state in these rat models, and/or the presence of myelin protein-reactive T-cells, confounds the interpretation of lesion pathology in terms of NMO mechanisms. In mice, direct intracerebral injection of AQP4-IgG and human complement produced the major pathological features found in human NMO, including loss of AQP4 and GFAP, inflammatory cell infiltration, loss of myelin, and perivascular deposition of activated complement (Saadoun et al., 2010). Development of NMO pathology required AQP4-IgG, complement and AQP4. Models based on direct intracerebral injection have been useful in elucidating the role of specific leukocyte types in NMO (Saadoun et al., 2012; Zhang and Verkman, 2013) and in testing of antibody blocking (Tradtrantip et al., 2013; Tradtrantip et al., 2012) and cell-targeted (Saadoun et al., 2012; Zhang and Verkman, 2013) therapeutics. However, mouse models require direct injection of human complement together with AQP4-IgG into brain, and hence do not closely recapitulate human NMO. Recently, mouse models of NMO optic neuritis (Asavapanumas et al., 2014a) and transverse myelitis (Zhang and Verkman, 2014) were developed, though, again, pathology required co-administration of AQP4-IgG and human complement directly into the CNS.
Improved animal models of NMO are thus needed. Attempts to immunize rodents with AQP4 peptides have so far been unsuccessful. As found in EAE models, mouse models involving B/T cell activation against myelin oligodendrocyte glycoprotein developed optic neuritis and spinal cord pathology reminiscent of NMO (Bettelli et al., 2006; Krishnamoorthy et al., 2006). There is active work in the development of transgenic mouse models of NMO involving AQP4-sensitized lymphocytes and expression of monoclonal NMO antibodies. So far, attempts to develop mouse models of NMO based on peripheral AQP4-IgG administration have not been successful, despite efficient access of serum AQP4-IgG to brain tissue in circumventricular organs (Ratelade et al., 2011). Also, we have been unable to produce NMO pathology in mice following intravenous administration of AQP4-IgG together with large amounts of human complement, both under baseline condition and using various approaches to permeabilize the blood–brain barrier (unpublished data).
We speculated that the weak activity of mouse complement, as reported previously (Bergman et al., 2000), might be responsible for the failures in attempted mouse models of NMO; however, the weak activity of mouse complement did not explain the failure of intravenous administration of AQP4-IgG and human complement. In investigating the intravenous model, we discovered that mouse serum is a strong inhibitor of AQP4-IgG-dependent CDC produced by complement from multiple species, including human. We found here that mouse serum contains inhibitor(s) of the classical complement pathway, and, unfortunately, even after their removal by affinity chromatography, the activity of the classical complement pathway is mouse serum remains very weak. We conclude that mice have limited utility as models of NMO.
2. Materials and methods
2.1. Reagents and antibodies
Complement/sera from mouse (CD1 and C57BL/6 strains), rat (Wistar strain), guinea pig and human were purchased from Innovative Research (Novi, MI). BUB/Bnj and Rag1 null mice (IgG-deficient) were purchased from the Jackson Laboratory (Bar Harbor, ME) and used for serum collections. For preparation of fresh mouse serum for CDC assays, blood was collected by cardiac puncture, clotted at room temperature for 30 min, and then centrifuged at 3000 g for 10 min. Serum was used directly in CDC assays or frozen at −80 °C. Mouse serum albumin and transferrin were purchased from Sigma–Aldrich (St Louis, MO), and mouse serum fibrinogen and fibronectin from Innovative Research. Serum from factor I-deficient mice was kindly provided by Dr Matthew Pickering (Imperial College, London, UK). Recombinant monoclonal human antibody rAb-53 (called AQP4-IgG), which recognizes extracellular epitope(s) on AQP4, was generated and characterized as described (Bennett et al., 2009; Crane et al., 2011). A chimeric AQP4-IgG (AQP4-IgGc), provided by Dr. Jeff Bennett (Univ. Colorado Denver), was generated by cloning the sequence of the variable region of heavy and light chains of rAb-53 upstream of the constant region of mouse IgG2a. For some experiments mouse serum was heat-treated for 30 min at 60 °C.
2.2. Cell culture and complement-dependent cytotoxicity assay
Chinese hamster ovary (CHO) cells stably expressing human AQP4-M23 (named CHO-AQP4 cells) were generated as described (Crane et al., 2011) and cultured at 37 °C in 5% CO2 95% air in F-12 Ham’s Nutrient Mixture medium supplemented with 10% fetal bovine serum, 200 μg/mL geneticin (selection marker), 100 U/mL penicillin and 100 μg/mL streptomycin. For assay of CDC, CHO-AQP4 cells were grown in 96-well plates until confluence and incubated with specified concentrations of AQP4-IgG and complement for 1 h at 23 °C, as described (Ratelade et al., 2013). Cells were then washed extensively and viability was measured by addition of 20% AlamarBlue (Invitrogen, Carlsbad, CA) for 1 h at 37 °C. Fluorescence was measured with a plate reader at excitation/emission wavelengths of 560/590 nm. Percentage cell viability was computed as: [(sample − 100% lysis)/(no lysis − 100% lysis)] × 100 where ‘100% lysis’ is fluorescence of cells incubated in 1% Triton X-100 and ‘no lysis’ is fluorescence of cells incubated with complement but no AQP4-IgG. In some experiments cell viability was measured by addition of 1 μM calcein-AM and 2 μM ethidiumhomodimer (Invitrogen) in PBS to stain live cells green and dead cells red.
2.3. Hemolysis assays
Hemolysis assays using IgM-coated sheep red blood cells (for assay of classical complement pathway) and uncoated rabbit red blood cells (for assay of alternative pathway) were performed according to manufacturers’ instructions (Complement Technology Inc., Tyler, TX). Briefly, 2.5 × 107 sheep red blood cells were incubated with specified concentrations of complement for 1 h at 37 °C in a total volume of 0.25 mL. 2.5 × 107 rabbit red blood cells were incubated with complement in Ca2+/Mg2+ free buffer containing 5 mM MgEGTA (total volume 0.25 mL) for 30 min at 37 °C. Cells were then centrifuged and absorbance of the supernatant was measured at 541 nm using a plate reader.
