

Abstract
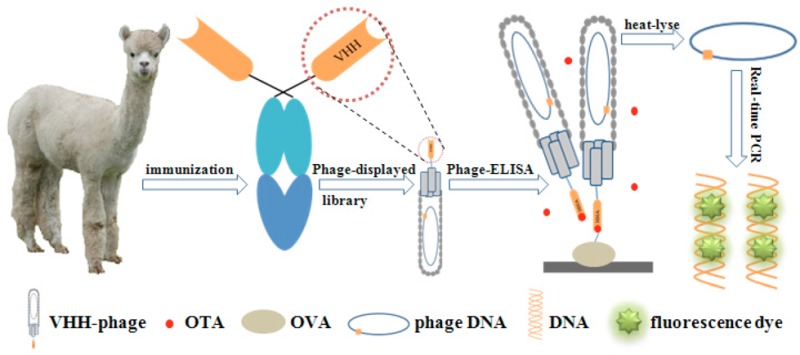
Phage display-mediated immuno-polymerase chain reaction (PD-IPCR) is an ultrasensitive detection technology that combines the advantages of immuno-PCR and phage display. The phage particle, which displayed antibody fragments including single-chain fragment variable (scFv), variable domain of heavy-chain antibodies (VHH), and antigen-binding fragment (Fab) on the surface can be directly used in IPCR, supplying both the detection antibody and deoxyribonucleic acid (DNA) template. In this work, we used ochratoxin A (OTA) as a model system to study the capacity of PD-IPCR in the detection of toxic small molecular weight compounds, especially mycotoxins. An alpaca-derived VHH library was constructed and subjected to four cycles of panning. In total, 16 clones with four unique sequences were selected by competitive binding with OTA. The clone VHH-28 resulted in the lowest 50% inhibitory concentration of 0.31 ng/mL in the phage enzyme-linked immunosorbent assay (ELISA) and was selected to develop the VHH phage-based real-time immuno-PCR (RT-IPCR). The detection limit of the VHH phage-based RT-IPCR was 3.7 pg/L, with a linear range of 0.01–1000 pg/mL. This method was compared with conventional ELISA, and validation results indicated the reliability of VHH phage-based RT-IPCR in the detection of OTA in cereal samples. This study provides a new idea for the ultrasensitive detection of mycotoxins and other toxic small molecular weight compounds.
Ochratoxin A (OTA), a secondary metabolite of several Aspergillus and Penicillium fungal species,1 is a common food contaminant that can enter the human body through the consumption of improperly stored food products. OTA is a potent toxin that can produce nephrotoxic, teratogenic, carcinogenic, neurotoxic, and immunosuppressive activity.2−6 In humans, OTA is classified as a possible carcinogen (group 2B) by the International Agency for Research on Cancer (IARC).7 OTA contamination occurs worldwide,8−12 which seriously threatens public health. Hence, there is an urgent need in the food industry for sensitive, specific, and rapid methods to monitor for the presence of OTA.
Many studies have been performed to develop methods for OTA determination, including immunoassay, instrumental analysis, and combined methods.13−16 Neverthless, instrumental methods are time-consuming and expensive for sample preparation and analysis. Alternatively, immunoassays, such as enzyme-linked immunosorbent assay (ELISA), are generally used to screen a mass of samples within a relatively short time. With the advantages of easy preparation and lack of toxicity, phage have been widely applied for mycotoxin detection as a reagent in ELISA.17−19 Phage have also been reported to be very suitable for immuno-PCR,20−23 a powerful technology combining the high specifity and ultrasensitivity of PCR.
Phage display-mediated immuno-polymerase chain rection (PD-IPCR), first described by Zhang and co-workers,20 is a highly promising technique for ultrasensitive analysis of antigens. The phage-displayed antibody fragment (VHH, scFv, Fab) and phage DNA can directly act as the detection antibody and PCR template, respectively. Therefore, it can avoid the time-consuming and costly preparation of a monoclonal antibody–reporter DNA conjugate, which is required in conventional IPCR. It has been reported that a noncompetitive format can be used for the detection of molecules in PD-IPCR, including noncompetitive phage anti-immunocomplex real-time (RT) PCR21 and phage-based open-sandwich immuno-PCR.24 However, there are no reports on the competitive format of PD-IPCR. In this study, we report the generation of OTA-specific VHH phage from an immunized alpaca VHH library and the application of VHH phage-based competitive RT-IPCR for ultrasensitive detection of OTA in cereal samples.
