

Abstract
Highly charged metal ions act as catalytic centers and structural elements in a broad range of chemical complexes. The nonbonded model for metal ions is extensively used in molecular simulations due to its simple form, computational speed, and transferability. We have proposed and parametrized a 12-6-4 LJ (Lennard-Jones)-type nonbonded model for divalent metal ions in previous work, which showed a marked improvement over the 12-6 LJ nonbonded model. In the present study, by treating the experimental hydration free energies and ion–oxygen distances of the first solvation shell as targets for our parametrization, we evaluated 12-6 LJ parameters for 18 M(III) and 6 M(IV) metal ions for three widely used water models (TIP3P, SPC/E, and TIP4PEW). As expected, the interaction energy underestimation of the 12-6 LJ nonbonded model increases dramatically for the highly charged metal ions. We then parametrized the 12-6-4 LJ-type nonbonded model for these metal ions with the three water models. The final parameters reproduced the target values with good accuracy, which is consistent with our previous experience using this potential. Finally, tests were performed on a protein system, and the obtained results validate the transferability of these nonbonded model parameters.
Introduction
Highly charged (trivalent and tetravalent) metal ions are of great interest in both supramolecular,1 biomolecular,2 and rare-earth chemistry.3 Some of these serve as the coordination center that performs structural, catalytic, or electron-transfer functions, while others are well-known biotoxins.4−6 For example, iron–sulfur clusters have biochemical functions involving the respiration and photosynthesis processes.7 Iron in hemoglobin is involved in the transport and transfers oxygen within organisms.8 Lanthanide series elements have attracted significant attention due to their specific electromagnetic and optical characteristics.9 Their complexes serve as luminescent probes because of their large Stokes shifts and emission lifetimes.2 The actinide series elements, such as Th, U, and Pu, are well known for their radioactivity.10 All of these can form highly charged metal ions, which pose a major challenge to computational modeling. Indeed, effective and accurate modeling of these ions will give insight into separation and recycling process aimed at reducing their harmfulness to the environment.
There are several theoretical methods to model metal ions: quantum mechanics (QM),11,12 molecular mechanics (MM),13−28 and the hybrid QM/MM method.29,30 Classical force fields, which use an analytical function to represent the relationship between the energy and configuration of a system, have a significant speed advantage over the quantum-based methods. It is the state-of-the-art tool to study systems at the molecular level when combined with molecular dynamics31,32 or Monte Carlo methods.33,34 For metal ions, there are several widely used models including the bonded model,19−24 nonbonded model,13−18 and cationic dummy model.25−27 The bonded model represents the interaction between the ion and its surrounding residues via the bond, angle, torsion, coulombic, and van der Waals (VDW) terms. Because of the harmonic approximation used in the bonded model, it does not simulate the processes involving chemical bond formation and dissociation.19 The nonbonded model usually places an integer charge on the metal ion and only uses the coulombic and VDW terms to represent the intermolecular interactions between the metal ion and surrounding particles. This simplification can result in a notable underestimation for modeling systems with strong covalent bonds.14 The dummy cationic model places the charge between the metal ions and the ligating atoms to mimic the covalent bond.25 Besides the models previously discussed, there are also some polarizable force fields that have been developed for metal ions in recent years.35−41
Even though more accurate models exist, the 12-6 Lennard-Jones nonbonded model is widely used due to its simple form, computational efficiency, and excellent transferability characteristics.13,15,16,42 However, in previous research, we found that the 12-6 Lennard-Jones (LJ) nonbonded model could not reproduce several experimental properties across a series of divalent metal ions due to the neglect of the ion-induced dipole interaction.18 For the divalent metal ions, on average, there is a 50 kcal/mol underestimation for the hydration free energy (HFE) if we want to reproduce the experimental ion–oxygen distance (IOD) values, while there is ∼0.27 Å reduction for the IOD values if we reproduce the experimental HFE values. In light of this, we proposed a 12-6-4 LJ-type nonbonded model to account for the charge-induced dipole interaction.18 After picking suitable parameters, it was demonstrated that it was possible to reproduce the experimental HFE, IOD, and coordination number (CN) values simultaneously for a series of divalent metal ions. Furthermore, it was shown the new nonbonded model was readily transferable to mixed systems such as salt solutions and nucleic acid systems.
In the present work, we have estimated the 12-6 LJ parameters for 24 highly charged metal ions (18 M(III) ions and 6 M(IV) ions) for three widely used water models (TIP3P,43 SPC/E,44 and TIP4PEW45), respectively. This illustrated that the 12-6 LJ nonbonded model has a much larger underestimation of the ion–water interactions for the highly charged ions than for the mono and dications. We next parametrized the 12-6-4 LJ-type nonbonded model for the 24 highly charged metal ions for the same three water models. In general, these parameters simultaneously reproduce both the experimental HFE, IOD, and CN values with good accuracy. Moreover, they are consistent with previous research.30,46−48 This work opens up new opportunities to simulate M(III) and M(IV) ions in aqueous solution using classical models. Furthermore, we carried out test simulations on a Fe(III)-containing protein system. Stable trajectories were obtained with the metal binding site being well-conserved, further supporting the excellent transferability of these parameters.
Method
Potential Form
In present work, we employed the nonbonded model in the AMBER force field49
 |
1 |
In eq 1, the Uij(rij) is the nonbonded interaction potential between atoms i and j at distance rij. It consists of electrostatic and VDW terms. Herein e represents the charge of the proton, while Qi and Qj are the partial charge of atoms i and j. The partial charge of metal ions is always treated as an integer number in the nonbonded model. The VDW interaction part uses a 12-6 Lennard-Jones (LJ) potential, in which there are two parameters (εij and Rmin,ij) that need to be determined. Using the geometric combining rule, the well depth of the LJ potential is
| 2 |
For the Rmin,ij parameters, there are two widely used combining rules
| 3 |
and
| 4 |
Equation 3 is employed in the AMBER50 and CHARMM51 force fields, while eq 4 is used in the OPLS force field.52 In the present work, we employed the Lorentz–Berthelot combining rules, which is the union of eqs 2 (Lorentz combining rule) and 3 (Berthelot combining rule).
For the 12-6-4 LJ-type potential, we employed the following expression
 |
5 |
An r–4 term was added to describe the ion-induced dipole interaction, which cannot be overlooked for highly charged systems. The parametrization work concentrated on the determination of the Rmin,i, εi for the different metal ions, and the C4ij term between the metal ion and oxygen atoms of the different water models. In the present work, we have determined the parameters for three widely used water models (TIP3P,43 SPC/E,44 and TIP4PEW45) independently. Previous work demonstrated that it is necessary to design different parameters for these water models due to their different geometries, charge distributions, and VDW parameters.14,18,53
HFE Calculation
The thermodynamic integration (TI) method54−57 was used to simulate the HFE values. TI calculates the free-energy change between two different states of a system. An initial-state/final-state mixing potential was used during the simulation, in which V0 and V1 represent the potential of the initial and final state, respectively, while λ governs the mixing between the two states. k determines whether the mixing is linear or of higher order (k = 1 is linear).
| 6 |
The ion solvation process is modeled as transfer of an isolated metal ion from the gas to liquid phase. In the present work, we employed the thermodynamic cycle depicted in Figure 1. To avoid the “end-point catastrophe”, we employed the linear scaling soft-core TI method58 to obtain the ΔGVDW term.
 |
7 |
Figure 1.

Thermodynamic cycle describing the determination of the hydration free energy of ions.
In eq 7, rij is the distance between the dummy atom and the surrounding particles, while σ is the distance at which the two particles’ VDW interaction is equal to zero. Here the ε is the well depth and α is a constant set to 0.5. The “end-point catastrophe” problem is largely avoided because there is limited energy penalty induced between the dummy atom and proximal particles when the VDW potential of the dummy atom is turned on.
| 8 |
| 9 |
As seen from eq 8, the free-energy of each process is obtained via the integration of the derivative of the potential with respect to λ. Herein we employed Gaussian quadrature59 (eq 9) to evaluate the integral in an efficient way.
As illustrated in Figure 1, we obtained the HFE values based on the free-energy changes associated with four processes: ΔGVDW, ΔGEle+Pol, −ΔGEle+Pol, and −ΔGVDW. Herein the HFE value is computed using HFE = 1/2 × (ΔGVDW + ΔGEle+Pol – (−ΔGEle+Pol – ΔGVDW)). The ΔGEle+Pol and −ΔGEle+Pol are ΔGEle and −ΔGEle respectively, when the 12-6 nonbonded model is employed. At first, a dummy atom was placed in the center of a cubic water box (with size ∼32 Å × 32 Å × 32 Å) with the closest water molecule ∼1.5 Å away from it. There are in total 722 water molecules in the TIP3P or SPC/E water boxes, while there are 732 water molecules in the TIP4PEW water box. Afterward 1000 steps of steepest descent minimization were performed, followed by 1000 steps conjugate gradient minimization. Then, a 500 ps heating procedure was performed to heat the system from 0 to 300 K in the NVT ensemble. Next, we equilibrated the system for 500 ps at 300 K and 1 atm using the NPT ensemble. The final snapshot from the equilibration simulation was used as the initial structure for the calculation of ΔGVDW. To balance accuracy and speed, we used the four-window linear soft-core scaling process to obtain the ΔGVDW value with λ values of 0.1127, 0.5, 0.88729, and 0.98, respectively. The simulation of last window was used to further equilibrate the system but was not used in the free-energy evaluation. The final snapshot was used to initiate the determination of ΔGEle+Pol. For the ΔGEle+Pol calculation process, a nine-window linear scaling scheme was utilized with λ values of 0, 0.2544, 0.12923, 0.29707, 0.5, 0.70292, 0.87076, 0.97455, and 1, respectively. Afterward, the −ΔGEle+Pol calculation process was carried out in a similar manner. In these simulations, the first and last windows (λ equal to 0 and 1) are not used in the final free-energy calculation but to further equilibrate the system. Finally, the −ΔGVDW simulation procedure was carried out using a three-window linear soft-core scaling process in which λ was set at 0.1127, 0.5, and 0.88729, respectively. For the determination of ΔGVDW and −ΔGVDW, each window was simulated for 300 ps, with the last 200 ps used for data collection. The ΔGEle+Pol and −ΔGEle+Pol simulations covered 200 ps, with the last 150 ps used for data collection in each window. All TI simulations were performed in the NPT ensemble.
