

Abstract
A better understanding of mechanisms governing receptor insertion to the plasma membrane (PM) requires an experimental approach with excellent spatial and temporal resolutions. Here we present a strategy that enables dynamic visualization of insertion events for dopamine D2 receptors into the PM. This approach includes tagging a pH-sensitive GFP, superecliptic pHluorin, to the extracellular domain of the receptor. By imaging pHluorin-tagged receptors under total internal reflection fluorescence microscopy (TIRFM), we were able to directly visualize individual receptor insertion events into the PM in cultured neurons. This novel imaging approach can be applied to both secreted proteins and many membrane proteins with an extracellular domain labeled with superecliptic pHluorin, and will ultimately allow for detailed dissections of the key mechanisms governing secretion of soluble proteins or the insertion of different membrane proteins to the PM.
Keywords: TIRF microscopy, Superecliptic pHluorin, Protein trafficking, Insertion, GPCR
INTRODUCTION
G-protein coupled receptors (GPCRs) compose the largest family of cell-surface receptors and are critical for mediating responses to a vast number of extracellular signaling molecules, including hormones, neurotrophic factors, chemokines, odorant molecules and light (Alberts et al., 1998). As a result, the internalization of GPCRs has been studied extensively to elucidate their roles in physiological and pathophysiological responses in order to pinpoint potential therapeutic targets (Drake et al., 2006; Filmore, 2004; Lappano and Maggiolini, 2011). However, few details are known about other contributing pathways such as the exocytosis or recycling pathways. Studying these pathways can complement existing information about key molecular mechanisms providing a more complete understanding of receptor trafficking and potentially a more diverse pool of available therapeutic targets.
The dopamine D2 receptor is one such GPCR in which dysregulation is proven to lead to multiple psychiatric disorders. It has been shown that low DRD2 functional levels in the NAc increase the risk for addiction, both for substances of abuse and for natural reward. In addition, lower DRD2 function increases the risk for attention deficit / hyperactivity disorder (ADHD) (Blum et al., 1996) and depression (Lawford et al., 2006; Park et al., 2005). Moreover, abnormally elevated function of DRD2 is thought to contribute to the positive symptoms of schizophrenia (Carlsson, 1988; Karam et al., 2010). These abnormal DRD2 functional levels associated with different psychiatric disorders are thought to originate, at least in part, from dysregulation of DRD2 trafficking. DRD2 function is characterized by its abundance on the plasma membrane (PM) (Lin, 2001;Kim and Lisman, 2001; Binda et al., 2002; Lin et al., 2002; Free et al., 2007; Tirotta et al., 2008; Genedani et al., 2010), it is then regulated by dynamic redistribution between intracellular compartments and the PM. This redistribution is determined by several dynamic trafficking processes, including forward trafficking from the Golgi apparatus to the PM, endocytosis from the PM, and recycling through the endosomal recycling pathways, as well as targeting towards the degradation pathways. These dynamic trafficking processes are finely balanced and ultimately determine both the number and functional level of DRD2 on the neuronal surface. Therefore, targeting DRD2 trafficking mechanisms to manipulate the number of DRD2 on the neuronal surface offers a potentially powerful therapeutic strategy for restoring the DRD2 functional level in addicted individuals. Moreover, elucidating the detailed molecular mechanisms governing dynamic trafficking of DRD2 represents an important first step toward this therapeutic goal. While some studies have underscored the importance of DRD2 endocytosis in de-sensitization of this receptor, and to some degree have identified molecular mechanisms governing DRD2 endocytosis (Kim and Lisman, 2001; Macey et al., 2004), little is known about the regulation of DRD2 recycling or insertion into the plasma membrane. The main reason for the lack of knowledge of DRD2 insertion is that current studies of DRD2 trafficking primarily use biochemical or immunocytochemical approaches, which lack sufficient spatial and/or temporal resolution and therefore do not allow detailed examination of the molecular mechanisms regulating dynamic trafficking of DRD2.
In this unit, we present a method that allows examination of receptor insertion to the PM with excellent spatial and temporal resolution in cultured neurons. Basic Protocol 1 briefly describes a method for preparing mouse glia culture for neuronal culture conditioning and details the preparation for an interface co-culture of mouse glia and mouse striatal medium spiny neurons. Basic Protocol 2 demonstrates how to transfect cultured mouse medium spiny neurons in the interface culture condition. Basic Protocol 3 and 4 presents methods for quantitative analysis of pHluorin labeled DRD2 dynamic insertion to the PM.
BASIC PROTOCOL 1: Interfacial Neuron-Glia Culture
To investigate the dynamic insertion of DRD2 in cultured medium spiny neurons, it is essential to use healthy neurons. Mixed cultures of neurons and glia on the same coverslip can provide healthy mouse neurons for live imaging purposes. However, neurons in a mixed culture are not ideal for total internal fluorescence (TIRF) imaging experiments, as under this culture condition, neurons tend to situate on top of glia, with their somata and dendritic processes beyond the evanescent field of TIRF microscopy. We therefore employed the interface culture method, which provides a low-density neuronal culture with minimal glial cells on the same coverslip. Below, we describe the critical parts of the glia culture method to support the neuron/glia interface culture.
Note: All protocols using live animals must follow officially approved procedures for the care and use of laboratory animals as well as be submitted for review and approval by an Institutional Animal Care and Use Committee (IACUC).
Materials
Purified Bovine Collagen Solution Type 1, 3mg/ml (Advanced Biomatrix #5005-B)
Dissection buffer (see recipe)
Glia culture medium (see recipe)
Neonatal mice
DNase I solution (see recipe)
Papain solution (see recipe)
PBS Buffer 10X (Life Technologies # AM9625)
0.05% Trypsin-EDTA (1X) (LifeTechnologies #25300-054)
70% Nitric acid
Poly-L-Lysine solution (see recipe)
Neuronal plating media (see recipe)
E15–18 pregnant mice
T75 culture flask
Dissection microscope
Fire-polished glass Pasteur pipette
70 µm cell-strainer
Culture inserts (Millipore PICM03050)
6-well plates
15 mL conical tube
25 mm Round Coverslip (Warner Instruments)
Thomas Cover Glass Staining Outfits (Thomas Scientific)
Isotemp Oven
Hemocytometer
Prepare the glia culture
Day 0
-
1.
Coat T75 flasks with 1 mg/ml of collagen solution (1:3 dilution of Purified Bovine Collagen Solution in ddH2O). Set the flasks upright and let dry overnight in a sterile tissue culture hood.
Note: If collagen does not dry completely, glia cells will not grow well.
-
2.
Prepare dissection buffer and glia media (see recipes) store at 4°C until ready for use.
Day 1
-
3.
On the day of glia culture, warm glia media for at least 30 minutes in a 37°C water bath before dissection of mouse brains. Euthanize neonatal mice by rapid decapitation. Dissect cerebral cortexes under a dissection microscope and place dissected tissue in a 15 mL conical tube with ice-cold dissection buffer.
-
4.
Prepare the papain solution (see recipe) and digest dissected brain tissues in 2 mL papain solution at 37°C for 20 minutes.
-
5.
Following papain digestion, rinse brain tissues with 10 mL of pre-warmed glia media. Add 2 mL of fresh glia media to the digested brain tissues, and triturate the tissues with a fire-polished glass Pasteur pipette.
Fully triturated tissues should appear as a slightly opaque solution. No cellular debris should be visible.
-
6.
Add 8 mL of fresh glia media to the dissociated tissues. Filter the dissociated tissue suspension with a 70µm cell-strainer. Seed cells into T75 flasks (usually 2–3 postnatal pups typically seed one T75 flask) and place into a 37°C cell culture incubator.