Materials and Methods
Chemicals and Ragents
All organic solvents and inorganic chemicals were of reagent grade. Ochratoxin A, fumonisin B1, aflatoxin B1, deoxynivalenol, and zearalenone standards were obtained from Fermentek Ltd. (Jerusalem, Israel). Ochratoxin B was from BioAustralis (Smithfield, NSW, AUS). Keyhole limpet hemocyanin (KLH) and ovalbumin (OVA) were purchased from Sigma Chemical Co. (St. Louis, MO). T4 DNA Ligase was obtained from New England Biolabs, Inc. (Beverly, MA). Human peripheral lymphocyte separation medium and glutaraldehyde 50% solution in water were purchased from Sangon Biotech (Shanghai, China). Horseradish peroxidase (HRP)-conjugated anti-M13 monoclonal antibody was obtained from GE Healthcare (Piscataway, NJ). The ochratoxin A ELISA test kit was purchased from Green Spring (Shenzhen, China). SfiI, NotI, first-strand cDNA synthesis kit, RNAiso Plus, PrimeSTAR HS DNA polymerase, and SYBR premix EX TaqII (Tli RNaseH Plus) were obtained from Takara Dalian (Dalian, China). TIANprep mini plasmid kit, TIANquick midi purification kit, and TIANgel midi purification kit were purchased from Tiangen (Beijing, China). MicroAmp Fast Optical 96-well reaction plate and primers used in this study were obtained from Life Technologies (Grand Island, NY). pHEN1 phagemid vector, helper phage M13KO7, and Escherichia coli TG1 cells were generous gifts from Dr. Wei-Jun Ma (Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences).
Alpaca Immunization
OTA–KLH conjugates (immunogen) and OTA–OVA conjugates (coating antigen) were prepared by covalently attaching the carboxylic acid of OTA to a carrier protein as described by Kawamura et al.25 A 3-year-old male alpaca was immunized by subcutaneous, lower back injection of OTA–KLH (250 μg) diluted in 0.5 mL of phosphate-buffered saline (PBS) mixed with 0.5 mL of Freund’s complete adjuvant. A second injection, 3 weeks after the first, was followed by three injections every 2 weeks for a total of five immunizations. Samples of fresh blood (20 mL) were collected 1 week after the fourth and fifth immunization, from which the peripheral blood lymphocytes were isolated with the human peripheral lymphocyte separation medium, respectively. The lymphocytes were stored at −80 °C until used for total RNA extraction.
Library Construction
Total RNA was extracted from the peripheral blood lymphocytes by use of the RNAiso Plus following the manufacturer’s instructions. First-strand complementary DNA (cDNA) was derived from total RNA by use of a first-strand cDNA synthesis kit. The heavy chains of the heavy-chain antibody (HCAb) were amplified by PCR using PrimeSTAR HS DNA polymerase with sense and antisense primers AlpVh-LD (CTT GGT GGT CCT GGC TGC) and CH2-R (GGT ACG TGC TGT TGA ACT GTT CC), respectively. After amplification, doublet PCR bands between 635 and 740 bp corresponding to HCAb were agarose-gel-purified. Gene-specific primers AlpVh-SfiI (TCG CGG CCC AGC CGG CCA TGG CCC AGK TGC AGC TCG TGG AGT CNG GNG G), AlpVHHR1-NotI (CGA GTG CGG CCG CGG GGT CTT CGC TGT GGT GCG), and AlpVHHR2-NotI (CGA GTG CGG CCG CTT GTG GTT TTG GTG TCT TGG G) were used to amplify the VHH gene and add SfiI and NotI restriction enzyme sites flanking the 5′ and 3′ termini of VHH coding sequences, respectively. The resulting PCR products were purified with TIANgel midi purification kit and digested with Sfi I and Not I restriction enzymes. Amplified VHH DNA were pooled and ligated into the pHEN1 vector at 3:1 molar ratio by use of T4 DNA ligase. The recombinant plasmid was introduced into competent cells of E. coli TG1 by electroporation and the cells were plated on 2YT–ampicillin agar plates. Transformants were scraped off the plates and stored in 2YT plus 25% glycerol at −80 °C until further use. The resulting library had an estimated size of 2 × 107 independent clones.
VHH Library Panning
The phage-displayed VHH library [100 μL, 2 × 1012 colony-forming units (cfu)/mL] was subjected to four cycles of panning in 96-well microtiter plates. The antigen well was incubated with 10.0, 7.5, 5.0, and 2.5 μg of OTA–OVA conjugate for cycles 1, 2, 3, and 4 overnight at 4 °C. Nonspecific binding was blocked by incubation with 300 μL of blocking buffer [3% (w/v) OVA in PBS] for 1 h at 37 °C. To avoid selection of OVA-binding VHH phages, the wells of a microtiter plate coated with 100 μL of blocking buffer was used for phage preabsorption for 1 h at 4 °C. Then the supernatant was transferred to the antigen wells and incubated for 1 h at 4 °C. After being washed 12 times with PBST, the bound VHH phages of the first and second round were eluted with 100 μL of elution buffer [0.2 M glycine hydrochloride (pH 2.2) and 1 mg/mL bovine serum albumin (BSA)] and neutralized with 15 μL of 1 M Tris-HCl (pH 9.1), while the bound VHH phages of the third and fourth round were competitively eluted with OTA diluted in 1.25% methanol (50 and 5 ng/mL) by 30 min incubation at 4 °C. The Tween-20 concentrations in PBST were 0.05%, 0.1%, 0.25%, and 0.5% for the four cycles of panning, respectively. Postabsorption of the eluted VHH phage was performed throughout panning under the same conditions. After each cycle of panning, eluted VHH phages were used to infect E. coli TG1 cells for subsequent amplification as described by Ghahroudi et al.26 Individual clones were picked randomly after four cycles of panning and tested against the OTA–OVA conjugate by competitive phage ELISA. Plasmid DNAs derived from the positive VHH phage clones determined by phage ELISA were extracted and sequenced by use of the universal primers M13-R (AGC GGA TAA CAA TTT CAC ACA GGA) and pHEN-R (GCC CAT TCA GAT CCT CTT C).