We have employed two different methods to evaluate the uncertainty of the simulated HFE values in the present work. These results are gathered in the Supporting Information (SI). The first method (Set 1) divided each sampling segment (ΔGVDW, ΔGEle+Pol, −ΔGEle+Pol, and −ΔGVDW) into two even portions and estimated the uncertainties for the VDW and electrostatic plus polarization free-energy determinations. For example, we have 200 ps of sampling for each window for the determination of ΔGVDW and −ΔGVDW. We used the first 100 ps of sampling to calculate the ΔGVDW-part 1 and −ΔGVDW-part 1 values, while we used the later 100 ps of sampling to obtain the ΔGVDW-part 2 and −ΔGVDW-part 2. Then, we assessed the uncertainty of the VDW free-energy determination by calculating the standard deviation based on these four values. The uncertainty of the electrostatic plus polarization free-energy determination was obtained in a similar manner with each fragment using 75 ps of sampling. Subsequently, we obtain the total uncertainty in the HFE value by adding the uncertainties of the VDW and electrostatic plus polarization free-energy determinations.
The second method (Set 2) uses eq 10, in which the τA is the autocorrelation time of observable A while (⟨A2⟩c)1/2 is the standard deviation of A. T is the sampling time of the simulation and δA is the final uncertainty of the observable A. The final HFE values given in the spreadsheets given in the SI are depicted as ⟨A⟩ ± σ. Again, the uncertainty of the VDW and electrostatic plus polarization free-energy determinations were evaluated separately, and the final uncertainty was treated as their sum. Herein we used a τA value equal to 500 fs for the VDW free-energy determinations, while we used 250 fs for the electrostatic plus polarization scaling part. We obtained these values based on test simulations, and they are consistent with previous work.60
| 10 |
The Set 1 approach yields uncertainties in the range of 0.1–7.5 kcal/mol with an average of ∼1.2 kcal/mol, while Set 2 gives uncertainties in the range of 1.1 to 2.0 kcal/mol with an average of ∼1.4 kcal/mol. On the basis of these analyses, we estimate that the HFE uncertainty is in the ±2.0 kcal/mol range, which is quite small given the magnitude of HFEs we are computing.
IOD and CN Calculation
A metal ion (with an integer +3 or +4 partial charge) was solvated in the center of a cubic water box (with the same size as described in the HFE simulation part). Then, 1000 steps of steepest descent and 1000 steps of conjugated gradient minimization were carried out to relax the initial structure. Afterward, a 500 ps “heating” simulation was performed in the NVT ensemble that took the system from 0 to 300 K. Next, 500 ps of equilibration, followed by 2 ns of sampling were performed at 300 K and 1 atm. Snapshots were stored every 0.5 ps (every 500 steps for 4000 snapshots in total) for the subsequent IOD and CN analysis. The radial distribution function (RDF) of the ion and water oxygen atom was then calculated based on the average volume of the entire trajectory in the range of 0−5.0 Å with a grid resolution of 0.01 Å. The IOD value was evaluated based on two quadratic fits of the RDF. The first quadratic fit was performed using the points within ±0.1 Å of the first peak of RDF. In this way, the apex value was obtained with an accuracy of 0.01 Å. The second quadratic fitting was done based on the points within ±0.1 Å of the apex obtained from the first fitting. In total, 21 points were used for each fit. The maximum given by the second fitting was treated as the final IOD value to two decimal places. The CN value was obtained by integrating from the origin to the first minimum of the RDF.
The AMBER 12 suite of programs49 was used to perform the simulations, while the Amber Tools suite of programs49 was utilized to carry out the data analysis. The particle mesh Ewald (PME)61−63 method and periodic boundary condition (PBC) were employed throughout. The time-step was 1 fs, while the cutoff was set to 10 Å. For the temperature control, the Langevin algorithm was utilized with a collision frequency equal to 5.0 ps–1. The isotropic pressure algorithm was used to control the pressure. The pressure relaxation time was set to 10 and 1 ps in the TI and standard MD simulations, respectively. The SHAKE64,65 algorithm was employed to restrain the bond lengths involving hydrogen atoms. Herein the “three-point” algorithm was used for the water molecules.65
Results and Discussion
Experimental Values
The HFE values for all M(III) and M(IV) ions investigated were taken from Marcus.66 They were determined based on using ΔhydG0[H+] = −1056 kJ/mol.66 It is one of the most complete databases regarding the thermodynamic properties of ions. The IOD and CN values for M(III) ions were taken from Marcus’ review,67 while the IOD and CN values for the M(IV) ions were taken from a number of sources.68−71 The experimental effective ionic radii were obtained from Shannon.72 On the basis of the IOD values and effective ionic radii, we estimated the effective radii of the coordinated water and display the data in Table 1. Some highly charged ions that readily hydrolyze water such as As3+, Sn4+, and Pb4+ ions30 were not considered in the present work.
Table 1. Experimental HFE and IOD Values of M(III) and M(IV) Metal Ions.
| metal ion | electron configuration | HFE (kcal/mol)a | IOD (Å)b | CNb | effective ion radii (Å) | first shell water radii (Å) |
|---|---|---|---|---|---|---|
| Al3+ | [Ne] | –1081.5 | 1.88 | 6 | 0.54 | 1.34 |
| Fe3+ | [Ar]3d5 | –1019.4 | 2.03 | 6 | 0.65 | 1.38 |
| Cr3+ | [Ar]3d3 | –958.4 | 1.96 | 6 | 0.62 | 1.34 |
| In3+ | [Kr]4d10 | –951.2 | 2.15 | 6 | 0.80 | 1.35 |
| Tl3+ | [Xe]4f145d10 | –948.9 | 2.23 | 4–6 | 0.89 | 1.34 |
| Y3+ | [Kr] | –824.6 | 2.36 | 8 | 0.90 | 1.46 |
| La3+ | [Xe] | –751.7 | 2.52 | 8.0–9.1 | 1.03 | 1.49 |
| Ce3+ | [Xe]4f1 | –764.8 | 2.55 | 7.5 | 1.01 | 1.54 |
| Pr3+ | [Xe]4f2 | –775.6 | 2.54 | 9.2 | 0.99 | 1.55 |
| Nd3+ | [Xe]4f3 | –783.9 | 2.47 | 8.0–8.9 | 0.98 | 1.49 |
| Sm3+ | [Xe]4f5 | –794.7 | 2.44 | 8.0–9.9 | 0.96 | 1.48 |
| Eu3+ | [Xe]4f6 | –803.1 | 2.45 | 8.3 | 0.95 | 1.50 |
| Gd3+ | [Xe]4f7 | –806.6 | 2.39 | 8.0–9.9 | 0.94 | 1.45 |
| Tb3+ | [Xe]4f8 | –812.6 | 2.40 | 8.0–8.2 | 0.92 | 1.48 |
| Dy3+ | [Xe]4f9 | –818.6 | 2.37 | 7.4–7.9 | 0.91 | 1.46 |
| Er3+ | [Xe]4f11 | –835.3 | 2.36 | 6.3–8.2 | 0.89 | 1.47 |
| Tm3+ | [Xe]4f12 | –840.1 | 2.36 | 8.1 | 0.88 | 1.48 |
| Lu3+ | [Xe]4f14 | –840.1 | 2.34 | 8 | 0.86 | 1.48 |
| Hf4+ | [Xe]4f14 | –1664.7 | 2.16c | 8c | 0.85 | 1.31 |
| Zr4+ | [Kr] | –1622.8 | 2.19c | 8c | 0.86 | 1.33 |
| Ce4+ | [Xe] | –1462.7 | 2.42d | 9d | 0.87 | 1.55 |
| U4+ | [Rn]6d15f1 | –1567.9 | 2.42e | 9–11e | 0.89 | 1.53 |
| Pu4+ | [Rn]5f4 | –1520.1 | 2.39f | 8f | 0.86 | 1.53 |
| Th4+ | [Rn] | –1389.8 | 2.45e | 9–11e | 0.94 | 1.51 |
Scanning Parameter Space
To balance time and accuracy, we performed parameter scanning using two parallel curves. One is for Rmin/2 from 0.9 to 2.3 Å with 0.1 Å intervals without the C4 term (namely using the 12-6 LJ nonbonded model), while the other is for the same Rmin/2 value sets with a constant C4 term equal to 500 kcal/mol·Å4. The ε values are obtained for each Rmin/2 value based on the noble gas curve (NGC), which was previously developed.14 The HFE, IOD, and CN values for each parameter point are collected in Tables SI.1 and SI.2 in the Supporting Information (SI). These data points might be useful to those who want to parametrize the 12-6 LJ or 12-6-4 LJ-type nonbonded model with different target values from the present work.
12-6 LJ Parameters Estimation
On the basis of the quadratic fitting of the data points from the parameter scans without the C4 term (see the SI), we estimated the HFE and IOD parameter sets. The two parameter sets are shown in Tables 2 and 3, while the estimated absolute and percent errors are shown in Table SI.3. Similar to our parametrization of divalent metal ions using the 12-6 LJ nonbonded model,14 the Rmin/2 parameters in the HFE parameter set are in excellent agreement with the VDW radii calculated using the quantum-mechanical scaling principle (QMSP) method.73 For the Al3+, Y3+, and La3+ ions, the estimated Rmin/2 values in the estimated HFE parameter set for the TIP3P water model are 0.981, 1.454, and 1.628 Å respectively. The calculated VDW radii are 1.046, 1.481, and 1.642 Å, respectively, based on the QMSP method. There is only a 6.2, 1.8, and 0.9% difference between these two sets of values. This further validates the physical meaningful of our parametrization work. Moreover, the estimated Rmin/2 values used in the 12-6 LJ parameter sets could be used as the VDW radii for RESP charge-fitting procedures. For example, in the work of Kuznetsov et al.,74 they used 1.4 Å as the VDW radius for the RESP charge fitting for both the Fe2+ and Fe3+ ions, while for the IOD parameter set the Fe2+ radius was determined to be 1.409 Å in our previous research.18 Herein, the IOD parameter sets for the Fe3+ ion estimated the radius as 1.386, 1.386, and 1.375 for the TIP3P, SPC/E, and TIP4PEW water models, respectively.