Day 2
-
7.
Aspirate the media from the T75 flasks. Rinse the flasks with 10mL PBS. Then add 10–15 mL of fresh glia media to each flask.
Glia in T75 should be confluent in approximately 12 days.
Prepare glial culture inserts
Day 12
-
8.
Place culture inserts into 6-well plates, and coat them with collagen (1:3 dilution of Purified Bovine Collagen Solution in ddH2O). Allow culture inserts to air-dry overnight in a biosafety cabinet.
This step can be performed a day in advance such that culture inserts are ready when glial cells reach confluency.
Plate glia on culture inserts
Day 13
-
9.
Following overnight drying, add 2 mL of glia culture media in each well of the 6-well plate underneath each culture insert.
-
10.
Rinse confluent glial cells in T75 flask with PBS, and add 5mL of 0.05% trypsin to each T75 flask. Incubate trypsin at 37°C for 5 minutes
-
11.
Once glial cells are dissociated from the T75 flasks, seed 2 mL onto each of the culture inserts (one T75 flask per 6-well plate, 2mL each insert).
These glia culture inserts will be used to condition the neuronal cultures.
Prepare for neuronal culture
Day 14
-
12.
Load dry 25mm round coverslips onto Thomas Cover Glass Staining Outfits, or comparable coverslip rack and soak coverslips in 70% Nitric acid in large glass dish under the chemical fume hood overnight.
Do not soak coverslip in water before soaking in Nitric acid.
Day 15
-
13.
Remove coverslips from nitric acid. Soak coverslips in sterile deionized water under a fume hood. Exchange water and repeat to wash coverslips at least three times.
When removing the coverslip rack from nitric acid suspend the rack over the acid container for a few minutes or until no discernable volumes of acid remain. Use small volumes of deionized water and more frequent washes to rinse remaining HNO3 from coverslips and rack. Be aware combining nitric acid with water results in an exothermic reaction that may generate heat and fumes.
-
14.
When finished rinsing coverslips, discard water and bake coverslips in Isotemp Oven at 200 °C overnight.
This will serve to sterilize coverslip as well as to remove any trace amounts of HNO3.
Day 16
-
15.
Take coverslips out of oven and air cool in culture hood. Place one coverslip per well of a six-well culture plate in tissue culture biosafety cabinet. Then pipette 1mL of 1mg/mL poly-L-lysine solution (PLL) to coat coverslip with PLL. Place the 6-well plate with coverslip/PLL in the tissue culture incubator overnight.
Make sure coverslip does not float on top of PLL solution.
Day 17
-
16.
Wash PLL coated coverslip at least 3 times with autoclaved deionized water in tissue culture biosafety cabinet. Subsequently, incubate coverslip in 2 mL neuronal plating media overnight in tissue culture incubator.
Prepare and plate neuronal culture
Day 18
-
17.
Obtain at least 12 mL of Neuronal plating media from stock (2 mL per coverslip). Add 2% (v/v) B27 supplement to the Neuronal plating media and warm to 37°C.
-
18.
Remove plating media in the six-well plate and add 2mL of fresh plating media with B-27 to each well containing a glass coverslip.
-
19.
Euthanize pregnant mice (E15–18) with CO2, and embryonic mice by decapitation. Dissect the striatum from the embryonic brain and place into a 15mL conical tube with ice-cold dissection buffer.
-
20.
Prepare the papain solution (see recipe) and digested dissected brain tissues in 2 mL of papain solution at 37°C for 20 minutes.
-
21.
Following papain digestion, rinse brain tissues with 10mL pre-warmed Neuronal Culture Media. Then add 2 mL of fresh Neuronal Culture Media to the digested brain tissues. Triturate the tissues with a fire-polished glass Pasteur pipette.
-
22.
Add 8 mL of fresh neuronal culture media to the dissociated tissues. Filter the dissociated tissue suspension with a 70 µm cell-strainer.
-
23.
Following dissociation, count the number of cells in the neuronal suspension with a hemocytometer. Seed neurons on each coverslip at a density of 0.34 × 106 cells per well of a six-well plate, and place in a 37°C incubator overnight.
Note: On the day of plating, neurons are considered days in vitro (DIV) 0.
-
24.
To prepare glia culture inserts for conditioning the neuronal cultures, remove the glia media from the glia culture inserts and add fresh plating medium: 2 mL in the insert and 2 mL underneath the insert).
Day 19
-
25.
The next day (DIV 1), move neuron coverslips into the 6-well plate containing the glia culture inserts. Place neuron coverslips underneath the glia culture inserts, one coverslip per glial insert in each well.
-
26.
Replace 1 mL of culture media from glial insert and 1 mL culture media from neuron chamber with fresh pre-warmed Neuronal Culture Media (with B-27) every 3–4 days until they are ready for transfection and imaging experiments.
Typically 7–8 days for striatal mouse neurons
BASIC PROTOCOL 2: Neuronal Transfection with Lipofectamine 2000 Using Plasmid cDNA
Neuronal cultures are transfected 24–72 hours before imaging using Lipofectamine™ 2000 reagent. Expected transfection efficiencies are usually low (~1%), but capable of consistently transfecting 5–10 neurons per coverslip. This is considered sufficient for studying live-cell insertion events.
Materials
Lipofectamine™ 2000 (LifeTechnologies #11668-019)
Neurobasal Medium (LifeTechnologies #21103-049)
plasmid cDNA superecliptic pHluorin Drd2
Neuronal culture media + B27 (see recipe)
Neuronal-Glia culture
50 mL conical tube
Eppendorf tube
Day 26
-
Dilute 2µl Lipofectamine™ 2000 in 100µl of fresh Neurobasal medium in an eppendorf tube, and 1µg of DNA in 100µl of fresh Neurobasal medium in a separate eppendorf tube. Then, carefully combine the dilute DNA with the dilute Lipofectamine™ 2000. Incubate the DNA and Lipofectamine mixture for 20 minutes at room temperature after mixing.
Note: Neuronal transfection is performed using Lipofectamine™ 2000. For each neuronal culture (one culture per coverslip), 2µl Lipofectamine™ 2000 and 1µg of DNA are used. Fresh Neurobasal medium with no supplement is used to dilute Lipofectamine and DNA. If transfecting multiple coverslips, scale up the amount of Lipofectamine™ 2000 and DNA accordingly.
While DNA and Lipofectamine mixture is incubating, transfer 1mL of the media from underneath each glia culture insert, and 1mL from inside each insert to a sterilized 50 mL conical tube. Add the same volume of fresh Neuronal Culture Media + B27 to the saved medium and incubate at 37°C. This medium will be used for neuronal culture following transfection.
-
Following the 20-minute Lipofectamine/DNA incubation, remove glia culture inserts momentarily from the 6-well plates. Add 200µl Lipofectamine/DNA mixture to each coverslip with neurons. Return the glia culture inserts to each well, above the neurons. Incubate neurons with the Lipofectamine/DNA mixture at 37°C for 2–4 hours.
Generation of bubbles under the membrane of the culture inserts should be avoided
After the 2–4 hour incubation period, remove glia inserts. Aspirate the old culture media with the Lipofectamine/DNA mixture from each well. Add 2 mL of the media saved in the 50 mL conical tube (step 2) to neurons (2 mL per well). Return glia culture inserts to each well. Replace 2 mL of the glia media from each insert with the media saved in the 50 mL conical tube (step 2).
Incubate neurons an additional 24–72 hours to allow expression of transfected cDNA before imaging.