2.4. Protein A-fraction purification
Serum was diluted twice in 0.15 M NaCl, 20 mM Na2HPO4, pH 7.4 (binding buffer) and incubated for 1 h at 4 °C with protein-A resin (Genscript, Piscataway, NY) (1:1, volume serum:settled resin). Protein A-resin was centrifuged and washed five times in binding buffer. Proteins (called prot-AF for protein-A fraction) were then eluted by incubating protein-A resin in 0.1 M glycine, pH 2.5 for 30 min at 23 °C. The eluate was then neutralized to pH 7.4 with 1 M Tris–HCl, pH 8.5 buffer. Prot-AF was concentrated using Amicon Ultra Centrifugal Filter Units (Millipore, Billerica, MA). In some experiments, the protein-A resin was eluted with binding buffer containing increasing NaCl concentrations (0.3–1 M NaCl, 10 min elution per buffer) before acidic elution. For depletion of prot-AF from mouse serum, serum was diluted twice in binding buffer and incubated for 1 h at 4 °C with protein-A resin. The protein-A resin was centrifuged, the supernatant was isolated, and the incubation with protein-A resin was repeated twice. Prot-AF-depleted serum was then concentrated twice.
2.5. Electrophoresis and immunoblot
Serum or prot-AF fractions (volume of 5 μL) were mixed in 1x NuPAGE sample reducing agent (Invitrogen) and 1× NuPAGE LDS sample buffer (Invitrogen) and loaded on NOVEX-NuPAGE 4–12% BT gels (Life technologies, Carlsbad, CA). Protein gels were then silver stained using the SilverQuest staining kit (Invitrogen). For immunoblot, proteins were blotted at 160 mA for 1.5 h onto polyvinylidene difluoride membranes (Millipore) using transfer buffer (Invitrogen). Membranes were blocked with 3% BSA and incubated with HRP-conjugated goat anti-mouse IgG antibody (Santa Cruz Biotechnology, Santa Cruz, CA) at 4 °C overnight. Membranes were then rinsed and labeled proteins were detected using the ECL Plus enzymatic chemiluminescence kit (GE Healthcare).
2.6. AQP4-IgG binding to AQP4
CHO-AQP4 cells were incubated with 10 μg/mL AQP4-IgG alone or supplemented with 10% mouse serum or 1 mg/mL mouse prot-AF for 1 h at 23 °C. Cells were then rinsed with PBS, fixed in 4% PFA for 15 min and permeabilized with 0.1% Triton X-100. Cells were blocked in 1% BSA and incubated for 30 min with 0.4 μg/mL polyclonal, C-terminal-specific rabbit anti-AQP4 antibody (Santa Cruz Biotechnology), then rinsed with PBS. Cells were then incubated for 30 min with 4 μg/mL goat anti-human IgG-conjugated Alexa Fluor 555 (to detect AQP4-IgG) and goat anti-rabbit IgG-conjugated Alexa Fluor 488 (to detect AQP4). AQP4-IgG binding to AQP4 was quantified as described (Crane et al., 2011).
2.7. Protein identification using reversed-phase liquid chromatography/electrospray tandem mass spectrometry (LC–MS/MS)
20 μl samples (0.1 μg/μl) were denatured with 8 M urea at pH 7.5 and reduced with 50 mM dithiothreitol at 60 °C for 30 min, followed by alkylation with 50 mM iodoacetamide at room temperature in the dark for 1 h. The sample was then incubated overnight with trypsin (1:20 weight/weight) at 37 °C. The peptides formed from the digestion were analyzed by on-line LC–MS/MS. The LC separation was performed using a NanoAcquity UPLC system (Waters, Milford, MA) on an Easy-Spray PepMap® column (75 μm × 15 cm, Thermo Scientific, Waltham, MA) and the MS/MS analysis was performed using a LTQ Orbitrap XL mass spectrometer (Thermo Scientific). The sequence included one survey scan in the FT mode in the Orbitrap with mass resolution of 30,000 followed by six CID scans in the LTQ, focusing on the six most intense peptide ion signals whose m/z values were not in the dynamically updated exclusion list. The analytical peak lists were generated from the raw data using in-house software, PAVA (Guan et al., 2011). The MS/MS data were searched against the UniProt database using an in-house search engine Protein Prospector (http://prospector.ucsf.edu/prospector/mshome.htm).
2.8. Statistics
Data are presented as mean ± S.E.M. Statistical comparisons were made using the non-parametric Mann–Whitney test when comparing two groups. Data presented here are representative of three independent experiments.
3. Results
3.1. Mouse serum has poor complement activity compared to human, rat and guinea pig sera
We first tested the cytotoxicity of mouse serum (from CD1 strain) in an AQP4-IgG-dependent CDC assay. In this assay CHO cells expressing AQP4-M23 (CHO-AQP4) were incubated with AQP4-IgG and 5% complement-containing serum for 1 h at 23 °C. Cell viability was then measured with AlamarBlue. Fig. 1A shows cytotoxicity of human, rat, guinea pig and mouse serum as a function of AQP4-IgG concentration. Little cytotoxicity was produced by mouse serum under conditions where marked cytotoxicity was produced by human, rat and guinea pig sera. Because AQP4-IgG is a human antibody, we considered the possibility that AQP4-IgG could not activate mouse complement as a consequence of inability of mouse C1q to bind to human Fc. To address this issue a chimeric AQP4-IgG (AQP4-IgGc) was generated containing the variable region of the human AQP4-IgG and the constant region of mouse IgG2a (Fig. 1B). Cytotoxicity of mouse serum was also near-zero for this chimeric antibody, but high for human, rat and guinea pig sera. Fig. 1C shows a live/dead cell staining of CHO-AQP4 cells after incubation with 10 μg/mL AQP4-IgG and 5% sera from different species. In agreement with the AlamarBlue data, mouse serum produced little cytotoxicity compared to human, rat and guinea pig sera.
Fig. 1.

