

Abstract
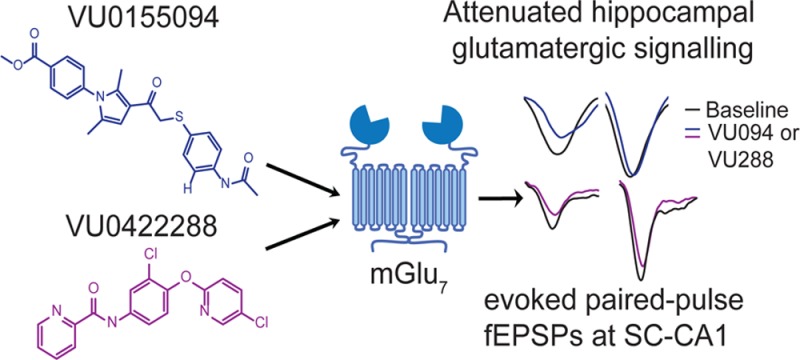
Metabotropic glutamate receptor 7 (mGlu7) is a member of the group III mGlu receptors (mGlus), encompassed by mGlu4, mGlu6, mGlu7, and mGlu8. mGlu7 is highly expressed in the presynaptic active zones of both excitatory and inhibitory synapses, and activation of the receptor regulates the release of both glutamate and GABA. mGlu7 is thought to be a relevant therapeutic target for a number of neurological and psychiatric disorders, and polymorphisms in the GRM7 gene have been linked to autism, depression, ADHD, and schizophrenia. Here we report two new pan-group III mGlu positive allosteric modulators, VU0155094 and VU0422288, which show differential activity at the various group III mGlus. Additionally, both compounds show probe dependence when assessed in the presence of distinct orthosteric agonists. By pairing studies of these nonselective compounds with a synapse in the hippocampus that expresses only mGlu7, we have validated activity of these compounds in a native tissue setting. These studies provide proof-of-concept evidence that mGlu7 activity can be modulated by positive allosteric modulation, paving the way for future therapeutics development.
Keywords: Allosteric modulator, metabotropic glutamate receptor, electrophysiology, hippocampus
Metabotropic glutamate receptor 7 (mGlu7), a member of the Class C G protein-coupled/7 transmembrane spanning receptor (GPCR/7TMR) superfamily, is widely expressed in the central nervous system (CNS) and is thought to play a critical role in modulating normal neuronal function and synaptic transmission. Studies with mGlu7 knockout (KO) animals have predicted therapeutic potential for mGlu7 manipulation in numerous neurological and psychiatric disorders including schizophrenia, depression, anxiety, and epilepsy.1 Numerous polymorphisms in GRM7 have now been linked to autism, depression, ADHD, and schizophrenia.2−5 mGlu7 is one of eight members of the mGlu family, of which there are three subgroups; mGlu4, mGlu6, mGlu7, and mGlu8 belong to group III. With the exception of mGlu6, which is restricted in expression to the retina, the group III receptors are located predominantly presynaptically in CNS neurons where they function to modulate both glutamate and γ-amino butyric acid (GABA) release. mGlu7 is a particularly interesting target, and its biology is predicted to be distinct from mGlu4 and mGlu8 due to its specific localization within the presynaptic active zones of glutamatergic and GABAergic synapses.6 In addition, the affinity of mGlu7 for glutamate is several orders of magnitude less than that of mGlu4 and mGlu8.7−9 Based on this low glutamate affinity, it has been proposed that mGlu7 may serve as an “emergency brake”, only becoming activated during intense synaptic activity when glutamate levels rise to high micromolar levels. Indeed, this hypothesis is supported by the fact that mGlu7 KO mice display both spontaneous and evoked seizures,10 suggesting a function for mGlu7 in modulating synaptic activity during periods of high synaptic glutamate levels.
All three widely expressed group III mGlu members are candidates for therapeutics development; however, due to the high conservation of the glutamate binding site across all of mGlu subtypes, it has been difficult to develop compounds with appropriate selectivity profiles to aid in performing proof-of-concept studies regarding the therapeutic potential of each receptor. We and others have recently made significant inroads into the understanding of mGlu4 biology and therapeutic potential by targeting allosteric sites on the receptor protein,11−16 resulting in the development of highly selective positive allosteric modulators (PAMs). These compounds bind within the more sequence-divergent transmembrane domains of the receptors, which are physically removed from the glutamate binding site in the large N-terminal domain of the Class C GPCRs. In the case of mGlu7, an allosteric agonist, AMN082, which interacts with the transmembrane spanning region of the receptor, has been reported (structure shown in Figure 1).17 In cell lines, this compound has been shown to directly activate mGlu7 via an allosteric binding site. Although AMN082 has been used to explore the biology and therapeutic potential of mGlu7 (for examples, see refs (18−24), among others), it is rapidly metabolized in vivo. Additionally, it exhibits significant off-target activities and is not active in every assay of mGlu7 function.25,26 In addition to AMN082, two transmembrane-binding negative allosteric modulators, MMPIP27 and ADX71743,28 as well as a selective antagonist that binds closer to the glutamate binding domain, XAP044,29 have recently been reported in the literature. Again, some of these compounds exhibit complex pharmacology; MMPIP, in particular, exhibits different effects in distinct cell backgrounds and in electrophysiological assessments.30 To date, no PAMs of mGlu7 have been reported. PAMs for this receptor would complement the existing tool kit of mGlu7 compounds and provide further validation of the role of this receptor in basic biology as well as pathophysiology. We report here two new compounds, VU0155094 and VU0422288, the first of which emerged from a high-throughput screening (HTS) campaign and the second derived from a chemical optimization program for mGlu4 PAMs. These compounds exhibit distinct pharmacological profiles in vitro and potentiate the activity of each of the group III mGlu’s, revealing novel insights into the interaction of orthosteric ligands with each receptor. Despite their lack of selectivity, we have found that VU0155094 and VU0422288 are highly useful for electrophysiology studies at the hippocampal Schaffer collateral-CA1 (SC-CA1) synapse, where an exclusive role for mGlu7 in modulating synaptic transmission has been previously reported26,31 and further validated here. Therefore, combining this synapse-specific expression with pharmacological studies has allowed us to exploit nonselective PAMs to explore mGlu7 biology, with a future goal of using these compounds to interrogate other disease models in which mGlu7 function might be altered at SC-CA1 synapses, such as models with deficits in hippocampal-based cognitive tasks.
Figure 1.

Structures of mGlu group III orthosteric and allosteric ligands.
Results
Discovery of VU0155094, a Group III mGlu Positive Allosteric Modulator
During our initial screening program, we were focused on the identification of novel PAMs for mGlu8. We screened the Molecular Libraries Program compound library, which contained approximately 100 000 compounds, as described32 using a HEK293 cell line expressing rat mGlu8 and human G protein inwardly rectifying potassium channels (GIRK) 1 and 2 and a thallium flux readout.33 This screening effort resulted in approximately 2000 primary hits, which were then counter-screened using an HEK293 cell line in which rat mGlu8 was coexpressed with the chimeric G protein Gqi9 as well as another cell line expressing GIRK 1/2 channels but no mGlu8. Compounds that were active in both mGlu8 assays, but did not induce responses in cells lacking mGlu8, were progressed for further study. Compounds active in GIRK-based assays independent of mGlu8 were followed up in a parallel program, resulting in the eventual development of ML297 (VU0456810), a selective GIRK channel activator.32
Further concentration–response curve studies revealed that only one compound, VU0155094, was validated as an mGlu8-active PAM that displayed concentration-dependent activity. This compound was distinct in structure from reported orthosteric agonists that are active at the group III receptors (Figure 1). VU0155094 exhibited a potency of 1.6 μM at rat mGlu8 when assessed using the cell line and assay employed for the primary HTS (Figure 2A), and did not exhibit agonist activity in this assay in the absence of glutamate (Figure 2B). Agonist activity in the GIRK-mediated thallium flux assay is commonly observed in our laboratories as increases in the baseline response, apparent in a “fold-shift” assay format.33 As shown in Figure 2B, the baselines of the glutamate concentration–response curves plus and minus 10 μM VU0155094 were not significantly different, indicating a lack of inherent agonism induced by the compound alone in this assay. VU0155094 was, as expected, able to shift the glutamate concentration–response curve to the left in this experiment (Figure 2B, 7.7-fold at 10 μM), suggesting that it acts as an mGlu8 PAM. Progressive fold-shift studies performed in another cell line in which rat mGlu8 was coexpressed with the promiscuous G protein, Gα15, again showed the expected profile for a PAM, with concentration-dependent increases in glutamate potency observed that approached a limit of cooperativity (Figure 2C). As an initial test for selectivity of this lead, the compound was profiled for fold-shift responses in rat mGlu2/Gα15-expressing cells and was unable to shift the glutamate concentration–response in any direction, indicating a lack of PAM or NAM activity at mGlu2 (Figure 2D).
Figure 2.

Identification and in vitro pharmacological characterization of VU0155094, a verified HTS lead. (A) Increasing concentrations of VU0155094 were applied to cells coexpressing rat mGlu8 and GIRK channels in the presence of an EC20 concentration of glutamate and thallium flux was measured. VU0155094 potentiated the glutamate response with a potency of 1.6 μM (pEC50 of 5.79 ± 0.07, N = three independent determinations performed in triplicate). (B) A 10 μM concentration of VU0155094 was applied to rat mGlu8/GIRK cells 2 minutes prior to the application of increasing concentrations of glutamate. The glutamate response was shifted 7.7 fold to the left. (C) Increasing concentrations of VU0155094 progressively left-shifted the glutamate concentration–response in cells expressing mGlu8 and the promiscuous G protein Gα15 (2.7-fold at 1 μM, 6.4-fold at 3 μM, 13.3-fold at 10 μM, and 21.4-fold at 30 μM; N = three independent determinations performed in duplicate). (D) VU0155094 was inactive in cells expressing rat mGlu2 and Gα15 (N = two independent determinations performed in duplicate).
This initial characterization suggested that VU0155094 represented a true mGlu8 PAM lead. Further selectivity profiling at a 10 μM concentration, however, showed that the compound potentiated responses of mGlu4, mGlu6 and mGlu7 in addition to mGlu8 but not the other mGlus, suggesting that VU0155094 is a pan-group III PAM (Supporting Information Figure S1). VU0155094 was further profiled with full potency determinations at mGlu4, mGlu7, and mGlu8 (Figure 3). These studies revealed that VU0155094 potentiated these receptors with potencies of 3.2 μM at mGlu4, 1.5 μM at mGlu7, and 900 nM at mGlu8. Additional studies profiling off-target activity at 68 targets using a Eurofins/PanLabs profile showed that VU0155094 was inactive at all targets with the exception of the norepinephrine transporter, where it induced a 50% inhibition of radioligand binding when tested at a 10 μM concentration (data not shown). These results suggest a very clean ancillary pharmacology profile for VU0155094.
Figure 3.

Potency determinations of VU0155094 at mGlu4, mGlu7, and mGlu8 reveal similar potency at all group III receptors. Increasing concentrations of VU0155094 were applied 2 min prior to the addition of an EC20 concentration of either glutamate (for mGlu4 and mGlu8) or L-AP4 (for mGlu7) in calcium mobilization assays utilizing receptor coexpression with either Gqi5 (mGlu4) or Gα15 (mGlu7 and mGlu8). N = three independent determinations performed in triplicate; mean ± SEM shown.
Structure–Activity Relationship (SAR) of the VU0155094 Scaffold
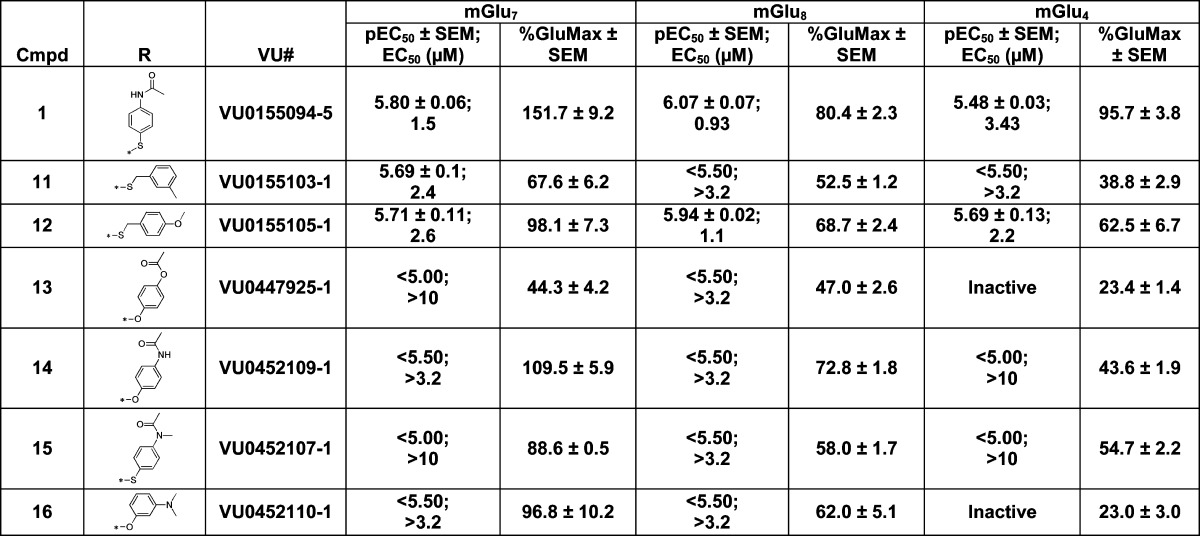
To explore structure–activity relationships within the VU0155094 scaffold, an SAR analysis at mGlu4, mGlu7, and mGlu8 was undertaken. These studies resulted in the generation of compounds 1–16, which examined the N-pyrrole substituents and the thio-aryl groups (Tables 1 and 2), via an SAR-by-catalog approach. Changing the ester group in VU0155094 to the primary sulfonamide, 2, led to small losses of potency for mGlu7 and mGlu8 (∼1–3-fold), but a larger loss of potency for mGlu4 (>10 μM) (Table 1). Removal of the ester or sulfonamide group was not well tolerated with the exception of (1) the 1,4-dioxane moiety, 4, for which the mGlu7/8 potency was similar to the sulfonamide but, again, induced loss of mGlu4 potency, and (2) the thiophene ethyl, 7, which retained potency at all three receptors. Moving of the ethyl, 9, or the difluoromethoxy phenyl group, 8, was not tolerated.