BASIC PROTOCOL 3: Total Internal Reflection Fluorescence Imaging
This protocol describes the general procedure to image the insertion of pHluorin labeled DRD2 (pH-DRD2) to the PM using an objective-based TIRF microscope. All steps of this protocol (post warm up) should be performed in a dark, humidity controlled (optional) room.
Note: All users should be properly trained in laser safety and other relevant safety procedures before using a laser based TIRF microscope.
Materials
Artificial Cerebrospinal Fluid (aCSF) (see recipe)
Lens cleaning solution
Immersion oil (n=1.52)
Lens paper
Transfected neurons
TIRF microscope set up
Inverted microscope (e.g., Zeiss AxioObserver D1)
-
Light source
488-nm excitation laser (e.g., Newport Excelsior 200mW)
Epi-fluorescent light source (e.g., Zeiss metal halide lamp HXP 120V)
Laser beam shutter and controller (e.g., Vincent Associates Uniblitz LS6 shutter, VCM–D1 controller)
-
Filter cubes in microscope (e.g., Chroma Technology Corporation)
EGFP: excitation filter (ET470/40×) dichroic mirror (Z488RDC), emission filter (ET525/50m )
A-Plan 100X Objective lens (e.g., Zeiss, NA 1.46)
sCMOS camera (e.g., Hamamatsu Photonics ORCA Flash 4.0 v2)
Image acquisition software (µManager )
Heating insert for live imaging, Specific model depends on microscope platform used.
Live Cell Imaging Chamber, Specific model depends on microscope platform used.
Note: The imaging system described here and in Figure 1 is the setup used by the authors’ laboratory. In principle, any commercial TIRF imaging platform with sufficient laser excitation and either a sCMOS or Electron Multiplying Charge Coupled Device (EMCCD) camera should provide comparable imaging performance.
Figure 1.
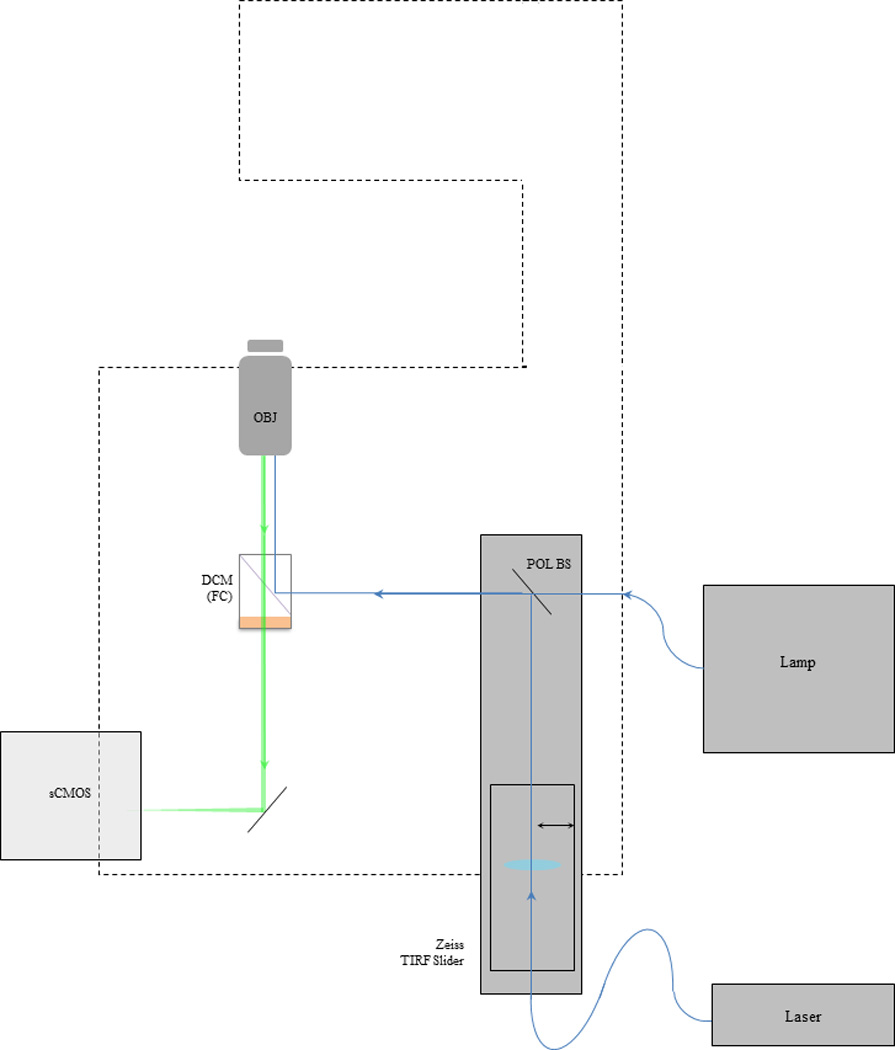
Hardware configuration for TIRFM. Any inverted fluorescence microscope can be adapted to perform total internal fluorescence microscopy. In this instance a manual Zeiss AxioObserver microscope was used as the main imaging platform for this TIRF microscope. An excitation laser (Newport Excelsior 488nm 200mW) was then coupled to a proprietary Zeiss TIRF slider via an optical fiber (KineFLEX–P–2–S–488–640–0.7–FCP–P2) in a laser port on the microscope body. The laser beam position is controlled via a fine adjustment knob then reflected out of the TIRF slider by a polarized beam splitter (POL BS) and onto the back aperture of a TIRF objective lens (Zeiss α–plan 100X objective lens N.A. = 1.46) by a dichroic mirror (DCM) inside a filter cube (FC). Resulting GFP fluorescence was selected with an emission filter mounted in the filter cube and relayed to a digital scientific complementary metal–oxide–semiconductor (sCMOS) camera (ORCA-Flash4.0 V2 Hamamatsu). In addition, a standard metal halide lamp (lamp) was coupled to the rear port on the microscope body for epi-fluorescence imaging. During epi-fluorescence imaging the excitation laser was blocked with a laser beam shutter (S) integrated between the laser head and fiber launcher.
Day 28
-
1.
At least 30 minutes prior to imaging experiments, warm the aCSF in a 37°C water bath. Place the heating insert for live imaging on the microscope stage and turn on the heating insert, laser, epi-fluorescence light source and camera to allow components to reach a stable operating state.
-
2.
Clean the TIRF objective and bottom of sample coverslip with lens paper and lens cleaning solution.
-
3.
Launch image acquisition software (µManager). Set camera exposure, binning, region of interest (ROI), and gain if applicable.
Typical settings for this experiment using a sCMOS camera were as follows:100 ms exposure.
-
3.
Confirm that the TIRF setup is properly aligned and that illumination is uniform.
A thin layer of fluorescent dye such as Alexa Fluor488 (LifeTechnologies) can be spread across a coverslip to check for uniformity. Uniform illumination should result in uniform fluorescence; compare pixel intensity across the field of view and adjust the critical angle as necessary.
-
4.
Assemble a coverslip with transfected neurons into the live-imaging chamber. Place pre-warmed aCSF in the chamber after the chamber is assembled.
Mounting of a neuronal coverslip into the live-imaging chamber will vary with chamber type. In this authors’ setup a custom perfusion chamber was used to facilitate assembly of the chamber. It is important that this step be completed as quickly as possible to avoid damage to live cells. In practice any imaging chamber with coverglass bottom will work for this purpose.
-
5.
Add a small drop of immersion oil to the objective lens. Place the sample chamber onto the heating insert on the microscope stage, and identify transfected neurons visually using an epi-fluorescent light source.
IMPORTANT NOTE: It is critical that no bubbles be present in the immersion oil for successful TIRFM.
-
6.
