

Abstract
Urea transporter (UT) proteins, which include isoforms of UT-A in kidney tubule epithelia and UT-B in vasa recta endothelia and erythrocytes, facilitate urinary concentrating function. Inhibitors of urea transporter function have potential clinical applications as sodium-sparing diuretics, or ‘urearetics,’ in edema from different etiologies, such as congestive heart failure and cirrhosis, as well as in syndrome of inappropriate antidiuretic hormone (SIADH). High-throughput screening of drug-like small molecules has identified UT-A and UT-B inhibitors with nanomolar potency. Inhibitors have been identified with different UT-A versus UT-B selectivity profiles and putative binding sites on UT proteins. Studies in rodent models support the utility of UT inhibitors in reducing urinary concentration, though testing in clinically relevant animal models of edema has not yet been done.
Keywords: Urea transporters, Diuretic, Kidney, High-throughput screening
Introduction
Urea transporter (UT) proteins facilitate the passive transport of urea across the plasma membrane in certain cell types. The involvement of UTs in the generation of concentrated urine by the kidney is the major role of UTs [3, 6, 7, 22, 30]. Urinary concentration involves a countercurrent multiplication mechanism, which is facilitated by aquaporins, the NKCC2 (Na+/K+/2Cl− cotransporter) in the thick ascending limb of the loop of Henle, and urea transporters in tubule epithelia and vasa recta endothelia [20, 24]. On theoretical grounds, loss of UT function is predicted to disrupt urinary concentrating ability [3, 30]
As reviewed in Chap. 5, epithelial cells in kidney tubules express isoforms of UT-A, encoded by the SLc14A2 gene, and endothelial cells in vasa recta express UT-B, encoded by the SLc14A1 gene [4, 10, 21, 25–28]. As diagramed in Fig. 9.1, UT-A1 and UT-A3 are expressed in kidney inner medullary collecting duct, with UT-A1 at the luminal membrane and UT-A3 at the basolateral membrane [10]. UT-A2 is expressed in thin descending limb of the loop of Henle [10]. Knockout mice lacking both UT-A1 and UT-A3 manifest a marked urinary concentrating defect, in large part because of impaired urea transport from tubular fluid in the inner medullary collecting duct to the medullary interstitium [8, 9]. Interestingly, urinary concentrating function is unimpaired in UT-A2 knockout mice [29] and in UT-A1/A3 knockout mice after transgenic replacement of UT-A1 [14]. Knockout mice lacking UT-B [2, 15, 31], and rare humans with loss of function mutations in UT-B [13, 23], which is the erythrocyte Jk antigen, manifest a relatively mild urinary concentrating defect.
This chapter is focused on small-molecule UT inhibitors. Applications of UT inhibitors include research tools and potential drug development candidates. Selective, potent UT inhibitors can be advantageous over gene knockout to study UT functions because of potential confounding compensatory in knockout mice, such as changes in expression of non-UT proteins. As discussed further below, UT inhibitors have potential clinical applications in edema and syndrome of inappropriate antidiuretic hormone (SIADH). Until recently, available UT inhibitors included the non-selective membrane-intercalating agent phloretin and chemical analogs of urea, such as dimethylthiourea, which have millimolar potency [19, 35]. The discovery and characterization of nanomolar-potency small-molecule UT inhibitors is reviewed in this chapter.
Methods to Assay Urea Transport
Older Assays of Urea Transport
Assays of urea transport rely on measurements of urea movement across cell membranes or cell layers, or secondary effects of urea movement on water transport and hence on cell or vesicle/liposome volume. For example, transport of urea across an epithelial cell monolayer grown on a porous filter has been measured from the kinetics of urea appearance on the trans-side of the filter following addition of urea to one side of the filter [11]. Urea concentration measurement requires fluid sampling and an enzymatic, urease-based colorimetic assay, as to date there is no optical indicator of urea concentration. Radiolabeled urea (14C-urea) can be used in place of chemical urea, as used in some older measurements [19, 32]. A similar approach can be used to measure urea transport across cell plasma membranes; however, the rapid urea equilibration time makes the separation of cells from the extracellular solution very challenging. Measurement of secondary cell volume changes in response to urea movement in water-permeable cells can be accomplished by a variety of methods [33], volume-dependent light scattering of small cells such as erythrocytes or membrane vesicles/liposomes being the most popular [11, 33]. Though some of these methods are quite accurate and quantitative, they are technically tedious and hence not suitable for automated highthroughput screening.