Table 2. Estimated HFE Parameter Set for M(III) and M(IV) Metal Ions Using the 12-6 LJ Nonbonded Potential.
| TIP3P |
SPC/E |
TIP4PEW |
||||
|---|---|---|---|---|---|---|
| Rmin/2 (Å) | ε (kcal/mol) | Rmin/2 (Å) | ε (kcal/mol) | Rmin/2 (Å) | ε (kcal/mol) | |
| Al3+ | 0.981 | 0.00000832 | 0.991 | 0.00001107 | 0.876 | 0.00000026 |
| Fe3+ | 1.082 | 0.00011017 | 1.091 | 0.00013462 | 0.984 | 0.00000907 |
| Cr3+ | 1.188 | 0.00089969 | 1.196 | 0.00103208 | 1.096 | 0.00015019 |
| In3+ | 1.202 | 0.00114198 | 1.209 | 0.00128267 | 1.110 | 0.00020260 |
| Tl3+ | 1.206 | 0.00122067 | 1.213 | 0.00136949 | 1.114 | 0.00022027 |
| Y3+ | 1.454 | 0.02639002 | 1.459 | 0.02759452 | 1.375 | 0.01205473 |
| La3+ | 1.628 | 0.09399072 | 1.629 | 0.09454081 | 1.553 | 0.05807581 |
| Ce3+ | 1.595 | 0.07688443 | 1.597 | 0.07786298 | 1.519 | 0.04525501 |
| Pr3+ | 1.568 | 0.06441235 | 1.571 | 0.06573030 | 1.492 | 0.03655251 |
| Nd3+ | 1.548 | 0.05605698 | 1.551 | 0.05726270 | 1.471 | 0.03064622 |
| Sm3+ | 1.522 | 0.04630154 | 1.526 | 0.04772212 | 1.445 | 0.02431873 |
| Eu3+ | 1.503 | 0.03994409 | 1.507 | 0.04122946 | 1.425 | 0.02014513 |
| Gd3+ | 1.495 | 0.03745682 | 1.499 | 0.03868661 | 1.417 | 0.01863432 |
| Tb3+ | 1.481 | 0.03336723 | 1.485 | 0.03450196 | 1.403 | 0.01619682 |
| Dy3+ | 1.468 | 0.02986171 | 1.472 | 0.03091095 | 1.389 | 0.01400886 |
| Er3+ | 1.431 | 0.02133669 | 1.436 | 0.02236885 | 1.350 | 0.00909668 |
| Tm3+ | 1.421 | 0.01937874 | 1.426 | 0.02034021 | 1.340 | 0.00808758 |
| Lu3+ | 1.421 | 0.01937874 | 1.426 | 0.02034021 | 1.340 | 0.00808758 |
| Hf4+ | 1.087 | 0.00012321 | 1.098 | 0.00015685 | 0.977 | 0.00000741 |
| Zr4+ | 1.139 | 0.00036479 | 1.149 | 0.00044254 | 1.031 | 0.00003240 |
| Ce4+ | 1.353 | 0.00941798 | 1.360 | 0.01020237 | 1.257 | 0.00270120 |
| U4+ | 1.209 | 0.00128267 | 1.218 | 0.00148497 | 1.105 | 0.00018227 |
| Pu4+ | 1.273 | 0.00339720 | 1.281 | 0.00379705 | 1.172 | 0.00067804 |
| Th4+ | 1.463 | 0.02858630 | 1.468 | 0.02986171 | 1.370 | 0.01141046 |
Table 3. Estimated IOD Parameter Set for M(III) and M(IV) Metal Ions Using the 12-6 LJ Nonbonded Potential.
| TIP3P |
SPC/E |
TIP4PEW |
||||
|---|---|---|---|---|---|---|
| Rmin/2 (Å) | ε (kcal/mol) | Rmin/2 (Å) | ε (kcal/mol) | Rmin/2 (Å) | ε (kcal/mol) | |
| Al3+ | 1.297 | 0.00471279 | 1.296 | 0.00465074 | 1.285 | 0.00401101 |
| Fe3+ | 1.386 | 0.01357097 | 1.386 | 0.01357097 | 1.375 | 0.01205473 |
| Cr3+ | 1.344 | 0.00848000 | 1.343 | 0.00838052 | 1.333 | 0.00743559 |
| In3+ | 1.461 | 0.02808726 | 1.461 | 0.02808726 | 1.450 | 0.02545423 |
| Tl3+ | 1.513 | 0.04321029 | 1.513 | 0.04321029 | 1.502 | 0.03962711 |
| Y3+ | 1.602 | 0.08034231 | 1.602 | 0.08034231 | 1.590 | 0.07447106 |
| La3+ | 1.718 | 0.15060822 | 1.718 | 0.15060822 | 1.707 | 0.14295367 |
| Ce3+ | 1.741 | 0.16721338 | 1.741 | 0.16721338 | 1.729 | 0.15845086 |
| Pr3+ | 1.733 | 0.16134811 | 1.734 | 0.16207614 | 1.722 | 0.15343866 |
| Nd3+ | 1.681 | 0.12564307 | 1.681 | 0.12564307 | 1.669 | 0.11803919 |
| Sm3+ | 1.659 | 0.11189491 | 1.659 | 0.11189491 | 1.647 | 0.10475707 |
| Eu3+ | 1.666 | 0.11617738 | 1.666 | 0.11617738 | 1.655 | 0.10948690 |
| Gd3+ | 1.623 | 0.09126804 | 1.623 | 0.09126804 | 1.612 | 0.08544204 |
| Tb3+ | 1.630 | 0.09509276 | 1.630 | 0.09509276 | 1.619 | 0.08912336 |
| Dy3+ | 1.609 | 0.08389240 | 1.609 | 0.08389240 | 1.597 | 0.07786298 |
| Er3+ | 1.602 | 0.08034231 | 1.602 | 0.08034231 | 1.590 | 0.07447106 |
| Tm3+ | 1.602 | 0.08034231 | 1.602 | 0.08034231 | 1.590 | 0.07447106 |
| Lu3+ | 1.588 | 0.07351892 | 1.588 | 0.07351892 | 1.577 | 0.06841702 |
| Hf4+ | 1.499 | 0.03868661 | 1.501 | 0.03931188 | 1.483 | 0.03393126 |
| Zr4+ | 1.519 | 0.04525501 | 1.521 | 0.04595090 | 1.503 | 0.03994409 |
| Ce4+ | 1.684 | 0.12758274 | 1.689 | 0.13084945 | 1.667 | 0.11679623 |
| U4+ | 1.684 | 0.12758274 | 1.689 | 0.13084945 | 1.667 | 0.11679623 |
| Pu4+ | 1.662 | 0.11371963 | 1.666 | 0.11617738 | 1.645 | 0.10359269 |
| Th4+ | 1.708 | 0.14364160 | 1.713 | 0.14710519 | 1.690 | 0.13150785 |
Combining the data with previous work on M(II) metal ions, we summarized the absolute and percent errors for the 12-6 LJ nonbonded model for M(II), M(III), and M(IV) ions in Tables 4 and 5. These results, taken as a whole, show that the underestimation of the 12-6 LJ nonbonded model increases dramatically as the charge on the metal ion increases. For example, for the TIP3P water model, the average absolute error goes from ∼50 kcal/mol for M(II) ions to ∼80 kcal/mol for M(III) and ∼240 kcal/mol for M(IV) ions for the IOD parameter set, while the average absolute error of the IOD values for the HFE parameter set increases from −0.27 Å for M(II) ions to −0.29 Å for M(III) ions and −0.58 Å for M(IV) ions. For some of the monovalent ions, it is possible to reproduce both the experimental HFE and IOD values at the same time;53 this underestimation of the 12-6 LJ nonbonded model is pretty small and can almost be neglected. Because there are significant errors associated with the 12-6 LJ nonbonded model for highly charged ions, we did not carry out further refinement work on the estimated 12-6 LJ parameters because the resultant parameters would be of limited usefulness.
Table 4. Estimated Average Absolute and Percent IOD Errors (in Brackets) for the 12-6 HFE Parameter Set against Experimental Values for Divalent, Trivalent, and Tetravalent Metal Ionsa.
| M(II) | M(III) | M(IV) | ||
|---|---|---|---|---|
| TIP3P | avg. IOD error | –0.27 (−12.4%) | –0.29 (−12.8%) | –0.58 (−25.0%) |
| IOD error SD | 0.14 (7.8%) | 0.14 (7.4%) | 0.15 (7.0%) | |
| SPC/E | avg. IOD error | –0.26 (−12.3%) | –0.28 (−12.4%) | –0.57 (−24.5%) |
| IOD error SD | 0.14 (7.6%) | 0.13 (7.2%) | 0.14 (6.6%) | |
| TIP4PEW | avg. IOD error | –0.36 (−16.8%) | –0.41 (−18.1%) | –0.74 (−32.0%) |
| IOD error SD | 0.17 (10.2%) | 0.16 (9.2%) | 0.17 (8.2%) |
Absolute IOD errors are in angstroms.