Table 1. SAR Evaluation of the Left-Hand Aryl Ester Moiety of VU0155094a.
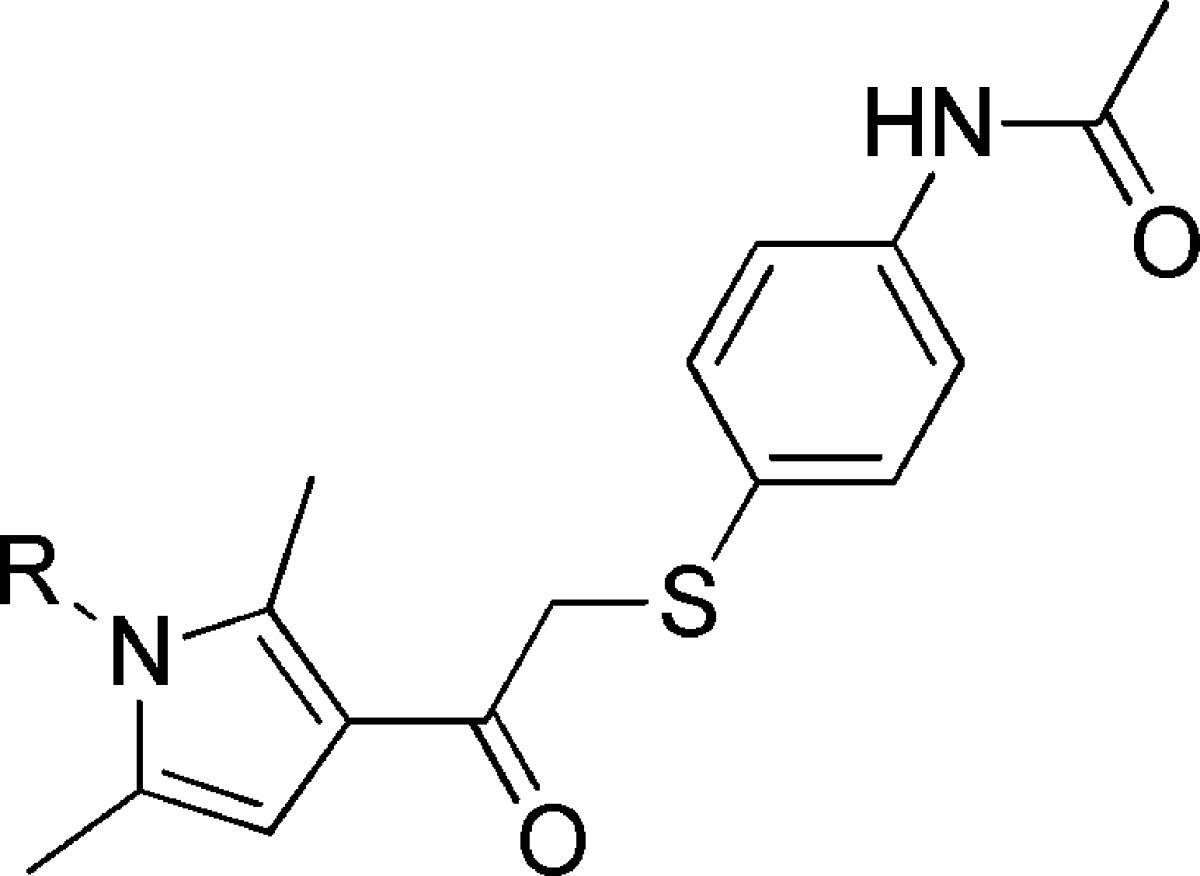
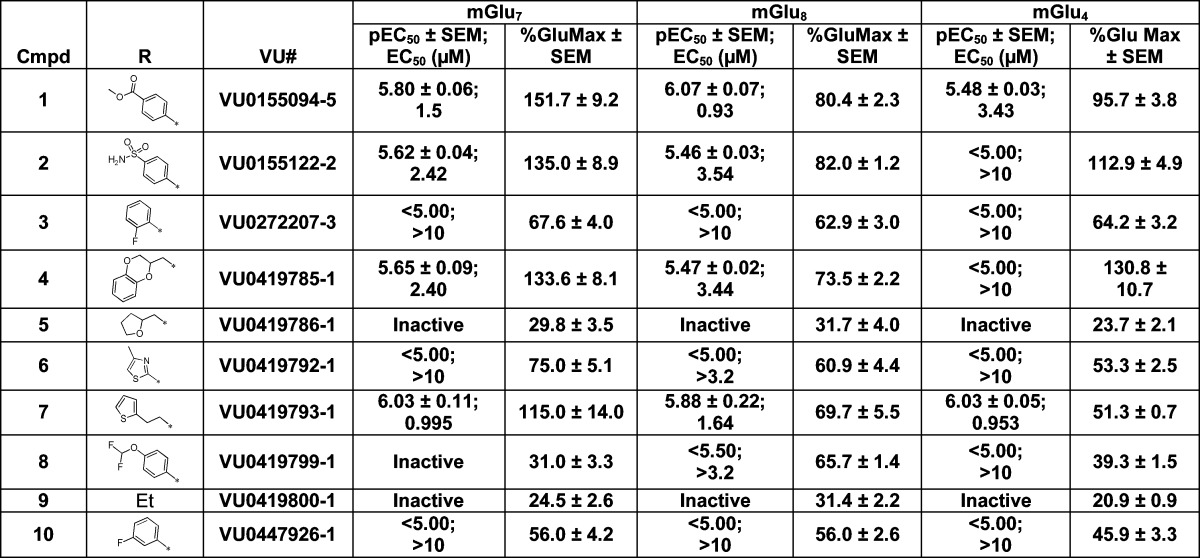
Data are composed of three independent experiments performed in triplicate (mean ± SEM values shown).
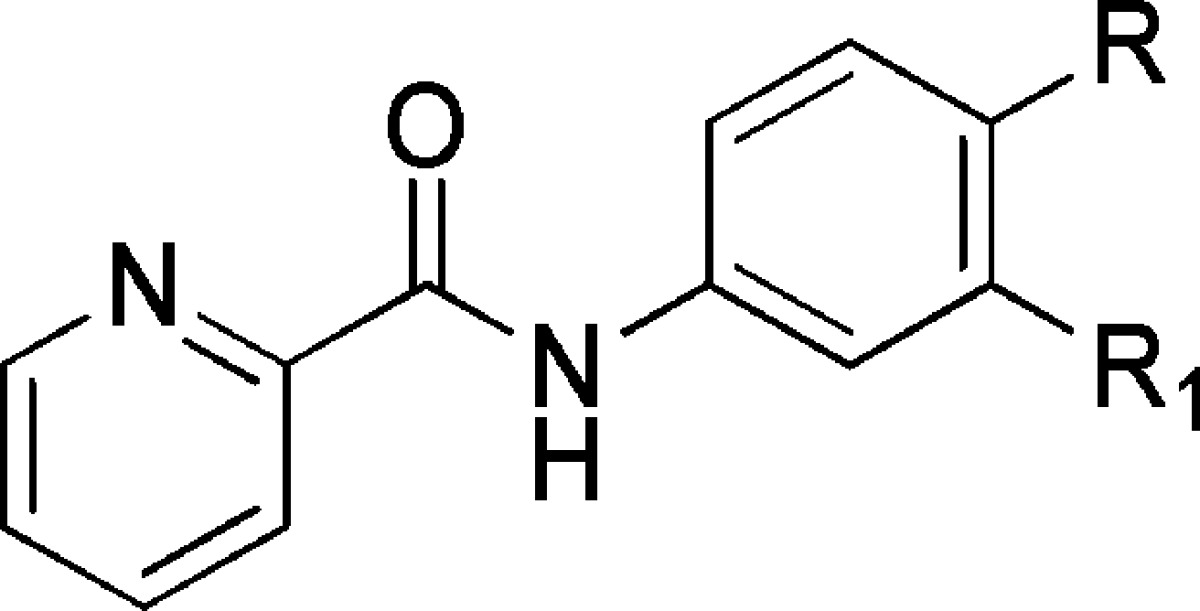
Table 2. SAR Evaluation of the Thioaryl Ether Moietya.
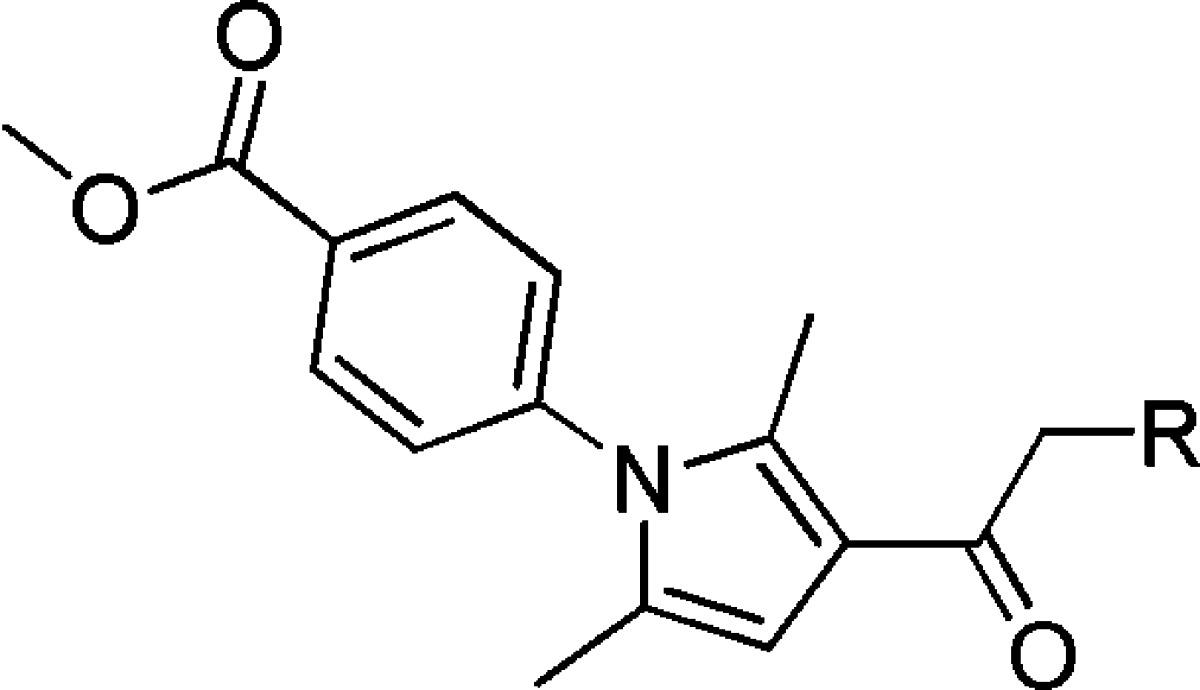
Data are composed of three independent experiments performed in triplicate (mean ± SEM values shown).
Next, the right-hand portion was evaluated by keeping the methyl ester moiety constant (Table 2). Eliminating the acetamide group and adding a methylene spacer was tolerated (11 and 12), although these compounds were not more potent than VU0155094. Replacing the thioether with the aryl ether, 14, was not as active, nor was alkylation of the acetamide nitrogen, 15. Overall, changes to this portion of the molecule did not produce any compounds with better potency or selectivity when compared to VU0155094.
