

Abstract

The mechanisms responsible for drug resistance in the Asn31 variant of the M2 protein of influenza A are not well understood. Molecular dynamics simulations were performed on wild-type (Ser31) and S31N influenza A M2 in the homotetramer configuration. After evaluation of 13 published M2 structures, a solid-state NMR structure with amantadine bound was selected for simulations, an S31N mutant structure was developed and equilibrated, and the native and mutant structures were used to determine the binding behavior of amantadine and the dynamics of water in the two channels. Amantadine is stable in the plugging region of wild-type M2, with the adamantane in contact with the Val27 side chains, while amantadine in S31N M2 has more variable movement and orientation, and spontaneously moves lower into the central cavity of the channel. Free energy profiles from umbrella sampling support this observation. In this configuration, water surrounds the drug and can easily transport protons past it, so the drug binds without blocking proton transport in the S31N M2 channel.
Introduction
The M2 proton channel plays an important role in transporting protons, both across the viral envelope and across the Golgi of the infected cell in influenza A or B. The structure and dynamics of water molecules in the channel is of fundamental importance for the transport of protons through the channel. Blocking the M2 channel arrests these important proton transport processes in the viral replication cycle and prevents infection.
Drugs such as amantadine and rimantadine, which were once effective at treating influenza A, have become obsolete due to widespread resistance.1 Influenza A with M2 featuring a mutation from Ser31 to Asn31 (S31N) has become prevalent worldwide,2 and no effective anti-M2 influenza medication is currently available for its treatment. Understanding why drugs lose efficacy after viral mutation can help inspire changes to existing drugs or lead to the invention of novel drugs adapted to existing and future mutations.
Molecular modeling and dynamics simulations have proven useful in studying biological systems, and many studies have been performed on M2 channels3−6 and its inhibitors.7−10 Some of the most salient recent observations include the general shape of the free energy profile for the passage of amantadine through wild-type (WT) and V27A M2,11 the three main amino positions observed for amine compounds in the WT channel,7 the C-ward amine configuration for amantadine in the WT channel and N-ward isoxazole configuration for a novel S31N M2 blocker in the S31N channel,12 and the agreement with experimental efficacies for relative binding energies calculated using free energy perturbation for a set of adamantane compounds in WT and S31N M2 channels.13 The important roles of luminal water in the binding cavity have been highlighted in these studies, all of which used TIP3P water to maintain consistency with the protein, lipid, and ion force fields. They all use 4-fold symmetric, homotetrameric M2-truncate structures. However, they vary in the precise M2 structures, which have been determined with different methods, at different temperatures, and in different lipidic or detergent environments; and which vary in M2 His37 and drug titration states. These important physical factors, among others (presence or absence of counterions in the channel, structure of the protein N- and C-termini, lipid characteristics, etc.), are still poorly established and are currently in the process of evaluation. Although recent FRET data indicate that the functional protein may be dimeric in cells,14 we continue to focus on the homotetrameric structure for this investigation to agree with earlier structural15,16 and functional studies.17−19 We describe our more comprehensive rationale for selection of the other physical factors mentioned. This study aims to use molecular dynamics simulations to compare binding behavior of amantadine when positioned inside WT or the primary amantadine-insensitive M2 (S31N) with the goal of identifying the mechanism of resistance.
Methodology
M2 channel structures used in the study were derived from the RCSB Protein Data Bank. The homotetrameric protein was embedded in a membrane (situated perpendicular to the z axis) comprised of 96 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) lipid molecules to agree with the lipid used for most solid-state NMR structure determinations. Periodic boundary conditions with unit cells of about 60 × 60 × 90 Å3 in x, y, and z dimensions were used for all systems, and TIP3P20 water molecules with ∼150 mM NaCl (net electroneutral system) encased the bilayer. Molecular dynamics packages CHARMM37,21 NAMD 2.9,22 and VMD 1.9.123 were used for simulations and analysis, and the CHARMM36 all-atom empirical force field was used, except as noted, to describe proteins and lipids.24−28
Unless otherwise noted, all simulations were performed with positively charged amantadine and neutral His37 residues. Despite solid-state NMR data that shows the pKa for the His37 tetrad is near 8.0 for the first two His37 protonations,29 we reasoned that each His37 side chain would be protonated only at the Nε site under conditions of neutral pH with positively charged amantadine bound.30,31 Although amantadine has a pKa of 10.1 in aqueous solution and has been shown by solid-state NMR to be neutral in the M2 channel at pH 8.0,32 we reasoned that the pK shift expected upon binding in the low dielectric region would probably not be sufficient to result in deprotonation of the drug under neutral, or at least under acidic, conditions.