Table 5. Estimated Average Absolute and Percent HFE Errors (in brackets) for the 12-6 IOD Parameter Set against Experimental Values for Divalent, Trivalent, and Tetravalent Metal Ionsa.
| M(II) | M(III) | M(IV) | ||
|---|---|---|---|---|
| TIP3P | avg HFE error | 51.1 (−11.5%) | 82.7 (−9.3%) | 244.3 (−15.7%) |
| HFE error SD | 25.2 (4.5%) | 42.7 (3.6%) | 62.7 (3.3%) | |
| SPC/E | avg HFE error | 51.9 (−11.7%) | 81.8 (−9.2%) | 244.9 (−15.8%) |
| HFE error SD | 24.3 (4.3%) | 41.6 (3.5%) | 61.8 (3.2%) | |
| TIP4PEW | avg HFE error | 67.2 (−15.2%) | 108.0 (−12.3%) | 283.0 (−18.2%) |
| HFE error SD | 27.5 (4.5%) | 46.4 (3.7%) | 65.1 (3.3%) |
Absolute HFE errors are in kilocalories per mole.
12-6-4 Parameter Determination
After initial parameter selection and subsequent fine-tuning, the final 12-6-4 parameters were determined. The final optimized 12-6-4 parameters are given in Table 6 while the simulated HFE, IOD, and CN values are shown in Table SI.4. These parameters reproduce the experimental HFE values by ±1 kcal/mol and the IOD values by ±0.01 Å for the M(III) ions, while they reproduce the HFE values by ±2 kcal/mol and the IOD values by ±0.01 Å for the M(IV) ions. Just as in the 12-6-4 parameter sets for divalent metal ions, the Rmin/2 terms are similar between the three water models, while the C4 term for TIP4PEW water is generally larger than for the other two water models for the same metal ion. This may due to the smaller dipole of the TIP4PEW water model (2.32 D) relative to the TIP3P (2.35 D) and SPC/E (2.35 D) water models. Figure 2 shows the accuracy comparison between the 12-6-4 parameter set and the 12-6 parameter sets for divalent, trivalent, and tetravalent metal ions. We can see that there is significant improvement in the accuracy using the 12-6-4 parameter set, which is able to reproduce the experimental HFE and IOD values simultaneously. While for the 12-6 LJ nonbonded model, if you want to reproduce the experimental HFE values, the error in the simulated IOD values would increase along with the formal charge of the metal ions. Vice versa, if you simulate the IOD values using the 12-6 LJ nonbonded model, the error of the calculated HFE would increase markedly with an increase in the oxidation state of the metal ion in question.
Table 6. Final Parameters for the 12-6-4 LJ-Type Nonbonded Model for Metal Ions in Three Water Models.
| TIP3P |
SPC/E |
TIP4PEW |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Rmin/2 (Å) | ε (kcal/mol) | C4 (kcal/mol·Å4) | Rmin/2 (Å) | ε (kcal/mol) | C4 (kcal/mol·Å4) | Rmin/2 (Å) | ε (kcal/mol) | C4 (kcal/mol·Å4) | |
| Al3+ | 1.369 | 0.01128487 | 399 | 1.375 | 0.01205473 | 406 | 1.377 | 0.01232018 | 488 |
| Fe3+ | 1.443 | 0.02387506 | 428 | 1.450 | 0.02545423 | 442 | 1.448 | 0.02499549 | 519 |
| Cr3+ | 1.415 | 0.01827024 | 258 | 1.414 | 0.01809021 | 254 | 1.408 | 0.01703790 | 322 |
| In3+ | 1.491 | 0.03625449 | 347 | 1.487 | 0.03507938 | 349 | 1.486 | 0.03478983 | 425 |
| Tl3+ | 1.571 | 0.06573030 | 456 | 1.569 | 0.06484979 | 455 | 1.564 | 0.06268139 | 535 |
| Y3+ | 1.630 | 0.09509276 | 216 | 1.624 | 0.09180886 | 209 | 1.624 | 0.09180886 | 294 |
| La3+ | 1.758 | 0.17997960 | 152 | 1.763 | 0.18380968 | 165 | 1.755 | 0.17769767 | 243 |
| Ce3+ | 1.782 | 0.19865859 | 230 | 1.786 | 0.20184160 | 242 | 1.776 | 0.19392043 | 315 |
| Pr3+ | 1.780 | 0.19707431 | 264 | 1.782 | 0.19865859 | 272 | 1.774 | 0.19235093 | 348 |
| Nd3+ | 1.724 | 0.15486311 | 213 | 1.735 | 0.16280564 | 235 | 1.720 | 0.15202035 | 297 |
| Sm3+ | 1.711 | 0.14571499 | 230 | 1.703 | 0.14021803 | 224 | 1.706 | 0.14226734 | 314 |
| Eu3+ | 1.716 | 0.14920231 | 259 | 1.721 | 0.15272873 | 273 | 1.711 | 0.14571499 | 345 |
| Gd3+ | 1.658 | 0.11129023 | 198 | 1.646 | 0.10417397 | 186 | 1.652 | 0.10769970 | 280 |
| Tb3+ | 1.671 | 0.11928915 | 235 | 1.666 | 0.11617738 | 227 | 1.665 | 0.11556030 | 313 |
| Dy3+ | 1.637 | 0.09900804 | 207 | 1.637 | 0.09900804 | 206 | 1.639 | 0.10014323 | 298 |
| Er3+ | 1.635 | 0.09788018 | 251 | 1.629 | 0.09454081 | 247 | 1.628 | 0.09399072 | 328 |
| Tm3+ | 1.647 | 0.10475707 | 282 | 1.633 | 0.09675968 | 262 | 1.638 | 0.09957472 | 356 |
| Lu3+ | 1.625 | 0.09235154 | 249 | 1.620 | 0.08965674 | 247 | 1.617 | 0.08806221 | 331 |
| Hf4+ | 1.600 | 0.07934493 | 827 | 1.592 | 0.07543075 | 810 | 1.599 | 0.07884906 | 956 |
| Zr4+ | 1.609 | 0.08389240 | 761 | 1.609 | 0.08389240 | 760 | 1.610 | 0.08440707 | 895 |
| Ce4+ | 1.766 | 0.18612361 | 706 | 1.761 | 0.18227365 | 694 | 1.761 | 0.18227365 | 835 |
| U4+ | 1.792 | 0.20665151 | 1034 | 1.791 | 0.20584696 | 1043 | 1.791 | 0.20584696 | 1183 |
| Pu4+ | 1.752 | 0.17542802 | 828 | 1.750 | 0.17392181 | 828 | 1.753 | 0.17618319 | 972 |
| Th4+ | 1.770 | 0.18922704 | 512 | 1.773 | 0.19156806 | 513 | 1.758 | 0.17997960 | 625 |
Figure 2.
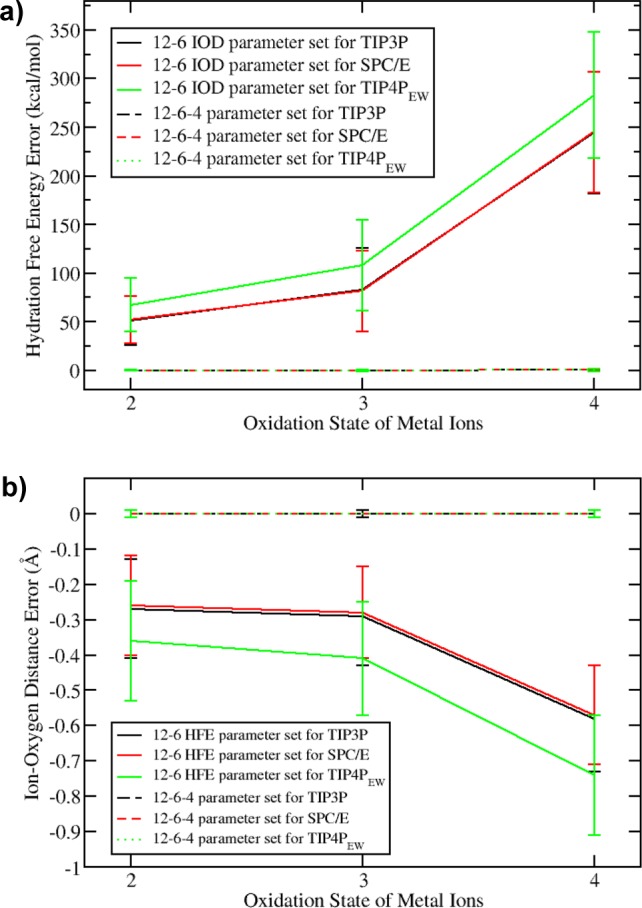
(a) HFE errors for the 12-6 IOD and 12-6-4 parameter sets for M(II), M(III), and M(IV) metal ions. (b) IOD errors for the 12-6 HFE and 12-6-4 parameter sets for the M(II), M(III), and M(IV) metal ions.
1. Trivalent Metal Ions
Main Group and Transition-Metal Ions
These metal ions have much stronger ion–water interactions than the Ln3+ ions. Some of them are extremely inert ions. They form a stable octahedral structure with water molecules in the first solvation shell. Data in Table 1 indicate that the average effective radius of the first solvation shell water is ∼1.35 Å for the first several metal ions, which is consistent with strong interactions between the coordinated water molecules and these metal ions. These values are close to previously proposed coordinated water radius (∼1.34 Å).75 The corresponding average values are ∼1.49 Å and ∼1.46 Å for the Ln3+ and the M(IV) metal ions, respectively, which implies a smaller electronic cloud overlap between the metal ion center and each of the coordinated water molecules.