Identification and SAR of VU0422288, Another Group III mGlu PAM with Properties Distinct from Those of VU0155094
In parallel to our HTS efforts, we also explored the selectivity profile of mGlu4 PAMs that had been developed within our program. These efforts led to the identification of the scaffold represented by VU0422288 (Figure 1).34 This compound was also nonselective among mGlu4, mGlu7, and mGlu8 but was more potent than VU0155094 (Figure 4, mGlu8 = 125 nM, mGlu7 = 146 nM, mGlu4 = 108 nM for VU0422288 versus ∼1–3 μM for VU0155094, Figure 3). Similar to VU0155094, we undertook an exploration of the VU0422288 scaffold to define moieties important for activity and selectivity. Initial SAR analysis centered around the right-hand aryl ether portion as well as the left-hand amide moiety. The synthesis of the compounds is outlined in Scheme 1, which follows a two-step protocol from the commercially available 4-aminophenols. Thus, 4-amino-3-halogenphenol, 17, was reacted with either substituted fluoropyridine or chloropyrimidine yielding the biaryl ethers, 18. Finally, reaction with an acid chloride under basic conditions led to the isolation of the final products, 19.
Figure 4.

Potency determinations of VU0422288 at mGlu4, mGlu7, and mGlu8 reveal similar potency at all group III receptors but enhanced potency compared to VU0155094. Increasing concentrations of VU0422288 were applied 2 min prior to the addition of an EC20 concentration of either glutamate (for mGlu4 and mGlu8) or L-AP4 (for mGlu7) in calcium mobilization assays utilizing receptor coexpression with either Gqi5 (mGlu4) or Gα15 (mGlu7 and mGlu8). N = three independent determinations performed in triplicate.
Scheme 1. Synthesis of Aryl Ether Picolinamide.

From this initial SAR examination, it was apparent that small structural changes greatly impacted potency; for example, removing the 4-chloro atom from 20 resulted in a ∼10–20-fold loss in potency in the unsubstituted pyrimidine, 21. A further loss of potency was observed by changing the pyrimidine ether, 21, to the pyrimidine amine, 22, which induced an additional >10-fold loss in potency at each receptor subtype (Table 3). However, the unsubstituted pyridine ether, 23, was of similar potency to the original hit, 20. Substitution of a trifluoromethyl group, 24, led to a slight loss in potency compared to 20. Changing the internal phenyl halogen from the chloro to fluoro with the most potent chloropyridine right-hand piece did not lead to an appreciable loss in potency (e.g., 20 vs 26). However, the fluoro substituent did lead to a significant loss of potency on the 2-pyrimidine right-hand analogue (e.g., 21 vs 27). It should be noted that compounds 21 and 23 have been disclosed in the patent literature; however, no specific potency data was reported for any receptor.35
Table 3. SAR of the Right-Hand Aryl Ethera.
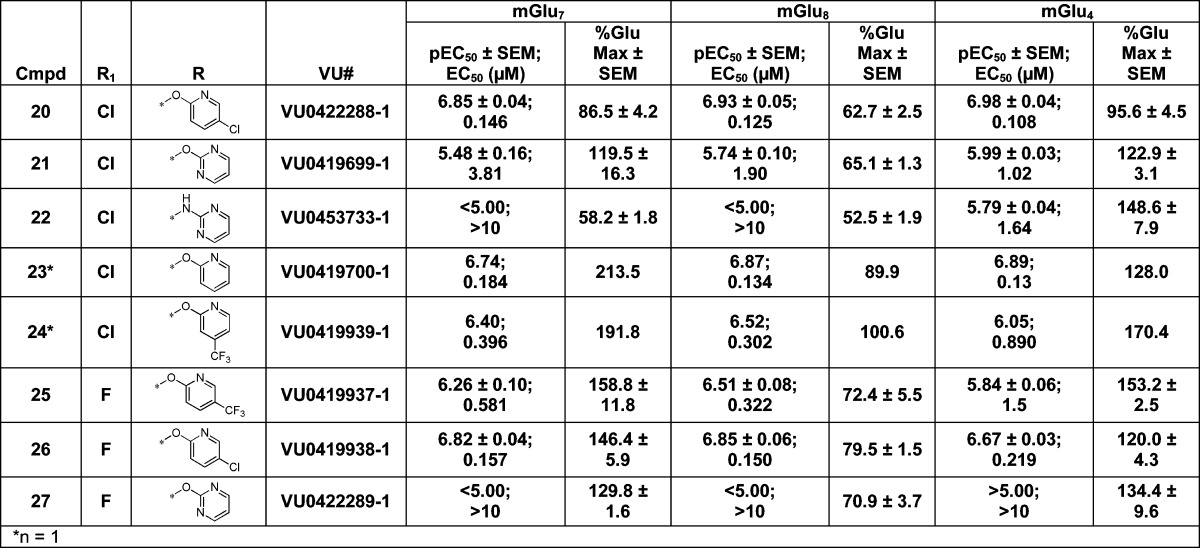
Data are composed of three independent experiments performed in triplicate (mean ± SEM values shown).
The SAR around the amide portion was centered on picolinamide replacements, as this amide has been shown to be favored by the mGlu4 receptor with little substitution tolerated34,36 (Table 4). Much like the case for mGlu4, little tolerance for substitution was observed for both mGlu7 and mGlu8. Addition of a chlorine atom to the 4-position, 28, led to a complete loss of activity at all three receptors. Replacing the picolinamide with isonicotinamide, 29, or nicotinamide, 30, also was not tolerated. Interestingly, the 2-chloropyrimidine-4-carboxamide, 31, retained potency at mGlu4, but lost activity at mGlu7/8 compared to the picolinamide. However, replacing the 2-chloro with 2-methyl, 32, destroyed the mGlu4 activity. Moving from 6-membered heterocyclic replacements to 5-membered replacements (33–39) did not lead to a more active compound than VU0422888. Again, minor modifications within these compounds led to significant differences in observed potencies. For example, N-methyl-pyrazole-3-carboxamide, 33, was nearly 3-fold less active at all three receptors when compared to the 2-methylthiazole-4-carboxamide derivative, 37. In addition to the heteroaryl replacements for the picolinamide, several cycloheteroalkanes (data not shown) were evaluated, with only the 2-tetrahydropyran derivative, 40, retaining potency. Lastly, substituted phenyl derivatives (data not shown) were also evaluated with only the unsubstituted phenyl, 41, showing any activity, although this compound did show some selectivity for mGlu7 and mGlu4. At the end of this exercise, we decided to further characterize VU0155094 and VU0422288 for use as group III mGlu PAMs. This decision was based on further profiling of VU0422288 in the Ricerca/Eurofins Lead Profiler panel of 68 different GPCRs, ion channels, and transporters; VU0422288 did not show activity at any of these targets.
Table 4. Evaluation of Picolinamide Replacementsa.
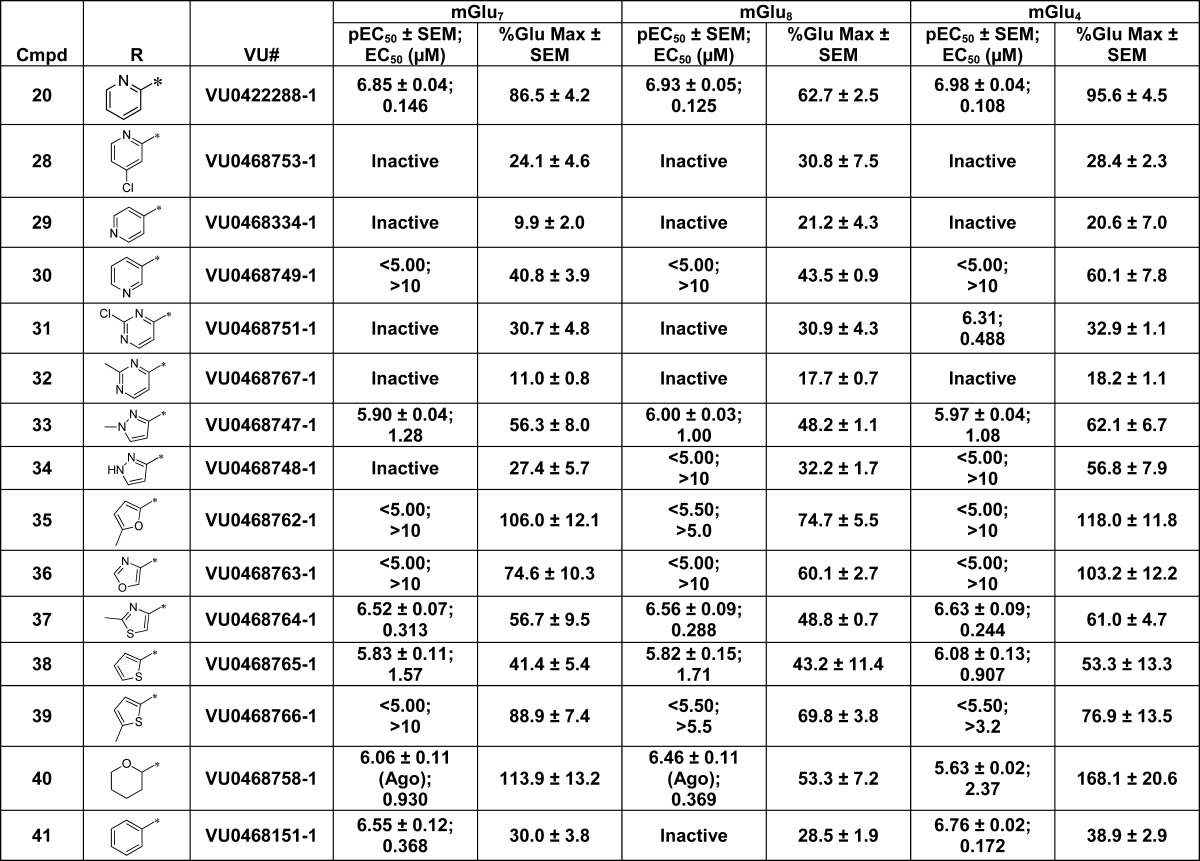
Data are composed of three individual experiments performed in triplicate (mean ± SEM values shown).
VU0155094 and VU0422288 Exhibit Distinct Properties in Their Interactions with mGlu4, mGlu7, and mGlu8
We next performed in vitro efficacy studies to further profile the interactions of VU0155094 and VU0422288 with each receptor. We began by performing agonist concentration–response curves in cells expressing mGlu4, mGlu7, or mGlu8 with the orthosteric agonists glutamate, L-AP4, and LSP4-2022 in two distinct assays37 (Supporting Information Figure S3), in which responses were profiled in cells coexpressing chimeric or promiscuous G proteins to couple the receptors to calcium mobilization or in cells coexpressing GIRK1/2 channels.33 These techniques allow for the examination of both Gα (calcium assay) and Gβγ regulated pathways downstream of receptor activation. The use of different agonists was employed to determine if there was any probe dependence, either between agonists at each receptor or when responses were potentiated by VU0155094 or VU0422288, as allosteric modulators can exhibit differential cooperativity with distinct agonists.38−41 Additionally, as glutamate will activate all mGlus and ionotropic glutamate receptors and is a substrate for glutamate transporters, studies in native tissue preparations require a more selective surrogate agonist to be used.
To understand changes in the activity of agonists in the presence of these PAMs, as well as to compare activities of these agonists themselves across receptors, we applied an operational model of agonism to orthosteric agonist concentration-response curves in the presence and absence of increasing concentration of VU0155094 or VU0422288 (shown conceptually in Figure 5 and data are shown in Supporting Information Figures S3–S5). As the orthosteric binding site of the mGlus is physically removed from the transmembrane domains, we initially fit the curves using eq 2 (Methods) and assumed no effect on orthosteric agonist affinity (i.e., α = 1) induced by the PAMs. As shown in Supporting Information Table 1, we observed significant differences in several of the agonist KA values in the presence of VU0155094 or VU0422288; additionally, probe dependence for this effect was noted. For example, the KA values for LSP4-2022 were right-shifted at each receptor when at least one modulator was present. At mGlu7, the affinity of glutamate was increased in the presence of VU0155094 but not significantly changed by VU0422288. At mGlu7 and mGlu8, we observed a decrease in affinity of L-AP4 induced by VU0422288; this was true for the mGlu8/L-AP4 combination with VU0155094 as well. These results clearly show distinct interactions between the orthosteric and allosteric sites on the different receptors.
Figure 5.
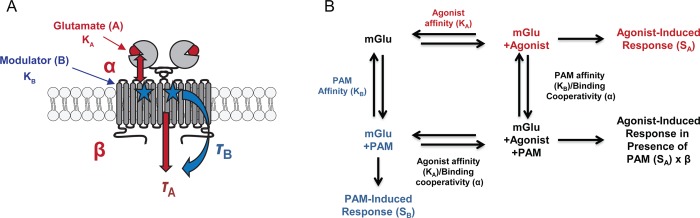
Schematic diagram of operational modeling parameters. (A) Schematic of an mGlu dimeric receptor. Glutamate or orthosteric agonists (red circle) bind in the large extracellular binding domain of the mGlus, and modulators (blue stars) bind in the transmembrane domains. KA represents orthosteric agonist affinity, while KB is allosteric modulator affinity. The results shown here demonstrate interactions between the allosteric and orthosteric sites in terms of affinity modulation. Affinity modulation is governed by the cooperativity factor α, and efficacy modulation is governed by β. The parameters τA and τB represent the ability of orthosteric agonist and allosteric ligands, respectively, to directly activate the receptor. (B) Interaction of allosteric parameters and effects on response shown using the allosteric ternary complex model. Adapted from ref (42).