After a healthy transfected neuron has been identified, photobleach the cell surface by switching to TIRF illumination for approximately one minute or until all pre-existing pHluorin fluorescence on the plasma membrane is eliminated and single insertion events are visible (Figure 2).
pHluorin-receptors on the plasma membrane exhibit significant fluorescence at the beginning of the TIRF imaging experiment, which interferes with visualization of individual insertion events.
-
7.
Once the pre-existing pHluorin fluorescence has been bleached, set the acquisition software experiment duration to capture the following insertion events in situ. Record as a series of tiff images or a tiff image stack.
In this experiment neurons were treated with 1µM dopamine and recordings were performed immediately following or 20 minutes after dopamine stimulation. Each recording was performed for 1 minute at a 10 Hz acquisition rate (100ms exposure) and contained a total of 600 images that were saved on a hard drive for analysis.
-
8.
Repeat as necessary on subsequent transfected cells until all neurons have been recorded or until cell viability becomes questionable.
Figure 2.

Healthy medium spiny neuron DIV 7. (A) Transfected neuron before photobleaching. (B) Transfected neuron after photobleaching. Note the improved contrast allowing visualization of insertion events. Examples of insertion events are boxed in red.
BASIC PROTOCOL 4: Manual Data Analysis for TIRFM Visualization of Insertion Events
The manual analysis of insertion recordings is simple and requires no additional resources, but takes a significant amount of user input and time. Consequently this method is not very practical for large sample experiments. The following protocol describes the procedure used to manually process, identify and analyze insertion events with ImageJ. Results inherently provide spatial information about the direction of insertion as well as calculated insertion frequency in events per unit area over time, and relative number of molecules involved in individual insertion events.
Materials
Recombinant GFP (Clontech)
Artificial Cerebrospinal Fluid (aCSF) (see recipe)
TIRF microscope set up (see protocol 3)
Computer
Image processing software (ImageJ)
Quantify fluorescence intensity of single recombinant GFP
-
1.
Dilute GFP (clontech) to approximately 100 ng/mL.
-
2.
Image single GFP molecules with acquisition settings identical to experimental conditions. See protocol 3 for TIRFM imaging.
-
3.
Open recordings with ImageJ by either dragging the dataset onto an open imageJ window or through the menu options (File > open or File > import… > image sequence). Select a region of interest around a single GFP molecule using the rectangle tool.
Single molecules should appear as diffraction limited spots or point spread functions (PSF) with a diameter of 200–250 nm. In addition, photobleaching is assumed to be a one-step process for single GFP molecules. Therefore a PSF can be considered a single molecule if its fluorescence intensity is eliminated rather than gradually reduced.
-
4.
In the menu options select Analyze > measure to calculate the area, integrated density (IntDen), and mean intensity of the region of interest (ROI).
A results window should appear displaying various measurements made within the ROI selected. If mean intensity or integrated density is not displayed, open ‘set measurements’ in Analyze > set measurements. Check box to select the missing measurement.
-
5.
Click and drag the center of the ROI region and move ROI to a new PSF.
It is important that the ROI remain the same area for the most accurate results. Resizing the rectangular selection can be avoided by zooming in on the image and only clicking the center of the selection box to drag it to a new area. It may be helpful to note the dimensions of the ROI in the event that a new region is accidentally selected.
-
6.
Calculate the average background IntDen for each single GFP. Drag the ROI to a background area and record several intensity measurements.
Be sure to measure background levels at the same time point as the insertion event since the background integrated density decreases as the cell is photobleached.
-
7.
Repeat measurements as necessary to generate a statistically sound dataset. Save results window as separate text file.
-
8.
Import results into Microsoft Excel. Subtract the background from the integrated density of each ROI. The fluorescence intensity for a single GFP is then the calculated average of the new background-subtracted IntDen values.
Identifying and Processing Insertion Events
-
9.
Open an experimental recording dataset in ImageJ.
Typically a 600 tif image stack
-
10.
Navigate the recording frame by frame and identify insertion events. Refer to figure 3.
-
11.
For each insertion event select a region of interest around an insertion event and plot fluorescence change over time (Image > Stacks > Plot Z-axis Profile). Use this plot to determine the time point (frame number) signifying the beginning of the event (peak maximum intensity).
In addition this plot can be used to verify an insertion event based on the peak shape. Note the maximum fluorescence at the beginning of the event exponentially decays over several seconds due to photobleaching.
-
12.
Measure both the background fluorescence level at the beginning of the insertion event and the insertion event. Subtract background IntDen from event IntDen. Store in a spreadsheet as amplitude.
Measure integrated density by selecting Analyze > measure. Avoid resizing the ROI selection between measurements.
Figure 3.

Identifying insertion events in medium spiny neuron. (A) Insertion events will appear as small bright bursts of fluorescence. (B) Fluorescence from insertion events is subsequently bleached within several seconds. Both the size and the time required for pHluorin tagged receptors to bleach are helpful parameters in identifying or verifying insertion events.
Calculate relative number of receptors for experimental recordings
-
13.
Divide integrated density of ROI for a single insertion event by the previously calculated average fluorescence intensity of a single GFP molecule.
The relative number of pHluorin labeled receptors present in any given insertion event is directly related to the intensity or amplitude (z profile) of the emitted fluorescence. In this experiment the intensity representing a single pHluorin labeled receptor is assumed to be approximately the same as the intensity of a single recombinant GFP.
Calculate insertion frequency of receptors for experimental recordings
-
14.
Once all the insertion events have been identified for a single recording, create two maximum intensity projections (Image > Stacks > Z project: max intensity).
One projection is for reference, the other will become a binary image to calculate total area.
-
15.
Create a mask image of the neuron by manually adjusting the threshold level (Image > Adjust > Threshold). Using the duplicate max intensity projection as a reference, adjust the minimum threshold level such that the red mask covers the neuron. The threshold maximum should be set to the highest possible integer.
-
16.
Measure the integrated density of the mask image (Analyze > measure) then divide by the pixel intensity value of the mask. Store the result as the total surface area in pixels.
Note that no ROI is needed to measure the entire image. Also note that the integrated density is equivalent to the sum of all the pixels since it is the product of the area measured and the mean pixel value.
-
17.
Normalized frequency (insertion events per unit area over time) can then be calculated by dividing the total number of events in a recording by the surface area.
ALTERNATE PROTOCOL 4: Data Analysis for TIRFM Visualization of Insertion Events with Wombat Software
Due to the long processing times associated with manual image analysis, a program was developed to automate the identification and characterization of insertion events. Results are presented as a text document. Note: Wombat insertion analysis program is only compatible with Mac operating systems.
Materials
Recombinant GFP (Clontech)
Artificial Cerebrospinal Fluid (aCSF) (see recipe)
TIRF microscope set up (see protocol 3)
Computer (Macintosh Operating System)
Image processing software (ImageJ)
Wombat insertion analysis software
Microsoft Excel or similar spreadsheet software (optional)
Quantify the fluorescence of a single GFP molecule (steps 1 through 8 in basic protocol 4)
-
Organize raw insertion recordings. All recordings to be analyzed should be organized into subfolders; one recording per folder. All subfolders should be located in a single batch analysis folder.
Wombat software preserves folder names and lists quantitative results with respect to each subfolder when analysis is complete. To avoid confusing datasets choose a descriptive name for each subfolder such as ‘treatment_cell#’.
-
Open Wombat software. Set the input directory by browsing to the batch analysis folder. Similarly choose the output directory to store results. Finally set the amplitude threshold above background intensity levels (figure 4).
The amplitude threshold is the main parameter used to identify insertion events by extracting intensity values greater than the threshold for further characterization. Note that lower amplitude thresholds may extend the processing time.