Al3+, In3+, and Tl3+ are group IV ions. For the C4 parameters derived herein, we obtained a sequence of Tl3+ > Al3+ > In3+. The Al3+ ion is the smallest M(III) ion, resulting in a relatively larger C4 term due to its strong covalent interaction with coordinated water molecules. Tl has two oxidation states, +1 and +3, and the HFE values of the Tl+ and K+ are almost the same in Marcus’ HFE set.66 Tl3+ could have very strong covalent interactions with the surrounding residues. The reduced electric potential of M(III) + 3e– = M is −1.67, −0.3382, and +0.72 eV for Al3+, In3+, and Tl3+, respectively.76 The positive reduction potential of Tl3+ makes it a very reactive species. It readily obtains electrons from its surroundings, which may be the reason for a strong charge-transfer effect between the Tl3+ ion and the surrounding water molecules. The 12-6-4 parameters of In3+ and Tl3+ ions gave an excellent prediction for the HFE and IOD values but overestimated the CN value (8 instead of 6), and this is mainly due to the lack of a correction for the water–water interactions in the first solvation shell during the simulations. The water–water interactions were parametrized to reproduce the pure liquid water properties in the original parameter design. However, the first solvation shell water molecules of the highly charged metal ions should more strongly repel one another due to their bigger charge separations. This effect is smaller for M(I) and M(II) metal ions, but it dramatically increases for the highly charged ions. Meanwhile, this kind of effect may decrease in protein systems due to the preorganization of the metal ion binding sites.
Fe3+ has a larger C4 term than Al3+, the smallest M(III) ion, which suggests that Fe3+ has a stronger interaction with its surrounding water molecules. This is consistent with quantum-mechanical charge-field molecular dynamics (QMCF-MD) simulations, which shows that the force constant between the ion and the oxygen of first solvation shell water molecules (kion-O) is 198 N/m for Fe3+ compared with 185 N/m for Al3+.30 This is a consequence of both electrostatic and covalent interactions. The Fe3+ ion has an average 1.85e47 charge (from a Mulliken analysis) in the QMCF simulation, while Al3+ ion has a corresponding value of 2.5 e,77 which implies that there is a stronger charge-transfer effect for the Fe3+ ion and its surrounding water molecules than for the Al3+ ion. There is a slight overestimation of the CN for Fe3+ ions. Also, as previously discussed, this may be due to the underestimation of the interactions between the first solvation water molecules. While this effect is operative in aqueous solution, it will likely be less of an issue in protein systems (see discussion below).
Y3+ and Ln3+ Ions
The +3 oxidation state is the typical oxidation state of the Ln elements, with the exception that Eu2+ and Ce4+ could also be observed. This is because Eu2+ has a half-filled 4f orbital while Ce4+ has the same electronic configuration as Xe. The interaction of the Ln3+ ions with surrounding water molecules would be expected to have more ionic character than the M(III) ions previously discussed. For example, the C4 terms between the Ln3+ ions and water molecules are between 152 and 282 kcal/mol·Å4, which is smaller than the 258–456 kcal/mol·Å4 range seen for the other +3 metal ions previously discussed. Previous simulations found that the kion-O values are much smaller for the Ln3+ ions; for example, La3+, Ce3+, Lu3+, and Er3+ have kion-O values ∼110 N/m, while the values for the Al3+ and Fe3+ ions are 185 and 203 N/m, respectively.46 The Ln3+ ions have effective ionic radii in the range of 0.86 to 1.03 Å and IOD values in the range of 2.34 to 2.55 Å. These values are similar to that of the Ca2+ ion (whose effective ionic radius and IOD value is 1.0072 and 2.46 Å,78 respectively). Therefore, they have been used as probes to investigate the role of Ca2+ ions in biological systems.79
From Table 6, we observe that the La3+ and Gd3+ ions have the smallest C4 terms among the Ln3+ ions. This may be because they have either totally empty or half-filled 4f orbitals, making them more likely to form isolated ions, which reduces the covalent character of their bonds with coordinated water molecules. It is easy to see the “lanthanide contraction” effect from Table 1. The effective ion radius decreases monotonically with an increase in the metal ions’ atomic number due to the poor shielding of the 4f electrons toward 5s and 5p orbitals.80 A similar tendency can also be seen for the HFE and IOD values along the series. Our final Rmin/2 parameters are consistent with this pattern as well. Meanwhile, the CN also decreases along the Ln3+ ion series. Previous work reached the conclusion that the lighter Ln3+ ions (La3+ to Nd3+) prefer a CN of ∼9 and the heavier ions (Gd3+ to Tb3+) prefer a CN of ∼8, while the middle ions such as Sm3+ and Eu3+ have CN between these two values.81−86 It was proposed that the former Ln3+ ions have a tricapped trigonal prism structure, which then shifts to a distorted bicapped trigonal prism structure for the heavier elements as one of the two capping water molecules leaves the first solvation shell.87 Generally speaking, there is a good agreement between the 12-6-4 parameters for the HFE, IOD, and CN values with experiment, with the exception that some of the CN values were slightly overestimated. The final parameters gave a CN in the range of 9 to 10 for Ln3+ ions rather than the range of 8 to 9 reported in the literature. As previously discussed, this may be due to the fact that there is no water–water interaction correction term in the present parametrization process. Moreover, the CN values given by Marcus (as shown in Table 1) likely also vary under different experimental conditions (counterions used solute concentration, etc.). Among all ions, Al3+, Y3+, and La3+ have the same electron configurations as the noble gas atoms Ne, Kr, and Xe, respectively. Using the TIP3P water model as an example, we can see their C4 values decrease from 399 to 216 and 152 kcal/mol·Å4, respectively.
2. Tetravalent Metal Ions
There are only a few M(IV) ions that exist in aqueous solution, while the others are readily hydrolyzed into polynuclear complexes in water.68−71 Table 1 shows the M(IV) ions examined herein. These ions exist in at least highly acidic solution. The CN values of these metal ions are greater than 8, with some of them being ∼10 according to experiment.68−71 Previous work found that Pu(IV), Th(IV), and U(IV) could strongly bind to transferrin, an iron-transport protein.88 Hence, the parameters developed herein might facilitate theoretical research on the biotoxicity of these M(IV) ions.
The Zr4+ and Hf4+ are in the IVB group. Even though Hf4+ has a larger atomic number than Zr4+, due to the “lanthanide contraction” effect, it has a smaller effective radius, smaller IOD, smaller HFE, and a bigger C4 term than Zr4+. These observations reflect its stronger interaction with the surrounding water molecules. In contrast, Ce4+ and Th4+ are in the same group where the larger atomic number (Th4+) has the bigger ionic radius, bigger HFE, and smaller C4 terms. This may be because they share the same electronic structure as Xe and Rn, respectively. Besides these ions, Zr4+ is another M(IV) ion that has a noble gas atom’s electronic structure (the same as Kr). There is also a single trend for the C4 terms of Zr4+, Ce4+, and Th4+ ions for each specific water model. For instance, the C4 terms for the TIP3P water model are 761, 706, and 512 kcal/mol·Å4, respectively.
Th, U, and Pu are in the An series and are the largest elements investigated in the present work. Their tetravalent metal ions exist only in highly acidic solutions. Canaval et al. investigated Th4+ in aqueous solutions using the QMCF-MD method. They found a stable nine-coordinate complex, and even the third layer of water molecules has a bigger mean residence time than that of pure water, implying they are stabilized by the highly charged Th4+ ion.48 U4+ fluoresces due to the electron transition between the 6d15f1 and 5f2 electronic configurations.200 The U4+ ion has the largest C4 term of all of the M(IV) metal ions investigated. Frick et al. investigated the U4+ ion in aqueous solution using the QMCF-MD method, and the CN value was characterized as 9, while the average charge of U4+ was predicted to be +2.68 from Mulliken population analyses.300 Odoh et al. simulated the Pu3+, Pu4+, PuO2+, and PuO22+ ions in water solution using the Car–Parrinello molecular dynamics method.89 They predicted that the pKa value for the first hydrolysis step for the Pu3+, Pu4+, PuO2+, and PuO22+ ions is 6.65, 0.17, 9.51, and 5.70, respectively, showing a general tendency that the larger the charge of the metal center, the lower the pKa value of the first hydrolysis reaction. Hf4+, Zr4+, and Pu4+ have relatively smaller IOD values among the tetravalent ions, where they all have experimental CNs of ∼8.71 Ce4+ was determined to have an experimental CN of ∼9,68 while U4+ and Th4+ have CNs between 9 and 11.71 Soderholm et al. proposed that counterions also play a key role in the first solvation shell structure, while the 9-, 10-, or 11-coordinated Th4+ have very small energy differences and are in a dynamic equilibration.90 The simulated HFE and IOD values of the 12-6-4 parameter set are in excellent agreement with the experiment. The simulated CN values of most of the M(IV) ions are ∼10, with Hf4+ having a CN ∼8 for the TIP4PEW and SPC/E water models. Herein, the TIP3P model always predicted a larger CN value than the other two water models, which may be because it has a smaller C12 term (∼582.0 × 105 kcal·Å12/mol) than that of the SPC/E (∼629.4 × 105 kcal·Å12/mol) and TIP4PEW (∼656.1 × 105 kcal·Å12/mol) models.
3. Redox Ion Pairs
Later, we analyze several redox pairs to explore the consistency of the C4 parameters we determined with respect to the behavior of these pairs in aqueous solution. We also calculated the relative HFE between each redox pair for the TIP3P water model (see below). The divalent metal ions’ 12-6-4 parameters are from previous work.18 A nine-windows TI simulation (50 ps of equilibration and 150 ps of sampling for each window) was performed forward and backward, respectively, to obtain the final results. The results further validate the method employed in the present work. For example, the simulated relative HFEs of Fe2+/Fe3+, Cr2+/Cr3+, and Ce3+/Ce4+ ion pairs were 580.2, 516.9, and 698.2 kcal/mol, while the experimental values are 579.6, 516.2, and 697.6 kcal/mol, respectively.66
Fe2+/Fe3+ Ion Pairs
The experimental IOD values shrink ∼0.08 Å from 2.11 Å of Fe2+ to 2.03 Å of Fe3+ ion. Moin et al. investigated ferrous and ferric ions in water using the QMCF-MD method. They obtained a force constant kion-O of 193 N/m for Fe3+, which is almost twice as strong as that of Fe2+ (93 N/m), while the effective charges (from a Mulliken population analysis) for the Fe2+ ion are in the range of 1.25 to 1.45 (with an average of 1.36), and for the Fe3+ ion it is in the range of 1.70 to 1.95 (with an average of 1.85).47 Fe2+/Fe3+ redox pairs exist broadly in biologically related system such as the Fe–S proteins and heme structures.7,91 Moreover, Fe2+/Fe3+ redox pairs play fundamental roles in many electron-transfer processes. We have determined the 12-6-4 parameters for Fe2+ ion with three different water models in previous work.18 For the TIP3P water model, the final optimized parameters were Rmin/2 = 1.457 Å, ε = 0.02710805 kcal/mol, and C4 = 163 kcal/mol·Å4.18 From the 12-6-4 parameters determined for the Fe3+ ion herein, we find that the Rmin/2 decreases slightly as the outer shell electron number decreases, while the C4 term increases by ∼2.5 times relative to Fe2+. This is consistent with a ratio between the C4 terms for a trivalent and divalent ion of [3/2]2 = 2.25, which is derived from the original ion-induced dipole equation (eq 11 in a prior publication18).