Using the Nosé–Hoover Langevin piston pressure control33,34 and Langevin dynamics, systems were kept, after initial heating, near 1 atm isotropically (i.e., zero surface tension) and 310 K to achieve the NPT ensemble. Long-range Coulombic interactions were calculated using the particle mesh Ewald (PME) algorithm.35 The Shake method36 was used to keep all bonds with hydrogen rigid at ideal lengths and angles, and short-range electrostatic and van der Waals forces were smoothed using a switching distance of 11 Å.
Channel Hydration and Initial Equilibration
RCSB Protein Data Bank structures 1NYJ,372H95,382KAD,392KIH,402KIX,412KQT,422KWX,432L0J,312LJB,442LJC,442LY0,122RLF,45 and 3LBW(16) were imported, residues beyond position 46 were truncated, and the transmembrane domains of the M2 channels were embedded in DMPC lipid in water, as described previously. Multiple phases of energy minimization with decreasing harmonic restraints were performed to bring the systems toward an energy minimum under the CHARMM force field. Following minimization, water molecules were inserted into channel pores in either excess (overhydrated) or low (underhydrated) amounts in separate systems to provide starting points for water to begin equilibration. With the protein backbone lightly restrained to the average backbone coordinates of each channel’s set of models (or, if the structure consists of just one model, to the backbone of that model), the systems were equilibrated for 6 ns without drug present to allow water to exit or fill the channel pore. Varying random-number-generator seeds for initial velocity assignments were used to produce three distinct runs of 6 ns each, which proved to be sufficient time to essentially equilibrate the overfilled channels. CHARMM was used to postprocess the trajectories for RMSD and water-content calculations.
Unrestrained Dynamics of Drugged WT and S31N Systems
The solid-state NMR amantadine-bound structure by Cady et al., 2KQT, was ultimately selected to model WT M2, and from the same structure, an S31N mutant homology model was created with Swiss-PdbViewer,46,47 which was subsequently hydrated and equilibrated as performed for the other channels. The M2 blocker, amantadine, was used in each system to test for an aqueous path for proton conductance with drug present for both WT and S31N M2 channels. Force field parameters for amantadine were generated using adamantane parameters from the CHARMM general force field (CGenFF) version 3648,49 using ParamChem.50,51
In the initial simulations with unrestrained drug molecules, protonated or deprotonated amantadine molecules were added to previously hydrated WT and S31N M2 channel systems. Drugs were inserted with adamantane carbon atoms positioned ≥2.2 Å away (in the C-terminal direction) from the Val27 side chain atoms and oriented with the amino group pointing toward the C-terminus in order to agree with previous findings.52 For electroneutrality, a chloride ion (Cl–) was added near the amine group in systems with protonated drug. Water molecules within 2.2 Å of amantadine were deleted, and several rounds of energy minimization were performed.
Each system was heated and equilibrated with five different initial random velocity sets and then independently simulated for 10.5 ns with a time step of 1.5 fs, using NAMD. Following simulations, center-of-mass positions were determined from the simulation trajectories, oriented frame-wise to the protein backbone, using CHARMM. Two-dimensional density profiles were produced using the VMD Density Profile Tool,53 three-dimensional volume densities were calculated using VMD’s VolMap plugin, and volume-density maps were processed from VolMap output using OpenDX.
Potential of Mean Force Computation
Umbrella sampling of protonated amantadine in hydrated WT and S31N M2 channels was carried out using CHARMM37. A harmonic restraint of 0.1 kcal/mol-Å2 was applied to hold each protein backbone atom to the highly colocalized protein backbone coordinates averaged over all 17 2KQT PDB models. These channel backbone restraints allow only modest fluctuations of the tetramer, such that the drug binding cavity is readily explored on a short time scale, at the obvious cost of not fully simulating the protein backbone mobility at each drug umbrella position. 2KQT was determined with amantadine in the binding site. There, and at most other sites, where the compound is smaller than the cavity, such fluctuations are not expected to impact the free energy significantly. A z-dimensional harmonic restraint of 10 kcal/mol-Å2 was applied to the center of mass of the drug’s adamantane cage as an umbrella potential.