Due to these apparent changes in KA values, we reanalyzed the calcium assay data shown in Supporting Information Figures S3–S5 using eq 2 by allowing the α value to be shared across data sets and setting the maximal response (Em) to the maximum response observed with any agonist/modulator combination. These revised data are shown in Supporting Information Table 2, which report KA and τA values for each PAM versus the responses seen in the presence of vehicle (DMSO). For all three agonists at mGlu4, and for L-AP4 and glutamate at mGlu7, both PAMs also increased the maximal response to agonist (Supporting Information Figures S4 and S5). In contrast, both PAMs left-shifted the responses to each agonist at mGlu8. This latter finding correlates with the agonist responses observed in Supporting Information Figure S3, in which all three agonists induce the same level of response for mGlu8. However, these data indicate that, for all orthosteric agonists employed, the mGlu4 response does not approach the maximal capacity of the system for calcium mobilization. In contrast, while responses to glutamate and L-AP4 were both left and upward shifted in mGlu7-expressing cells, the response to LSP4-2022 was only left-shifted without an increase in the maximal response, suggesting that LSP4-2022 acts as a full agonist in this assay. Interestingly, the efficacy of L-AP4 (τA) was only significantly increased at mGlu4 and mGlu8 in the presence of VU0422288, and not VU0155094. Likewise, glutamate and LSP4-2022 were similarly unaffected at mGlu4, while the efficacy of LSP4-2022, but not glutamate, was enhanced by both PAMs at mGlu8.
We then used operational modeling to predict the affinity (KB), cooperativity (α and/or β), and efficacy (τB) of the PAMs (Tables 5 and 6).42 At mGlu4 with L-AP4 or LSP4-2022 as the agonist, VU0155094 mediated the majority of its activity via efficacy (β) rather than affinity (α) modulation, whereas the reverse was true for these same agonists at mGlu8 (Table 5). This may be due to the lack of a change in Em observed with agonists at mGlu8 relative to other receptors. Furthermore, apparent agonist and/or modulator affinity and cooperativity values differed with distinct agonists at mGlu7. For example, when L-AP4 was used as the agonist, the affinity of VU0155094 was 4-fold greater than that when glutamate was used. VU0155094 exhibited neutral efficacy (β) cooperativity with L-AP4 and LSP-2022 at mGlu7, whereas both affinity and efficacy modulation contributed to changes in the glutamate response and the greater degree of overall cooperativity observed. This suggests that VU0155094 exhibits probe dependence that is distinct with various orthosteric agonists and at the different group III mGlu subtypes. When compared to VU0155094, VU0422288 exhibited 30–100-fold higher affinity at each receptor (Table 6). At mGlu7 and mGlu8, VU0422288 positively modulated both affinity and efficacy of all three orthosteric agonists. In contrast, at mGlu4 the manifestation of cooperativity was unique to each agonist, and there was a strong potentiation of the efficacy of L-AP4 (β = 10.4) coupled with a reduction of affinity (α = 0.33).
Table 5. Predicted Affinities and Cooperativities for VU0155094 across the Group III mGlus Using Calcium Assays with Chimeric G Proteins Reveals Distinct Receptor–PAM Interactionsa.
| mGlu4 |
mGlu7 |
mGlu8 |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Glu | L-AP4 | LSP4-2022 | Glu | L-AP4 | LSP4-2022 | Glu | L-AP4 | LSP4-2022 | |
| pKB | 4.88 ± 0.03 | 4.88 ± 0.05d | 4.97 ± 0.10 | 4.77 ± 0.18c | 5.38 ± 0.22cd | 5.10 ± 0.17e | 5.07 ± 0.37 | 4.83 ± 0.07d | 4.85 ± 0.08e |
| KB (μM) | 13.3 | 13.2 | 10.6 | 17 | 4.2 | 8.0 | 8.6 | 14.7 | 14.0 |
| log τB | n.a | n.a | n.a | –0.17 ± 0.03 | –0.09 ± 0.04 | –0.22 ± 0.06 | –0.12 ± 0.03 | –0.14 ± 0.04 | –0.10 ± 0.06 |
| τB | n.a | n.a | n.a | 0.81 | 1.16 | 0.61 | 0.75 | 0.73 | 0.80 |
| log β | 0.16 ± 0.08fh | 0.67 ± 0.27fi | 0.33 ± 0.07j | 0.68 ± 0.12gh | 0.07 ± 0.03gi | –0.10 ± 0.10gj | 0.08 ± 0.02h | 0.05 ± 0.03i | 0.05 ± 0.06j |
| β | 1.4 | 4.7 | 2.1 | 4.8 | 1.2 | 0.8 | 1.2 | 1.1 | 1.1 |
| log α | 0.28 ± 0.07 | –0.15 ± 0.30k | 0.17 ± 0.13l | 0.58 ± 0.13 | 0.39 ± 0.21 | 0.82 ± 0.26l | 0.61 ± 0.39 | 0.92 ± 0.08k | 0.93 ± 0.06l |
| α | 1.9 | 0.71 | 1.5 | 3.8 | 2.4 | 6.6 | 4.1 | 8.3 | 8.6 |
| n | 6.2 ± 1.8 | 2.0 ± 0.2 | 2.2 ± 0.03 | 2.3 ± 0.4 | 6.7 ± 3.1 | 2.9 ± 1.2 | 4.6 ± 2.5 | 2.9 ± 0.4 | 3.0 ± 0.3 |
| basal | 6.9 ± 1.8 | 7.1 ± 0.7 | 7.6 ± 0.6 | 0.4 ± 2.3 | 3.3 ± 2.4 | 3.8 ± 0.8 | 3.3 ± 0.4 | 2.4 ± 0.3 | 2.3 ± 0.5 |
| Emb | 195.7 | 195.7 | 195.7 | 377.2 | 377.2 | 377.2 | 102.3 | 102.3 | 102.3 |
Data were fitted from curves shown in Figure S4 (Supporting Information) using eq 2. The α value was shared between data sets, and the Em was constrained to the maximal level of potentiation observed for that subtype between any combination of ligands in a given experiment. For statistical analyses, pKB, log α, and log β values were compared between agonists at one receptor and then separately between receptors for a given agonist. All statistical tests were one-way ANOVA with a Tukey’s post-test to compare all columns. Data are comprised of three individual experiments performed in duplicate (mean ± SD values shown).
Em value constrained to the maximal level of potentiation observed for that subtype between any combination of ligands in a given experiment; the mean is reported. All p values <0.05.
pKB between mGlu7/glutamate versus mGlu7/L-AP4.
pKB between mGlu7/L-AP4 versus mGlu4/L-AP4 and mGlu8/L-AP4.
pKB between mGlu7/LSP4-2022 versus mGlu8/LSP4-022.
Log β between mGlu4/glutamate versus mGlu4/L-AP4.
Log β between mGlu7/glutamate versus mGlu7/L-AP4 and mGlu7/LSP4-2022.
Log β between mGlu7/glutamate versus mGlu4/glutamate and mGlu8/glutamate.
Log β between mGlu4/L-AP4 versus mGlu7/L-AP4 and mGlu8/L-AP4.
Log β between mGlu4/LSP4-2022 versus mGlu7/LSP4-2022 and mGlu8/LSP4-2022.
Log α of mGlu4/L-AP4 versus mGlu8/L-AP4.
Log α of mGlu4/LSP4-2022 versus mGlu7/LSP4-2022 and mGlu8/LSP4-2022.
Table 6. Predicted Affinities and Cooperativities for VU0422288 across the Group III mGlus Using Calcium Assays with Chimeric G Proteins or Thallium Flux Assays for GIRK Channel Activationa.
| mGlu4 |
mGlu7 |
mGlu8 |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Glu | L-AP4 | LSP4-2022 | Glu | L-AP4 | LSP4-2022 | Glu | L-AP4 | LSP4-2022 | |
| calcium | |||||||||
| pKB | 6.96 ± 0.18l | 6.98 ± 0.20l | 7.06 ± 0.18l | 6.84 ± 0.20 | 6.84 ± 0.17l | 6.95 ± 0.10l | 6.69 ± 0.18 | 6.66 ± 0.15 | 6.80 ± 0.21 |
| KB (nM) | 110 | 105 | 88.0 | 146 | 143 | 112 | 203 | 218 | 157 |
| log β | 0.42 ± 0.09c | 1.02 ± 0.21cd | 0.72 ± 0.34e | 0.31 ± 0.08d | 0.19 ± 0.12 | 0.26 ± 0.21 | 0.21 ± 0.11 | 0.22 ± 0.05d | 0.13 ± 0.06e |
| β | 2.7 | 10.4 | 5.3 | 2.0 | 1.6 | 1.8 | 1.6 | 1.7 | 1.3 |
| log α | –0.03 ± 0.03f | –0.48 ± 0.20g | –0.21 ± 0.32 | 0.31 ± 0.06f | 0.27 ± 0.06g | 0.12 ± 0.17 | 0.18 ± 0.09f | 0.20 ± 0.07g | 0.35 ± 0.14 |
| α | 0.94 | 0.33 | 0.62 | 2.0 | 1.9 | 1.3 | 1.5 | 1.6 | 2.2 |
| n | 2.3 ± 0.1 | 1.3 ± 0.05 | 1.6 ± 0.2 | 2.6 ± 0.6 | 2.4 ± 0.7 | 2.1 ± 0.4 | 2.7 ± 0.5 | 2.1 ± 0.5 | 2.1 ± 0.06 |
| basal | 7.1 ± 1.8 | 7.2 ± 0.9 | 7.8 ± 0.4 | 1.3 ± 2.4 | 0.4 ± 1.8 | 3.6 ± 1.4 | 2.4 ± 0.5 | 1.9 ± 0.2 | 1.8 ± 0.4 |
| Emb | 195.7 | 195.7 | 195.7 | 377.2 | 377.2 | 377.2 | 102.3 | 102.3 | 102.3 |
| GIRK | |||||||||
| pKB | 6.51 ± 0.11l | 6.52 ± 0.10l | 6.57 ± 0.03l | 6.69 ± 0.55 | 6.34 ± 0.15l | 6.59 ± 0.11l | 6.20 ± 0.25 | 6.61 ± 0.19 | 6.45 ± 0.27 |
| KB (nM) | 306 | 304 | 268 | 205 | 462 | 255 | 626 | 243 | 352 |
| log αβ | 0.54 ± 0.06i | 0.58 ± 0.04j | 0.54 ± 0.003k | 0.15 ± 0.04hi | 0.36 ± 0.06hj | 0.34 ± 0.07h | 0.36 ± 0.05ik | 0.37 ± 0.03j | 0.41 ± 0.06 |
| αβ | 3.5 | 3.8 | 3.5 | 1.4 | 2.3 | 2.2 | 2.3 | 2.3 | 2.6 |
Data were fitted from curves shown in Supporting Information Figure S5 (calcium assay) using eq 2 and Supporting Information Figure S6 (GIRK assay) using the four parameter logistical eq 3. For calcium data, the α value was shared between datasets and the Em was constrained to the maximal level of potentiation observed for that subtype between any combination of ligands in a given experiment and to a value just above the largest agonist max value for GIRK assays. For statistical analyses, pKB, log α, and log β values were compared between agonists at one receptor and then separately between receptors for a given agonist. All statistical tests were one-way ANOVA with a Tukey’s post-test to compare all columns with the exception of comparisons of pKB values between assays for each condition, which were assessed using an unpaired Student’s t test. Data are composed of three individual experiments performed in duplicate (mean ± SD values shown).
Em value constrained to the maximal level of potentiation observed for that subtype between any combination of ligands in a given experiment; the mean is reported. All p values <0.05.
Log β of mGlu4/glutamate versus mGlu4/L-AP4.
Log β of mGlu4/L-AP4 versus mGlu7/L-AP4 and mGlu8/L-AP4.
Log β of mGlu4/LSP4-2022 versus mGlu8/LSP4-2022.
Log α of mGlu4/glutamate versus mGlu7/glutamate and mGlu8/glutamate.
Log α of mGlu4/L-AP4 versus mGlu7/L-AP4 and mGlu8/L-AP4.
Log αβ of mGlu7/glutamate versus mGlu7/L-AP4 and mGlu7/LSP4-2022.
Log αβ of all glutamate conditions are significantly different from each other.
Log αβ of mGlu4/L-AP4 versus mGlu7 L-AP4 and mGlu8 L-AP4.