Poor cytotoxicity produced by mouse serum in an assay of AQP4-IgG-dependent CDC. (A) AQP4-IgG-dependent CDC in presence of mouse (CD1 strain), human, guinea pig or rat serum. CHO-AQP4 cells were incubated for 1 h at 23 °C with 5% serum and increasing concentrations of human AQP4-IgG. Cell viability was measured with AlamarBlue (mean ± S.E., n = 3). (B) Cell viability as in A but in presence of the chimeric AQP4-IgGc (containing mouse constant region) (mean ± S.E., n = 3). (C) Live (green)/dead (red) cell staining of CHO-AQP4 cells incubated with 10 μg/mL AQP4-IgG and 5% mouse, human, guinea pig or rat serum. (D) Cell viability in CHO-AQP4 cells incubated with 10 μg/mL AQP4-IgG and 5% human serum or serum from CD1, BUB/Bnj (BUB) or C57BL/6 (C57) mice (mean ± S.E., n = 3). *P < 0.01 compared to mouse sera. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
Earlier studies reported differences in complement activity between different strains of mice, with the BUB/Bnj mice having 2–10-fold greater complement activity than other strains (Ong and Mattes, 1989). Fig. 1D shows low or absent cytotoxicity of sera from CD1, C57BL/6 and BUB/Bnj mice, with data for human serum as positive control. Therefore, the poor AQP4-IgG-dependent CDC produced by mouse complement is not strain-specific. Subsequent experiments were done with serum from CD1 mice.
The cytotoxicity of mouse serum was then tested in AQP4-IgG-independent assays of CDC. The commonly used hemolysis assay was done with IgM-coated sheep erythrocytes as a measure of activity of the classical complement pathway. Fig. 2A shows cell killing of sheep erythrocytes as a function of serum concentration. Mouse serum had very low cytotoxicity compared to human serum under these experimental conditions, which is consistent with the results for AQP4-IgG-dependent CDC which involves the classical complement pathway (Phuan et al., 2013). Fig. 2B shows lysis of uncoated rabbit erythrocytes in Ca2+/Mg2+-free conditions (preventing C1 complex formation) as a measure of activity of the alternative complement pathway. Mouse serum had moderate cytotoxic activity in this assay, albeit reduced compared to human serum.
Fig. 2.
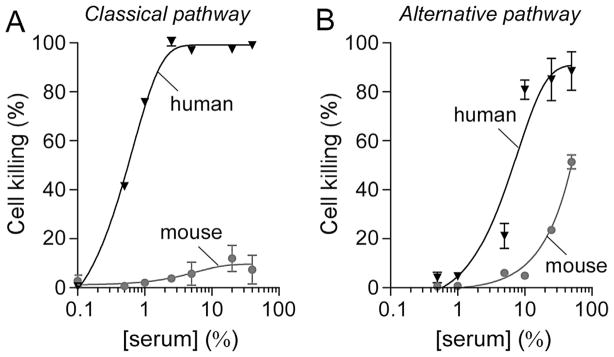
Low classical complement pathway activity of mouse serum. (A) Classical complement pathway activity of mouse and human serum. IgM-coated sheep erythrocytes were incubated for 1 h at 37 °C with increasing concentrations of mouse or human serum. Cells were then centrifuged and cell killing was quantified by measuring absorbance of the supernatant at 541 nm (mean ± S.E., n = 3). (B) Alternative complement pathway activity of mouse and human serum. Uncoated rabbit erythrocytes were incubated for 30 min at 37 °C with increasing concentrations of mouse or human serum. Cell killing was measured as in A (mean ± S.E., n = 3).
3.2. Mouse serum inhibits complement-mediated cytotoxicity of human, rat and guinea pig sera
Because peripheral administration of AQP4-IgG and human complement in mice failed to produce NMO pathology in circumventricular organs (Ratelade et al., 2011), we investigated the effects of mouse serum on complement activation. Fig. 3A shows cell viability of CHO-AQP4 cells after incubation with 10 μg/mL AQP4-IgG, 5% human, rat or guinea pig serum, and increasing concentrations of mouse serum. Remarkably, mouse serum inhibited CDC produced by human, rat and guinea pig complement, with near 100% inhibition at 10% mouse serum. The inhibition of CDC by mouse serum was confirmed with the live/dead cell staining assay in which cells were incubated with 10 μg/mL AQP4-IgG and 5% human serum, with or without 10% mouse serum (Fig. 3B). The presence of mouse serum completely prevented cytotoxicity produced by human serum. Heat-treatment of mouse serum at 60 °C for 30 min reduced its inhibition activity by more than 10-fold (Fig. 3C), suggesting that the complement inhibitor(s) are proteins.
Fig. 3.

Inhibition of AQP4-IgG-dependent CDC by mouse serum. (A) Cell viability of CHO-AQP4 cells, measured as in Fig. 1A, after incubation for 1 h at 23 °C with 10 μg/mL AQP4-IgG, 5% human, rat or guinea pig serum, and increasing concentrations of mouse serum (mean ± S.E., n = 3). (B) Live/dead cell staining of CHO-AQP4 cells incubated for 1 h at 23 °C with 10 μg/mL AQP4-IgG and 5% human serum (HS) and/or 10% mouse serum. (C) Cell viability of CHO-AQP4 cells after incubation with 10 μg/mL AQP4-IgG, 5% human serum, and increasing concentrations of mouse serum (with or without heat-treatment at 60 °C for 30 min) (mean ± S.E., n = 3).
3.3. Complement inhibitor(s) in mouse serum are contained in a protein A-fraction
We next attempted to identify the complement inhibitor(s) present in mouse serum. The most abundant proteins in mouse serum were first tested. Fig. 4A shows that purified mouse albumin, fibrinogen or transferrin did not inhibit cytotoxicity in CHO-AQP4 cells incubated with 10 μg/mL AQP4-IgG and 5% human serum. For comparison with mouse serum the protein concentrations plotted on the abscissa correspond to their concentrations in mouse serum. A prior study reported that mouse fibronectin inhibits human complement (Hitsumoto et al., 1999); however, as seen in Fig. 4A mouse fibronectin did not inhibit cytotoxicity of human complement in the AQP4-IgG-dependent CDC assay. Other studies reported that a mouse complement inhibitor, factor I, could inhibit human complement (Mandle et al., 1977); however, this was not the case as the inhibition activity of serum from factor I-deficient mice was the same as that of serum from wild-type mice (Fig. 4B).
Fig. 4.