Langevin dynamics were used to bring each system to 310 K, after which the Nosé–Hoover thermostat and the Verlet leapfrog algorithm54 were used to equilibrate each system at constant pressure and temperature for 1.5 ns with a 1.5 fs time step. The effects of initial amantadine orientation were explored by inserting amantadine at each umbrella window with its amine group oriented either toward the positive-z dimension or negative-z dimension along the simulation z axis, after which any overlapping water was removed. The preliminary studies showed that Cl– would fit in the channel with amantadine, but we reasoned that the probability of Cl– being in the channel at the time of amantadine entry, or entering the channel in conjunction with amantadine, would be low, so we excluded Cl– from the channel for these studies. Two runs with randomized initial velocity assignments were performed for each amantadine orientation. Windows were sampled at 0.25 Å intervals from −4 to 12 Å along the simulation z axis for a total of 65 windows. For each trajectory frame, adamantane center-of-mass positions were extracted and processed with the weighted histogram analysis program, WHAM,55 for the potential of mean force (PMF) calculation. A convergence tolerance of 1 × 10–6 and 750 bins were used to produce the PMF for each channel, orientation, and starting velocity. After examining the results and finding substantial amantadine orientation sampling, the PMFs from the four runs, each referenced to its global minimum, were averaged and then re-referenced to the global minimum of the average PMF for each channel.
Results and Discussion
Preliminary Equilibration
The number of water molecules and the root-mean-square deviation (RMSD) in protein atom position for the set of 13 aforementioned M2 channels from the RCSB Protein Data Bank were studied (see Table S1 in the Supporting Information). Each channel was placed in DMPC lipid, hydrated, and allowed to equilibrate. The average amount of water contained in a sphere spanning protein residues 27–41 over the last 2 ns of simulation, the average backbone RMSD of residues 26–43 over the last 2 ns of simulation, and snapshot comparisons between initial PDB structures and structures from final, equilibrated trajectory frames were determined for each structure (Figure 1).
Figure 1.
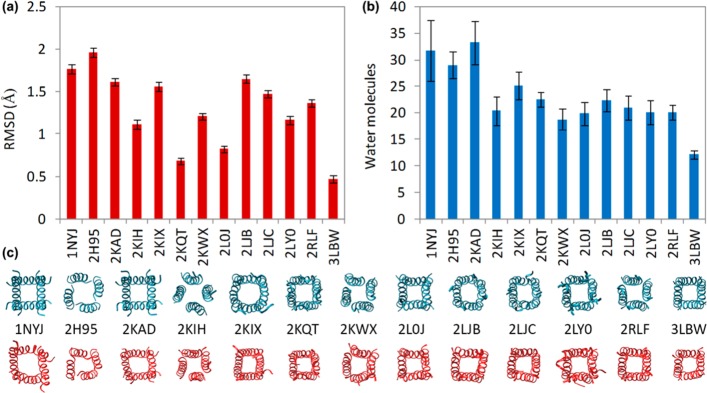
Averages over the last 2 ns of three 6 ns simulations with light backbone constraints, here using the CHARMM27 force field. Each M2 channel’s (a) average protein-backbone RMSD of residues 26–43 from model 1 of its PDB structure, (b) average water content between residues 27–41, and (c) initial/final trajectory frames are shown. The end-view snapshots of the initial (blue) and final (red) structures below demonstrate the relative stability of the tilted helix bundle. Side views better show the variations between PDB models,56 but the end-view was chosen here to demonstrate the volume of the central cavities.
The M2 crystal structure, 3LBW, had the smallest average RMSD when equilibrated but had significantly less water content (about 12 lumenal molecules) than all of the other structures, each of which have over 18 water molecules in the channel lumen. After filling and equilibrating the solid-state NMR M2 protein (see Figure S1 in the Supporting Information), 2KQT had the next-lowest average RMSD and water content (about 22 lumenal molecules) more closely matched the water content of the other structures tested. For these reasons, and because the M2 structure was determined and refined in liquid crystal bilayer conditions with amantadine present, 2KQT was used for the remainder of the study and as the template structure for the creation of an S31N homology model.
Amantadine Protonation and Orientation
With neutral amantadine positioned in the hydrated WT M2, unrestrained dynamics simulations showed that the amine nitrogen of amantadine flipped between facing the C-terminus and the N-terminus and back in three out of five 10.5 ns simulations of WT M2 protein (see Figure S2 in the Supporting Information). In contrast, with protonated amantadine and a Cl– in the channel, in none of the five 10.5 ns simulations did the amine nitrogen flip to face the N-terminus.
During simulations, the Cl– generally stayed near the amine group of amantadine. Apparently, the ionic interaction between the Cl– in the channel and the protonated amino group of amantadine was strong enough to prevent amantadine from reversing direction and Cl– from leaving the channel. When Cl– was excluded and the amine group left deprotonated, amantadine was more prone to reverse orientation toward the N-terminus.