Preparation of VHH Phages
The recombinant E. coli TG1 containing the pHEN1-VHH plasmid was inoculated into 2YT medium containing 2% (v/v) glucose and 100 μg/mL ampicillin, followed by incubation and shaking (220 rpm) at 37 °C until OD600 of the medium reached 0.5. After being superinfected with helper phage M13K07 by incubation with no shaking at 37 °C for 15 min, the culture was subjected to incubation and shaking (220 rpm) at 37 °C for 45 min. Cells were harvested by centrifugation and transferred to 2YT medium containing 100 μg/mL ampicillin and 50 μg/mL kanamycin. VHH phages were produced by growing the culture overnight with shaking (220 rpm) at 30 °C and titered by determining the colony-forming units (cfu).
VHH Phage-Based Competitive Real-Time Immuno-PCR
Wells of a 96-well PCR plate were treated with 20 μL of 0.8% glutaraldehyde in Milli-Q water for 5 h at 37 °C and washed six times with Milli-Q water. The wells were coated with 20 μL of OTA–OVA conjugate in 0.05 M carbonate buffer (pH 9.6) overnight at 4 °C. After the wells were blocked with PBS containing 3% (w/v) OVA at 37 °C for 1 h, 10 μL of VHH phage supernatant (4 × 107 cfu) and 10 μL of serial concentrations (0, 0.01, 0.1, 1, 10, 100, and 1000 pg/mL) of OTA in 2.5% methanol–water were added into the plate and incubated for 1 h at 37 °C. Then the plate was washed six times with PBST followed by six washings with Milli-Q water to remove unbound VHH phages. The bound phages were finally quantified by real-time immuno-PCR (RT-IPCR).
RT-IPCR was performed directly in a 96-well PCR plate by use of the 7900HT fast real-time PCR system. Each PCR reaction (20 μL) consisted of 2× SYBR* Premix Ex TaqII (Tli RNaseH Plus), 50× ROX reference dye, 10 μM of each primer, forward primer phage-F1 (GGG CCG ATT CAC CAT CTC) and reverse primer phage-R1 (GCG GAC GTA ACA GTA ATA CAC G), VHH phage, and sterilized water. A dissociation-curve analysis was performed at 95 °C for 15 s, 60 °C for 1 min, and 95 °C for 15 s. The thermal cycle conditions included 95 °C for 10 min, followed by 40 cycles of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s. The 103-bp amplicon encoding part of the VHH DNA was confirmed by agarose gel electrophoresis.
We established a VHH phage-based competitive RT-IPCR standard curve using the optimized concentration of coating antigen (4 μg/mL), input of VHH phage (4 × 107 cfu/well), and 10-fold serial dilutions of OTA standard (0.01, 0.1, 1, 10, 100, and 1000 pg/mL) (Figure 4B). Fluorescence was detected during PCR amplification by the 7900HT fast real-time PCR system (Life Technologies, Grand Island, NY), and amplification curves of 10-fold serial dilutions of OTA were also obtained from it.
Figure 4.

VHH phage-based competitive RT-IPCR assay of OTA. (A) OTA amplification curves of VHH phage-based competitive RT-IPCR assay. Curves b–g represent serial 10-fold dilutions of OTA from 0.01 to 1000 pg/mL. Curves a and h are the negative control (no OTA) and blank control (no VHH phage), respectively. (B) OTA standard curve.
Sample Preparation
Samples of corn, wheat, and rice collected from a local grocery store were finely ground and stored in the freezer at −20 °C before analysis. Sample preparation for measurement with VHH phage-based competitive RT-IPCR was performed according to the OTA ELISA kit manufacturer’s instructions. Briefly, 5 g of each sample was weighed into a 50 mL centrifuge tube and extracted with 25 mL of 60% methanol in deionized water and 2 mL of petroleum ether by shaking vigorously for 5 min. Then the mixture was centrifuged at 8000g for 5 min and the middle liquid layer was diluted 24 times for RT-IPCR analysis and 2 times for conventional ELISA analysis, respectively.