High-Throughput Assay of Urea Transport for UT-B Inhibitor Identification
The first high-throughput screening assay for identification of UT inhibitors utilized erythrocytes, which natively express UT-B [16]. The assay, as diagramed in Fig. 11.1a, involves a single-time-point readout of erythrocyte lysis by near-infrared light absorbance. Erythrocytes are preloaded with the urea analog acetamide, which is transported by UT-B at a rate such that the equilibration time for acetamide transport is comparable to that of osmotic water transport [35]. In the assay, imposing a large, outwardly directed gradient of acetamide causes cell swelling, which is limited by UT-B-facilitated acetamide efflux. UT-B inhibition slows acetamide efflux and increases cell lysis. However, the Imax values from the erythrocyte lysis assay are not absolute inhibition rates of urea transport, because of nonlinearity between acetamide permeability and percentage erythrocyte lysis rate, and differences between acetamide and urea in their transport by UT-B [35].
Fig. 11.1.

Assays for high-throughput identification of small-molecule UT inhibitors. a Erythrocyte osmotic lysis assay for UT-B inhibitor discovery. Erythrocytes expressing water and urea channels (AQP1 and UT-B) are preloaded with the urea analog acetamide. Following replacement of the external buffer with urea/acetamide-free isomolar solution, water entry results in cell swelling, which is limited by UT-B-mediated urea/acetamide efflux. Under optimized assay conditions, UT-B-facilitated urea/acetamide prevents osmotic lysis, whereas UT-B inhibition impairs urea/acetamide exit resulting in substantial lysis. (Bottom) Biphasic cell volume changes in the lysis assay. Increased erythrocyte volume beyond a threshold results in lysis. The dashed curve shows the hypothetical time course of erythrocyte volume if lysis had not occurred. b (Top) Stopped-flow measurements of urea transport in human erythrocytes. Concentration–inhibition curves for indicated compounds determined by light scattering in response to a 100 mM inwardly directed urea gradient. (Bottom) Numerically simulated inhibitor concentration dependence used to determine IC50 from stopped-flow experiments. The inverse of normalized cell volume, Vo/V(t), is plotted to approximate light scattering data at indicated percentages of urea transport inhibition. c (Left) Assay for UT-A1 inhibitors. MDCK cells stably expressing UT-A1, AQP1, and YFP-H148Q/V163S were subjected to an 800 mM inwardly directed urea gradient. A rapid decrease in cell volume (reduced fluorescence) due to the water efflux through AQP1 is followed by cell reswelling (increased fluorescence) due to urea and water influx. The UT-A1 inhibitor phloretin alters curve shape. (Right) UT-A1 and AQP1 immunofluorescence of the triply transfected cells, shown with YFP fluorescence. Adapted from [5, 16]
Conditions were optimized for high-throughput screening to give a robust assay with high sensitivity and a low false-positive rate, including empirical selection of the optimal acetamide loading concentration, mixing conditions, and wavelength for absorbance readout. Greater erythrocyte lysis is seen as reduced optical density at 710 nm (O.D.710). The goodness of the optimized assay was evaluated by screening plates containing positive and negative controls (0 and 100 % lysis), which gave a good statistical Z′-factor of ~0.6 for the screen. As discussed further below, the erythrocyte lysis assay has been used successfully to identify inhibitors of human and rodent UT-B.
Stopped-flow light scattering is the gold standard for secondary analysis of UT-B inhibition and quantitative determination of IC50 values. A suspension of erythrocytes is mixed rapidly (<1 ms) with a urea-containing solution to create an inwardly directed urea gradient. A non-saturating concentration of urea is used to minimize competitive effects that would confound IC50 interpretation. The inwardly directed urea gradient causes initial osmotic water efflux and erythrocyte cell shrinking, which is followed by coupled urea and water influx. Scattered light intensity at 90° provides a quantitative measure of cell volume. Figure 11.1b (top) shows light scattering curves for different concentrations of a UT-B inhibitor, showing progressive slowing of the phase of decreasing light scattering with increased inhibitor concentration. To deduce the percentage inhibition from the light scattering data, the results are compared with computational prediction that involves numerical integration of the Kedem–Katchalsky flux equations for coupled erythrocyte water and urea transport (Fig. 11.1b, bottom), as described [33].