Amantadine Localization: WT vs S31N Channel
Starting from the hydrated, equilibrated amantadine+-WT and amantadine+-S31N systems, unrestrained simulations of 10.5 ns were performed using NAMD. Adamantane center-of-mass positions were compared from the simulation trajectories (Figure 2a). In WT M2 simulations, amantadine stayed relatively stable between 6 and 7.5 Å along the z axis. In simulations of S31N M2, amantadine rapidly drifted toward the C-terminus (in some cases before the end of the heating phase) and, throughout the five separate simulations, appeared to be less stable, with positions ranging from 2 to 6 Å along the z axis. Mass-density profiles of the adamantane amino carbon (C1) averaged over the five simulations for each channel confirm this result (Figure 2b).
Figure 2.
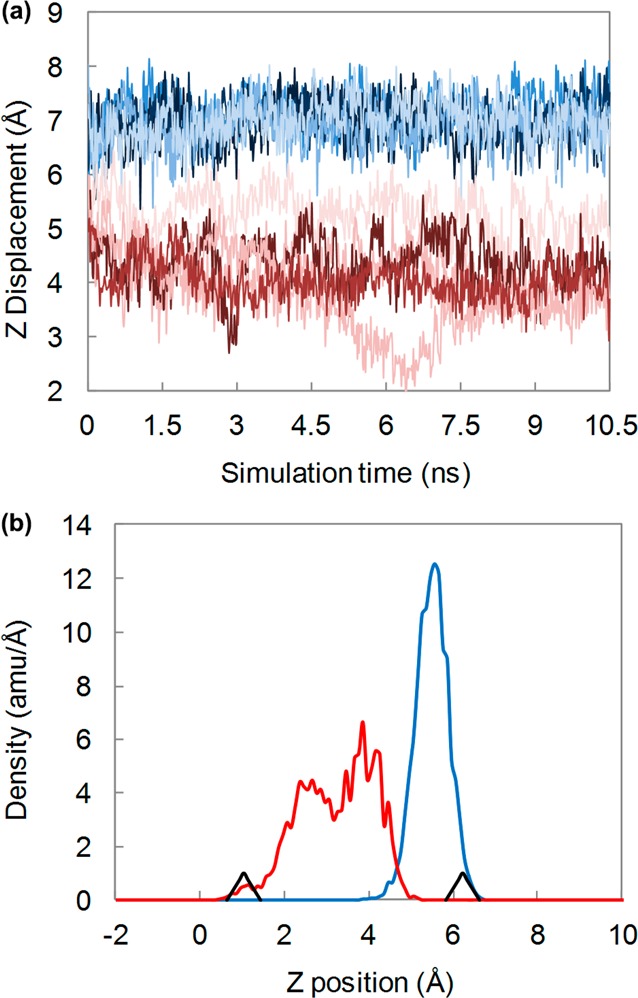
Amantadine positions during five unrestrained simulations of WT (blue) and S31N (red) M2. (a) Superimposed adamantane center of mass z-dimension trajectories for five runs. (b) Carbon mass-density profile of the adamantane C1 averaged over all five simulations. Black arrowheads in part b denote the mean α-carbon positions for Gly34 (left) and Ser/Asn31 (right).
The carbon mass-density profile and center-of-mass positions indicate significantly different binding-site behavior between WT and S31N M2 channels in the presence of amantadine. When placed in WT M2, amantadine stayed very stable within just a few angstroms of the Val27 residues (Figure 2a and b, blue). However, when placed in S31N M2, amantadine drifted toward the C-terminus, with greater diversity between runs and throughout each simulation (Figure 2a and b, red). Thus, it appears that the S31N mutation alters the binding site of M2, preventing amantadine stability within the narrower region of the channel between residue positions 27 and 31.
Water Density
To determine whether drug position affects the location of water molecules and the potential to form water-wires for Grotthus transport of protons in M2, linear water densities throughout all simulations were calculated (Figure 3).
Figure 3.

Water density, averaged over the five unrestrained simulations of Figure 2. WT (blue) and S31N (red) M2 channels with protonated amantadine and Cl– in the central cavity. Black arrowheads denote the mean α-carbon positions for Gly34 (left) and Ser/Asn31 (right).
The water-density profiles show that the WT M2 channel has very low water density from 4 to 6 Å on the z axis, and in S31N M2, significantly more water density is observed in the same region. Cross-sectional volume-density maps confirm this finding: WT M2 shows very little water density around and above the drug in the channel pocket (Figure 4a), where S31N M2 shows significant water density in this region (Figure 4b).
Figure 4.

Amantadine (red) and water (blue) cross-sectional volume densities are shown for both (a) WT and (b) S31N M2 channels averaged over five simulations. Trajectory snapshots of two M2 monomers (green ribbons) are superimposed, with stick figures for Val27, Ser/Asn31, His37, and Trp41 side chains from top to bottom, respectively.