Complement inhibitor(s) in mouse serum interact with mouse IgG. (A) Cell viability of CHO-AQP4 cells, measured as in Fig. 1A, after incubation for 1 h at 23 °C with 10 μg/mL AQP4-IgG, 5% human serum, and increasing concentrations of mouse serum (%) or indicated purified mouse serum proteins (expressed as serum-equivalent concentrations on the x-axis) (mean ± S.E., n = 3). (B) Cell viability as in A as a function of serum concentration of sera from wild-type (WT) or factor I-deficient (FI−/−) mice (mean ± S.E., n = 3). (C) Mouse or human serum was incubated with a protein-A resin and the eluted proteins called ‘protein-A fraction’ (prot-AF). Cell viability as in A as a function of human or mouse prot-AF concentration (mean ± S.E., n = 3). (D) Inhibition of complement activity as in A by serum from wild-type (WT) or IgG-deficient (IgG−) mice. Inset, immunoblot for mouse IgG on sera from WT and IgG− mice (HC, heavy chain; LC, light chain). (E) Graph shows viability of CHO-AQP4 cells incubated with 10 μg/mL AQP4-IgG, 5% human serum and/or 1 mg/mL prot-AF from WT or IgG− serum (mean ± S.E., n = 3). Gel on the right shows protein content of prot-AF from WT and IgG− sera.
To investigate the possibility that mouse IgG, the second most abundant serum protein, is the complement inhibitor, mouse IgG was purified using a protein-A resin. The fraction containing IgG and IgG-bound proteins isolated from serum with protein-A is called the ‘protein-A fraction’ (prot-AF). Fig. 4C shows inhibition of human complement cytotoxicity by mouse prot-AF but not by human prot-AF, with a 100% inhibition at 1 mg/mL prot-AF.
As IgG is by far the major constituent of prot-AF, we investigated the possibility that mouse IgG was the complement inhibitor by testing serum from IgG-deficient mice on human complement activity. The absence of IgG in serum of IgG-deficient mice was confirmed by immunoblot (Fig. 4D). Surprisingly, IgG-deficient serum produced the same inhibition of complement activity as serum from wild-type mice. Therefore, mouse IgG is not responsible for the complement inhibition effect of mouse prot-AF.
We isolated prot-AF from IgG-deficient mouse serum and found that at 1 mg/mL it did not inhibit human complement activity, compared with the 100% inhibition produced by prot-AF from sera of wild-type mice (Fig. 4E). Fig. 4E (right) shows the protein content of prot-AF from wild-type and IgG-deficient serum. Several proteins are present with IgG in prot-AF from wild-type serum, whereas prot-AF from IgG-deficient serum contains little protein. The complement inhibitor(s) in prot-AF are thus protein(s) that bind to mouse IgG rather than to protein-A.
3.4. Identification of candidate complement inhibitors
To attempt to identify the complement inhibitor(s) in mouse serum, we performed serial mild elutions of the protein-A column bound with mouse serum using increasing salt concentrations to separate the inhibitor(s) from mouse IgG. Fig. 5A shows the complement inhibition activity of the elution fractions. Nearly all of the inhibition activity was present in the fraction eluted with the 0.3 M salt buffer, as seen by comparison with the last lane (‘acid elution total’) obtained by direct acidic elution. No inhibition activity remained in the 0.5 M fraction. Following the NaCl elutions, an acidic elution was performed to remove remaining IgGs and remaining proteins from protein-A. No inhibition activity remained in this fraction. Fig. 5B shows the protein content of the elution fractions. There was much less protein in the 0.3 M fraction compared to prot-AF, and nearly all mouse IgG was removed. Fig. 5C shows that prot-AF-depleted mouse serum had greatly reduced complement inhibition activity compared to normal serum.
Fig. 5.

Identification of candidate complement inhibitors in mouse serum. (A) Mouse serum was incubated with a protein-A resin for 1 h at 4 °C. Protein-A resin was incubated with buffers with increasing salt concentrations (from 0.3 to 1 M), and finally with an acidic buffer. Graph shows viability of CHO-AQP4 cells incubated with 10 μg/mL AQP4-IgG, 5% human serum (HS), and 10% mouse serum or an equivalent volume of indicated elution fractions from the protein-A resin (mean ± S.E., n = 3). ‘Acid elution total’ represents the protein fraction eluted directly from protein-A resin with the acidic buffer. (B) Protein content of the fractions generated in A. Five μL of each fraction was resolved by SDS-PAGE gel and silver stained. (C) Cell viability as in A of cells incubated with normal mouse serum or serum depleted of prot-AF (mean ± S.E., n = 3).
We performed mass spectrometry on the 0.3 and 0.5 M fractions, reasoning that the inhibitor(s) would be much more abundant in the active 0.3 M fraction than the inactive 0.5 M fraction. Table 1 list the proteins identified by mass spectrometry present in the 0.3 and absent in the 0.5 M fraction or greatly enriched in the 0.3 M fraction compared to 0.5 M fraction. The most abundant proteins are serum albumin and transferrin, which were already ruled out as the complement inhibitors (see Fig. 4A). A few proteins involved in the classical complement pathway activation were identified, several of which are known to interact with IgGs, including components of the C1 complex (C1q, C1r, C1s). Some proteins are involved in the lectin complement pathway (Mbl1, Mbl2, Masp1, Masp2) or in the alternative pathway activation (C3, factor B, properdin). Interestingly, several members of the serpin superfamily (serpin-a1, serpin-a3, serpin-c1) were identified. These proteins are serine protease inhibitors and are candidate complement inhibitors. One member of this family, serpin-g1, also called C1-inhibitor, inhibits the activity of serine proteases C1s and C1r that are required for activation of classical complement pathway (Ricklin et al., 2010).
Table 1.
Candidate complement inhibitors identified by mass spectrometry.