High-Throughput Assay of Urea Transport for UT-A Inhibitor Identification
The challenges in the development of a high-throughout assay to identify UT-A inhibitors included the lack of easily obtained cell lines that natively express UT-A, the rapidity of UT-A-facilitated urea equilibration across cell membranes, and the difficulty in robust measurement of cell volume using an automated screening platform. As diagramed in Fig. 11.1c, our assay for identification of UT-A1 inhibitors involved measurement of cell volume changes in response to a rapidly imposed gradient of urea in MDCK cells stably expressing UT-A1 [5, 11]. Cell volume was followed using the chloride-sensing, genetically encoded fluorescent protein YFP-H148Q/V163S, which was developed previously for chloride channel screening [12]. Reduced cell volume concentrates intracellular chloride, producing a near-instantaneous reduction in YFP fluorescence. The cells also stably express a water channel (AQP1), so that osmotic water equilibration time is much faster than urea equilibration time. Figure 11.1c (right) shows fluorescence micrographs of the triply transfected cells used for screening, showing plasma membrane expression of AQP1 (red) and UT-A1 (blue), and cytoplasmic YFP expression (green). Addition of urea to the extracellular solution in a platereader format drives osmotic water efflux and cell shrinking, which is followed by urea (and water) entry with return to the original cell volume. A urea concentration gradient of 800 mM was chosen empirically to produce a robust fluorescence signal for screening. Original data from 96-well plates in Fig. 11.1c (center) show a robust difference in fluorescence curve shape with UT-A1 inhibition by phloretin. The same assay paradigm can be used for other UT isoforms, and because the fluorescence signal comes only from transfected cells, a transient transfection approach can be used in which AQP1-expressing cells are costably transfected with vectors encoding UT and YFP.
UT-B Inhibitors
An initial screen of 50,000 diverse, small-molecule drug-like compounds was done using human erythrocytes based on UT-B-facilitated acetamide transport as described in Fig. 11.1a. Primary screening yielded ~30 UT-B inhibitors belonging to the phenylsulfoxyoxozole (Fig. 11.2a), benzenesulfonanilide, phthalazinamine, and aminobenzimidazole chemical classes [16]. Analysis of ~700 chemical analogs of these four scaffolds gave many active compounds, the most potent of which inhibited UT-B urea transport with IC50 ~10 nM, with ~100 % inhibition at higher concentrations. The compounds were characterized and used to confirm water transport through UT-B, which was reported in our earlier studies using erythrocytes from UT-B and UT-B/AQP1 knockout mice [33]. Though the potency of the best compound was excellent, it was not further developed because of its (i) relatively low potency for rodent UT-B, precluding testing in rodent models; (ii) high UT-B versus UT-A specificity, which is predicted to produce relatively minor benefit in vivo; and (iii) its rapid metabolism in hepatic microsomes, making it difficult to maintain therapeutic concentrations in vivo.
Fig. 11.2.

UT-B inhibitor identification by high-throughput screening. a Chemical structures of two classes of UT-B inhibitors identified from screens. b Concentration–inhibition data with fit to single-site inhibition model. c Docking of UTBinh-14 in a homology structural model of human UT-B showing UTBinh-14 binding at the cytoplasmic surface. The computed site of urea analog methylurea (a urea analog) is shown. (Inset) Zoomed-in view of UTBinh-14 bound in a groove at the UT-B channel region. d Urine osmolality in wild-type mice following dDAVP (1 μg/kg) and UTBinh-14 (300 μg) (or vehicle) (mean ± S.E., 6 mice per group, * P < 0.01). Same protocol as in B, but in UT-B knockout mice. Adapted from [1, 34]
In a follow-on study, 100,000 compounds were screened using mouse erythrocytes with the goal of identifying potent inhibitors of rodent (and human) UT-B [34]. The screen produced triazolothienopyrimidine UT-B inhibitors, with the most potent compound being 3-(4-ethyl-benzenesulfonyl)-thieno[2,3-e][1,2,3]triazolo[1,5-a]pyrimidin-5-yl]-thiophen-2-ylmethylamine (UTBinh-14, Fig. 11.2a). A 5-step synthesis procedure for UTBinh-14 was developed to generate highly pure compound for analytical and in vivo studies. UTBinh-14 fully and reversibly inhibited urea transport with IC50 of ~10 nM for human UT-B and ~25 nM for mouse UT-B (Fig. 11.2b). UTBinh-14 was highly selective against UT-B versus UT-A isoforms. Competition studies showed reduced inhibition potency with increasing urea concentration, suggesting that UTBinh-14 bound to the UT-B protein near the urea binding site, which was supported by homology modeling and molecular docking computations (Fig. 11.2c).