Log αβ of mGlu4/LSP4-2022 versus mGlu7 LSP4-2022.
pKB differs between assays (i.e., mGlu4/Glu/pKB/calcium is significantly different from mGlu4/Glu/pKB/GIRK).
We then attempted to assess the affinity and cooperativity of these compounds in the GIRK assay; unfortunately, we were not able to obtain acceptable curve fits for VU0155094 due to apparent nonspecific inhibition of GIRK channels induced by this compound at high concentrations. For VU0422288, we were able to fit data by applying a modified version of the allosteric ternary complex model (eq 3) to the shifts in agonist potency in the presence of modulator,43 which allowed us to assess affinity and cooperativity as a combined αβ parameter while setting the Em value to a level just above the maximal response observed. As shown in Table 6, VU0422288 exhibited similar affinities for all agonists used and across each receptor subtype; however, the KB values for the compound between assays (calcium versus GIRK) were different for mGlu4 and mGlu7, regardless of agonist, with the exception of the mGlu7/glutamate combination. When glutamate was used, VU0422288 exhibited distinct cooperativities at each receptor (mGlu7 < mGlu8 < mGlu4) when assessed using the GIRK assay. Cooperativity of VU0422288 was significantly lower with glutamate compared to the other two agonists at mGlu7, whereas, at the other subtypes, cooperativity was similar for all agonists. Overall, these studies provide evidence that VU0155094 and VU0422288 functionally act as group III mGlu PAMs with distinct properties that potentiate the activity of multiple orthosteric agonists across two pathways. However, this potentiation is textured and reveals distinct degrees of probe dependence, which is a new finding for the group III mGlus.
In addition to evaluating the effects of the PAMs, operationally fitting the data also allowed us to directly compare the affinities and efficacies of orthosteric agonists at the group III receptors. These studies revealed new insight into the interaction and signaling bias induced by group III mGlu orthosteric agonists (Figure S3 and Table 3 in the Supporting Information). In these studies, L-AP4 exhibited 26-fold higher affinity for mGlu4, 6-fold higher for mGlu7 and 15-fold higher for mGlu8 when compared to glutamate. In comparison, LSP4-2022 showed 9-fold higher affinity for mGlu4, 70-fold higher for mGlu7 and 10-fold lower affinity for mGlu8. In GIRK assays, L-AP4 again showed higher affinity for mGlu4 and mGlu8 (93-fold and 80-fold, respectively) but similar affinity for mGlu7 when compared to glutamate. LSP4-2022 was higher affinity for mGlu4 (72-fold) and mGlu7 (23-fold), but not mGlu8 when compared to glutamate. Additionally, we assessed potential for orthosteric ligand bias by calculating the correlation of efficacy/affinity (log τ/KA) for the two assays; these studies revealed a strong correlation (Supporting Information Figure S7A). However, when concentration–response curve data were plotted using a bias plot, which allows for comparison of compound activity in two assays at the same concentrations, we noted that glutamate was biased toward calcium over GIRK at mGlu4 and mGlu8, whereas the reverse was true for mGlu7 (Supporting Information Figure S7B–D). Comparison of log τ/KA values relative to glutamate revealed that L-AP4 and LSP4-2022 bias mGlu4 and mGlu8 signaling to GIRK over calcium. In contrast, at mGlu7, L-AP4 and LSP4-2022 bias signaling to calcium over GIRK. These results suggest, for the first time, that there is texture in agonist signaling bias downstream of group III mGlu activation, and that different agonists may engage specific pathways in a distinct manner.
VU0155094 and VU0422288 potentiate mGlu7-Modulated Changes in Synaptic Transmission in the Hippocampus
With two structurally distinct group III receptor PAMs in hand, we sought to further validate their activity in a native tissue setting. Due to the nonselective nature of the compounds, we focused our studies on the Schaffer collateral-CA1 (SC-CA1) synapse of the hippocampus. This synapse has been described as undergoing a developmental switch at the presynaptic level, with mGlu8 being expressed at early developmental stages and mGlu7 activation being responsible for modulation of synaptic transmission in adult rats.26,31 To further validate selective mGlu7 expression in adult animals, we recorded evoked field excitatory postsynaptic potentials (fEPSPs) at SC-CA1 synapses at baseline and then in the presence of specific concentrations of three distinct agonists, LSP4-2022, LSP1-2111, an mGlu4-preferring orthosteric agonist (Figure 1),44 and (S)-3,4-DCPG (Figure 1), an mGlu8-preferring agonist. While LSP4-2022 is more potent at mGlu4 than mGlu7,37 the inclusion of the LSP1-2111 control should allow detection of an mGlu4-mediated effect. These studies showed that only LSP4-2022 was able to significantly reduce fEPSPs (Figure 6), suggesting that activity was mediated by mGlu7. This, along with previous data showing that very high concentrations of L-AP4 were required to reduce transmission at this synapse in adult animals and that mGlu4 PAMs had no effect at this location,26 further suggest that mGlu7 is the only group III mGlus mediating inhibition of fEPSPs at the SC-CA1 synapse. As shown in Figure 6B and D, LSP4-2022 also increased the paired-pulse ratio, suggesting that it modulates fEPSPs via a presynaptic mechanism of action, which is consistent with the expression pattern of mGlu7 at these synapses.
Figure 6.

SC-CA1 synaptic transmission is modulated by LSP4-2022 but not the mGlu4 agonist LSP1-2111 or the mGlu8 agonist, (S)-3,4-DCPG. (A) Application of either a selective mGlu8 agonist, DCPG, or an mGlu4 selective agonist, LSP1-2111, has no effect on fEPSP slope. Application of 30 μM LSP4-2022, a concentration that activates both mGlu4 and mGlu7, causes a decrease in the fEPSP slope. (B) Sample traces showing paired-pulses during the baseline recording (1) and during each drug addition (2). Scale bars represent 0.4 mV by 5 ms. (C) Quantification of slope change in response to 3 μM LSP1-2111, 1 μM DCPG, or 30 μM LSP4-2022. Data are normalized as percent of baseline slope averaged over the entire drug addition (N = 3–4). ***p < 0.001 vs baseline, +++p < 0.001 vs LSP4-2022. (D) Application of 30 μM LSP4-2022 causes an increase in paired-pulse ratio. **p < 0.01 vs baseline. Paired-pulse ratios were calculated as described in the Methods section.
To determine if our newly identified PAMs could potentiate LSP4-2022-induced reductions in fEPSP slopes, we preapplied either vehicle, 1 μM VU0422288, or 30 μM VU0155094 to slices for 5 min and then coapplied vehicle or PAM with LSP4-2022. These studies showed that both compounds were able to potentiate LSP4-2022-induced reductions in the fEPSP slope (Figure 7A, C). Additionally, the compounds caused an increase in the paired-pulse ratio, suggesting that they are acting presynaptically (Figure 7B, D). Coupled with the selectivity studies shown in Figure 6, our results suggest that both compounds are potentiating the effects of LSP4-2022 at presynaptically expressed mGlu7 receptors. Thus, the combination of tools, coupled with restricted expression of only mGlu7 at this synapse, has allowed us to validate the PAM mechanism for the manipulation of mGlu7 function in a neuronal population.
Figure 7.

VU0155094 and VU0422288 potentiate fEPSP slopes induced by LSP4-2022. (A) Pretreatment of slices with either 1 μM VU0422288 or 30 μM VU0155094 caused a significant decrease in the fEPSP slope compared to application of 30 μM LSP4-2022 alone. (B) Sample traces of baseline fEPSP slopes (1) or in the presence of either VU0155094 or VU0422288 in combination with 30 μM LSP4-2022 (2). Scale bars represent 0.4 mV by 5 ms. (C) Quantification of slope change in response to LSP4-2022 application, VU0155094 or VU0422288 alone, and VU0155094 or VU0422288 and LSP4-2022 application. Data are normalized as percent of baseline slope averaged over either PAM alone or PAM and LSP4-2022 addition (N = 4). **p < 0.01 vs baseline, ***p < 0.001 vs baseline, +p < 0.05 vs LSP4-2022, ++p < 0.01 vs LSP4-2022 (D) Application of VU0155094 or VU0422288 and LSP4-2022 increases the paired-pulse ratio. *p < 0.05 vs baseline.
Discussion
As the mGlus belong to a highly druggable class of targets, the GPCRs, much attention has been focused on this receptor family not only to better understand and explore their biology but also to target them for therapeutics development. Among the group III mGlus, mGlu7 is the most widely distributed presynaptic subtype, acting as an auto- and heteroreceptor to regulate glutamate and GABA release from presynaptic terminals during periods of intense synaptic activity. In addition, mGlu7 has been implicated in multiple psychiatric and neurological conditions by both animal and human studies,2−5,10 making it an untapped target for drug development.
We now report the discovery of two new group III mGlu PAMs, VU0155094 and VU0422288. Although highly selective PAMs for the mGlu4 subtype of the family are available and have been used in vivo and in native tissues11,15,45 and there is one reported PAM targeting mGlu8,46 there are no reported PAMs for mGlu7. While not selective among the group III receptors, VU0155094 and VU0422288 represent important advances in the development of pharmacological tools to dissect the role of these receptors in vitro and in the context of appropriate native tissue settings.
Mechanistically, both VU01550994 and VU0422288 increase the efficacy of orthosteric agonists for the group III receptors. Modeling of the PAM interactions with each receptor using the operational model of allosterism revealed interesting distinctions in the ways these ligands interact with each group III receptor in the presence of different agonists. For example, VU0155094 showed similar predicted affinity values for mGlu4 and mGlu8 regardless of the agonist used (Table 5). In contrast, the affinity of VU0155094 for mGlu7 differed depending upon which agonist was studied, with the compound showing a higher affinity with L-AP4 compared to glutamate. Additionally, the contributions of affinity and efficacy cooperativity for VU0155094 in potentiating mGlu7 responses were very distinct depending upon the agonist, with significantly increased efficacy cooperativity (log β) seen with glutamate compared to essentially neutral efficacy cooperativity with L-AP4 and LSP4-2022 (Table 5). In the case of VU0422288, the compound exhibited similar affinities across all receptors with all agonists tested in calcium assays (Table 6), but the affinity was generally lower at mGlu4 and mGlu7 when GIRK activity was used as a readout. In calcium assays with mGlu4, the relative contributions of affinity and efficacy modulation to the overall potentiation of VU0422288 distinctly differed. For example, with glutamate VU0422288 was neutral with respect to affinity (α = 0.94, calcium assay) but positive for efficacy (β = 2.7, calcium assay), whereas with L-AP4 affinity modulation was negative (α = 0.33); however, this was offset by stronger positive efficacy cooperativity (β = 10.4). Conversely, in the GIRK assay, VU0422288 also showed differential cooperativity dependent on the agonist at mGlu7; however, this was lower for glutamate relative to the other two agonists. While examples of probe dependence are routinely observed for other GPCRs when interacting with allosteric modulators, most notably the family A receptors,40,41 probe dependence for the mGlus is less explored and has been assumed to be more limited due to the physical separations between the binding sites of the orthosteric ligands (which bind in the large N-terminal domain of the receptor) and the allosteric sites, which are generally within the 7-transmembrane spanning domain. Our findings clearly show that there are interactions between orthosteric and allosteric sites for the group III mGlus. Additionally, we identified clear signal bias between pathways that differed for each receptor with distinct agonists (Supporting Information Figure S7). Interestingly, LSP4-2022 robustly activates mGlu7 and its improved efficacy is apparent in vitro, where it induces a higher maximal response than glutamate in both calcium and GIRK assays (Supporting Information Figures S3–S5). While the mechanism for this distinction at mGlu7 is not known, it has been proposed that the enhanced potency and selectivity of LSP4-2022 at mGlu4 is generated by occupation of an additional binding pocket in the N-terminal domain.37 This would suggest that LSP4-2022 binds to an overlapping, yet structurally distinct, site when compared to glutamate or L-AP4, which could contribute to the probe dependence observed with VU0155094 and VU0422288 potentiation.
VU0155094 and VU0422288 represent valuable tool compounds for multiple areas of group III mGlu biology. For example, the ability of these structurally distinct ligands to bind to all of the group III receptors and not groups I and II suggests that there is at least one common allosteric site shared among the group III receptors. This site could be further explored using in vitro-based structure–function studies to aid in pharmacological tool design and future drug discovery. VU0422288 is particularly interesting, in that it has quite high predicted affinity across the group III receptors. For this reason, VU0422288 may represent a tool that could be radiolabeled for saturation and competition binding studies, thereby expanding the tool box to profile compound–receptor interactions.