| Accession number | Gene | Number of unique peptides | Coverage % seq | MW | Protein |
|---|---|---|---|---|---|
| Q921I1 | Tf | 25 | 36.9 | 76.7 | Serotransferrin |
| Q566I6 | C1ra | 13 | 24.9 | 80.0 | Complement component 1, r subcomponent |
| P41317 | Mbl2 | 11 | 36.9 | 25.9 | Mannose-binding protein C |
| Q14DT6 | C1s | 10 | 19.9 | 77.4 | Complement component 1, s subcomponent |
| Q61838 | A2m | 9 | 10.6 | 16.5 | Alpha-2-macroglobulin |
| Q8CFG8 | C1sb | 8 | 16.3 | 76.7 | Complement C1s-B subcomponent |
| P26262 | Klkb1 | 8 | 18.8 | 71.3 | Plasma kallikrein |
| Q61147 | Cp | 7 | 11.3 | 12.1 | Ceruloplasmin |
| P01027 | C3 | 6 | 5.1 | 18.6 | Complement C3 |
| P07759 | Serpina3k | 5 | 17.0 | 46.8 | Serine protease inhibitor A3 K |
| P07758 | Serpina1a | 5 | 16.2 | 46.0 | Alpha-1-antitrypsin 1-1 |
| P11680 | Cfp | 5 | 12.7 | 50.3 | Properdin |
| Q02105 | C1qc | 5 | 17.1 | 25.9 | Complement C1q subcomponent subunit C |
| Q6PJA7 | Igh | 4 | 10.6 | 52.2 | Igh protein |
| P22599 | Serpina1b | 4 | 11.9 | 45.9 | Alpha-1-antitrypsin 1-2 |
| Q00897 | Serpina1d | 4 | 11.9 | 45.9 | Alpha-1-antitrypsin 1-4 |
| Q58EV2 | Apoa1 | 4 | 22.2 | 23.0 | Apoa1 protein |
| O08677 | Kng1 | 4 | 5.7 | 73.1 | Kininogen-1 |
| P98064 | Masp1 | 4 | 10.5 | 79.9 | Mannan-binding lectin serine protease 1 |
| P21614 | Gc | 3 | 12.4 | 53.6 | Vitamin D-binding protein |
| P14106 | C1qb | 3 | 11.1 | 26.7 | Complement C1q subcomponent subunit B |
| P23953 | Ces1c | 3 | 4.3 | 61.0 | Carboxylesterase 1C |
| P04186 | Cfb | 3 | 5.0 | 85.0 | Complement factor B |
| P06330 | 2 | 28.0 | 12.9 | Ig heavy chain V region AC38 205.12 | |
| Q569X1 | Ighg | 2 | 7.4 | 52.1 | Ighg protein |
| Q8R3H6 | Ighg | 2 | 7.4 | 51.7 | Ighg protein |
| Q91Z05 | Ighg | 2 | 7.4 | 51.9 | Ighg protein |
| P33622 | Apoc3 | 2 | 27.3 | 10.9 | Apolipoprotein C-III |
| Q91WP0 | Masp2 | 2 | 5.0 | 75.5 | Mannan-binding lectin serine protease 2 |
| P19221 | F2 | 2 | 4.4 | 70.2 | Prothrombin |
| Q19LI2 | A1bg | 2 | 5.7 | 56.5 | Alpha-1B-glycoprotein |
| P01592 | Igj | 1 | 9.4 | 18.0 | Immunoglobulin J chain |
| Q9QWK4 | Cd5l | 1 | 3.1 | 38.8 | CD5 antigen-like |
| Q8CIR9 | Masp1 | 1 | 4.2 | 37.1 | Serine protease MASP3 (Fragment) |
| P39039 | Mbl1 | 1 | 5.9 | 25.3 | Mannose-binding protein A |
| P01746 | 1 | 10.0 | 15.5 | Ig heavy chain V region 93G7 | |
| P20918 | Plg | 1 | 3.3 | 90.8 | Plasminogen |
| P02762 | Mup6 | 1 | 12.8 | 20.6 | Major urinary protein 6 |
| P01680 | 1 | 8.5 | 13.8 | Ig kappa chain V-IV region S107B | |
| P06909 | Cfh | 1 | 0.6 | 13.9 | Complement factor H |
| P32261 | Serpinc1 | 1 | 2.4 | 52.0 | Antithrombin-III |
| P98086 | C1qa | 1 | 5.7 | 25.9 | Complement C1q subcomponent subunit A |
| P09813 | Apoa2 | 1 | 14.7 | 11.3 | Apolipoprotein A-II |
| Q6S9I0 | Kng2 | 1 | 4.2 | 47.8 | Kng2 protein |
| P07724 | Alb | 68/6 | 86.8/13.2 | 68.6 | Serum albumin |
| I6L985 | Igh | 19/11 | 36/21.1 | 51.9 | Igh protein |
| Q9ESB3 | Hrg | 11/1 | 23.2/2.9 | 59.1 | Histidine-rich glycoprotein |
| Q9DCD9 | Gm16844 | 7/1 | 20/3.8 | 45.8 | Putative uncharacterized protein |
| Q58E61 | Gm16844 | 7/1 | 18.1/3.3 | 52.6 | Igh protein |
| P01872 | Igh-6 | 4/1 | 12.3/5.7 | 49.9 | Ig mu chain C region secreted form |
Proteins were identified by mass spectrometry in the 0.3 M salt fraction that were not detected in the 0.5 M fraction, or enriched in the 0.3 M compared to 0.5 M fraction (last 6 rows of the table). Table entries include accession numbers (Uniprot database), gene name, number of peptides detected for each protein, coverage of the protein sequence by the detected peptides (in %), molecular weight (in kDa), and name of the protein. In the last 6 rows, the number of peptides and coverage are listed for the 0.3 and 0.5 M fractions.
3.5. IgG-bound complement inhibitor(s) in mouse serum act on the classical pathway
To investigate the utility of mice for studies of diseases involving the alternative complement pathway, as has been reported (Holers, 2008), we determined whether the IgG-bound complement inhibitor(s) present in prot-AF acted on both the classical and alternative complement pathways. It was first verified that mouse serum did not interfere with AQP4-IgG binding to AQP4, which could have explained the results found earlier. Fig. 6A shows binding of AQP4-IgG (red fluorescence) to CHO-AQP4 cells in presence of mouse serum or prot-AF. Total AQP4 was stained with a C-terminal antibody (green fluorescence), and binding of AQP4-IgG to AQP4 was quantified as a red-to-green fluorescence ratio (as done previously, Crane et al., 2011). AQP4-IgG binding to AQP4 was not affected by mouse serum or prot-AF.
Fig. 6.

Complement inhibitor in mouse serum inhibits the classical pathway. (A) CHO-AQP4 cells were incubated with 10 μg/mL AQP4-IgG alone or supplemented with 10% mouse serum or 1 mg/mL mouse prot-AF for 1 h at 23 °C. Cells were then washed and fixed. AQP4-IgG binding was detected using a secondary anti-human antibody (red) and AQP4 was stained with a C-terminus specific antibody (green). Graph on the right shows AQP4-IgG binding quantified as red-to-green fluorescence ratio (mean ± S.E., n = 5). (B) Percentage of intact sheep (classical pathway) or rabbit (alternative pathway) erythrocytes incubated with 1% (classical) or 10% (alternative) human serum and increasing amounts of mouse prot-AF (mean ± S.E., n = 3). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
Hemolysis assays, as done in Fig. 2, were used to investigate the effect of prot-AF on the classical and alternative pathways. Fig. 6B shows that prot-AF inhibited complement activation by classical pathway (left, IgM-coated sheep erythrocytes), but not by the alternative pathway (right, uncoated rabbit erythrocytes).