-
Run Wombat insertion analysis.
Depending on the number of recordings being analyzed, and number of insertion events present, Wombat insertion analysis may take several hours to complete. When finished the Wombat log will read ‘idle’ and the progress bar will disappear. A ‘Batchfoldername_results.txt’ text document will be saved in the folder selected as output directory.
Import (Mac) or open results from Wombat into Microsoft Excel.
Calculate relative number of receptors for experimental recordings. Divide integrated density of ROI for a single insertion event by the previously calculated average fluorescence intensity of a single GFP molecule.
Perform statistical calculations on raw data to determine effects, if any, from treatments.
Figure 4.
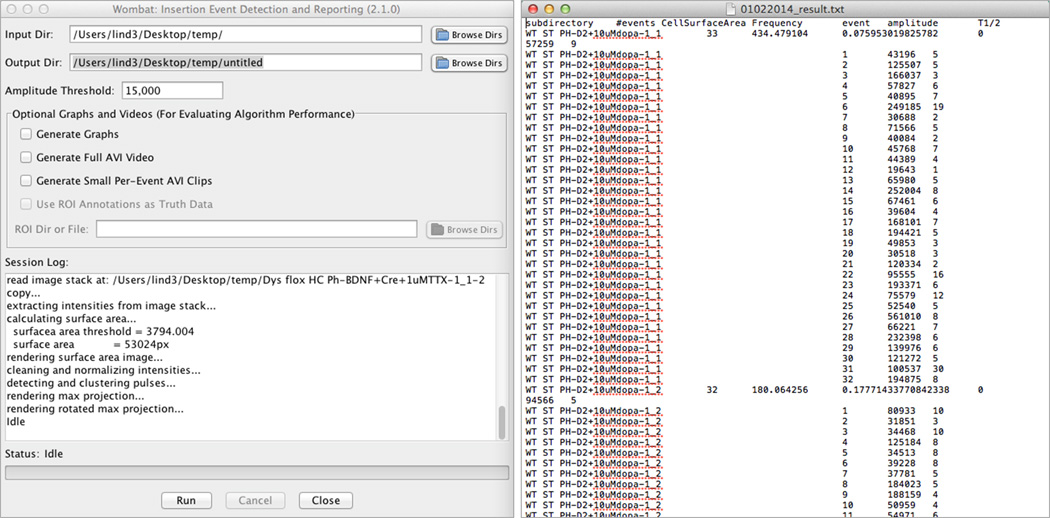
Wombat insertion analysis software and representative results. (Left) Wombat user interface post analysis. (Right) Raw results reported by Wombat are saved to a text document which includes the number of events in a recording, cell surface area, normalized frequency, amplitude for each event, and the half-life (frame) of each event.
REAGENTS AND SOLUTIONS
Use deionized, distilled water in all recipes and protocol steps unless otherwise specified
Artificial Cerebrospinal Fluid (aCSF)
119 mM NaCl
2.5 mM KCl
30 mM glucose
2 mM CaCl2
1 mM MgCl2
25 mM HEPES (LifeTechnologies #15630-080)
Adjust pH to 7.4
Filter sterilize using a 0.22 µm filter
Store up to 6 months at 4°C
Dissection Buffer
100 mL 10X HBSS (LifeTechnologies #14185-052)
10 mL Pen/Strep (LifeTechnologies #15140-122)
10 mL 100mM Na Pyruvate (LifeTechnologies #11360-070)
30 mL 1M Glucose
Adjust pH to 7.4
Filter sterilize using a 0.22-um filter
Store up to 6 months at 4°C
DNase I Solution
10 mg/ 1mL dissection buffer
Aliquot 20 µL into storage tubes
Store up to 12 months at −20°C
Glia Culture Medium
500 mL DMEM (LifeTechnologies #10313-021)
5 mL Pen/Strep (LifeTechnologies #15140-122)
5 mL 100X GlutaMAX™ (LifeTechnologies #35050-061)
50 mL fetal bovine serum; FBS (Biowest #US1520)
Filter sterilize using a 0.22-um filter
Store up to 6 months at 4°C
Neuronal Culture Media
500 mL Neurobasal Media (LifeTechnologies #21103-049)
5 mL (100X) Pen-Strep (LifeTechnologies #15140-122)
5 mL 100X GlutaMAX™ (LifeTechnologies #35050-061)
Filter sterilize using a 0.22-um filter
- Aliquot into 50 mL tubes
- Store up to 6 months at 4°C
Add 2% B-27 (v/v) immediately before use in culture
Store up to 7 days at 4°C
Neuronal Plating Media
Neuronal Culture media
5% HI Horse Serum (v/v) (LifeTechnologies #26050-070)
Store up to 6 months at 4°C
Papain solution
100µl Papain, 20mg/ml slurry; (Worthington Biochemical Corp.)
20µl 1% DNase I (Sigma #DN25-10MG)
2mL dissection buffer
Prepare fresh for each use, storage of solution not recommended. Discard after use.
Poly-L-lysine solution
1 mg/ml poly-L-lysine (Sigma #P2636-1G)
Dissolve in 1L of 100mM Borate Buffer (pH = 8.5)
Filter sterilize using a 0.22-um filter
Aliquot into 25mL tubes
Store up to 12 months at −20°C
COMMENTARY
Background Information
Internalization of G-protein coupled receptors has traditionally been characterized by radioligand binding assays. While these methods yield powerful quantitative data and are capable of high-throughput measurements they do not reveal the spatial or temporal information necessary to fully deduce molecular mechanisms nor are they easily applied to studying internal molecular pathways such as recycling or insertion to the plasma membrane (PM) (Evans, 2004).
Total internal reflection fluorescence microscopy (TIRFM) as an optical method provides excellent spatial and temporal information that can be used to dynamically study insertion of G protein-coupled receptors (GPCRs) to the PM when imaging a superecliptic pHluorin labeled receptor. As a pH-sensitive GFP derivative, superecliptic pHluorin is non-fluorescent at pH <6.0 and thus non-fluorescent when traveling within a vesicle lumen which has a resting potential at ~5.6 pH. In addition, superecliptic pHluorin exhibits strong green fluorescence upon increases in pH during events such as fusion to the plasma membrane or other disruptions to the vesicular membrane (Miesenbock et al. 1998;(Sankaranarayanan et al., 2000). A steady irradiation of 488-nm light can therefore allow active visualization of insertion events in vitro when single vesicles contain superecliptic pHluorin labeled receptors.
TIRFM is particularly well suited to studying live-cell GPCR insertion and offers several distinct advantages over other forms of fluorescence microscopy. First, the low penetration depth of this imaging technique reduces the background fluorescence to less than 2000-fold of a normal epi-fluorescence image increasing the signal-to-noise ratio and consequently the ability to identify the receptor of interest (Funatsu et al., 1995). In addition, because the illuminated optical section is thin (maximum ~200nm), virtually all the emission fluorescence collected is in focus. Finally, cells are exposed to significantly less light than they would be in an epi-fluorescence illumination scheme resulting in reduced phototoxcity. (Axelrod, 2001b)
Unlike confocal microscopy however, TIRF’s thin slice-like illumination is unable to reach depths greater than ~200 nm. While TIRF may offset this limitation with improved resolution at the PM, it is ultimately restricted in its usefulness for assessing molecular dynamics beyond this regime. To fully comprehend the potential uses and misuses of TIRF it is helpful to understand the theory behind it.