It is also noticeable in Figure 4 that the amantadine density is more distributed in the binding site in the S31N channel, with only modest density N-ward from the Ser/Asn31 side chains, in contrast to WT M2. Furthermore, amantadine density appears to favor one side of the channel, allowing water to fill the available space laterally. It appears that amantadine is repelled off of the channel axis by water molecules attracted into the space, as there are no obvious attractive forces in the walls of the symmetrical channel that would pull it away from the axis of the channel.
The difference in water density is readily observed when visualizing the trajectories as movies. It is rare to see any water molecules approach the upper portion of amantadine in WT M2 (see snapshot in Figure 5a); however, in S31N M2, amantadine is often oriented in such a way that continuous strands of water can be observed to form at different times throughout the simulations (Figure 5b).
Figure 5.

Trajectory snapshots showing water molecules, two adjacent protein monomers (with the N- and C-termini arranged from top to bottom, respectively), and amantadine (shown in red) in (a) WT and (b) S31N M2 systems.
The results illustrate that WT M2 with amantadine present has much less water density near the Val27 residues or lateral to the amantadine. S31N M2 has much more water density above amantadine, especially as it drifts toward the C-terminus, as well as a visible amount of water density surrounding the drug in all simulations. The region between Ser/Asn31 and Gly34, according to volume densities and adamantane-cage position plots, seems to allow for amantadine to be more flexible in its amine orientation and lateral motion, thus preventing consistent block of proton conductance.
Umbrella Sampling of WT and S31N Channels
To investigate the reasons behind the difference in binding behavior of amantadine to WT and S31N M2, free energy profiles of amantadine through each channel were computed via umbrella sampling. The potential of mean force (PMF) for each channel was calculated by analyzing the position of the adamantane cage of amantadine, the center of mass of which was constrained to specific z coordinates at 0.25 Å intervals, in independent simulations (Figure 6).
Figure 6.

Free energy landscapes of amantadine in WT (blue) and S31N (red) M2 channels. The free energy minimum of each channel was set to 0 kcal/mol. One standard deviation between runs is shown in dotted lines of the corresponding color above and below each trace. Gly34 and Ser/Asn31 mean α-carbon positions (black arrowheads) are shown for reference from left to right, respectively.
The WT PMF shows deep wells at around 0 and 4 Å, with a broad, low shelf at around 6 Å along the z axis. In contrast, the S31N structure produces a steady decline in the free energy profile as amantadine moves from the Val27 entryway, followed by a very steep decline between 6 and 4 Å. For the WT, the shelf at 6 Å represents a level energy landscape where amantadine can dwell with its adamantane center of mass positioned near the Ser31 residues. Here, the adamantane cage is plugged into the Val27 sphincter, filling the lumen as in the simulations of Figure 2b. The deeper well at 3.5 Å indicates a somewhat lower free energy for amantadine sunk deeper into the central cavity by a few Å. However, the modest difference between the two free energy levels suggests that the amantadine can frequently and rapidly return to the blocking position at 6 Å. Furthermore, water molecules below the Ser31 region in WT are more ordered, and are consequently less likely to be displaced.7 In contrast, in the S31N construct, the free energy profile would drive amantadine relentlessly to the deeper region of the central cavity, as observed in the unconstrained drug simulations above.
Additional unconstrained 50 ns simulations with amantadine started at the lower position in WT M2 (and a Cl– nearby) also show that amantadine can return to the blocking position at 6 Å from the lower free energy well at 3 Å. In two out of five runs, amantadine returned spontaneously to the blocking position at 6 Å within a few ns and remained stable (see Figure S3 in the Supporting Information).
The S31N PMF in Figure 6 exhibits a sharp rise in free energy between 4 and 7 Å. The plugging site at 6 Å is clearly inhospitable compared with deeper positions in the central cavity, as observed in the unrestrained trajectories. A significant free energy well at 4 Å, with steep barriers on either side, indicates a strong binding preference for protonated amantadine in this region. The lack of a shelf or well-defined local minimum for the adamantane center of mass near the Asn31 residues is a key indicator of its instability in that region, compared to amantadine in WT M2.