4. Discussion
Motivated by the desire to establish mouse models that closely recapitulate human NMO disease in which CDC is a major pathogenic mechanism, we studied the cytotoxicity of mouse complement in AQP4-IgG-dependent CDC assays. We found here that: (i) activity of classical complement pathway in mouse serum ex vivo is very low compared to sera from human, rat and guinea pig; (ii) mouse serum contains inhibitor(s) of complement activation that bind to IgG; and (iii) the inhibitor(s) act mainly on the classical complement pathway. These results have important implications for generation of mouse models of NMO and other diseases involving the classical complement pathway.
The low activity of mouse serum in producing cytotoxicity by the classical complement pathway has been reported previously. Bergman and colleagues reported very low cytotoxicity of mouse complement compared to human and rat complement in a CDC assay involving monoclonal antibody targeting of a surface antigen on tumor cells (Bergman et al., 2000). The classical pathway was the main cytotoxicity mechanism in their assay as addition of EDTA or EGTA (that chelate Ca2+ and Mg2+ ions, inhibiting formation of the C1 complex) abolished cytotoxicity. Another early study reported heterogeneity in complement activity among different mouse strains, with the BUB/Bnj strain having the highest activity of the classical complement pathway (Ong and Mattes, 1989). However, the activity of BUB/Bnj complement was 10-fold lower compared to human complement. Here, the cytotoxicity of BUB/Bnj complement was near-zero, as seen for complement of other mouse strains in the AQP4-IgG-dependent CDC assay. The low cytotoxicity of mouse serum may be the consequence of very low concentrations of complement proteins in mouse serum compared to rat or human sera (Ong and Mattes, 1989). Mouse and rat sera have similar concentrations of C1, but much lower concentrations of C2 and C4, and some other complement proteins. Another study reported that mouse C4 lacks classical pathway C5 convertase subunit activity (Ebanks and Isenman, 1996). An additional issue is that the mouse complement pathway is known to be relatively unstable (Lachmann, 2010). Recognizing this concern, here we followed recommended precautions to preserve complement activity (Lachmann, 2010), and in most studies fresh serum was used in CDC assays. Finally, mouse complement has been reported to be sexually dimorphic, with sera from male mice having higher complement activity than from females (Baba et al., 1984). We found near zero activity of the classical complement pathway in sera from both male and female mice.
Data in humans and experimental rodent models implicate a central role of the complement pathway in the initiation of the NMO pathology. Perivascular deposition of IgG and complement is found in human NMO lesions (Lucchinetti et al., 2002). In mice, intracerebral injection of AQP4-IgG and human complement produces NMO-like pathology (Saadoun et al., 2010). Intracerebral injection in mice of human complement and a modified AQP4-IgG deficient in C1q binding did not produce NMO pathology (Ratelade et al., 2013). Also, co-administration with a C1q-targeted monoclonal antibody prevented the development of NMO pathology in mice (Phuan et al., 2013). Intracerebral injection of AQP4-IgG alone did not produce pathology, nor did peripheral administration of AQP4-IgG even though binding to AQP4 in circumventricular organs was observed (Ratelade et al., 2011). From these previous results and the data here, we conclude that mice are not suitable for NMO models requiring an active endogenous classical complement pathway. We reported recently that intracerebral injection of AQP4-IgG (without added complement) in rats is sufficient to produce NMO pathology (Asavapanumas et al., 2014b), which is in agreement with the finding that rat and human sera have similar high complement activity.
We found here modest activity of the alternative complement pathway in mice, supporting the utility of mice to study diseases involving the alternative pathway. Several mouse models of human diseases involving alternative pathway activation have been reported, including models of rheumatoid arthritis, atypical hemolytic–uremic syndrome, type II membrano-proliferative glomerulonephritis, and macular degeneration (Holers, 2008). Phenotypes of C1q-deficient mice (Botto et al., 1998), such as development of glomerulonephritis, are due to other functions of C1q such as removal of apoptotic cells or clearance of immune complexes rather than defective classical complement cytotoxicity (Nayak et al., 2010). These observations underscore the poor utility of mice in studying pathologies involving the classical complement pathway.
The major finding here is that mouse serum protected AQP4-expressing cells from AQP4-IgG-mediated CDC in presence of human, rat and guinea pig complement. Hemolysis assays showed that the inhibitor(s) act on the classical complement pathway and not on the alternative pathway, suggesting that the inhibitor(s) targets complement proteins of the early classical cascade, including C1, C2 and/or C4. Complement inhibition by mouse serum was reported in an early study and proposed to result from complement inhibitor factor I (Mandle et al., 1977). However, using serum from factor I-deficient mice (Rose et al., 2008) here we found comparable complement inhibition activity with serum from wild-type mice in the AQP4-IgG-dependent CDC assay. In addition, we found that factor I-deficient serum had the same low cytotoxicity as serum from wildtype mice (not shown). Another early study reported inhibition of human complement by mouse fibronectin (Hitsumoto et al., 1999); however, here we found that purified mouse fibronectin did not inhibit human complement. Mass spectrometry allowed us to identify several potential candidates for the mouse inhibitor, among which included members of the serpin (serine protease inhibitor) superfamily such as C1-inhibitor and alpha-1-antitrypsin. The next steps in identification of the specific protein(s) responsible for complement inhibition by mouse serum would be challenging, requiring the isolation or expression of each mouse protein and generation of knockout mice. Though we conclude that mice are not useful models of NMO where active serum complement is required, our results raise the possibility of using mice whose sera are depleted of complement(s) inhibitors and supplemented with human complement. However, the technical challenges and concerns with such a model are difficult to justify as rats already have an active complement system.
5. Conclusions
Though NMO pathology in mice can be produced by direct injection of AQP4-IgG and human complement into the central nervous system, the low activity of the classical complement pathway and the presence of complement inhibitor(s) in mouse serum preclude the use of mice in genetic and other experimental models of NMO where serum complement activity is needed.