Another exciting area in which these compounds might be exploited is in the study of mGlu heterodimerization. Recent studies have shown that mGlus within groups II and III can coassemble in vitro,47,48 and these findings have recently been extended to rodent brain tissue.49 These studies have shown that, in the context of mGlu2 and mGlu4 heterodimerization, mGlu4 PAMs that bind to a specific binding site are no longer efficacious when mGlu2 is present;48,49 other PAMs retain or exhibit enhanced potentiation in the presence of mGlu2.49 Both of these new PAMs can now be used to probe different heterodimer combinations and to determine if occupation of binding sites on both receptors (for example, mGlu7 and mGlu8) results in retained, enhanced, or lost potentiation when compared to a homodimer. It also will be interesting to examine whether VU0422288 and VU0155094 show differential efficacy when tested in the context of a heteromeric assembly of group III and group II mGlu (for example, mGlu8 and mGlu2). As we have shown that heterodimers appear to be functional in CNS, the present studies lay valuable groundwork to explore mGlu pharmacology in detail and may allow for the development of tools to probe different heterodimer combinations in native tissue settings where a pair of receptors is coexpressed. For example, both mGlu4 and mGlu8 have been shown to be coexpressed and functional at piriform cortex-lateral olfactory tract synapses,50 and comparison of the activity of an mGlu4/8 active PAM versus an mGlu4-selective PAM could provide important insight into the role of a potential mGlu4/8 heterodimer expressed at this synaptic location.
mGlu7 has been shown to be expressed presynaptically at SC-CA1 synapses in adult animals. This synapse undergoes a “developmental switch” in which the dominant presynaptic mGlu expressed in these presynaptic terminals shifts from mGlu8 at early developmental stages to mGlu7 in adult rats.26,31 Our data confirm these previous reports and suggest that there is no other functional presynaptic group III mGlu at SC-CA1 synapses in adult animals. This restricted expression of mGlu7, along with the increased potency of LSP4-2022 for mGlu7, allowed us to use nonselective PAMs to validate a role for mGlu7 in modulating excitatory synaptic transmission at these synapses. Preapplication of either VU0155094 or VU0422288 potentiated LSP4-2022-induced reductions in fEPSP slopes and also increased the paired-pulse ratio, confirming presynaptic actions of the compounds. These data suggest that the PAM mechanism may be a viable path forward for manipulating mGlu7 function in vivo and opens new avenues for exploring this mechanism in pathophysiological states in which mGlu7 may underlie disease etiology or be engaged for therapeutic benefit.
In summary, we have identified and characterized VU0155094 and VU0422288, two group III mGlu PAMs. These compounds exhibit distinctions in their interactions with the receptor subtypes and can be used to probe group III receptor function in vitro and in native tissues. By exploiting a synapse that selectively expresses mGlu7, we have shown that potentiation of mGlu7 function in the hippocampus induces relevant and robust effects in excitatory synaptic transmission, which will aid in the exploration of this widely expressed, glutamate-regulated receptor in a variety of physiological and pathological states.
Methods
Cell Lines and Cell Culture
Establishment and culture of the human mGlu4 (hmGlu4)/Gqi5/CHO has been described.51 All GIRK cell lines were prepared and cultured as reported.33 Rat mGlu7/Gα15/HEK cells, rat mGlu8/Gα15/HEK cells, and rat mGlu8/Gqi9 HEK cells were obtained by stable coexpression of Gα15/pCMV with rat mGlu7 or mGlu8 receptor cDNA that had been cloned into a pIRESpuro3 vector (Invitrogen). mGlu4 cells were cultured in 90% DMEM, 10% dialyzed FBS, 100 U/mL penicillin/streptomycin, 20 mM HEPES, 1 mM sodium pyruvate, 2 mM l-glutamine, 20 μg/mL proline (Sigma-Aldrich, Inc., St. Louis, MO), 20 μg/mL puromycin (Sigma-Aldrich, Inc., St. Louis, MO), and 400 μg/mL G418 sulfate (Mediatech, Inc., Herndon, VA). mGlu7 and mGlu8 polyclonal cells were grown in 90% DMEM, 10% FBS, 100 units/mL penicillin/streptomycin, 20 mM HEPES, 1 mM sodium pyruvate, 2 mM l-glutamine, 1× nonessential amino acids, 700 μg/mL G418, and 0.6 μg/mL puromycin. All cell culture reagents were purchased from Invitrogen (Carlsbad, CA) unless otherwise noted.
In Vitro Pharmacology
Thallium Flux Assays
Thallium flux assays were performed according to methods described,51 with minor modifications. For dye loading, media was exchanged with Assay Buffer (Hanks balanced salt solution (HBSS) containing 20 mM HEPES, pH 7.4) using an ELX405 microplate washer (BioTek), leaving 20 μL/well, followed by addition of 20 μL/well 2× FluoZin-2 AM (330 nM final) indicator dye (Life Technologies, prepared as a DMSO stock and mixed in a 1:1 ratio with pluronic acid F-127) in Assay Buffer. After 1 h incubation at room temperature, dye was exchanged with Assay Buffer, leaving 20 μL/well. Thallium flux was measured at room temperature using a Functional Drug Screening System 7000 (FDSS 7000, Hamamatsu). Baseline readings were taken (2 images at 1 Hz; excitation, 470 ± 20 nm; emission, 540 ± 30 nm), and test compounds (2×) were added in a 20 μL volume and incubated for 140 s before the addition of 10 μL of Thallium Buffer with or without agonist (5×). Data were collected for an additional 2.5 min and analyzed using Excel (Microsoft Corp, Redmond, WA) as previously described,51 and the concentration–response curves were fitted to a four-parameter logistic equation to determine potency estimates using GraphPad Prism (La Jolla, CA):
| 1 |
where A is the molar concentration of the compound; bottom and top denote the lower and upper plateaus of the concentration–response curve, respectively; HillSlope is the Hill coefficient that describes the steepness of the curve; and EC50 is the molar concentration of compound required to generate a response halfway between the top and bottom.
Calcium Assays
Human mGlu4/Gqi5/CHO cells (30 000 cells/20 μL/well), rat mGlu7/Gα15/HEK cells (15 000 cells/20 μL/well), and rat mGlu8/Gα15/HEK cells (15 000 cells/20 μL/well) were plated in black-walled, clear-bottomed, TC-treated, 384-well plates (Greiner Bio-One, Monroe, NC) in DMEM containing 10% dialyzed FBS, 20 mM HEPES, 100 units/mL penicillin/streptomycin, and 1 mM sodium pyruvate (Plating Medium). The cells were grown overnight at 37 °C in the presence of 5% CO2. The next day, the medium was removed and replaced with 20 μL of 1 μM Fluo-4, AM (Life Technologies, Thermo Fisher Scientific, Grand Island, NY) prepared as a 2.3 mM stock in DMSO and mixed in a 1:1 ratio with 10% (w/v) pluronic acid F-127 and diluted in Assay Buffer (Hank’s balanced salt solution, 20 mM HEPES and 2.5 mM Probenecid (Sigma-Aldrich, St. Louis, MO)) for 45 min at 37 °C. Dye was removed and replaced with 20 μL of Assay Buffer. For concentration–response curve experiments, compounds were serially diluted 1:3 into 10 point concentration response curves in DMSO, transferred to daughter plates using an Echo acoustic plate reformatter (Labcyte, Sunnyvale, CA), and diluted in Assay Buffer to a 2× final concentration. Calcium flux was measured using the Functional Drug Screening System 7000 (FDSS7000, Hamamatsu, Japan). After establishment of a fluorescence baseline for 4 s (4 images at 1 Hz; excitation, 470 ± 20 nm; emission, 540 ± 30 nm), 20 μL of test compounds was added to the cells, and the response was measured. After 142 s, 10 μL (5×) of an EC20 concentration of glutamate was added to the cells, and the response of the cells was measured; after an additional 120 s, 12 μL (5×) of an EC80 concentration of agonist was added and readings taken for an additional 40 s. Calcium fluorescence was recorded as fold over basal fluorescence, and raw data were normalized to the maximal response to glutamate. Potency (EC50) and maximum response (% Glu Max) for compounds were determined using a four parameter logistical equation in GraphPad Prism (La Jolla, CA). For efficacy experiments, a constant amount of compound was applied prior to the addition of a full glutamate concentration–response curve and the left shift of the EC50 of the curves was calculated as “fold-shift”.
Primary screening and initial follow-up assays were performed as reported.32 Selectivity testing across the mGlus was performed as described.45 Eurofins/PanLabs selectivity screening was performed using the LeadProfilingScreen (Catalog #68) as described: https://www.eurofinspanlabs.com/Catalog/Products/ProductDetails.aspx?prodId=0aCrd3Mu4RA%3D.
Operational Modeling of Allosterism
Shifts of agonist concentration–response curves by allosteric modulators were globally fitted to an operational model of allosterism:42
| 2 |
where A is the molar concentration of the orthosteric agonist; B is the molar concentration of the allosteric modulator; KA is the equilibrium dissociation constant of the orthosteric agonist, and KB is the equilibrium dissociation constant of allosteric modulator. Affinity modulation is governed by the cooperativity factor α, and efficacy modulation is governed by β. The parameters τA and τB relate to the ability of orthosteric agonist and allosteric ligands, respectively, to directly activate the receptor. Basal, Em, and n represent the basal system response, maximal possible system response, and the transducer function that links occupancy to response, respectively. For the quantification of PAM effects, no values in the curve fit were constrained with the exception of τB, which was set to −100 for VU0155094 as there was no evidence of allosteric agonist activity, and α, which was set to 1 as we assumed affinity modulation was not operative. In a separate analysis, agonist concentration–response curves were directly fitted to the operational model of agonism52 with Em constrained to the maximal level of potentiation observed for that subtype, to define KA and τA independent of allosteric modulators.
For GIRK data sets, we could not apply an operational model of allosterism due to the fact that at high concentrations of modulator decreases in agonist Emax were observed. In these instances, shifts in agonist potency were fitted to a simpler allosteric ternary complex model as reported.43 In this equation,
| 3 |
pEC50 is the negative logarithm of the agonist EC50 in the presence of the allosteric modulator, d is the agonist EC50 in the absence of modulator as estimated from the global analysis, pKB is the negative logarithm of the dissociation constant of the allosteric modulator, and αβ is the cooperativity between the allosteric modulator and orthosteric agonist. This equation integrates modulator effects on orthosteric ligand affinity and efficacy into a single cooperativity parameter.
Electrophysiology
Animals
All of the animals used in the present studies were group housed with food and water available ad libitum. Animals were kept under a 12 h light/dark cycle with lights on from 6:00 AM to 6:00 PM and were tested during the light phase. All of the experimental procedures were approved by the Vanderbilt University Animal Care and Use committee and followed the guidelines set forth by the Guide for the Care and Use of Laboratory Animals.
Extracellular Field Potential Recordings
Six week old male C57BL6/J mice (Jackson Laboratories, Bar Harbor, ME) were anesthetized with isofluorane, and the brains were removed and submerged in ice-cold cutting solution (in mM: 230 sucrose, 2.5 KCl, 8 MgSO4, 0.5 CaCl2, 1.25 NaH2PO4, 10 d-glucose, 26 NaHCO3). Coronal slices containing the hippocampus were cut at 400 μm using a compresstome (Precisionary Instruments, Greenville, NC). Slices were transferred to a holding chamber containing NMDG-HEPES recovery solution (in mM: 93 NMDG, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 d-glucose, 5 sodium ascorbate, 2 thiourea, 3 sodium pyruvate, 10 MgSO4, 0.5 CaCl2, pH 7.3, 305 mOsm) for 15 min at 32 °C. Slices were then transferred to a room temperature holding chamber for at least 1 h containing ACSF (in mM: 126 NaCl, 1.25 NaH2PO4, 2.5 KCl, 10 d-glucose, 26 NaHCO3, 2 CaCl2, 1 MgSO4) supplemented with 600 μM sodium ascorbate for slice viability. All buffers were continuously bubbled with 95% O2/5% CO2. Subsequently, slices were transferred to a submersion recording chamber where they were perfused with 32 °C ACSF at a rate of 2 mL/min. Borosilicate glass electrodes were pulled using a Flaming/Brown micropipet puller (Sutter Instruments, Novato, CA) and had a resistance of 3–5 MΩ when filled with ACSF. Evoked paired-pulse field excitatory post synaptic potentials (fEPSPs) were recorded by placing a glass recording electrode in the stratum radiatum of CA1 and a concentric bipolar stimulating electrode near the CA3-CA1 border. Paired-pulse fEPSPs were evoked using 100 μs duration stimulations spaced 20 ms apart. Data were digitized using a Multiclamp 700B, Digidata 1322A, and pClamp 10 software (Molecular Devices, Sunnyvale, CA). Input–output curves were generated to determine the stimulation intensity that produced 40–50% of the maximum response before each experiment, which was used as the baseline stimulation. Baseline stimulation was applied at 0.05 Hz. For drug experiments, drugs were diluted in ACSF and added for 5 or 10 min after 10 min of stable baseline was recorded. Recordings then continued for an additional 15 min after the drug was removed from the slice. Sampled data were analyzed offline using Clampfit 10.2 (Molecular Devices, Sunnyvale, CA). Paired-pulse ratios were calculated by dividing the slope of the second response by the slope of the first response. The slopes from three sequential sweeps were averaged and then normalized to the average slope calculated during the predrug period (percent of baseline). Statistical significance was analyzed using Graphpad Prisim 5.0. Significance in the drug effect on fEPSP slope was assessed using a one-way ANOVA with Bonferroni’s multiple comparison post-test. A paired-Student’s t test was used to assess differences in the paired-pulse ratios between baseline and LSP4-2022 alone. A one-way repeated measures ANOVA with Dunnett’s multiple comparisons was used to asses differences in paired-pulse ratios between baseline, LSP4-2022, and each respective PAM. All averaged data are presented as mean ± SEM.