Statistical Analysis
A standard curve was produced by plotting the average threshold cycle (CT) values of duplicates against OTA concentrations with Origin version 8.0 (OriginLab Corp., Northampton, MA) and used for OTA quantification in the cereal samples. The limit of detection (LOD) was obtained as the estimated OTA concentrations that are euquivalent to the mean blank value plus 3 standard deviations (SD) as described by Ueda and co-workers.24
Results and Discussion
Selection of OTA-Specific VHH Phages
An alpaca-derived VHH immuno library of 2 × 107 individuals was constructed and used to select OTA-selective VHH phages through four cycles of panning. The number of phage output in each cycle of panning is shown in Figure 1. In order to enrich VHH phage clones of increasing selectivity for OTA, the bound phages were eluted by glycine hydrochloride in the first two cycles of panning. The number of phage output in the second panning was approximately 3 orders of magnitude higher than in the first panning. This result indicated the effective enrichment of specific clones in the first two cycles of panning. It has been reported that competitive selection could be applied for isolation of high-affinity antibodies.27,31 Hence, we performed competitive elution using low concentrations of free OTA by adding 100 μL of 50 and 5 ng/mL OTA in 2.5% methanol–PBS and incubating for 20 min at room temperature in the third and fourth cycles of panning, respectively. The number of phage output of the third panning decreased compared to the second panning, showing a very limited amount of VHH phages with high affinity in the second panning eluate. In total, 48 clones randomly selected from the third and fourth cycle of panning were rescued and their capacity to bind OTA by competitive phage ELISA was determined. We found that the 48 phage clones all showed clear but different degrees of binding inhibition by free OTA (data not shown), indicating that our panning strategy was effective in isolating positive VHH phage clones with high affinity to the coating antigen. Briefly, we performed four cycles of panning with adjustments of OTA–OVA conjugate concentration (100, 75, 50, and 25 μg/mL), Tween-20 concentration (v/v) in PBST (0.05%, 0.1%, 0.25%, and 0.5%), elution model (acid elution and competitive elution), OTA concentration for competitive elution (50 and 5 ng/mL), and preabsorption of phage binding OVA.
Figure 1.
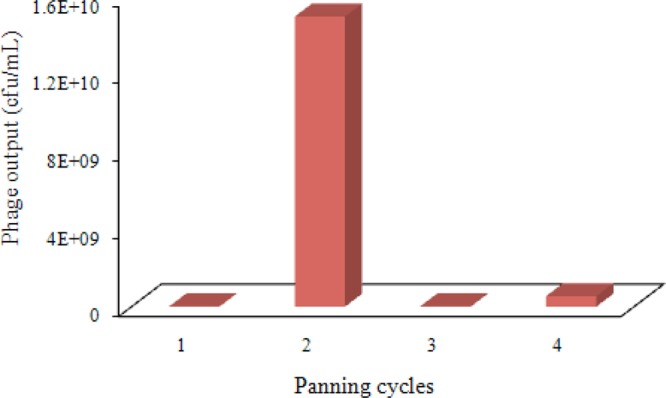
Phage output number: 10 μL of the phage output was diluted in sterile LB medium from each panning cycle and mixed with 100 μL of E. coli TG1 cells with OD600 = 0.3. The mixture was incubated for 15 min at 37 °C without shaking and then plated on a LB plate with ampicillin (100 μg/mL). The number of colonies was counted the next day.
Sequence Alignment of Isolated VHH Phages to OTA
A total of 3 clones after the third cycle of panning and 13 clones from the fourth cycle of panning that showed complete inhibition of binding to the coating antigen by free OTA were sequenced (Figure 2). In total, four unique sequences (och1, och2, och3, and och4) were obtained from the 16 VHH phage clones (Figure 3). Interestingly, VHH-43 and VHH-44 show much lower sensitivity in phage ELISA than the rest of clones in och1 do, while VHH-28 (och2) and VHH-32 (och3) with different sequences have very similar sensitivity in phage ELISA. It has been reported that the vast majority of the rescued phage particles that display pIII–antibody fusions will contain only a single copy.28 Besides, it has been demonstrated that monovalent display is very essential for selecting high-affinity antibodies.29 So it can be inferred that multicopy pIII–VHH fusions are displayed on the particle surface of VHH-43 and VHH-44, resulting in lower sensitivity in phage ELISA. Affinity is a binding strength between an antigenic determinant and an antigen-binding site, and complementarity-determining regions (CDRs) account for formation of the antibody binding pocket.30 It has been reported that VHHs with different sequences that were highly homologous in CDRs had similar sensitivity in ELISA.31 Since VHH-28 (och1) and VHH-32 (och2) have good similarity in CDRs, they show very similar sensitivity in phage ELISA.
Figure 2.