Acknowledgments
This work was supported by a grant from the Guthy-Jackson Charitable Foundation, and grants EY13574, DK35124, EB00415 and DK72517 from the National Institutes of Health. We thank Dr. Jeffrey Bennett (Univ. Colorado Denver, Aurora, CO) for providing recombinant monoclonal NMO antibody. Mass spectrometry was provided by the Bio-Organic Biomedical Mass Spectrometry Resource at UCSF (A.L. Burlingame, Director) supported by the Biomedical Technology Research Centers program of the NIH National Institute of General Medical Sciences, NIH NIGMS 8P41GM103481.
Abbreviations
- ADCC
antibody-dependent cellular cytotoxicity
- AQP4
aquaporin-4
- AQP4-IgG
AQP4 immunoglobulin G autoantibody
- AQP4-IgGc
chimeric AQP4-IgG
- CDC
complement-dependent cytotoxicity
- CHO-AQP4 cells
Chinese hamster ovary cells expressing human AQP4-M23
- CNS
central nervous system
- NMO
neuromyelitis optica
- prot-AF
protein-A fraction
Footnotes
URL: http://www.ucsf.edu/verkman (A.S. Verkman).
Authors’ contribution
JR performed the experiments. JR and ASV conceived the study and wrote the manuscript.
References
- de Seze J, Stojkovic T, Ferriby D, Gauvrit JY, Montagne C, Mounier-Vehier F, Verier A, Pruvo JP, Hache JC, Vermersch P. Devic’s neuromyelitis optica: clinical, laboratory, MRI and outcome profile. J Neurol Sci. 2002;197:57–61. doi: 10.1016/s0022-510x(02)00043-6. [DOI] [PubMed] [Google Scholar]
- Jacob A, McKeon A, Nakashima I, Sato DK, Elsone L, Fujihara K, de Seze J. Current concept of neuromyelitis optica (NMO) and NMO spectrum disorders. J Neurol Neurosurg Psychiatry. 2013;84:922–930. doi: 10.1136/jnnp-2012-302310. [DOI] [PubMed] [Google Scholar]
- Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol. 2007;6:805–815. doi: 10.1016/S1474-4422(07)70216-8. [DOI] [PubMed] [Google Scholar]
- Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202:473–477. doi: 10.1084/jem.20050304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujihara K. Neuromyelitis optica and astrocytic damage in its pathogenesis. J Neurol Sci. 2011;306:183–187. doi: 10.1016/j.jns.2011.02.018. [DOI] [PubMed] [Google Scholar]
- Jarius S, Wildemann B. AQP4 antibodies in neuromyelitis optica: diagnostic and pathogenetic relevance. Nat Rev Neurol. 2010;6:383–392. doi: 10.1038/nrneurol.2010.72. [DOI] [PubMed] [Google Scholar]
- Papadopoulos MC. Verkman AS: Aquaporin 4 and neuromyelitis optica. Lancet Neurol. 2012;11:535–544. doi: 10.1016/S1474-4422(12)70133-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett JL, Lam C, Kalluri SR, Saikali P, Bautista K, Dupree C, Glogowska M, Case D, Antel JP, Owens GP, Gilden D, Nessler S, Stadelmann C, Hemmer B. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann Neurol. 2009;66:617–629. doi: 10.1002/ana.21802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratelade J, Asavapanumas N, Ritchie AM, Wemlinger S, Bennett JL, Verkman AS. Involvement of antibody-dependent cell-mediated cytotoxicity in inflammatory demyelination in a mouse model of neuromyelitis optica. Acta Neuropathol. 2013;126:699–709. doi: 10.1007/s00401-013-1172-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent T, Saikali P, Cayrol R, Roth AD, Bar-Or A, Prat A, Antel JP. Functional consequences of neuromyelitis optica-IgG astrocyte interactions on blood–brain barrier permeability and granulocyte recruitment. J Immunol. 2008;181:5730–5737. doi: 10.4049/jimmunol.181.8.5730. [DOI] [PubMed] [Google Scholar]
- Pohl M, Fischer MT, Mader S, Schanda K, Kitic M, Sharma R, Wimmer I, Misu T, Fujihara K, Reindl M, Lassmann H, Bradl M. Pathogenic T cell responses against aquaporin 4. Acta Neuropathol. 2011;122:21–34. doi: 10.1007/s00401-011-0824-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradl M, Misu T, Takahashi T, Watanabe M, Mader S, Reindl M, Adzemovic M, Bauer J, Berger T, Fujihara K, Itoyama Y, Lassmann H. Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann Neurol. 2009;66:630–643. doi: 10.1002/ana.21837. [DOI] [PubMed] [Google Scholar]
- Kinoshita M, Nakatsuji Y, Kimura T, Moriya M, Takata K, Okuno T, Kumanogoh A, Kajiyama K, Yoshikawa H, Sakoda S. Neuromyelitis optica: passive transfer to rats by human immunoglobulin. Biochem Biophys Res Commun. 2009;386:623–627. doi: 10.1016/j.bbrc.2009.06.085. [DOI] [PubMed] [Google Scholar]
- Kimura A, Hsu M, Seldin M, Verkman AS, Scharfman HE, Binder DK. Protective role of aquaporin-4 water channels after contusion spinal cord injury. Ann Neurol. 2010;67:794–801. doi: 10.1002/ana.22023. [DOI] [PubMed] [Google Scholar]
- Saadoun S, Waters P, Bell BA, Vincent A, Verkman AS, Papadopoulos MC. Intracerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain. 2010;133:349–361. doi: 10.1093/brain/awp309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadoun S, Waters P, Macdonald C, Bell BA, Vincent A, Verkman AS, Papadopoulos MC. Neutrophil protease inhibition reduces neuromyelitis optica-immunoglobulin G-induced damage in mouse brain. Ann Neurol. 2012;71:323–333. doi: 10.1002/ana.22686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Verkman AS. Eosinophil pathogenicity mechanisms and therapeutics in neuromyelitis optica. J Clin Invest. 2013;123:2306–2316. doi: 10.1172/JCI67554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tradtrantip L, Ratelade J, Zhang H, Verkman AS. Enzymatic deglycosylation converts pathogenic neuromyelitis optica anti-aquaporin-4 immunoglobulin G into therapeutic antibody. Ann Neurol. 2013;73:77–85. doi: 10.1002/ana.23741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tradtrantip L, Zhang H, Saadoun S, Phuan PW, Lam C, Papadopoulos MC, Bennett JL, Verkman AS. Anti-Aquaporin-4 monoclonal antibody blocker therapy for neuromyelitis optica. Ann Neurol. 2012;71:314–322. doi: 10.1002/ana.22657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asavapanumas N, Ratelade J, Papadopoulos MC, Bennett JL, Levin MH, Verkman AS. Experimental mouse model of optic neuritis with inflammatory demyelination produced by passive transfer of neuromyelitis optica-immunoglobulin G. J Neuroinflamm. 2014a;11:16. doi: 10.1186/1742-2094-11-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Verkman AS. Longitudinally extensive NMO spinal cord pathology produced by passive transfer of NMO-IgG in mice lacking complement inhibitor CD59. J Autoimmun. 2014 doi: 10.1016/j.jaut.2014.02.011. http://dx.doi.org/10.1016/j.jaut.2014.02.011. [DOI] [PMC free article] [PubMed]