Compounds
(S)-3,4-Dicarboxyphenylglycine (DCPG) was purchased from Tocris Bioscience. LSP1-2111 was synthesized at Vanderbilt University within the Vanderbilt Center for Neuroscience Drug Discovery. LSP4-2022 was synthesized partially by Francine Acher’s lab at Université Paris Descartes and at Vanderbilt University within the Vanderbilt Center for Neuroscience Drug Discovery according to published methods (ref (53) and patent WO2012/156931). VU0155094 was identified as a screening hit and additional compound, and analogues around this scaffold were purchased in powder form from ChemDiv. Synthetic methods and characterization for VU0422288 and analogues are included in the Supporting Information.
Glossary
Abbreviations
- ACSF
artificial cerebrospinal fluid
- ADX71743
(+)-6-(2,4-dimethylphenyl)-2-ethyl-6,7-dihydrobenzo[d]oxazol-4(5H)-one
- AMN082
N,N′-dibenzhydrylethane-1,2-diamine dihydrochloride
- NMDG
N-methyl d-glucamine
- ADHD
attention deficit hyperactivity disorder
- CNS
central nervous system
- DCPG
(S)-3,4-dicarboxyphenylglycine
- fEPSP
field excitatory postsynaptic potential
- GIRK
G protein inwardly rectifying potassium
- GPCR
G protein-coupled receptor
- HTS
high-throughput screening
- L-AP4
l-2-amino-4-phosphonobutyric acid
- LSP1-2111
(2S)-2-amino-4-[hydroxy[hydroxy(4-hydroxy-3-methoxy-5-nitro-phenyl)methyl]phosphoryl]butanoic acid
- LSP4-2022
(2S)-2-amino-4-(((4-(carboxymethoxy)phenyl)(hydroxy)methyl)(hydroxy)phosphoryl)butanoic acid
- MMPIP
6-(4-methoxyphenyl)-5-methyl-3-pyridin-4-ylisoxazolo[4,5-c]pyridin-4(5H)-one
- mGlu
metabotropic glutamate receptor
- NAM
negative allosteric modulator
- PAM
positive allosteric modulator
- SAR
structure–activity relationship
- SC-CA1
Schaffer collateral-CA1
- VU0155094
methyl 4-(3-(2-((4-acetamidophenyl)thio)acetyl)-2,5-dimethyl-1H-pyrrol-1-yl)benzoate
- VU0422288
N-(3-chloro-4-((5-chloropyridin-2-yl)oxy)phenyl)picolinamide
- XAP044
7-hydroxy-3-(4-iodophenoxy)-4H-chromen-4-one
Supporting Information Available
Supplemental Methods, Supplemental Table 1 (group III mGlu PAMs VU0155094 and VU0422288 affect the affinity of orthosteric agonists), Supplemental Table 2 (revised curve fitting reveals differential interactions of VU0155094 and VU0422288 with distinct group III mGlus), Supplemental Table 3 (summary of agonist affinity and efficacy estimates across each group III mGlu subtype), Supplemental Figure S1 (selectivity profiling of VU0155094 reveals that VU0155094 is a pan group III mGlu PAM), Supplemental Figure S2 (selectivity profiling of VU0422288 reveals that VU0422288 is a pan group III mGlu PAM), Supplemental Figure S3 (agonist concentration–response curves for mGlu4, mGlu7, and mGlu8 compared across assays), Supplemental Figure S4 (VU0155094 exhibits efficacy as a PAM for mGlu4, mGlu7, and mGlu8 as assessed by calcium mobilization), Supplemental Figure S5 (VU0422288 exhibits efficacy as a PAM for mGlu4, mGlu7, and mGlu8 as assessed by calcium mobilization), Supplemental Figure S6 (VU0422288 exhibits efficacy as a PAM for mGlu4, mGlu7, and mGlu8 as assessed by thallium flux through GIRK channels), and Supplemental Figure S7 (orthosteric agonists show distinctions in interactions with mGlu4, mGlu7, and mGlu8 that are assay dependent). This material is available free of charge via the Internet at http://pubs.acs.org/.
Author Contributions
# N.J.-S. and J.R.F. contributed equally to this work.
# N.J-S., J.R.F., R.Z., P.J.C., and C.M.N. designed, performed and analyzed in vitro pharmacology experiments. R.K., A.G.W., P.J.C., and Z.X. designed and analyzed data from electrophysiology experiments. M.E.M., D.W.E., S.R.B., M.H., D. R., F.A., B.J.M., M.R.W, C.W.L., and C.R.H. designed and performed chemical synthesis. K.J.G. and C.M.N. performed operational modeling. C.D.W, E.L.D., L.M.L., T.J.U., and C.M.N. performed and analyzed data from the high-throughput screening campaign. C.R.H., R.K., K.J.G., and C.M.N. wrote the manuscript with input from all authors.
Supported by NIH Grant NS078262, an International Rett Syndrome Foundation Basic Research Award, and an Autism Speaks Treatment Award (to C.M.N.) and NS031373 (P.J.C.). R.K. was partially supported through the Howard Hughes Medical Institute/Vanderbilt University Medical Center Certificate Program in Molecular Medicine (HHMI/VUMC CPMM) and a Weatherstone Predoctoral Fellowship from Autism Speaks. Primary high throughput screening was supported by Grant NS053536 (C.M.N.) at the Vanderbilt Molecular Libraries Screening Centers Network, supported by U54 MH074427 (C.D.W. and P.J.C.). Vanderbilt is a Specialized Chemistry Center within the Molecular Libraries Probe Centers Networks, supported by U54 MH084659 (C.W.L.). M.E.M. was supported via T32 MH093366. K.J.G. is supported by NHMRC (Australia) Overseas Biomedical Postdoctoral training fellowship. A.G.W. was supported by T32 NS007491 and a PhRMA Foundation Postdoctoral Fellowship.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Niswender C. M.; Conn P. J. (2010) Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol 50, 295–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton S. P. (2011) A new lead from genetic studies in depressed siblings: assessing studies of chromosome 3. Am. J. Psychiatry 168, 783–789. [DOI] [PubMed] [Google Scholar]
- Breen G.; Webb B. T.; Butler A. W.; van den Oord E. J.; Tozzi F.; Craddock N.; Gill M.; Korszun A.; Maier W.; Middleton L.; Mors O.; Owen M. J.; Cohen-Woods S.; Perry J.; Galwey N. W.; Upmanyu R.; Craig I.; Lewis C. M.; Ng M.; Brewster S.; Preisig M.; Rietschel M.; Jones L.; Knight J.; Rice J.; Muglia P.; Farmer A. E.; McGuffin P. (2011) A genome-wide significant linkage for severe depression on chromosome 3: the depression network study. Am. J. Psychiatry 168, 840–847. [DOI] [PubMed] [Google Scholar]
- Ganda C.; Schwab S. G.; Amir N.; Heriani H.; Irmansyah I.; Kusumawardhani A.; Nasrun M.; Widyawati I.; Maier W.; Wildenauer D. B. (2009) A family-based association study of DNA sequence variants in GRM7 with schizophrenia in an Indonesian population. Int. J. Neuropsychopharmacol. 12, 1283–1289. [DOI] [PubMed] [Google Scholar]
- Mick E.; Neale B.; Middleton F. A.; McGough J. J.; Faraone S. V. (2008) Genome-wide association study of response to methylphenidate in 187 children with attention-deficit/hyperactivity disorder. Am. J. Med. Genet., Part B 147B, 1412–1418. [DOI] [PubMed] [Google Scholar]
- Dalezios Y.; Lujan R.; Shigemoto R.; Roberts J. D.; Somogyi P. (2002) Enrichment of mGluR7a in the presynaptic active zones of GABAergic and non-GABAergic terminals on interneurons in the rat somatosensory cortex. Cereb. Cortex 12, 961–974. [DOI] [PubMed] [Google Scholar]
- Wright R. A.; Arnold M. B.; Wheeler W. J.; Ornstein P. L.; Schoepp D. D. (2000) Binding of [3H](2S,1′S,2′S)-2-(9-xanthylmethyl)-2-(2′-carboxycyclopropyl) glycine ([3H]LY341495) to cell membranes expressing recombinant human group III metabotropic glutamate receptor subtypes. Naunyn-Schmiedeberg's Arch. Pharmacol. 362, 546–554. [DOI] [PubMed] [Google Scholar]
- Wu S.; Wright R. A.; Rockey P. K.; Burgett S. G.; Arnold J. S.; Rosteck P. R. Jr.; Johnson B. G.; Schoepp D. D.; Belagaje R. M. (1998) Group III human metabotropic glutamate receptors 4, 7 and 8: molecular cloning, functional expression, and comparison of pharmacological properties in RGT cells. Brain Res. Mol. Brain Res. 53, 88–97. [DOI] [PubMed] [Google Scholar]
- Makoff A.; Pilling C.; Harrington K.; Emson P. (1996) Human metabotropic glutamate receptor type 7: molecular cloning and mRNA distribution in the CNS. Brain Res. Mol. Brain Res. 40, 165–170. [DOI] [PubMed] [Google Scholar]
- Sansig G.; Bushell T. J.; Clarke V. R.; Rozov A.; Burnashev N.; Portet C.; Gasparini F.; Schmutz M.; Klebs K.; Shigemoto R.; Flor P. J.; Kuhn R.; Knoepfel T.; Schroeder M.; Hampson D. R.; Collett V. J.; Zhang C.; Duvoisin R. M.; Collingridge G. L.; van Der Putten H. (2001) Increased seizure susceptibility in mice lacking metabotropic glutamate receptor 7. J. Neurosci. 21, 8734–8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Poul E.; Bolea C.; Girard F.; Poli S.; Charvin D.; Campo B.; Bortoli J.; Bessif A.; Luo B.; Koser A. J.; Hodge L. M.; Smith K. M.; DiLella A. G.; Liverton N.; Hess F.; Browne S. E.; Reynolds I. J. (2012) A potent and selective metabotropic glutamate receptor 4 positive allosteric modulator improves movement in rodent models of Parkinson’s disease. J. Pharmacol. Exp. Ther. 343, 167–177. [DOI] [PubMed] [Google Scholar]
- Ossowska K.; Wardas J.; Berghauzen-Maciejewska K.; Glowacka U.; Kuter K.; Pilc A.; Zorn S. H.; Doller D. (2014) Lu AF21934, a positive allosteric modulator of mGlu4 receptors, reduces the harmaline-induced hyperactivity but not tremor in rats. Neuropharmacology 83C, 28–35. [DOI] [PubMed] [Google Scholar]
- Slawinska A.; Wieronska J. M.; Stachowicz K.; Marciniak M.; Lason-Tyburkiewicz M.; Gruca P.; Papp M.; Kusek M.; Tokarski K.; Doller D.; Pilc A. (2013) The antipsychotic-like effects of positive allosteric modulators of metabotropic glutamate mGlu4 receptors in rodents. Br. J. Pharmacol. 169, 1824–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slawinska A.; Wieronska J. M.; Stachowicz K.; Palucha-Poniewiera A.; Uberti M. A.; Bacolod M. A.; Doller D.; Pilc A. (2013) Anxiolytic- but not antidepressant-like activity of Lu AF21934, a novel, selective positive allosteric modulator of the mGlu(4) receptor. Neuropharmacology 66, 225–235. [DOI] [PubMed] [Google Scholar]
- Bennouar K. E.; Uberti M. A.; Melon C.; Bacolod M. D.; Jimenez H. N.; Cajina M.; Kerkerian-Le Goff L.; Doller D.; Gubellini P. (2013) Synergy between L-DOPA and a novel positive allosteric modulator of metabotropic glutamate receptor 4: implications for Parkinson’s disease treatment and dyskinesia. Neuropharmacology 66, 158–169. [DOI] [PubMed] [Google Scholar]
- Fallarino F.; Volpi C.; Fazio F.; Notartomaso S.; Vacca C.; Busceti C.; Bicciato S.; Battaglia G.; Bruno V.; Puccetti P.; Fioretti M. C.; Nicoletti F.; Grohmann U.; Di Marco R. (2010) Metabotropic glutamate receptor-4 modulates adaptive immunity and restrains neuroinflammation. Nat. Med. 16, 897–902. [DOI] [PubMed] [Google Scholar]
- Mitsukawa K.; Yamamoto R.; Ofner S.; Nozulak J.; Pescott O.; Lukic S.; Stoehr N.; Mombereau C.; Kuhn R.; McAllister K. H.; van der Putten H.; Cryan J. F.; Flor P. J. (2005) A selective metabotropic glutamate receptor 7 agonist: activation of receptor signaling via an allosteric site modulates stress parameters in vivo. Proc. Natl. Acad. Sci. U.S.A. 102, 18712–18717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahi A.; Fizia K.; Dietz M.; Gasparini F.; Flor P. J. (2012) Pharmacological modulation of mGluR7 with AMN082 and MMPIP exerts specific influences on alcohol consumption and preference in rats. Addict Biol. 17, 235–247. [DOI] [PubMed] [Google Scholar]