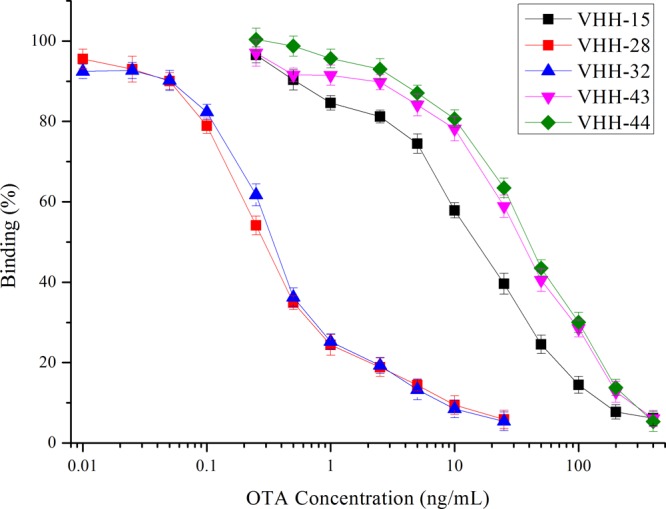
Competitive phage ELISA with VHH phage clones from the third and fourth cycles of panning. The 96-well microtiter plate was coated with 100 μL of OTA–OVA (5 μg/mL) in PBS per well overnight at 4 °C. The mixture of VHH phage and OTA was added, and then binding was detected with HRP/anti-M13 monoclonal conjugate. IC50 levels varied from 0.31 ng/mL for VHH-28 (red squares) to 42.01 ng/mL for VHH-44 (green diamonds). Clones (VHH-17, 20, 25, 27, 31, 34, 35, 36, 39, 47, and 48) that have very similar sensitivity as VHH-15 in phage ELISA are not shown. Error bars are standard deviations of the mean with three well replicates.
Figure 3.

Alignment of amino acid sequences of selected VHH phage clones. Sixteen clones are divided into four groups: och1 (VHH-15, 17, 20, 25, 27, 31, 34, 35, 39, 43, 44, 47, and 48), och2 (VHH-28), och3 (VHH-32), and och4 (VHH-36). The deduced amino acid sequences of the four clones are given in the single-letter code. (∼) Absence of amino acid residues; (.) residues identical to those of clones in och1.
It is clear that the framework regions are highly conserved among the four sequences. We can distinguish four sequences of VHH phages according to the composition of CDRs. The CDR3 region of och1 contains 15 amino acids, which is the longest in the four sequences. och2 and och3 both exhibit excellent sensitivity in the competitive phage ELISA and are highly homologous in the CDR2 region but show significant amino acid variation in CDR3 region. Since VHH-28 in och2 has the lowest IC50 in the phage ELISA, we chose VHH-28 for further research.
VHH Phage-Based Competitive Real-Time Immuno-PCR
VHH phage-based RT-PCR competitive standard curves were produced from serial concentrations of OTA standard. The threshold cycle (CT) was determined by setting a fluorescence threshold in the exponential phase of the amplification curves, reading out the fractional cycle number at which the amplication curve crossed the threshold (Figure 4A). Along with the standard samples, a negative control containing all assay components (without OTA) was run. A blank control containing the RT-PCR reagents only (without VHH phage) was also included. All samples were run in triplicate.
It is obvious that the mean CT values for 10-fold serial dilutions of OTA standard from 0.01 pg/mL to 1000 pg/mL increase as the increase of OTA concentration (Figure 4B). The standard curve exhibited an excellent limit of detection of 3.7 pg/L and displayed a correlation coefficient of 0.996, and this quantification proved to be linear over a wide range of OTA standard concentrations (from 0.01 to 1000 pg/mL).
Cross-Reactivity
The specificity of the assay was tested for the phage clone VHH-28 by use of five common mycotoxins: ochratoxin B (OTB), fumonisin B1 (FB1), deoxynivalenol (DON), aflatoxin B1 (AB1), and zearalenone (ZEN) (Figure 5). The cross-reactivity of the assay was obtained by comparing 50% inhibitory concentrations (IC50).32 Negligible cross-reactivity was observed, except for OTB, which showed 3.5% cross-reactivity, indicating the excellent selectivity of VHH-28 in VHH phage-based competitive RT-IPCR for OTA.
Figure 5.

Cross-reactivity of VHH phage-based competitive RT-IPCR. The 96-well PCR plates were coated with 20 μL/well of OTA–OVA conjugate (4 μg/mL). Serial 10-fold dilutions of (a) ochratoxin A or (b–f) tested compounds (ochratoxin B, fumonisin B1, deoxynivalenol, aflatoxin B1, and zearalenone) in 2.5% methanol–PBS were mixed with an equal volume of VHH-28 (4 × 109 cfu/mL) in PBS. The mixture (20 μL/well) was then added to wells and incubated at 37 °C for 1 h. The bound phage were detected by RT-PCR.