Physical Basis of Total Internal Reflection Fluorescence Microscopy
The concept of TIRF microscopy is founded upon the principles governing the refraction of light. When a light beam propagating through a transparent medium with a high refractive index (RI), such as an objective lens, encounters an interface with a lower refractive index (aqueous sample), the beam is bent towards the medium with the higher index of refraction. Total internal reflection (TIR) occurs when the incident angle of the light beam at the coverslip, θ, is greater than the ‘critical angle’ completely reflecting the excitation beam back towards the objective lens. The critical angle is defined by rearranging Snell’s law below;
| Equation 1 |
where n1 and n2 are the refractive indices of the sample and the coverslip, respectively (Axelrod, 2001b). Experimentally TIR can be achieved by focusing the incident beam radially off axis at the back aperture of the objective lens. The farther the beam is positioned off axis, the larger the corresponding incident angle will be ( figure 5). When the incident angle is greater than the critical angle the light reflected off the coverslip generates a thin electromagnetic field, the evanescent field, capable of exciting fluorophores at the interface.
Figure 5.

Schematic illustrating TIR illumination. (A) Wide field illumination used in epi-fluorescence. The excitation beam travels directly through the coverslip-sample interface exciting all of the fluorophores directly. (B) Highly-inclined illumination. Adjusting the light beam partially off axis at the back aperture of the objective (small angles of incidence) results in a compromise between the contrast provided by TIR and penetration depth present in widefield illumination. The excitation beam travels through the sample at an angle dependent on n1 (coverslip), n2 (sample) and the incident angle. (C) TIRF. The excitation beam enters the objective off axis from the right resulting in an incident angle greater than the critical angle. The light beam is reflected off the coverslip-sample interface and an evanescent field is produced. Only fluorophores within the evanescent field are excited.
The depth or thickness of the evanescent field, defined as the distance from the coverslip where the intensity decays to 37% of the initial intensity, is a function of excitation wavelength (λθ), the refractive indices of the sample and the coverslip (n1) and (n2) respectively, and the incident angle of the excitation light on the coverslip (θincident) modeled by the following equation;(Axelrod, 2013)
| Equation 2 |
Since the excitation wavelength and refractive indices of the coverslip and sample are generally fixed parameters in any given experiment, the depth of the evanescent wave can be varied by adjusting the incident angle of the excitation light beyond the critical angle. Larger incident angles correspond with lower penetration depths when θcritical < θincident < θmaximum. The maximum angle of incident light that can be generated (θm) is solely dependent on the numerical aperture of the objectives lens and is given by:
| Equation 3 |
Critical Parameters and Troubleshooting
The Sample
Co-culture your neurons
Starting with a relatively low density of healthy neurons is a key aspect in this experiment. A low density ensures that the neurons are securely attached to the coverslip and are therefore within the evanescent field of TIRF illumination. Ideally the neuron density should be as low as possible without affecting the survival rate. This will be dependent on the type of neuron used for experiments. For example, we found that the minimum seeding density for striatal neurons is approximately 300,000 cells per well, cortical mouse neurons 200,000 cells per well, and hippocampal neurons typically need only 100,000 cells per well to remain healthy. However, mouse neurons often have difficulty surviving in sparse populations without additional support via glial cells, which naturally aid and support the growth of neurons through secreted metabolites and growth factors. Thus co-culturing mouse neurons with glia results in enhanced neuronal survival and improved transfection efficiency. (Millet and Gillette, 2012)
In addition to providing glial support, the interfacial co-culture format is important for TIRF imaging as it significantly reduces the possibility of clusters or layering. Normally in mixed cultures neurons will tend to be situated above glia and beyond the evanescent field of TIRF illumination. Using the interfacial co-culture method reduces the possibility of layering by limiting direct contact between glia and neurons while allowing the exchange of metabolites and growth factor. Despite the physical separation, glial cells will be present on the coverslip, though at much lower densities.
Transfection
Insuring that the superecliptic pHluorin is suitably expressed with the receptor of interest is one of the most critical aspects in this experiment as it ultimately allows the visualization of insertion events. Many factors can influence the success of a chemically based transfection including neuron age or maturity, density, overall health, amount of transfection reagent, and the environmental composition of the media. The transfection protocol in this unit provides helpful tips and details that should eliminate most problems concerning the transfection of superecliptic pHluorin into striatal mouse neurons; however, conditions may differ based on the type of neuron cultured.
In general, when performing a transient transfection, balancing neuron maturity, transfection efficiency, and peak expression can quickly become a concern. Transfection efficiency is highly dependent on neuron age where juvenile neurons show significantly higher transfection efficiencies than mature neurons. However, it is more important to promote the best physiological representations possible to assess accurate trafficking mechanisms. Reduced transfection efficiency may also be caused by the following: insufficient amounts of DNA through degradation, or dilution, sub-optimal transfection reagent-DNA ratios, incomplete complex formation during incubation, and inhibition due to media composition. Following the transfection reagent instructions and making sure all reagents are fresh will eliminate most of these potential problems.
Other common problems concerning transfection include reduced cell viability post procedure. Transfection reagents like Lipofectamine can produce cytotoxic reactions in low density neuronal cultures. If this is the case, try seeding at several slightly higher densities to increase the survival rate and assess the correct parameters for your sample. Keep in mind that clustered or layered cells cannot be imaged under TIRF illumination. As an alternative solution, Optifect™ transfection reagent (Invitrogen) has been formulated to successfully transfect low density cultures and proven in mouse striatal neurons (Lin et al., 2013; Luoma et al., 2011).
TIRF Equipment
Compatible Objective Lens for TIRF Imaging
Currently there are a variety of optical arrangements employed in TIRF imaging. The two most common methods for inducing an evanescent wave at the sample include a through-the-objective TIRF configuration (as shown in figure 1 Authors setup) or a prism-based set up. For the purpose of this article we will consider only the critical parameters associated with a prism-less configuration. For additional resources concerning alternative TIRFM arrangements we refer readers to Axelrod 2001b.
For through-the-objective TIRFM the numerical aperture (NA) of the objective lens is the most important characteristic in the system. The NA, a measure of the resolving power of the objective, determines both the critical angle necessary to achieve TIR and the maximum incident angle possible, which in turn determines the depth of evanescent field. TIRF theory asserts the numerical aperture must be greater than the refractive index of sample in order to refract the excitation light off the coverslip and generate an evanescent field. This is no problem for an interface with water n = 1.33 and a NA =1.4 objective. Yet, the same objective will only barely achieve TIR when used to view a biological specimen with refractive indices reaching up to 1.38. In this case the range of incident angles available for achieving TIR will be very small (~1.9 degrees of freedom), significantly increasing the difficulty in aligning the system. The evanescent field in this case will also be deep, and cellular organelles may convert the evanescent field into scattered propagating light consequently reducing contrast (Axelrod, 2001a). For best imaging quality a TIRFM objective should have an NA substantially larger than the RI of the specimen. Today a variety of objectives from numerous manufacturers are designed for TIRF imaging with this technical challenge in mind. The highest NA objective to date is a 100X NA=1.65 manufactured by Olympus. However, the objective requires specialized and expensive coverslips (RI = 1.78) as well as volatile immersion oil. As a general rule any objective with an NA ranging from 1.45–1.49 should provide quality TIRF illumination with the ability to adjust the depth of the evanescent field. Some objectives may also come with variable collars to compensate for coverslip thickness or temperature further improving image quality.
Image acquisition settings
Image acquisition settings should be attuned to the experimental conditions to maximize spatial and temporal image quality while minimizing harmful photo-toxic events. The most critical acquisition settings for live cell TIRF imaging consist of the camera exposure time, experimental duration, and laser power density.