- Fendt M.; Schmid S.; Thakker D. R.; Jacobson L. H.; Yamamoto R.; Mitsukawa K.; Maier R.; Natt F.; Husken D.; Kelly P. H.; McAllister K. H.; Hoyer D.; van der Putten H.; Cryan J. F.; Flor P. J. (2008) mGluR7 facilitates extinction of aversive memories and controls amygdala plasticity. Mol. Psychiatry 13, 970–979. [DOI] [PubMed] [Google Scholar]
- Pelkey K. A.; Yuan X.; Lavezzari G.; Roche K. W.; McBain C. J. (2007) mGluR7 undergoes rapid internalization in response to activation by the allosteric agonist AMN082. Neuropharmacology 52, 108–117. [DOI] [PubMed] [Google Scholar]
- Wieronska J. M.; Stachowicz K.; Acher F.; Lech T.; Pilc A. (2012) Opposing efficacy of group III mGlu receptor activators, LSP1-2111 and AMN082, in animal models of positive symptoms of schizophrenia. Psychopharmacology (Berlin, Ger.) 220, 481–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konieczny J.; Lenda T. (2013) Contribution of the mGluR7 receptor to antiparkinsonian-like effects in rats: a behavioral study with the selective agonist AMN082. Pharmacol. Rep. 65, 1194–1203. [DOI] [PubMed] [Google Scholar]
- Bradley S. R.; Uslaner J. M.; Flick R. B.; Lee A.; Groover K. M.; Hutson P. H. (2012) The mGluR7 allosteric agonist AMN082 produces antidepressant-like effects by modulating glutamatergic signaling. Pharmacol., Biochem. Behav. 101, 35–40. [DOI] [PubMed] [Google Scholar]
- Dolan S.; Gunn M. D.; Biddlestone L.; Nolan A. M. (2009) The selective metabotropic glutamate receptor 7 allosteric agonist AMN082 inhibits inflammatory pain-induced and incision-induced hypersensitivity in rat. Behav. Pharmacol. 20, 596–604. [DOI] [PubMed] [Google Scholar]
- Sukoff Rizzo S. J.; Leonard S. K.; Gilbert A.; Dollings P.; Smith D. L.; Zhang M. Y.; Di L.; Platt B. J.; Neal S.; Dwyer J. M.; Bender C. N.; Zhang J.; Lock T.; Kowal D.; Kramer A.; Randall A.; Huselton C.; Vishwanathan K.; Tse S. Y.; Butera J.; Ring R. H.; Rosenzweig-Lipson S.; Hughes Z. A.; Dunlop J. (2011) The metabotropic glutamate receptor 7 allosteric modulator AMN082: a monoaminergic agent in disguise?. J. Pharmacol. Exp. Ther. 338, 345–352. [DOI] [PubMed] [Google Scholar]
- Ayala J. E.; Niswender C. M.; Luo Q.; Banko J. L.; Conn P. J. (2008) Group III mGluR regulation of synaptic transmission at the SC-CA1 synapse is developmentally regulated. Neuropharmacology 54, 804–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki G.; Tsukamoto N.; Fushiki H.; Kawagishi A.; Nakamura M.; Kurihara H.; Mitsuya M.; Ohkubo M.; Ohta H. (2007) In vitro pharmacological characterization of novel isoxazolopyridone derivatives as allosteric metabotropic glutamate receptor 7 antagonists. J. Pharmacol. Exp. Ther. 323, 147–156. [DOI] [PubMed] [Google Scholar]
- Kalinichev M.; Rouillier M.; Girard F.; Royer-Urios I.; Bournique B.; Finn T.; Charvin D.; Campo B.; Le Poul E.; Mutel V.; Poli S.; Neale S. A.; Salt T. E.; Lutjens R. (2013) ADX71743, a potent and selective negative allosteric modulator of metabotropic glutamate receptor 7 (mGlu7): In vitro and in vivo characterization. J. Pharmacol. Exp. Ther. 344, 624–636. [DOI] [PubMed] [Google Scholar]
- Gee C. E.; Peterlik D.; Neuhauser C.; Bouhelal R.; Kaupmann K.; Laue G.; Uschold-Schmidt N.; Feuerbach D.; Zimmermann K.; Ofner S.; Cryan J. F.; van der Putten H.; Fendt M.; Vranesic I.; Glatthar R.; Flor P. J. (2014) Blocking Metabotropic Glutamate Receptor Subtype 7 (mGlu7) via the Venus Flytrap Domain (VFTD) Inhibits Amygdala Plasticity, Stress, and Anxiety-related Behavior. J. Biol. Chem. 289, 10975–10987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender C. M.; Johnson K. A.; Miller N. R.; Ayala J. E.; Luo Q.; Williams R.; Saleh S.; Orton D.; Weaver C. D.; Conn P. J. (2010) Context-dependent pharmacology exhibited by negative allosteric modulators of metabotropic glutamate receptor 7. Mol. Pharmacol. 77, 459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskys A.; Malenka R. C. (1991) Agonists at metabotropic glutamate receptors presynaptically inhibit EPSCs in neonatal rat hippocampus. J. Physiol. 444, 687–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann K.; Romaine I.; Days E.; Pascual C.; Malik A.; Yang L.; Zou B.; Du Y.; Sliwoski G.; Morrison R. D.; Denton J.; Niswender C. M.; Daniels J. S.; Sulikowski G. A.; Xie X. S.; Lindsley C. W.; Weaver C. D. (2013) ML297 (VU0456810), the first potent and selective activator of the GIRK potassium channel, displays antiepileptic properties in mice. ACS Chem. Neurosci. 4, 1278–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender C. M.; Johnson K. A.; Luo Q.; Ayala J. E.; Kim C.; Conn P. J.; Weaver C. D. (2008) A novel assay of Gi/o-linked G protein coupled receptor coupling to potassium channels provides new insights into the pharmacology of the group III metabotropic glutamate receptors. Mol. Pharmacol. 73, 1213–1224. [DOI] [PubMed] [Google Scholar]
- Engers D. W.; Niswender C. M.; Weaver C. D.; Jadhav S.; Menon U. N.; Zamorano R.; Conn P. J.; Lindsley C. W.; Hopkins C. R. (2009) Synthesis and evaluation of a series of heterobiarylamides that are centrally penetrant metabotropic glutamate receptor 4 (mGluR4) positive allosteric modulators (PAMs). J. Med. Chem. 52, 4115–4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolea C., and Addex Pharma S. A. (2009) Preparation of amido derivatives and their use as positive allosteric modulators of metabotropic glutamate receptors. Patent WO2009010454 A2, p 67.
- Jones C. K.; Bubser M.; Thompson A. D.; Dickerson J. W.; Turle-Lorenzo N.; Amalric M.; Blobaum A. L.; Bridges T. M.; Morrison R. D.; Jadhav S.; Engers D. W.; Italiano K.; Bode J.; Daniels J. S.; Lindsley C. W.; Hopkins C. R.; Conn P. J.; Niswender C. M. (2012) The metabotropic glutamate receptor 4-positive allosteric modulator VU0364770 produces efficacy alone and in combination with L-DOPA or an adenosine 2A antagonist in preclinical rodent models of Parkinson’s disease. J. Pharmacol. Exp. Ther. 340, 404–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudet C.; Vilar B.; Courtiol T.; Deltheil T.; Bessiron T.; Brabet I.; Oueslati N.; Rigault D.; Bertrand H. O.; McLean H.; Daniel H.; Amalric M.; Acher F.; Pin J. P. (2012) A novel selective metabotropic glutamate receptor 4 agonist reveals new possibilities for developing subtype selective ligands with therapeutic potential. FASEB J. 26, 1682–1693. [DOI] [PubMed] [Google Scholar]
- Kenakin T. (2008) Functional selectivity in GPCR modulator screening. Comb. Chem. High Throughput Screening 11, 337–343. [DOI] [PubMed] [Google Scholar]
- Kenakin T. (2010) G protein coupled receptors as allosteric proteins and the role of allosteric modulators. J. Recept. Signal Transduct Res. 30, 313–321. [DOI] [PubMed] [Google Scholar]
- Valant C.; Felder C. C.; Sexton P. M.; Christopoulos A. (2012) Probe dependence in the allosteric modulation of a G protein-coupled receptor: implications for detection and validation of allosteric ligand effects. Mol. Pharmacol. 81, 41–52. [DOI] [PubMed] [Google Scholar]
- Suratman S.; Leach K.; Sexton P.; Felder C.; Loiacono R.; Christopoulos A. (2011) Impact of species variability and ’probe-dependence’ on the detection and in vivo validation of allosteric modulation at the M4 muscarinic acetylcholine receptor. Br. J. Pharmacol. 162, 1659–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach K.; Sexton P. M.; Christopoulos A. (2007) Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology. Trends Pharmacol. Sci. 28, 382–389. [DOI] [PubMed] [Google Scholar]
- Leach K.; Wen A.; Cook A. E.; Sexton P. M.; Conigrave A. D.; Christopoulos A. (2013) Impact of clinically relevant mutations on the pharmacoregulation and signaling bias of the calcium-sensing receptor by positive and negative allosteric modulators. Endocrinology 154, 1105–1116. [DOI] [PubMed] [Google Scholar]
- Beurrier C.; Lopez S.; Revy D.; Selvam C.; Goudet C.; Lherondel M.; Gubellini P.; Kerkerian-LeGoff L.; Acher F.; Pin J. P.; Amalric M. (2009) Electrophysiological and behavioral evidence that modulation of metabotropic glutamate receptor 4 with a new agonist reverses experimental parkinsonism. FASEB J. 23, 3619–3628. [DOI] [PubMed] [Google Scholar]
- Johnson K. A.; Jones C. K.; Tantawy M. N.; Bubser M.; Marvanova M.; Ansari M. S.; Baldwin R. M.; Conn P. J.; Niswender C. M. (2013) The metabotropic glutamate receptor 8 agonist (S)-3,4-DCPG reverses motor deficits in prolonged but not acute models of Parkinson’s disease. Neuropharmacology 66, 187–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvoisin R. M.; Pfankuch T.; Wilson J. M.; Grabell J.; Chhajlani V.; Brown D. G.; Johnson E.; Raber J. (2010) Acute pharmacological modulation of mGluR8 reduces measures of anxiety. Behav. Brain Res. 212, 168–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doumazane E.; Scholler P.; Zwier J. M.; Trinquet E.; Rondard P.; Pin J. P. (2011) A new approach to analyze cell surface protein complexes reveals specific heterodimeric metabotropic glutamate receptors. FASEB J. 25, 66–77. [DOI] [PubMed] [Google Scholar]
- Kammermeier P. J. (2012) Functional and pharmacological characteristics of metabotropic glutamate receptors 2/4 heterodimers. Mol. Pharmacol. 82, 438–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin S.; Noetzel M. J.; Johnson K. A.; Zamorano R.; Jalan-Sakrikar N.; Gregory K. J.; Conn P. J.; Niswender C. M. (2014) Selective actions of novel allosteric modulators reveal functional heteromers of metabotropic glutamate receptors in the CNS. J. Neurosci. 34, 79–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P. J.; Xiang Z.; Conn P. J. (2008) Metabotropic glutamate receptors mGluR4 and mGluR8 regulate transmission in the lateral olfactory tract-piriform cortex synapse. Neuropharmacology 55, 440–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender C. M.; Johnson K. A.; Weaver C. D.; Jones C. K.; Xiang Z.; Luo Q.; Rodriguez A. L.; Marlo J. E.; de Paulis T.; Thompson A. D.; Days E. L.; Nalywajko T.; Austin C. A.; Williams M. B.; Ayala J. E.; Williams R.; Lindsley C. W.; Conn P. J. (2008) Discovery, characterization, and antiparkinsonian effect of novel positive allosteric modulators of metabotropic glutamate receptor 4. Mol. Pharmacol. 74, 1345–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black J. W.; Leff P. (1983) Operational models of pharmacological agonism. Proc. R Soc. London, Ser. B 220, 141–162. [DOI] [PubMed] [Google Scholar]
- Selvam C.; Oueslati N.; Lemasson I. A.; Brabet I.; Rigault D.; Courtiol T.; Cesarini S.; Triballeau N.; Bertrand H. O.; Goudet C.; Pin J. P.; Acher F. C. (2010) A virtual screening hit reveals new possibilities for developing group III metabotropic glutamate receptor agonists. J. Med. Chem. 53, 2797–2813. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.