Cr2+/Cr3+ Ion Pairs
Cr3+ forms a stable Cr(H2O)63+ complex in the aqueous phase. The Cr2+ and Cr3+ ions have [Ar]3d4 and [Ar]3d5 electronic structures, respectively, where the Cr(H2O)62+ complex has a strong Jahn–Teller effect while the Cr(H2O)63+ molecule has a standard octahedral configuration. The IOD values decreases from 2.08 to 1.96 Å for the Cr2+ and Cr3+ ions. Using the TIP3P water model as a representative example, we observe that the Rmin/2 parameter decreases from 1.431 to1.405 Å, while the C4 term increases from ∼137 to ∼258 kcal/mol·Å4 for the Cr2+ and Cr3+ ions (the Cr2+ parameters are reported in reference (18)).
Ce3+/Ce4+ Ion Pairs
Cerium has both +3 and +4 oxidation states. Ce4+ is the most stable state because it shares the same electronic configuration with Xe. Just like the two oxidation pairs previously discussed we find that the Rmin/2 value decreases while the C4 term increases significantly with increasing charge. For example, for the parameters determined for the TIP3P water model, the Rmin/2 decreased ∼0.03 Å, while the C4 term increased by ∼480 kcal/mol·Å4.
Validation on a Protein System
PDB entry 4BV1 was used to obtain the starting coordinates for this modeling exercise. It is a superoxide reductase (SOD) found in Nanoarchaeum equitans. It is a protein tetramer with each monomer having a metal site containing an Fe3+ ion. The structure has been determined by using X-ray crystallography to a resolution of 1.90 Å. The tetramer structure is shown in Figure 3, while Chain C with its metal site is shown in Figure 4. The metal site contains four histidine groups, one cysteine group, and one water molecule. By treating Chain C as the initial structure, we performed three simulations with different parameter sets (the HFE, IOD, and 12-6-4 parameter sets). For the 12-6-4 parameter set, the C4 terms between the Fe3+ ion and atom types other than water oxygen were evaluated using eq 11. The TIP3P water model was employed during the simulations. Details of the simulation procedures and the polarizability of each atom type are given in the SI.
| 11 |
Figure 3.

PDB entry 4BV1. Water molecules are not shown in the Figure, the ferric ion is shown as a sliver sphere. This picture was created by VMD.92
Figure 4.
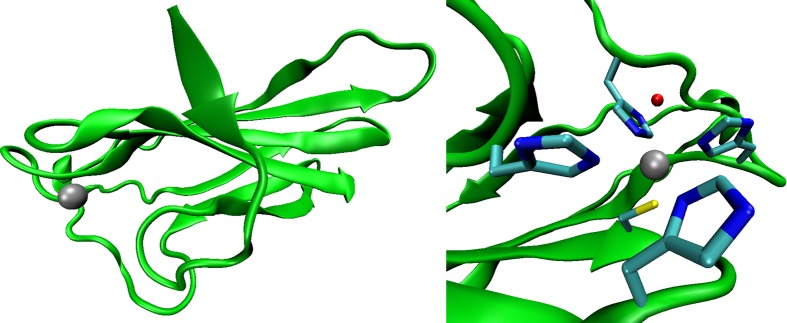
Chain C in PDB entry 4BV1 (left) and a close up of the metal site in Chain C (right). The ferric ion is represented as a silver sphere, and it is coordinated by one Cys, four His, and one water molecule. The figures were made using VMD.92
A total of 10 ns of sampling was performed during the simulation, and snapshots were stored after each 500 fs. The HFE parameter set prefers a smaller CN (of 4), and the metal ion moves out from the binding pocket, while stable metal complex structures were obtained for the simulations using the IOD and 12-6-4 parameter sets. An RMSD analysis was performed over the heavy atoms of the backbone and the metal site for the simulations by treating the initial structure (experimental structure) as reference. The results are depicted in Figure 5. The RMSD of the heavy backbone atoms fluctuated around ∼1.2 Å, while the RMSD of the metal site was ∼0.5 Å. These values illustrate that the metal binding site is stable during the course of the simulations.
Figure 5.
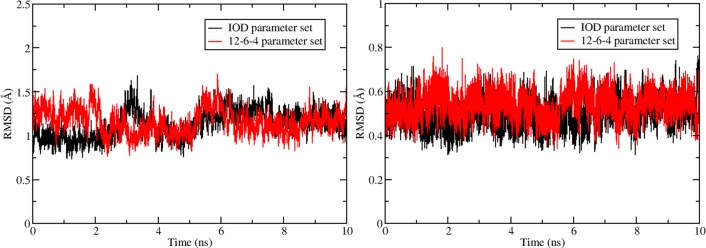
RMSD of heavy atoms of backbone (left) and the metal site (right, including the binding water molecule) for simulations with IOD and 12-6-4 parameter sets using the initial structure (experimental structure) as reference.
We have also performed an RMSF analysis of the backbone heavy atoms for each residue together with the oxygen atom in the metal site binding water. The results are shown in Figure 6. From this Figure it can be seen that the metal site residues: residue His10 (residue number 11), His 35 (residue number 36), His 41 (residue number 42), Cys 97 (residue number 98), and His 100 (residue number 101), all have relatively small RMSF values (∼0.5 Å). The metal site binding water (residue number 115, is not shown in the Figure since the protein ends at residue number 112) has a RMSF of ∼0.6 Å for both the simulations with the IOD and the 12-6-4 parameter sets. These results further validated that the metal ion site is stable over the course of the simulations.
Figure 6.
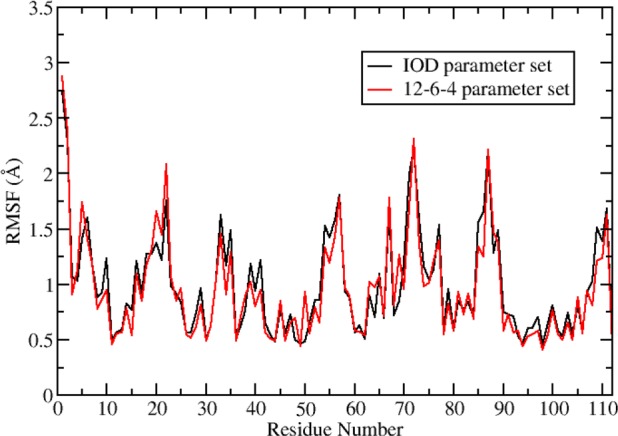
RMSF of heavy atoms of the protein residues in the simulations using IOD and 12-6-4 parameter sets.
Conclusions
In this work, we have estimated the 12-6 LJ parameters and determined the 12-6-4 LJ-type parameters for 24 highly charged metal ions (18 M(III) ions and 6 M(IV) ions) with three water models (TIP3P, SPC/E and TIP4PEW) based on a parameter scanning protocol. We have shown that with the increasing charge of the metal ions there is a notable decrease in the accuracy of the 12-6 LJ nonbonded model. Using TIP3P as an example, the average underestimation of the HFE values increases from ∼50 kcal/mol for M(II) ions to ∼80 kcal/mol for M(III) ions and to ∼240 kcal/mol for M(IV) ions when trying to reproduce the experimental IOD values. The average underestimation of the IOD values increases from −0.27 to −0.29 Å and −0.58 Å for the M(II), M(III), and M(IV) ions, respectively, when trying to reproduce the experimental HFE values.
The 12-6-4 LJ-type nonbonded model, which we previously described, addresses this problem in a consistent manner. It improves the accuracy of the 12-6 LJ nonbonded model remarkably with just a slight increase in computational cost. This parameter set, derived in this work, reproduced several experimental values (HFE, IOD, and CN) with good accuracy. They reproduce the HFE within ±1 kcal/mol for the M(III) ions and ±2 kcal/mol for M(IV) ions while reproducing the experimental IOD values to within ±0.01 Å. Moreover, excellent quantitative and qualitative agreement with previous experimental and computational work supports the validity of the 12-6-4 LJ-type nonbonded model. Testing in a protein system also revealed good transferability of the parameters determined herein.
Acknowledgments
We thank Dr. David Case (Rutgers) and Dr. Jason Swails (Rutgers) for help with programming the 12-6-4 model in AMBER and Shuai Wang (UF) for helping us write analysis code. We acknowledge the financial support from the United States National Institutes of Health (RO1’s GM044974 and GM066859), and computing support from the High Performance Computing Center of the University of Florida and High Performance Computing Center of Michigan State University.