To study in vivo effects of UTBinh-14 on urinary concentrating function, compound administration dose, route, and timing were established, from liquid chromatography/mass spectrometry assays, to maintain predicted therapeutic compound concentrations in blood, kidney, and urine [34]. Following intraperitoneal administration of UTBinh-14 in mice to achieve therapeutic concentrations in kidney, urine osmolality following dDAVP in UTBinh-14-treated mice was ~700 mosm/kg H2O lower than in vehicle-treated mice (Fig. 11.2d). UTBinh-14 did not significantly reduce urine osmolality in UT-B knockout mice, as expected. UTBinh-14 also increased urine output and reduced osmolality in mice given free access to water. Though these data provided proof of concept for the potential utility of UT-B inhibition to reduce urinary concentration in a high-vasopressin state, the reduction in urine osmolality was relatively modest and similar to that conferred by UT-B gene deletion, supporting the greater importance of UT-A versus UT-B in urinary concentrating function.
Two medicinal chemistry studies were done to further optimize UTBinh-14 properties, with focused investigation of structure–activity relationships with the goal of increasing UTBinh-14 metabolic stability [1, 18]. Systematic chemical analysis indicated a major role of CH2 hydroxylation in the ethyl substituent in UTBinh-14 metabolic stability. By replacing the CH2 hydrogens with fluorines in {3-[4-(1,1-difluoroethyl)-benzenesulfonyl]-thieno[2,3-e][1,2,3]triazolo[1,5-a]pyrimidin-5-yl}-thiophen-2-ylmethylamine, metabolic stability was ~40-fold improved with little effect on UT-B inhibition potency. The optimized UT-B inhibitor accumulated in kidney and urine in mice, and reduced maximum urinary concentration.
UT-A Inhibitors
The UT-A inhibition assay described in Fig. 11.1c was used to screen 100,000 synthetic small molecules for UT-A1 inhibition [5]. The screen was done on UT-A1 because it is predicted that this UT-A isoform is of the greatest importance for urinary concentration as it is the rate-limiting step in apical membrane urea transport in the inner medullary collecting duct and hence required to establish the hypermolar renal medullary interstitium. The initial screen produced four classes of compounds with low micromolar IC50, with an example of a dose–response study in Fig. 11.3a and chemical structures shown in Fig. 11.3b. Interestingly, the class D compounds have the same triazolothienopyrimidine scaffold as in UTBinh-14. Approximately 500 analogs were tested to establish structure–activity relationships.
Fig. 11.3.