The forces underlying the steep free energy well inside the S31N channel need to be further investigated. From the simulations presented here, it appears likely that attraction of the more polar Asn31 side chains to water molecules may be the driving force, as well as decreased stability of amantadine in proximity to the bulkier Asn31 side chains, as other studies have suggested.9,10
Conclusions
Molecular dynamics simulations on the influenza A M2 channels provide an explanation for why amantadine blocks WT M2 and fails to block S31N M2, as observed experimentally. Amantadine’s stability and narrow region of occupancy near the V27 sphincter of the M2 channel appears to result in a very dry region in which proton conductance by water-wire formation or hydronium-ion diffusion cannot occur. Amantadine’s instability and unusual binding behavior in the S31N M2 channel, as well as the steep free energy drop between the plugging site and the deeper noninhibitory binding site, suggest that interactions between the channel and water molecules drive the drug too deep into the channel. Hence, in the S31N channel, even if the drug can bind, it cannot block proton conductance.
Acknowledgments
The authors are grateful to Curtis Evans for several preliminary simulations and to Antonios Kolocouris and Harris Ioannidis for helpful discussions. The work was supported by grants from NIH (AI 23007 to Timothy Cross) and from the BYU Office of Research and Creative Activities. Simulations and analyses were performed at the Fulton Supercomputing Lab at Brigham Young University.
Supporting Information Available
Hydration study table, as well as figures of 2KQT RMSD and water content, amantadine orientation, and amantadine position in WT with non-blocking initial position. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Bright R. A.; Shay D.; Bresee J.; Klimov A.; Cox N.; Ortiz J. High Levels of Adamantane Resistance among Influenza A (H3N2) Viruses and Interim Guidelines for use of Antiviral Agents. United States, 2005–2006 Influenza Season. Morbidity and Mortality Weekly Report 2006, 55, 44–46. [PubMed] [Google Scholar]
- Bright R. A.; Shay D. K.; Shu B.; Cox N. J.; Klimov A. I. Adamantane Resistance among Influenza A Viruses Isolated Early During the 2005–2006 Influenza Season in the United States. J. Am. Med. Assoc. 2006, 295, 891–894. [DOI] [PubMed] [Google Scholar]
- Sansom M. S.; Kerr I. D. Influenza Virus M2 Protein: A Molecular Modelling Study of the Ion Channel. Protein Eng. 1993, 6, 65–74. [DOI] [PubMed] [Google Scholar]
- Wu Y.; Voth G. A. A Computational Study of the Closed and Open States of the Influenza A M2 Proton Channel. Biophys. J. 2005, 89, 2402–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa M.; Henderson D. J.; Busath D. D. Free-Energy Profiles for Ions in the Influenza M2-TMD Channel. Proteins 2009, 76, 794–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C.; Pohorille A. Activation and Proton Transport Mechanism in Influenza A M2 Channel. Biophys. J. 2013, 105, 2036–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Ma C.; Fiorin G.; Carnevale V.; Wang T.; Hu F.; Lamb R. A.; Pinto L. H.; Hong M.; Klein M. L.; et al. Molecular Dynamics Simulation Directed Rational Design of Inhibitors Targeting Drug-Resistant Mutants of Influenza A Virus M2. J. Am. Chem. Soc. 2011, 133, 12834–12841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhadeff R.; Assa D.; Astrahan P.; Krugliak M.; Arkin I. T. Computational and Experimental Analysis of Drug Binding to the Influenza M2 Channel. Biochim. Biophys. Acta 2014, 1838, 1068–1073. [DOI] [PubMed] [Google Scholar]
- Qin G.; Yu K.; Shi T.; Luo C.; Li G.; Zhu W.; Jiang H. How Does Influenza Virus A Escape from Amantadine?. J. Phys. Chem. B 2010, 114, 8487–8493. [DOI] [PubMed] [Google Scholar]
- Khurana E.; Devane R. H.; Dal Peraro M.; Klein M. L. Computational Study of Drug Binding to the Membrane-Bound Tetrameric M2 Peptide Bundle from Influenza A Virus. Biochim. Biophys. Acta 2011, 1808, 530–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonov H.; Astrahan P.; Krugliak M.; Arkin I. T. How Do Aminoadamantanes Block the Influenza M2 Channel, and How Does Resistance Develop?. J. Am. Chem. Soc. 2011, 133, 9903–9911. [DOI] [PubMed] [Google Scholar]
- Wang J.; Wu Y.; Ma C.; Fiorin G.; Pinto L. H.; Lamb R. A.; Klein M. L.; Degrado W. F. Structure and Inhibition of the Drug-Resistant S31N Mutant of the M2 Ion Channel of Influenza A Virus. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 1315–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkeka P.; Eleftheratos S.; Kolocouris A.; Cournia Z. Free Energy Calculations Reveal the Origin of Binding Preference for Aminoadamantane Blockers of Influenza A/M2TM Pore. J. Chem. Theory Comput. 2013, 9, 1272–1281. [DOI] [PubMed] [Google Scholar]