Solvent Effect
OTA is a relatively nonpolar chemical that has low solubility in water, so methanol is commonly used to prepare OTA standards and to extract OTA from cereal samples. Since methanol can affect the antigen–antibody interaction, we performed a study to optimize the concentration of methanol in the final assay buffer. A series of concentrations of OTA was diluted in 2.5%, 5%, 10%, and 20% methanol–PBS. The LOD values generated from each dilution buffer were compared. As shown in Figure 6, there were slight differences among 5%, 10%, and 20% methanol–PBS; however, the standard curve of VHH phage-based competitive RT-IPCR in 2.5% methanol–PBS exhibited the widest linear range and displayed the lowest LOD. So in the following analysis, 2.5% methanol–PBS was selected to dilute the ochratoxin A standard, and the sample extract was also diluted to a final concentration of 2.5% methanol.
Figure 6.
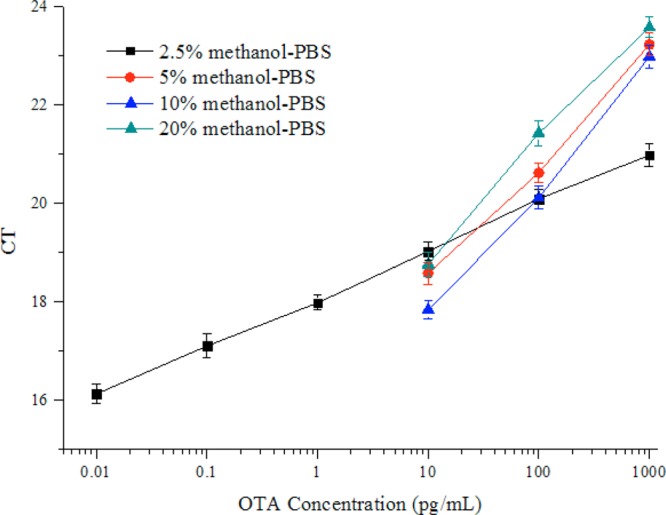
Performance of the VHH phage-based competitive RT-IPCR in 2.5% (■), 5% (red circles), 10% (blue triangles), and 20% (green triangles) methanol–PBS. Data are represented as an average ± standard deviation of six replicates. Serial 10-fold dilutions of ochratoxin A in 2.5%, 5%, 10%, and 20% methanol–PBS were mixed with equal volumes of VHH-28 (4 × 109 cfu/mL) in PBS. The mixture (20 μL/well) was added to wells and analyzed by VHH phage-based competitive RT-IPCR.
Assay Validation
A spike-and-recovery analysis was carried out to validate the assay with the newly developed VHH phage-based competitive RT-IPCR. Corn, wheat, and rice samples spiked with known concentrations of ochratoxin A were tested. Recoveries of 80–126% were obtained (Table 1).
Table 1. Recoveries of OTA Added to Corn, Wheat, and Rice Samplesa.
| VHH phage-based
RT-IPCR (n = 3) |
conventional
ELISA (n = 3) |
|||
|---|---|---|---|---|
| OTA added (μg/kg) | OTA recovered (μg/kg) | CV (%) | OTA recovered (μg/kg) | CV (%) |
| Corn | ||||
| 0.1 | 0.09 ± 0.01 | 11.1 | 0.08 ± 0.01 | 12.5 |
| 1 | 0.93 ± 0.09 | 9.7 | 0.89 ± 0.10 | 11.2 |
| 10 | 12.6 ± 1.07 | 8.5 | 10.94 ± 1.15 | 10.5 |
| 100 | 100 ± 9.80 | 9.8 | 96.3 ± 10.20 | 10.6 |
| Rice | ||||
| 0.1 | 0.12 ± 0.01 | 8.3 | 0.11 ± 0.01 | 9.1 |
| 1 | 1.03 ± 0.12 | 11.7 | 0.94 ± 0.09 | 9.6 |
| 10 | 8.75 ± 0.89 | 10.2 | 8.22 ± 0.98 | 11.9 |
| 100 | 93.9 ± 11.50 | 12.2 | 91.5 ± 10.70 | 11.7 |
| Wheat | ||||
| 0.1 | 0.08 ± 0.01 | 12.5 | 0.08 ± 0.01 | 12.5 |
| 1 | 0.94 ± 0.08 | 8.5 | 0.86 ± 0.09 | 10.5 |
| 10 | 9.12 ± 0.96 | 10.5 | 9.44 ± 1.05 | 11.1 |
| 100 | 116 ± 10.40 | 9.0 | 103 ± 12.60 | 12.2 |
Determinations were performed by VHH phage-based competitive RT-IPCR and conventional ELISA.