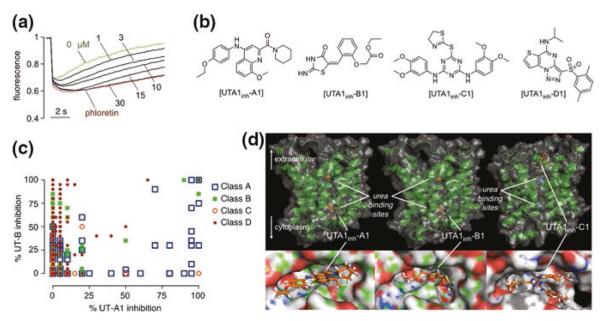
UT-A1 inhibitor identification by high-throughput screening. a Concentration-dependent inhibition of UT-A1 urea transport by compound UTA1inh-A1, using the primary screening assay described in Fig. 13.2c. b Chemical structures of UT-A1 inhibitors of four chemical classes. c UT-A1/UT-B selectivity. Percentage UT-B inhibition (y-axis) and UT-A1 inhibition (x-axis) for active compounds of four chemical classes tested at 25 μM. d Computational modeling of urea transporter—inhibitor interaction. Putative inhibitor binding sites in rat UT-A1 showing zoomed-in and zoomed-out representations of UTA1inh-A1 (left) and UTA1inh-B1 (center) bound to the UT-A1 cytoplasmic domain, and UTA1inh-C1 (right) bound to the extracellular domain. Positions of putative urea binding sites deduced from homology modeling are indicated. Adapted from [5]
Each of the four chemical classes contained multiple active compounds with drug-like properties, including the presence of multiple hydrogen bond acceptors, as well as favorable molecular weight, aLogP, and topological polar surface areas. UT-A1 inhibition by each of the compound classes was reversible. Inhibition by compounds of classes A, B, and C was non-competitive, as judged from the minimal effect of urea concentration on apparent IC50, whereas class D compounds showed partial competition with urea. Inhibition by compounds of classes A, B, and D occurred over several minutes, suggesting an intracellular site of action on UT-A1, whereas inhibition by class C compounds was very rapid, suggesting an extracellular site of action. An interesting finding was the identification of compounds with a wide range of UT-A1 versus UT-B selectivities, as shown in Fig. 11.3c. Even in the same chemical class, relatively minor chemical modifications produced compounds with very high selectivity and others with little UT-A1 versus UT-B selectivity.
Homology modeling and computational docking was done to investigate potential inhibitor binding sites and selectivity mechanisms. UT-A homology modeling was done using the homologous bovine UT-B bound to selenourea that was solved at 2.5 Å solution (PDB = 4EZD) [5]. Fig. 11.3d shows putative intracellular binding sites of class A and B inhibitors and an extracellular binding site for the class C inhibitors. While binding information based on homology models should be interpreted with caution, we note that docking computations were successful in identifying the most active inhibitors (comparing to tested analogs) for each of the class A, B, and D compounds, and the predicted order of class B compounds for UT-A1 inhibition potency from computation was in reasonable agreement with experimental data.
In Vivo Rat Studies
A compound (called PU-14) with thienoquinolin core structure was found to have inhibition activity on both UT-A and UT-B [17]. Figure 11.4a shows the chemical structure of the compound PU-14, 1-(3-amino-6-methylthieno [2, 3-b] quinolin-2-yl) ethanone. PU-14 inhibited human, rat, and mouse UT-B (Fig. 11.4b) as determined by the erythrocyte lysis assay. PU-14 did not change erythrocyte lysis rate in UT-B null mouse erythrocytes, as expected. PU-14 also inhibited UT-A1 (Fig. 11.4c) in an MDCK cell assay [11].
Fig. 11.4.

Urea transporter inhibition and diuretic activity of thienoquinolin (PU-14). a Chemical structure of PU-14. b Dose-dependent UT-B inhibition of PU-14, determined by the erythrocyte osmotic lysis assay. c PU-14 of rat UT-A1 measured in stably transfected MDCK cells. Where indicated, phloretin (0.7 mM) or PU-14 (4 μM) was present. d Diuretic activity of PU-14. Rats were subcutaneously injected with indicated amounts of PU-14 just after a 2-h urine collection (time 0). Urine samples were collected every 2 h. e Long-term diuretic effect of PU-14. Rats were subcutaneously injected with PU-14 at 50 mg/kg for 7 days. f Concentration of osmoles, urea, and non-urea solutes in the inner medulla of rats without (control) or with PU-14 treatment fed water ad libitum. Adapted from [17]
To evaluate in vivo activity of PU-14 on urinary concentrating function, rats fed ad libitum were studied in metabolic cages. Urine output significantly increased in a dose-dependent manner in rats subcutaneously administered PU-14 at 12.5, 50, and 100 mg/kg (Fig. 11.4d). Urinary osmolality and urea concentration were significantly decreased. The peak changes of urine output, urinary osmolality, and urinary urea concentration occurred betwn 2 and 4 h after PU-14 administration, with values returning to baseline by 10 h.