- Kawano K.; Yano Y.; Matsuzaki K. A Dimer Is the Minimal Proton-Conducting Unit of the Influenza A Virus M2 Channel. J. Mol. Biol. 2014, 426, 2679–2691. [DOI] [PubMed] [Google Scholar]
- Salom D.; Hill B. R.; Lear J. D.; DeGrado W. F. Ph-Dependent Tetramerization and Amantadine Binding of the Transmembrane Helix of M2 from the Influenza A Virus. Biochemistry 2000, 39, 14160–14170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acharya R.; Carnevale V.; Fiorin G.; Levine B. G.; Polishchuk A. L.; Balannik V.; Samish I.; Lamb R. A.; Pinto L. H.; DeGrado W. F.; et al. Structure and Mechanism of Proton Transport through the Transmembrane Tetrameric M2 Protein Bundle of the Influenza A Virus. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 15075–15080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi T.; Tu Q.; Pinto L. H.; Lamb R. A. The Active Oligomeric State of the Minimalistic Influenza Virus M2 Ion Channel Is a Tetramer. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 5000–5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugrue R. J.; Hay A. J. Structural Characteristics of the M2 Protein of Influenza A Viruses: Evidence That It Forms a Tetrameric Channel. Virology 1991, 180, 617–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holsinger L. J.; Lamb R. A. Influenza Virus M2 Integral Membrane Protein Is a Homotetramer Stabilized by Formation of Disulfide Bonds. Virology 1991, 183, 32–43. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar]
- Brooks B. R.; Brooks C. L. 3rd; Mackerell A. D. Jr.; Nilsson L.; Petrella R. J.; Roux B.; Won Y.; Archontis G.; Bartels C.; Boresch S.; et al. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kalé L.; Schulten K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K.. Vmd: Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33-8, 27-8. [DOI] [PubMed] [Google Scholar]
- MacKerell A. D.; Bashford D.; Bellott; Dunbrack R. L.; Evanseck J. D.; Field M. J.; Fischer S.; Gao J.; Guo H.; Ha S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- Mackerell A. D. Jr.; Feig M.; Brooks C. L. 3rd Extending the Treatment of Backbone Energetics in Protein Force Fields: Limitations of Gas-Phase Quantum Mechanics in Reproducing Protein Conformational Distributions in Molecular Dynamics Simulations. J. Comput. Chem. 2004, 25, 1400–1415. [DOI] [PubMed] [Google Scholar]
- Merz K. M.; Roux B.. Biological Membranes: A Molecular Perspective from Computation and Experiment; Birkhäuser: Boston, MA, 1996. [Google Scholar]
- Feller S. E.; Yin D.; Pastor R. W.; MacKerell A. D. Jr. Molecular Dynamics Simulation of Unsaturated Lipid Bilayers at Low Hydration: Parameterization and Comparison with Diffraction Studies. Biophys. J. 1997, 73, 2269–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauda J. B.; Venable R. M.; Freites J. A.; O’Connor J. W.; Tobias D. J.; Mondragon-Ramirez C.; Vorobyov I.; MacKerell A. D.; Pastor R. W. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114, 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J.; Fu R.; Nishimura K.; Zhang L.; Zhou H.-X.; Busath D. D.; Vijayvergiya V.; Cross T. A. Histidines, Heart of the Hydrogen Ion Channel from Influenza A Virus: Toward an Understanding of Conductance and Proton Selectivity. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 6865–6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams J. K.; Tietze D.; Wang J.; Wu Y.; DeGrado W. F.; Hong M. Drug-Induced Conformational and Dynamical Changes of the S31N Mutant of the Influenza M2 Proton Channel Investigated by Solid-State NMR. J. Am. Chem. Soc. 2013, 135, 9885–9897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M.; Yi M.; Dong H.; Qin H.; Peterson E.; Busath D. D.; Zhou H.-X.; Cross T. A. Insight into the Mechanism of the Influenza A Proton Channel from a Structure in a Lipid Bilayer. Science 2010, 330, 509–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M.; Li C.; Busath D. D.; Zhou H. X.; Cross T. A. Drug Sensitivity, Drug-Resistant Mutations, and Structures of Three Conductance Domains of Viral Porins. Biochim. Biophys. Acta 2011, 1808, 538–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martyna G. J.; Tobias D. J.; Klein M. L. Constant Pressure Molecular Dynamics Algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar]
- Feller S. E.; Zhang Y.; Pastor R. W.; Brooks B. R. Constant Pressure Molecular Dynamics Simulation: The Langevin Piston Method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G.; Smooth A. Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar]