A total of 38 domestic cereal samples were analyzed with a commercial ELISA kit and the VHH phage-based competitive RT-IPCR, respectively. Among the 38 samples, seven (C8–C12, C14, and C15) of 18 corn samples, five (W1–W5) of 10 wheat samples, and eight (R1–R8) of 10 rice samples were OTA-positive as detected by VHH phage-based competitive RT-IPCR (Table 2). In addition, OTA was detected in 12 samples (C10, C11, C15, W1–W4, and R4–R8) by the ELISA kit. Many more samples were determined by the VHH phage-based RT-IPCR to be OTA-positive because of the lower LOD (3.7 pg/L) compared to the ELISA kit (20 pg/mL). The results show that the recovery and reproducibility of the proposed method are satisfactory.
Table 2. Analysis of OTA Content in Cereal Samplesa.
| sample | VHH phage-based RT-IPCR (μg/kg, n = 3) | CV (%) | commercial ELISA kit (μg/kg, n = 3) | CV (%) |
|---|---|---|---|---|
| Corn | ||||
| C8 | 0.10 ± 0.03 | 30 | ND | |
| C9 | 0.12 ± 0.05 | 41 | ND | |
| C10 | 0.47 ± 0.11 | 23 | 0.51 ± 0.14 | 27 |
| C11 | 0.75 ± 0.13 | 17 | 0.63 ± 0.21 | 33 |
| C12 | 0.11 ± 0.05 | 45 | ND | |
| C14 | 0.14 ± 0.02 | 14 | ND | |
| C15 | 0.41 ± 0.15 | 36 | 0.55 ± 0.12 | 21 |
| Wheat | ||||
| W1 | 1.27 ± 0.13 | 10 | 0.93 ± 0.16 | 17 |
| W2 | 0.90 ± 0.08 | 9 | 0.92 ± 0.14 | 15 |
| W3 | 0.53 ± 0.05 | 9 | 0.72 ± 0.08 | 11 |
| W4 | 0.58 ± 0.07 | 12 | 0.78 ± 0.15 | 19 |
| W5 | 0.13 ± 0.02 | 15 | ND | |
| Rice | ||||
| R1 | 0.20 ± 0.05 | 25 | 0.28 ± 0.11 | 39 |
| R2 | 0.21 ± 0.03 | 14 | 0.26 ± 0.07 | 26 |
| R3 | 0.31 ± 0.06 | 19 | 0.27 ± 0.05 | 18 |
| R4 | 0.38 ± 0.09 | 23 | 0.30 ± 0.10 | 33 |
| R5 | 0.44 ± 0.12 | 27 | 0.34 ± 0.11 | 32 |
| R6 | 0.17 ± 0.04 | 23 | ND | |
| R7 | 0.16 ± 0.05 | 31 | ND | |
| R8 | 0.12 ± 0.03 | 25 | ND | |
Determinations were performed by VHH phage-based competitive RT-IPCR and commercial ELISA kit. The limits of quantitation were 0.44 ng/kg and 0.23 μg/kg, respectively.
Conclusions
In this study, we constructed an immunized alpaca VHH library and four unique sequences of OTA-specific recombinant VHH fragments were selected by four cycles of panning. It has been reported that PD-IPCR can greatly improve the detection sensitivity, combining the advantages of IPCR and phage display technology.20,21,23 The phage VHH-28, which showed the highest sensitivity in phage ELISA, was chosen and applied to VHH phage-based competitive RT-IPCR for the detection of OTA. We have shown that OTA can be detected by VHH phage-based competitive RT-IPCR at concentrations ranging from 0.01 to 1000 pg/mL, with a detection limit of 3.7 pg/L. Validation results from conventional ELISA and VHH phage-based RT-IPCR were in good agreement with each other. To our knowledge, this is the most sensitive assay reported to date for the detection of OTA. This study indicates that VHH phage-based competitive RT-PCR has great potential in ultrasensitive detection of mycotoxins and other toxic small molecular compounds.