The long-term effect of PU-14 on urinary concentrating ability was also studied. PU-14 at 50 mg/kg was subcutaneously injected every 6 h. As shown in Fig. 11.4e, the 24-h urine output in PU-14-treated rats was significantly higher than that in vehicle control rats. Urinary osmolality and urea concentration in PU-14-treated rats were significantly lower than in vehicle control rats. However, the excretion of osmoles, urea, and non-urea solutes was similar in PU-14-treated and vehicle control rats, suggesting that PU-14 caused a urea-selective diuresis without disturbing electrolyte metabolism. The progressively greater diuretic effect of PU-14 over days may be due to PU-14 accumulation in kidney.
Figure 11.4f shows the compositions of the aqueous component of the inner medulla as measured on supernatants of centrifuged homogenates. Total osmolalities were significantly lower in inner medullary tissue of PU-14-treated rats than those in vehicle control rats, which was primarily because of the reduced inner medullary urea concentration. The concentration of non-urea solutes was similar with those in vehicle control rats. There was no significant difference in blood Na+, K+, Cl−, urea, glucose, total cholesterol, and triglyceride after 7-day treatment with PU-14. These data suggest that PU-14 produces urea-selective diuresis without disturbing electrolyte excretion and metabolism.
Potential Clinical Indicators of Urea Transport Inhibitors
UT inhibitors, particularly of UT-A1, have several potential clinical indications. Urea transport inhibitors have a different mechanism of action from conventional diuretics, which block salt transport across kidney tubule epithelial cells. Diuretics such as furosemide are used widely to increase renal salt and water clearance in conditions associated with total body fluid overload, including congestive heart failure, cirrhosis, and nephrotic syndrome. By disrupting countercurrent mechanisms and intrarenal urea recycling, urea transport inhibitors, alone or in combination with conventional diuretics, may induce a diuresis in states of refractory edema where conventional diuretics are ineffective.
Summary and Future Directions
High-throughput screening has produced multiple chemical classes of small-molecule inhibitors of mammalian UTs, some with low nanomolar potency and UT-isoform selectivity. The UT inhibitors that have emerged from screening have many orders of magnitude greater inhibition potency than prior inhibitors. They should be useful as research tools to study the role of UTs in urinary concentrating function and in extrarenal tissues where they are expressed, and as drug development candidates. However, many challenges remain in the clinical development of UT inhibitors, including demonstration of efficacy in clinically relevant models of refractory edema and SIADH, and in medical chemistry in the selection of inhibitors with appropriate pharmacological properties.
Acknowledgments
Supported by National Institutes of Health grants DK101373, DK035124, DK072517, EB000415, and EY013574 (ASV), and National Natural Science Foundation of China grants 30500171, 30870921, 31200869, 81261160507, and 81170632, Drug Discovery Program grant 2009ZX09301-010-30, the Research Fund for the Doctoral Program of Higher Education 20100001110047, the 111 project, International Science & Technology Cooperation Program of China 2012DFA11070 (BY).
References
- 1.Anderson MO, Zhang J, Liu Y, Yao C, Phuan PW, Verkman AS. Nanomolar potency and metabolically stable inhibitors of kidney urea transporter UT-B. J Med Chem. 2012;55:5942–5950. doi: 10.1021/jm300491y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bankir L, Chen K, Yang B. Lack of UT-B in vasa recta and red blood cells prevents urea-induced improvement of urinary concentrating ability. Am J Physiol Renal Physiol. 2004;286:F144–F151. doi: 10.1152/ajprenal.00205.2003. [DOI] [PubMed] [Google Scholar]