- Ryckaert J.-P.; Ciccotti G.; Berendsen H. J. C. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of N-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar]
- Nishimura K.; Kim S.; Zhang L.; Cross T. A. The Closed State of a H+ Channel Helical Bundle Combining Precise Orientational and Distance Restraints from Solid State NMR. Biochemistry 2002, 41, 13170–13177. [DOI] [PubMed] [Google Scholar]
- Hu J.; Asbury T.; Achuthan S.; Li C.; Bertram R.; Quine J. R.; Fu R.; Cross T. A. Backbone Structure of the Amantadine-Blocked Trans-Membrane Domain M2 Proton Channel from Influenza A Virus. Biophys. J. 2007, 92, 4335–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cady S. D.; Mishanina T. V.; Hong M. Structure of Amantadine-Bound M2 Transmembrane Peptide of Influenza A in Lipid Bilayers from Magic-Angle-Spinning Solid-State NMR: The Role of Ser31 in Amantadine Binding. J. Mol. Biol. 2009, 385, 1127–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pielak R. M.; Schnell J. R.; Chou J. J. Mechanism of Drug Inhibition and Drug Resistance of Influenza A M2 Channel. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 7379–7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Pielak R. M.; McClintock M. A.; Chou J. J. Solution Structure and Functional Analysis of the Influenza B Proton Channel. Nat. Struct. Mol. Biol. 2009, 16, 1267–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cady S. D.; Schmidt-Rohr K.; Wang J.; Soto C. S.; Degrado W. F.; Hong M. Structure of the Amantadine Binding Site of Influenza M2 Proton Channels in Lipid Bilayers. Nature 2010, 463, 689–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pielak R. M.; Chou J. J. Solution NMR Structure of the V27A Drug Resistant Mutant of Influenza A M2 Channel. Biochem. Biophys. Res. Commun. 2010, 401, 58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pielak R. M.; Oxenoid K.; Chou J. J. Structural Investigation of Rimantadine Inhibition of the AM2-BM2 Chimera Channel of Influenza Viruses. Structure 2011, 19, 1655–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell J. R.; Chou J. J. Structure and Mechanism of the M2 Proton Channel of Influenza A Virus. Nature 2008, 451, 591–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guex N.; Peitsch M. C. Swiss-Model and the Swiss-PDBViewer: An Environment for Comparative Protein Modeling. Electrophoresis 1997, 18, 2714–2723. [DOI] [PubMed] [Google Scholar]
- Schwede T.; Kopp J.; Guex N.; Peitsch M. C. Swiss-Model: An Automated Protein Homology-Modeling Server. Nucleic Acids Res. 2003, 31, 3381–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; Hatcher E.; Acharya C.; Kundu S.; Zhong S.; Shim J.; Darian E.; Guvench O.; Lopes P.; Vorobyov I.; et al. CHARMM General Force Field: A Force Field for Drug-Like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W.; He X.; Vanommeslaeghe K.; MacKerell A. D. Jr. Extension of the CHARMM General Force Field to Sulfonyl-Containing Compounds and Its Utility in Biomolecular Simulations. J. Comput. Chem. 2012, 33, 2451–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; MacKerell A. D. Jr. Automation of the CHARMM General Force Field (CGenFF) II: Bond Perception and Atom Typing. J. Chem. Inf. Model. 2012, 52, 3144–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; Raman E. P.; MacKerell A. D. Jr. Automation of the Charmm General Force Field (Cgenff) Ii: Assignment of Bonded Parameters and Partial Atomic Charges. J. Chem. Inf. Model. 2012, 52, 3155–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cady S. D.; Wang J.; Wu Y.; DeGrado W. F.; Hong M. Specific Binding of Adamantane Drugs and Direction of Their Polar Amines in the Pore of the Influenza M2 Transmembrane Domain in Lipid Bilayers and Dodecylphosphocholine Micelles Determined by NMR Spectroscopy. J. Am. Chem. Soc. 2011, 133, 4274–4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgino T. Computing 1-D Atomic Densities in Macromolecular Simulations: The Density Profile Tool for VMD. Comput. Phys. Commun. 2014, 185, 317–322. [Google Scholar]
- Allen P.; Tildesley D. J.. Computer Simulation of Liquids; Clarendon Press: Oxford, U.K., 1989. [Google Scholar]
- Grossfield A.Wham: The Weighted Histogram Analysis Method, 2.0.9. Available: http://membrane.urmc.rochester.edu/content/wham.
- Zhou H. X.; Cross T. A. Modeling the Membrane Environment Has Implications for Membrane Protein Structure and Function: Influenza A M2 Protein. Protein Sci. 2013, 22, 381–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.