In this particular experiment the goal is to record and easily identify individual insertion events over a set period of time often to compare the effects of differential treatments on live-cells. Recent studies suggest receptor insertions to the PM and internalization events tend to vary in duration from seconds to several minutes depending on the receptor of interest and cell type (Li et al., 2012); (Brady et al., 2010) (Kotowski et al., 2011). The exposure time must then be set at shorter time scales than the insertion rate in order to accurately record events. It is also important to note that as the exposure time on a camera is reduced (or conversely as the framerate is increased) the shot noise caused by photons incident on the camera chip is increased. At very high framerates shot noise can add significantly to the background noise reducing contrast and image quality. To visualize pH-DRD2 we found a camera exposure time of 100 milliseconds provided excellent temporal resolution with little to no effect on image quality.
In addition, the duration of the experiment needs to be optimized to retain cell viability. Despite the live cell heated chamber insert, neuron health will deteriorate during the experiment due to unfavorable environmental conditions. To ensure we are studying physiologically realistic neurons approximately 30 minutes was allotted for imaging each coverslip. This time constraint, as well as the transfection efficiency, subsequently dictates the duration of each recording event. Note that live cell incubators, although costly, offer a CO2 controlled environment which can sustain cell viability for extended periods of time and thus circumvent the suggested 30 minute time constraint.
Finally, the laser power density (W/cm2) is directly related to the intensity of the evanescent field used to excite fluorophores and thus fluorescent emission of the pHluorin. In general higher laser powers will result in brighter fluorescence, but may also trigger photo-toxic events in biological structures. Since TIRF only illuminates a thin section of sample, the probability of causing irreparable damage to entire cell is low. Since some photobleaching must be done prior to recording insertion events in order to generate a baseline (eliminate extraneous pHluorins from neuronal surface) it is important to reduce unnecessary photobleaching to retain the health of cell during imaging experiments. One compromise to obtain high contrast and low probability of photobleaching is to use relatively short recording times per neuron at slightly higher laser powers. For this application, the laser power should be set to a level such that one minute of illumination will bleach a majority of pre-existing instances of pHluorin on the cell surface. For our set up we found that approximately 30–40mW at the back aperture of the objective lens was sufficient. Note that there can be significant power loss through an objective depending on the magnification and NA. Acceptable laser power levels should be identified and confirmed through experimental procedures.
Other acquisition settings such as binning can be adjusted to optimize spatial resolution. The Nyquist criterion, which states that the desired sampling rate should be twice the resolution, can be used to optimize the spatial resolution through objective magnification and or pixel binning. Since optical resolution is diffraction limited to 200–250 nm, the best image quality will theoretically occur at a desired sampling rate or effective pixel size of approximately 100 nm where effective pixel size is equal to camera pixel size divided by the total magnification (Piston, 1998). For instance, a 100X objective relaying an image to a sCMOS camera with a pixel size of 6 square microns would result in an effective pixel size of 60 × 60 nm. Noise characteristics are typically unrelated to effective pixel size however the smaller a pixel is the less likely a photon will be incident on that region (photons are now incident on across several pixels). Binning pixels can be used to help achieve ideal effective pixel size providing a larger area to collect photons thus improving maximizing signal collection. In our example applying a 2×2 bin would serve to double the effective pixel size to 120×120 nm and significantly boost the signal to noise ratio without losing resolution.
Additional TIRF related troubleshooting
Correct laser alignment is critical for TIRFM to work. If total internal reflection cannot be achieved, make sure that the beam path is not obstructed and that it is still being targeted to the sample. If the background appears abnormally high it is possible that the incident angle is less than the critical angle resulting in highly inclined illumination. In both cases consult the system manual about alignment procedures. Alternatively for protocols concerning the alignment of custom TIRF systems we refer readers to (Jaiswal and Simon, 2003; Johnson et al., 2012).
Other sources of trouble result in image heterogeneity. Common causes for image irregularities such as blotches or fringes are typically indicative of a dirty objective lens, coverslip, or bubbles in the immersion oil. Make sure both the objective and coverslip bottom are cleaned prior to imaging and that the refractive index of the oil, coverslip and objective match. Other disturbances in image homogeneity may be a result of poor cell attachment to the coverslip, or non-uniform illumination. The uniformity of TIRF illumination can be assessed using a fluorescent dye on coverslip. Focus on the coverslip and compare intensity values in different regions. Lower intensity at the edges of the field can be tolerated if those regions are systematically avoided in the analysis.
Anticipated Results
Raw data depicting superecliptic pHluorin labeled GPCR insertion events are in the form of tiff images or tiff image stacks. Insertion events appear as small clusters that suddenly exhibit a strong burst of fluorescence then rapidly disperse (Figure 3). Analysis procedures described in basic protocols 3 and 4 transform raw data to standardized measurements of insertion frequency in events per unit area over time, and relative number of molecules involved in individual insertion events. Quantitative results coupled with live cell recordings should provide powerful insights to molecular mechanisms concerning protein insertions to the plasma membrane.
Time Considerations
Glial and Neuronal interfacial co-culture and transfection
It typically takes 1–2 hours to dissociate the cerebral cortex of mouse pups to single cells used to seed T75 flasks for glial culture. Depending on the seed size, 13 days of growth will yield a confluent culture of glial cells. Preparation of the neuronal coverslips can be started on day 8 or 9 to save time. Dissociating the cerebral cortex to prepare neuronal culture from embryonic mice should also only take 1–2 hours. Once the neurons are plated onto coverslips expect to spend approximately 10 minutes (per 6-well plate) every 3–4 days changing culture media until cells are ready for transfection (typically 7–8 days for striatal neurons). The transfection procedure typically takes 3–4 hours to complete. Finally, neurons should be co-cultured for an additional 24–72 hours, depending on the receptor of interest, to allow expression of transfected cDNA.
TIRF Imaging and Data Analysis
The TIRF system should be turned on 30 minutes prior to imaging to allow the heating insert, laser, and camera to reach a stable operating state. This time can also be used to warm the aCSF to 37°C. The amount of time spent imaging is dependent on the number of samples prepared. During our experiments we spend about 2 minutes total bleaching then immediately imaging transfected neurons. Each coverslip typically takes 20–30 minutes to image all the positively transfected neurons. It is not advisable to spend more than 1 hour per coverslip as the health of the neuron slowly deteriorates (note this time may be extended if CO2 levels are also managed). Manual analysis of the image stacks for insertion events is a lengthy process and may take up to a week for a full day of data collection. A program was developed to accelerate the analysis process to approximately 2 hours.
ACKNOWLEDGEMENT
This work was supported by The Jackson Laboratory and NIDA IRP. We would like to thank Keith Sheppard and Glen Beane from the Computational Group of The Jackson Laboratory for the development of Wombat insertion analysis software.