- Bettelli E, Baeten D, Jager A, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T and B cells cooperate to induce a Devic-like disease in mice. J Clin Invest. 2006;116:2393–2402. doi: 10.1172/JCI28334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamoorthy G, Lassmann H, Wekerle H, Holz A. Spontaneous opticospinal encephalomyelitis in a double-transgenic mouse model of autoimmune T cell/B cell cooperation. J Clin Invest. 2006;116:2385–2392. doi: 10.1172/JCI28330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratelade J, Bennett JL, Verkman AS. Intravenous neuromyelitis optica autoantibody in mice targets aquaporin-4 in peripheral organs and area postrema. PLoS ONE. 2011;6:e27412. doi: 10.1371/journal.pone.0027412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman I, Basse PH, Barmada MA, Griffin JA, Cheung NK. Comparison of in vitro antibody-targeted cytotoxicity using mouse, rat and human effectors. Cancer Immunol Immunother. 2000;49:259–266. doi: 10.1007/s002620000120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane JM, Lam C, Rossi A, Gupta T, Bennett JL, Verkman AS. Binding affinity and specificity of neuromyelitis optica autoantibodies to aquaporin-4 m1/m23 isoforms and orthogonal arrays. J Biol Chem. 2011;286:16516–16524. doi: 10.1074/jbc.M111.227298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan S, Price JC, Prusiner SB, Ghaemmaghami S, Burlingame AL. A data processing pipeline for mammalian proteome dynamics studies using stable isotope metabolic labeling. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M111.010728. M111 010728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong GL, Mattes MJ. Mouse strains with typical mammalian levels of complement activity. J Immunol Methods. 1989;125:147–158. doi: 10.1016/0022-1759(89)90088-4. [DOI] [PubMed] [Google Scholar]
- Phuan PW, Zhang H, Asavapanumas N, Leviten M, Rosenthal A, Tradtrantip L, Verkman AS. C1q-targeted monoclonal antibody prevents complement-dependent cytotoxicity and neuropathology in in vitro and mouse models of neuromyelitis optica. Acta Neuropathol. 2013;125:829–840. doi: 10.1007/s00401-013-1128-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitsumoto Y, Okada M, Makino H. Inhibition of human and mouse complement-dependent hemolytic activity by mouse fibronectin. Immunopharmacology. 1999;42:203–208. doi: 10.1016/s0162-3109(99)00030-2. [DOI] [PubMed] [Google Scholar]
- Mandle RJ, Jr, McConnell-Mapes JA, Nilsson UR. Inhibition of complement by mouse serum: selective inactivation of the fourth component of human complement. J Immunol. 1977;119:180–186. [PubMed] [Google Scholar]
- Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holers VM. The spectrum of complement alternative pathway-mediated diseases. Immunol Rev. 2008;223:300–316. doi: 10.1111/j.1600-065X.2008.00641.x. [DOI] [PubMed] [Google Scholar]
- Ebanks RO, Isenman DE. Mouse complement component C4 is devoid of classical pathway C5 convertase subunit activity. Mol Immunol. 1996;33:297–309. doi: 10.1016/0161-5890(95)00135-2. [DOI] [PubMed] [Google Scholar]
- Lachmann PJ. Preparing serum for functional complement assays. J Immunol Methods. 2010;352:195–197. doi: 10.1016/j.jim.2009.11.003. [DOI] [PubMed] [Google Scholar]
- Baba A, Fujita T, Tamura N. Sexual dimorphism of the fifth component of mouse complement. J Exp Med. 1984;160:411–419. doi: 10.1084/jem.160.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchinetti CF, Mandler RN, McGavern D, Bruck W, Gleich G, Ransohoff RM, Trebst C, Weinshenker B, Wingerchuk D, Parisi JE, Lassmann H. A role for humoral mechanisms in the pathogenesis of Devic’s neuromyelitis optica. Brain. 2002;125:1450–1461. doi: 10.1093/brain/awf151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asavapanumas N, Ratelade J, Verkman AS. Unique neuromyelitis optica pathology produced in naive rats by intracerebral administration of NMO-IgG. Acta Neuropathol. 2014b;127:539–551. doi: 10.1007/s00401-013-1204-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botto M, Dell’Agnola C, Bygrave AE, Thompson EM, Cook HT, Petry F, Loos M, Pandolfi PP, Walport MJ. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet. 1998;19:56–59. doi: 10.1038/ng0598-56. [DOI] [PubMed] [Google Scholar]
- Nayak A, Ferluga J, Tsolaki AG, Kishore U. The non-classical functions of the classical complement pathway recognition subcomponent C1q. Immunol Lett. 2010;131:139–150. doi: 10.1016/j.imlet.2010.03.012. [DOI] [PubMed] [Google Scholar]
- Rose KL, Paixao-Cavalcante D, Fish J, Manderson AP, Malik TH, Bygrave AE, Lin T, Sacks SH, Walport MJ, Cook HT, Botto M, Pickering MC. Factor I is required for the development of membranoproliferative glomerulonephritis in factor H-deficient mice. J Clin Invest. 2008;118:608–618. doi: 10.1172/JCI32525. [DOI] [PMC free article] [PubMed] [Google Scholar]