Glossary
Abbreviations
- VDW
van der Waals
- LJ
Lennard-Jones
- PME
particle mesh Ewald
- HFE
hydration free energy
- TI
thermodynamic integration
- RDF
radial distribution function
- IOD
ion–oxygen distance
- CN
coordination number
Supporting Information Available
Simulated HFE, IOD, and CN values obtained using parameter scanning and the final determined 12-6-4 parameters; estimated percentage errors of the 12−6 HFE and IOD parameter sets; and procedures used in the protein simulations may be accessed in file (jp505875v_si_003.pdf). Uncertainty of each simulated HFE value may be viewed in files (jp505875v_si_001.xlsx and jp505875v_si_002.xlsx). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Steed J. W.; Atwood J. L.. Supramolecular Chemistry; John Wiley & Sons: Hoboken, NJ, 2009. [Google Scholar]
- Elbanowski M.; Ma̧kowska B. The Lanthanides as Luminescent Probes in Investigations of Biochemical Systems. J. Photochem. Photobiol., A 1996, 99285–92. [Google Scholar]
- Gschneidner K. A. Jr.; Eyring L.; Lander G. H.. Handbook on the Physics and Chemistry of Rare Earths; Elsevier: New York, 2001; Vol. 32. [Google Scholar]
- Pyle A. Metal Ions in the Structure and Function of RNA. J. Biol. Inorg. Chem. 2002, 77–8679–690. [DOI] [PubMed] [Google Scholar]
- Holm R. H.; Kennepohl P.; Solomon E. I. Structural and Functional Aspects of Metal Sites in Biology. Chem. Rev. 1996, 9672239–2314. [DOI] [PubMed] [Google Scholar]
- Stohs S.; Bagchi D. Oxidative Mechanisms in the Toxicity of Metal Ions. Free Radical Biol. Med. 1995, 182321–336. [DOI] [PubMed] [Google Scholar]
- Stephens P.; Jollie D.; Warshel A. Protein Control of Redox Potentials of Iron-sulfur Proteins. Chem. Rev. 1996, 9672491–2514. [DOI] [PubMed] [Google Scholar]
- Maton A.Human Biology and Health; Prentice Hall: Needham, MA, 1997. [Google Scholar]
- Bünzli J.-C. G. Benefiting from the Unique Properties of Lanthanide Ions. Acc. Chem. Res. 2006, 39153–61. [DOI] [PubMed] [Google Scholar]
- Miller J.Nuclear and Radiochemistry; John Wiley & Sons: Weinheim, Germany, 1981. [Google Scholar]
- Dudev T.; Lim C. Principles Governing Mg, Ca, and Zn Binding and Selectivity in Proteins. Chem. Rev. 2003, 1033773–788. [DOI] [PubMed] [Google Scholar]
- Dudev T.; Lim C. Competition Among Metal Ions for Protein Binding Sites: Determinants of Metal Ion Selectivity in Proteins. Chem. Rev. 2014, 1141538–556. [DOI] [PubMed] [Google Scholar]
- Åqvist J. Ion-water interaction Potentials Derived from Free Energy Perturbation Simulations. J. Phys. Chem. 1990, 94218021–8024. [Google Scholar]
- Li P.; Roberts B. P.; Chakravorty D. K.; Merz K. M. Rational Design of Particle Mesh Ewald Compatible Lennard-Jones Parameters for +2 Metal Cations in Explicit Solvent. J. Chem. Theory Comput. 2013, 9, 2733–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merz K. M. Carbon Dioxide Binding to Human Carbonic Anhydrase II. J. Am. Chem. Soc. 1991, 1132406–411. [Google Scholar]
- Stote R. H.; Karplus M. Zinc Binding in Proteins and Solution: A Simple but Accurate nonbonded Representation. Proteins: Struct., Funct., Bioinf. 1995, 23112–31. [DOI] [PubMed] [Google Scholar]
- Joung I. S.; Cheatham T. E. III. Molecular Dynamics Simulations of the Dynamic and Energetic Properties of Alkali and Halide Ions using Water-model-specific Ion Parameters. J. Phys. Chem. B 2009, 1134013279–13290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P.; Merz K. M. Taking into Account the Ion-induced Dipole Interaction in the Nonbonded Model of Ions. J. Chem. Theory Comput. 2014, 101289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters M. B.; Yang Y.; Wang B.; Füsti-Molnár L. s.; Weaver M. N.; Merz K. M. Structural Survey of Zinc-Containing Proteins and Development of the Zinc AMBER Force Field (ZAFF). J. Chem. Theory Comput. 2010, 692935–2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravorty D. K.; Wang B.; Lee C. W.; Giedroc D. P.; Merz K. M. Simulations of Allosteric Motions in the Zinc Sensor CzrA. J. Am. Chem. Soc. 2011, 13473367–3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ucisik M. N.; Chakravorty D. K.; Merz K. M. Jr. Structure and Dynamics of the N-Terminal Domain of the Cu (I) Binding Protein CusB. Biochemistry 2013, 52396911–6923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts B. P.; Miller B. R. III; Roitberg A. E.; Merz K. M. Jr. Wide-open flaps are Key to Urease Activity. J. Am. Chem. Soc. 2012, 134249934–9937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minkara M. S.; Ucisik M. N.; Weaver M. N.; Merz K. M. Jr. Molecular Dynamics Study of Helicobacter pylori Urease. J. Chem. Theory Comput. 2014, 1051852–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravorty D.; Wang B.; Lee C.; Guerra A.; Giedroc D.; Merz K. Jr. Solution NMR Refinement of a Metal Ion Bound Protein using Metal Ion Inclusive Restrained Molecular Dynamics Methods. J. Biomol. NMR 2013, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang Y.-P. Successful Molecular Dynamics Simulation of Two Zinc Complexes Bridged by a Hydroxide in Phosphotriesterase using the Cationic Dummy Atom Method. Proteins: Struct., Funct., Bioinf. 2001, 453183–189. [DOI] [PubMed] [Google Scholar]
- Duarte F.; Bauer P.; Barrozo A.; Amrein B. A.; Purg M.; Aqvist J.; Kamerlin S. C. L. Force-Field Independent Metal Parameters Using a Non-Bonded Dummy Model. J. Phys. Chem. B 2014, 118164351–4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena A.; Sept D. Multisite Ion Models that Improve Coordination and Free Energy Calculations in Molecular Dynamics Simulations. J. Chem. Theory Comput. 2013, 983538–3542. [DOI] [PubMed] [Google Scholar]
- Chakravorty D. K.; Wang B.; Ucisik M. N.; Merz K. M. Insight into the Cation−π Interaction at the Metal Binding Site of the Copper Metallochaperone CusF. J. Am. Chem. Soc. 2011, 1334819330–19333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R.; Hu P.; Wang S.; Cao Z.; Zhang Y. Flexibility of Catalytic Zinc Coordination in Thermolysin and HDAC8: a Born–Oppenheimer ab initio QM/MM Molecular Dynamics Study. J. Chem. Theory Comput. 2009, 61337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer T. S.; Weiss A. K.; Randolf B. R.; Rode B. M. Hydration of Highly Charged Ions. Chem. Phys. Lett. 2011, 5124139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Y.; Kollman P. A. Pathways to a Protein Folding Intermediate Observed in a 1-microsecond Simulation in Aqueous Solution. Science 1998, 2825389740–744. [DOI] [PubMed] [Google Scholar]
- Shaw D. E.; Maragakis P.; Lindorff-Larsen K.; Piana S.; Dror R. O.; Eastwood M. P.; Bank J. A.; Jumper J. M.; Salmon J. K.; Shan Y. Atomic-level Characterization of the Structural Dynamics of Proteins. Science 2010, 3306002341–346. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Ravimohan C. Monte Carlo Simulation of Differences in free Energies of Hydration. J. Chem. Phys. 1985, 8363050–3054. [Google Scholar]
- Jorgensen W. L.; Duffy E. M. Prediction of Drug Solubility from Monte Carlo Simulations. Bioorg. Med. Chem. Lett. 2000, 10111155–1158. [DOI] [PubMed] [Google Scholar]
- Wu J. C.; Piquemal J.-P.; Chaudret R.; Reinhardt P.; Ren P. Polarizable Molecular Dynamics Simulation of Zn (II) in Water using the AMOEBA Force Field. J. Chem. Theory Comput. 2010, 672059–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Yang W.; Piquemal J.-P.; Ren P. Modeling Structural Coordination and Ligand Binding in Zinc Proteins with a Polarizable Potential. J. Chem. Theory Comput. 2012, 841314–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H.; Whitfield T. W.; Harder E.; Lamoureux G.; Vorobyov I.; Anisimov V. M.; MacKerell A. D.; Roux B. t. Simulating Monovalent and Divalent Ions in Aqueous Solution Using a Drude Polarizable Force Field. J. Chem. Theory Comput. 2010, 63774–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakharov D. V.; Lim C. Zn Protein Simulations Including Charge Transfer and Local Polarization Effects. J. Am. Chem. Soc. 2005, 127134921–4929. [DOI] [PubMed] [Google Scholar]
- Sakharov D. V.; Lim C. Force Fields Including Charge Transfer and Local Polarization Effects: Application to Proteins Containing Multi/heavy Metal Ions. J. Comput. Chem. 2009, 302191–202. [DOI] [PubMed] [Google Scholar]
- Smith D. E.; Dang L. X. Computer Simulations of NaCl Association in Polarizable Water. J. Chem. Phys. 1994, 10053757–3766. [Google Scholar]
- Dang L. X. Development of Nonadditive Intermolecular Potentials using Molecular Dynamics: Solvation of Li+ and F– ions in Polarizable Water. J. Chem. Phys. 1992, 9696970–6977. [Google Scholar]
- Jorgensen W. L. Quantum and Statistical Mechanical Studies of Liquids. 10. Transferable Intermolecular Potential functions for Water, Alcohols, and Ethers. Application to Liquid Water. J. Am. Chem. Soc. 1981, 1032335–340. [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 792926–935. [Google Scholar]