- 3.Bankir L, Yang B. New insights into urea and glucose handling by the kidney, and the urine concentrating mechanism. Kidney Int. 2012;81:1179–1198. doi: 10.1038/ki.2012.67. [DOI] [PubMed] [Google Scholar]
- 4.Doran JJ, Klein JD, Kim YH, Smith TD, Kozlowski SD, Gunn RB, Sands JM. Tissue distribution of UT-A and UT-B mRNA and protein in rat. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1446–R1459. doi: 10.1152/ajpregu.00352.2004. [DOI] [PubMed] [Google Scholar]
- 5.Esteva-Font C, Phuan PW, Anderson MO, Verkman AS. A small molecule screen identifies selective inhibitors of urea transporter UT-A. Chem Biol. 2013;20:1235–1244. doi: 10.1016/j.chembiol.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fenton RA. Urea transporters and renal function: lessons from knockout mice. Curr Opin Nephrol Hypertens. 2008;17:513–518. doi: 10.1097/MNH.0b013e3283050969. [DOI] [PubMed] [Google Scholar]
- 7.Fenton RA. Essential role of vasopressin-regulated urea transport processes in the mammalian kidney. Pflugers Arch. 2009;458:169–177. doi: 10.1007/s00424-008-0612-4. [DOI] [PubMed] [Google Scholar]
- 8.Fenton RA, Chou CL, Stewart GS, Smith CP, Knepper MA. Urinary concentrating defect in mice with selective deletion of phloretin-sensitive urea transporters in the renal collecting duct. Proc Natl Acad Sci USA. 2004;101:7469–7474. doi: 10.1073/pnas.0401704101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fenton RA, Flynn A, Shodeinde A, Smith CP, Schnermann J, Knepper MA. Renal phenotype of UT-A urea transporter knockout mice. J Am Soc Nephrol. 2005;16:1583–1592. doi: 10.1681/ASN.2005010031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fenton RA, Stewart GS, Carpenter B, Howorth A, Potter EA, Cooper GJ, Smith CP. Characterization of mouse urea transporters UT-A1 and UT-A2. Am J Physiol Renal Physiol. 2002;283:F817–F825. doi: 10.1152/ajprenal.00263.2001. [DOI] [PubMed] [Google Scholar]
- 11.Frohlich O, Klein JD, Smith PM, Sands JM, Gunn RB. Regulation of UT-A1-mediated transepithelial urea flux in MDCK cells. Am J Physiol Cell Physiol. 2006;291:C600–C606. doi: 10.1152/ajpcell.00413.2005. [DOI] [PubMed] [Google Scholar]
- 12.Galietta LJ, Haggie PM, Verkman AS. Green fluorescent protein-based halide indicators with improved chloride and iodide affinities. FEBS Lett. 2001;499:220–224. doi: 10.1016/s0014-5793(01)02561-3. [DOI] [PubMed] [Google Scholar]
- 13.Klein JD, Blount MA, Sands JM. Molecular mechanisms of urea transport in health and disease. Pflugers Arch. 2012;464:561–572. doi: 10.1007/s00424-012-1157-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klein JD, Frohlich O, Mistry AC, Kent KJ, Martin CF, Sands JM. Transgenic mice expressing UT-A1, but lacking UT-A3, have intact urine concentration ability. FASEB J. 2013;27:1111.17. (Experimental Biology abstract) [Google Scholar]
- 15.Lei T, Zhou L, Layton AT, Zhou H, Zhao X, Bankir L, Yang B. Role of thin descending limb urea transport in renal urea handling and the urine concentrating mechanism. Am J Physiol Renal Physiol. 2011;301:F1251–F1259. doi: 10.1152/ajprenal.00404.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levin MH, de la Fuente R, Verkman AS. Urearetics: a small molecule screen yields nanomolar potency inhibitors of urea transporter UT-B. FASEB J. 2007;21:551–563. doi: 10.1096/fj.06-6979com. [DOI] [PubMed] [Google Scholar]
- 17.Li F, Lei T, Zhu J, Wang W, Sun Y, Chen J, Dong Z, Zhou H, Yang B. A novel small-molecule thienoquinolin urea transporter inhibitor acts as a potential diuretic. Kidney Int. 2013;83:1076–1086. doi: 10.1038/ki.2013.62. [DOI] [PubMed] [Google Scholar]