Acknowledgments
This work was financially supported by grants from National Basic Research Program of China (Grant 2013CB127804), National Science Foundation of China (Grants NSFC-31171696, NSFC-31301479, NSFC-31360386, and NSFC-31201360), Research Program of State Key Laboratory of Food Science and Technology, Nanchang University (Project SKLF-22A-201302), and in part by the National Institute of Occupational Safety and Health, 2U50OH007550, and the NIEHS Superfund Research Program, P42ES04699. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Anfossi L.; Giovannoli C.; Giraudi G.; Biagioli F.; Passini C.; Baggiani C. J. Agric. Food Chem. 2012, 60, 11491–11497. [DOI] [PubMed] [Google Scholar]
- Woo C.; Partanen H.; Myllynen P.; Vähäkangas K.; El-Nezami H. Toxicol. Lett. 2012, 208, 92–99. [DOI] [PubMed] [Google Scholar]
- Klarić M.; Rašić D.; Peraica M. Toxins 2013, 5, 1965–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfohl-Leszkowicz A.; Manderville R. Chem. Res. Toxicol. 2012, 252252–262. [DOI] [PubMed] [Google Scholar]
- Mosesso P.; Cinelli S.; Pinñero J.; Bellacima R.; Pepe G. Chem. Res. Toxicol. 2008, 21, 1235–1243. [DOI] [PubMed] [Google Scholar]
- Manderville R. Chem. Res. Toxicol. 2005, 18, 1091–1097. [DOI] [PubMed] [Google Scholar]
- Ochratoxin A. In Some Naturally Occurring Substances: Food Items and Constituents, Heterocyclic Aromatic Amines and Mycotoxins; IARC: Lyon, France, 1993; Vol. 56, pp 489–521. [Google Scholar]
- Copetti M.; Iamanaka B.; Nester M.; Efraim P.; Taniwaki M. Food Chem. 2013, 1361100–104. [DOI] [PubMed] [Google Scholar]
- Kumar R.; Ansari K.; Saxena N.; Dwivedi P.; Jain S.; Das M. Food Control 2012, 26163–67. [Google Scholar]
- Santos L.; Marín S.; Ramos A. Food Chem. 2010, 1223826–830. [Google Scholar]
- Chiotta M.; Ponsone M.; Sosa D.; Combina M.; Chulze S. Food Microbiol. 2013, 362182–190. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Ye Z.; Jin Y.; Wang S.; Zhang L.; Pei X. World Mycotoxin J. 2014, 7153–62. [Google Scholar]
- He Z.; He Q.; Xu Y.; Li Y.; Liu X.; Chen B.; Lei D.; Sun C. Anal. Chem. 2013, 852110304–10311. [DOI] [PubMed] [Google Scholar]
- He Q.; Xu Y.; Wang D.; Kang M.; Huang Z.; Li Y. Food Chem. 2012, 1341507–512. [Google Scholar]
- Mohammad H.; Amoli-Diva M.; Pourghazi K. J. Chromatogr. A 2013, 1320, 17–26. [DOI] [PubMed] [Google Scholar]
- Hun X.; Liu F.; Mei Z.; Ma L.; Wang Z.; Luo X. Biosens. Bioelectron. 2013, 391145–151. [DOI] [PubMed] [Google Scholar]
- Liu R.; Yu Z.; He Q.; Xu Y. Food Control 2007, 18, 872–877. [Google Scholar]
- He Q.; Xu Y.; Huang Y.; Liu R.; Huang Z.; Li Y. Food Chem. 2011, 126, 1312–1315. [Google Scholar]
- Liu X.; Xu Y.; He Q.; He Z.; Xiong Z. J. Agric. Food Chem. 2013, 61, 4765–4770. [DOI] [PubMed] [Google Scholar]
- Guo Y.; Zhou Y.; Zhang X.; Zhang Z.; Qiao Y.; Bi L.; Wen J.; Liang M.; Zhang J. Nucleic Acids Res. 2006, 348e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.; McCoy M.; Gee S.; Gonzalez-Sapienza G.; Hammock B. Anal. Chem. 2011, 83, 246–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Xu Y.; Huang Q.; Yi C.; Xiao T.; Li Q. Chem. Commun. 2013, 49, 3778–3780. [DOI] [PubMed] [Google Scholar]
- Yu X.; Burgoon M.; Shearer A.; Gilden D. J. Immunol. Methods 2007, 326, 33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J.; Hasan S.; Fujioka Y.; Ueda H. J. Immunol. Methods 2012, 377, 1–7. [DOI] [PubMed] [Google Scholar]
- Kawamura O.; Sato S.; Kajii H.; Nagayama S.; Ohtani K.; Chiba J.; Ueno Y. Toxin 1989, 278887–897. [DOI] [PubMed] [Google Scholar]
- Ghahroudi M.; Desmyter A.; Wyns L.; Hamers R.; Muyldermanns S. FEBS Lett. 1997, 414, 521–526. [DOI] [PubMed] [Google Scholar]
- Rosa S.; Rossotti M.; Carleiza C.; Carrión F.; Pritsch O.; Ahn K.; Last J.; Hammock B.; González-Spaienza G. Anal. Chem. 2011, 83, 7213–7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogenboom H.; Bruïne A.; Hufton S.; Hoet R.; Arends J.; Roovers R. Immunotechnology 1998, 4, 1–20. [DOI] [PubMed] [Google Scholar]
- Lowman H.; Bass S.; Simpson N.; Wells J. Biochemistry 1991, 30, 10832–10838. [DOI] [PubMed] [Google Scholar]
- Wilson I.; Stanfield R. Curr. Opin. Struct. Biol. 1993, 3, 113–118. [Google Scholar]
- Kim H.; McCoy M.; Majkova Z.; Dechant J.; Gee S.; Rosa S.; González-Spaienza G.; Hammock B. Anal. Chem. 2012, 84, 1165–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.; Zhuang H. Microchim. Acta 2011, 172, 233–239. [Google Scholar]