Footnotes
INTERNET RESOURCES (optional)
µManager: open source microscopy software
ImageJ: Image processing and analysis in Java
Wombat Insertion analysis software is available upon request
LITERATURE CITED
- Alberts B, Bray D, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Cell Communication. In: Robertson M, editor. Essential Cell Biology: An Introduction to the molecular biology of the cell. New York, NY: Garland Publishing Inc.; 1998. pp. 481–512. [Google Scholar]
- Axelrod D. Selective imaging of surface fluorescence with very high aperture microscope objectives. J Biomed Opt. 2001a;6:6–13. doi: 10.1117/1.1335689. [DOI] [PubMed] [Google Scholar]
- Axelrod D. Total internal reflection fluorescence microscopy in cell biology. Traffic. 2001b;2:764–774. doi: 10.1034/j.1600-0854.2001.21104.x. [DOI] [PubMed] [Google Scholar]
- Axelrod D. Evanescent excitation and emission in fluorescence microscopy. Biophys J. 2013;104:1401–1409. doi: 10.1016/j.bpj.2013.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binda AV, Kabbani N, Lin R, Levenson R. D2 and D3 dopamine receptor cell surface localization mediated by interaction with protein 4.1N. Mol Pharmacol. 2002;62:507–513. doi: 10.1124/mol.62.3.507. [DOI] [PubMed] [Google Scholar]
- Blum K, Sheridan PJ, Wood RC, Braverman ER, Chen TJ, Cull JG, Comings DE. The D2 dopamine receptor gene as a determinant of reward deficiency syndrome. J R Soc Med. 1996;89:396–400. doi: 10.1177/014107689608900711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady RJ, Damer CK, Heuser JE, O’Halloran TJ. Regulation of Hip1r by epsin controls the temporal and spatial coupling of actin filaments to clathrin-coated pits. J Cell Sci. 2010;123:3652–3661. doi: 10.1242/jcs.066852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson A. The current status of the dopamine hypothesis of schizophrenia. Neuropsychopharmacology. 1988;1:179–186. doi: 10.1016/0893-133x(88)90012-7. [DOI] [PubMed] [Google Scholar]
- Drake MT, Shenoy SK, Lefkowitz RJ. Trafficking of G protein-coupled receptors. Circ Res. 2006;99:570–582. doi: 10.1161/01.RES.0000242563.47507.ce. [DOI] [PubMed] [Google Scholar]
- Evans N. Methods of measuring internalization of G protein-coupled receptors: v. Chapter 12, p. Unit 12.6. Curr Protoc Pharmacol. 2004 doi: 10.1002/0471141755.ph1206s24. [DOI] [PubMed] [Google Scholar]
- Filmore D. It’s a GPCR World. Modern Drug Discovery. 2004;7:24–28. [Google Scholar]
- Free RB, Hazelwood LA, Cabrera DM, Spalding HN, Namkung Y, Rankin ML, Sibley DR. D1 and D2 dopamine receptor expression is regulated by direct interaction with the chaperone protein calnexin. J Biol Chem. 2007;282:21285–21300. doi: 10.1074/jbc.M701555200. [DOI] [PubMed] [Google Scholar]
- Funatsu T, Harada Y, Tokunaga M, Saito K, Yanagida T. Imaging of single fluorescent molecules and individual ATP turnovers by single myosin molecules in aqueous solution. Nature. 1995;374:555–559. doi: 10.1038/374555a0. [DOI] [PubMed] [Google Scholar]
- Genedani S, Carone C, Guidolin D, Filaferro M, Marcellino D, Fuxe K, Agnati LF. Differential sensitivity of A2A and especially D2 receptor trafficking to cocaine compared with lipid rafts in cotransfected CHO cell lines. Novel actions of cocaine independent of the DA transporter. J Mol Neurosci. 2010;41:347–357. doi: 10.1007/s12031-010-9328-y. [DOI] [PubMed] [Google Scholar]
- Jaiswal JK, Simon SM. Total internal reflection fluorescence microscopy for high-resolution imaging of cell-surface events: v. Chapter 4, p. Unit 4.12. Curr Protoc Cell Biol. 2003 doi: 10.1002/0471143030.cb0412s20. [DOI] [PubMed] [Google Scholar]
- Johnson DS, Jaiswal JK, Simon S. Total internal reflection fluorescence (TIRF) microscopy illuminator for improved imaging of cell surface events: v. Chapter 12, p. Unit 12.29. Curr Protoc Cytom. 2012 doi: 10.1002/0471142956.cy1229s61. [DOI] [PubMed] [Google Scholar]
- Karam CS, Ballon JS, Bivens NM, Freyberg Z, Girgis RR, Lizardi-Ortiz JE, Markx S, Lieberman JA, Javitch JA. Signaling pathways in schizophrenia: emerging targets and therapeutic strategies. Trends Pharmacol Sci. 2010;31:381–390. doi: 10.1016/j.tips.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CH, Lisman JE. A labile component of AMPA receptor-mediated synaptic transmission is dependent on microtubule motors, actin, and N-ethylmaleimide-sensitive factor. J Neurosci. 2001;21:4188–4194. doi: 10.1523/JNEUROSCI.21-12-04188.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotowski SJ, Hopf FW, Seif T, Bonci A, von Zastrow M. Endocytosis promotes rapid dopaminergic signaling. Neuron. 2011;71:278–290. doi: 10.1016/j.neuron.2011.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappano R, Maggiolini M. G protein-coupled receptors: novel targets for drug discovery in cancer. Nat Rev Drug Discov. 2011;10:47–60. doi: 10.1038/nrd3320. [DOI] [PubMed] [Google Scholar]
- Lawford BR, Young R, Noble EP, Kann B, Ritchie T. The D2 dopamine receptor (DRD2) gene is associated with co-morbid depression, anxiety and social dysfunction in untreated veterans with post-traumatic stress disorder. Eur Psychiatry. 2006;21:180–185. doi: 10.1016/j.eurpsy.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Li Y, Roy BD, Wang W, Zhang L, Sampson SB, Yang Y, Lin DT. Identification of two functionally distinct endosomal recycling pathways for dopamine D receptor. J Neurosci. 2012;32:7178–7190. doi: 10.1523/JNEUROSCI.0008-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JT, Chang WC, Chen HM, Lai HL, Chen CY, Tao MH, Chern Y. Regulation of feedback between protein kinase A and the proteasome system worsens Huntington’s disease. Mol Cell Biol. 2013;33:1073–1084. doi: 10.1128/MCB.01434-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Canfield V, Levenson R. Dominant negative mutants of filamin A block cell surface expression of the D2 dopamine receptor. Pharmacology. 2002;66:173–181. doi: 10.1159/000065531. [DOI] [PubMed] [Google Scholar]
- Luoma JI, Kelley BG, Mermelstein PG. Progesterone inhibition of voltage-gated calcium channels is a potential neuroprotective mechanism against excitotoxicity. Steroids. 2011;76:845–855. doi: 10.1016/j.steroids.2011.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macey TA, Gurevich VV, Neve KA. Preferential Interaction between the dopamine D2 receptor and Arrestin2 in neostriatal neurons. Mol Pharmacol. 2004;66:1635–1642. doi: 10.1124/mol.104.001495. [DOI] [PubMed] [Google Scholar]
- Millet LJ, Gillette MU. Over a century of neuron culture: from the hanging drop to microfluidic devices. Yale J Biol Med. 2012;85:501–521. [PMC free article] [PubMed] [Google Scholar]
- Park HJ, Kim ST, Yoon DH, Jin SH, Lee SJ, Lee HJ, Lim S. The association between the DRD2 TaqI A polymorphism and smoking cessation in response to acupuncture in Koreans. J Altern Complement Med. 2005;11:401–405. doi: 10.1089/acm.2005.11.401. [DOI] [PubMed] [Google Scholar]
- Piston DW. Choosing objective lenses: the importance of numerical aperture and magnification in digital optical microscopy. Biol Bull. 1998;195:1–4. doi: 10.2307/1542768. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S, De Angelis D, Rothman JE, Ryan TA. The use of pHluorins for optical measurements of presynaptic activity. Biophys J. 2000;79:2199–2208. doi: 10.1016/S0006-3495(00)76468-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirotta E, Fontaine V, Picetti R, Lombardi M, Samad TA, Oulad-Abdelghani M, Edwards R, Borrelli E. Signaling by dopamine regulates D2 receptors trafficking at the membrane. Cell Cycle. 2008;7:2241–2248. doi: 10.4161/cc.7.14.6307. [DOI] [PubMed] [Google Scholar]