- 18.Liu Y, Esteva-Font C, Yao C, Phuan PW, Verkman AS, Anderson MO. 1,1-Difluoroethyl-substituted triazolothienopyrimidines as inhibitors of a human urea transport protein (UT-B): new analogs and binding model. Bioorg Med Chem Lett. 2013;23:3338–3341. doi: 10.1016/j.bmcl.2013.03.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mayrand RR, Levitt DG. Urea and ethylene glycol-facilitated transport systems in the human red cell membrane. Saturation, competition, and asymmetry. J Gen Physiol. 1983;81:221–237. doi: 10.1085/jgp.81.2.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pannabecker TL. Comparative physiology and architecture associated with the mammalian urine concentrating mechanism: role of inner medullary water and urea transport pathways in the rodent medulla. Am J Physiol Regul Integr Comp Physiol. 2013;304:R488–R503. doi: 10.1152/ajpregu.00456.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sands JM. Renal urea transporters. Curr Opin Nephrol Hypertens. 2004;13:525–532. doi: 10.1097/00041552-200409000-00008. [DOI] [PubMed] [Google Scholar]
- 22.Sands JM. Critical role of urea in the urine-concentrating mechanism. J Am Soc Nephrol. 2007;18:670–671. doi: 10.1681/ASN.2006121314. [DOI] [PubMed] [Google Scholar]
- 23.Sands JM, Gargus JJ, Frohlich O, Gunn RB, Kokko JP. Urinary concentrating ability in patients with Jk(a-b-) blood type who lack carrier-mediated urea transport. J Am Soc Nephrol. 1992;2:1689–1696. doi: 10.1681/ASN.V2121689. [DOI] [PubMed] [Google Scholar]
- 24.Sands JM, Layton HE. The physiology of urinary concentration: an update. Semin Nephrol. 2009;29:178–195. doi: 10.1016/j.semnephrol.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shayakul C, Clémençon B, Hediger MA. The urea transporter family (SLC14): physiological, pathological and structural aspects. Mol Aspects Med. 2013;34:313–322. doi: 10.1016/j.mam.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 26.Shayakul C, Hediger MA. The SLC14 gene family of urea transporters. Pflugers Arch. 2004;447:603–609. doi: 10.1007/s00424-003-1124-x. [DOI] [PubMed] [Google Scholar]
- 27.Smith CP. Mammalian urea transporters. Exp Physiol. 2009;94:180–185. doi: 10.1113/expphysiol.2008.043042. [DOI] [PubMed] [Google Scholar]
- 28.Tsukaguchi H, Shayakul C, Berger UV, Tokui T, Brown D, Hediger MA. Cloning and characterization of the urea transporter UT3: localization in rat kidney and testis. J Clin Invest. 1997;99:1506–1515. doi: 10.1172/JCI119313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Uchida S, Sohara E, Rai T, Ikawa M, Okabe M, Sasaki S. Impaired urea accumulation in the inner medulla of mice lacking the urea transporter UT-A2. Mol Cell Biol. 2005;25:7357–7363. doi: 10.1128/MCB.25.16.7357-7363.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang B, Bankir L. Urea and urine concentrating ability: new insights from studies on mice. Am J Physiol Renal Physiol. 2005;288:F881–F896. doi: 10.1152/ajprenal.00367.2004. [DOI] [PubMed] [Google Scholar]
- 31.Yang B, Bankir L, Gillespie A, Epstein CJ, Verkman AS. Urea-selective concentrating defect in transgenic mice lacking urea transporter UT-B. J Biol Chem. 2002;277:10633–10637. doi: 10.1074/jbc.M200207200. [DOI] [PubMed] [Google Scholar]
- 32.Yang B, Verkman AS. Urea transporter UT3 functions as an efficient water channel. Direct evidence for a common water/urea pathway. J Biol Chem. 1998;273:9369–9372. doi: 10.1074/jbc.273.16.9369. [DOI] [PubMed] [Google Scholar]
- 33.Yang B, Verkman AS. Analysis of double knockout mice lacking aquaporin-1 and urea transporter UT-B. Evidence for UT-B-facilitated water transport in erythrocytes. J Biol Chem. 2002;277:36782–36786. doi: 10.1074/jbc.M206948200. [DOI] [PubMed] [Google Scholar]
- 34.Yao C, Anderson MO, Zhang J, Yang B, Phuan PW, Verkman AS. Triazolothienopyrimidine inhibitors of urea transporter UT-B reduce urine concentration. J Am Soc Nephrol. 2012;23:1210–1220. doi: 10.1681/ASN.2011070751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao D, Sonawane ND, Levin MH, Yang B. Comparative transport efficiencies of urea analogues through urea transporter UT-B. Biochim Biophys Acta. 2007;1768:1815–1821. doi: 10.1016/j.bbamem.2007.04.010. [DOI] [PubMed] [Google Scholar]