- Berendsen H. J. C.; Grigera J. R.; Straatsma T. P. The Missing Term in Effective Pair Potentials. J. Phys. Chem. 1987, 91246269–6271. [Google Scholar]
- Horn H. W.; Swope W. C.; Pitera J. W.; Madura J. D.; Dick T. J.; Hura G. L.; Head-Gordon T. Development of an Improved Four-site Water Model for Biomolecular Simulations: TIP4P-Ew. J. Chem. Phys. 2004, 120209665–9678. [DOI] [PubMed] [Google Scholar]
- Canaval L. R.; Sakwarathorn T.; Rode B. M.; Messner C. B.; Lutz O. M.; Bonn G. n. K. Erbium (III) in Aqueous Solution: An Ab Initio Molecular Dynamics Study. J. Phys. Chem. B 2013, 1174815151–15156. [DOI] [PubMed] [Google Scholar]
- Moin S. T.; Hofer T. S.; Pribil A. B.; Randolf B. R.; Rode B. M. A Quantum Mechanical Charge Field Molecular Dynamics Study of Fe2+ and Fe3+ ions in Aqueous Solutions. Inorg. Chem. 2010, 49115101–5106. [DOI] [PubMed] [Google Scholar]
- Canaval L. R.; Weiss A. K.; Rode B. M. Structure and Dynamics of the Th4+-ion in Aqueous Solution–An ab initio QMCF-MD study. Comput. Theor. Chem. 2013, 1022, 94–102. [Google Scholar]
- Case D. A.; Cheatham T. E.; Darden T.; Gohlke H.; Luo R.; Merz K. M.; Onufriev A.; Simmerling C.; Wang B.; Woods R. J. The Amber Biomolecular Simulation Programs. J. Comput. Chem. 2005, 26161668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornell W. D.; Cieplak P.; Bayly C. I.; Gould I. R.; Merz K. M.; Ferguson D. M.; Spellmeyer D. C.; Fox T.; Caldwell J. W.; Kollman P. A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117195179–5197. [Google Scholar]
- MacKerell A. D.; Bashford D.; Bellott M.; Dunbrack R.; Evanseck J.; Field M. J.; Fischer S.; Gao J.; Guo H.; Ha S. a. All-atom Empirical Potential for Molecular Modeling and DynamicsStudies of Proteins. J. Phys. Chem. B 1998, 102183586–3616. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Maxwell D. S.; Tirado-Rives J. Development and Testing of the OPLS all-atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 1184511225–11236. [Google Scholar]
- Joung I. S.; Cheatham T. E. Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations. J. Phys. Chem. B 2008, 112309020–9041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezei M. The Finite Difference Thermodynamic Integration, Tested on Calculating the Hydration free Energy Difference between Acetone and Dimethylamine in Water. J. Chem. Phys. 1987, 86127084–7088. [Google Scholar]
- Mitchell M. J.; McCammon J. A. Free Energy Difference Calculations by Thermodynamic Integration: Difficulties in Obtaining a Precise Value. J. Comput. Chem. 1991, 122271–275. [Google Scholar]
- Kollman P. Free Energy Calculations: Applications to Chemical and Biochemical Phenomena. Chem. Rev. 1993, 9372395–2417. [Google Scholar]
- Simonson T.; Carlsson J.; Case D. A. Proton Binding to Proteins: pKa Calculations with Explicit and Implicit Solvent Models. J. Am. Chem. Soc. 2004, 126134167–4180. [DOI] [PubMed] [Google Scholar]
- Steinbrecher T.; Mobley D. L.; Case D. A. Nonlinear Scaling Schemes for Lennard-Jones Interactions in Free Energy Calculations. J. Chem. Phys. 2007, 12721214108–13. [DOI] [PubMed] [Google Scholar]
- Hummer G.; Szabo A. Calculation of Free-energy Differences from Computer Simulations of Initial and Final States. J. Chem. Phys. 1996, 10552004–2010. [Google Scholar]
- Shirts M. R.; Pitera J. W.; Swope W. C.; Pande V. S. Extremely Precise free Energy Calculations of Amino Acid Side Chain Analogs: Comparison of Common Molecular Mechanics Force Fields for Proteins. J. Chem. Phys. 2003, 119115740–5761. [Google Scholar]
- Darden T.; York D.; Pedersen L. Particle mesh Ewald: An N [center-dot] log(N) method for Ewald sums in Large Systems. J. Chem. Phys. 1993, 981210089–10092. [Google Scholar]
- Cheatham T. E. III; Miller J. L.; Fox T.; Darden T. A.; Kollman P. A. Molecular Dynamics Simulations on Solvated Biomolecular Systems: The Particle Mesh Ewald Method Leads to Stable Trajectories of DNA, RNA, and Proteins. J. Am. Chem. Soc. 1995, 117144193–4194. [Google Scholar]
- Petersen H. G. Accuracy and Efficiency of the Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 3668. [Google Scholar]
- Ryckaert J.-P.; Ciccotti G.; Berendsen H. J. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-alkanes. J. Comput. Phys. 1977, 233327–341. [Google Scholar]
- Miyamoto S.; Kollman P. A. SETTLE: an Analytical Version of the SHAKE and RATTLE algorithm for Rigid Water Models. J. Comput. Chem. 1992, 138952–962. [Google Scholar]
- Marcus Y. Thermodynamics of Solvation of Ions. Part 5.-Gibbs Free Energy of Hydration at 298.15 K. J. Chem. Soc., Faraday Trans. 1991, 87182995–2999. [Google Scholar]
- Marcus Y. Ionic Radii in Aqueous Solutions. Chem. Rev. 1988, 8881475–1498. [Google Scholar]
- Sham T. Electronic Structure of Hydrated Ce4+ ions in Solution: An X-ray-Absorption Study. Phys. Rev. B 1989, 4096045. [DOI] [PubMed] [Google Scholar]
- Ankudinov A.; Conradson S.; de Leon J. M.; Rehr J. Relativistic XANES Calculations of Pu Hydrates. Phys. Rev. B 1998, 57137518. [Google Scholar]
- Moll H.; Denecke M.; Jalilehvand F.; Sandström M.; Grenthe I. Structure of the aqua ions and Fluoride Complexes of uranium (IV) and thorium (IV) in Aqueous Solution an EXAFS study. Inorg. Chem. 1999, 3881795–1799. [DOI] [PubMed] [Google Scholar]
- Hagfeldt C.; Kessler V.; Persson I. Structure of the Hydrated, Hydrolysed and Solvated zirconium (IV) and hafnium (IV) ions in Water and Aprotic Oxygen Donor Solvents. A Crystallographic, EXAFS Spectroscopic and large angle X-ray Scattering Study. Dalton Trans. 2004, 14, 2142–2151. [DOI] [PubMed] [Google Scholar]
- Shannon R. Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr. 1976, 325751–767. [Google Scholar]
- Stokes R. The van der Waals radii of Gaseous Ions of the Noble Gas Structure in Relation to Hydration Energies. J. Am. Chem. Soc. 1964, 866979–982. [Google Scholar]
- Kuznetsov A.; Zueva E.; Masliy A.; Krishtalik L. Redox Potential of the Rieske iron-sulfur protein quantum-chemical and Electrostatic Study. Biochim. Biophys. Acta 2010, 17973347–359. [DOI] [PubMed] [Google Scholar]
- Beattie J. K.; Best S. P.; Skelton B. W.; White A. H. Structural Studies on the Caesium alums, CsMIII [SO4] 2· 12H2O. J. Chem. Soc., Dalton Trans. 1981, 10, 2105–2111. [Google Scholar]
- Bard A. J.; Parsons R.; Jordan J.. Standard Potentials in Aqueous Solution; CRC Press: Boca Raton, FL, 1985; Vol. 6. [Google Scholar]
- Hofer T. S.; Randolf B. R.; Rode B. M. Al (III) Hydration Revisited. an ab initio Quantum Mechanical Charge Field Molecular Dynamics Study. J. Phys. Chem. B 2008, 1123711726–11733. [DOI] [PubMed] [Google Scholar]
- Jalilehvand F.; Spångberg D.; Lindqvist-Reis P.; Hermansson K.; Persson I.; Sandström M. Hydration of the Calcium Ion. An EXAFS, Large-Angle X-ray Scattering, and Molecular Dynamics Simulation Study. J. Am. Chem. Soc. 2000, 1233431–441. [DOI] [PubMed] [Google Scholar]
- Martin B.; Richardson F. S. Lanthanides as Probes for Calcium in Biological Systems. Q. Rev. Biophys. 1979, 1202181–209. [DOI] [PubMed] [Google Scholar]
- Cotton S.Lanthanide and Actinide Chemistry; John Wiley & Sons: Hoboken, NJ, 2007; Vol. 27. [Google Scholar]
- Habenschuss A.; Spedding F. H. The Coordination (hydration) of Rare Earth Ions in Aqueous Chloride Solutions from X ray Diffraction. I. TbCl3,DyCl3, ErCl3,TmCl3,and LuCl3. J. Chem. Phys. 1979, 7062797–2806. [Google Scholar]
- Habenschuss A.; Spedding F. H. The Coordination (hydration) of Rare Earth Ions in Aqueous Chloride Solutions from X-ray Diffraction. III. SmCl3, EuCl3, and Series Behavior. J. Chem. Phys. 1980, 731442–450. [Google Scholar]
- Habenschuss A.; Spedding F. H. The Coordination (hydration) of Rare Earth Ions in Aqueous Chloride Solutions from x-ray Diffraction. II. LaCl3, PrCl3, and NdCl3a). J. Chem. Phys. 1979, 7083758–3763. [Google Scholar]
- Yamaguchi T.; Nomura M.; Wakita H.; Ohtaki H. An extended x-ray Absorption fine Structure Study of Aqueous Rare Earth Perchlorate Solutions in Liquid and Glassy States. J. Chem. Phys. 1988, 8985153–5159. [Google Scholar]
- Kanno H.; Hiraishi J. Raman Spectroscopic Evidence for a Discrete change in Coordination Number of Rare Earth aquo-ions in the Middle of the Series. Chem. Phys. Lett. 1980, 753553–556. [Google Scholar]
- D’Angelo P.; Zitolo A.; Migliorati V.; Chillemi G.; Duvail M.; Vitorge P.; Abadie S.; Spezia R. Revised ionic radii of lanthanoid (III) ions in Aqueous Solution. Inorg. Chem. 2011, 50104572–4579. [DOI] [PubMed] [Google Scholar]
- D’Angelo P.; Zitolo A.; Migliorati V.; Persson I. Analysis of the Detailed Configuration of Hydrated Lanthanoid (III) Ions in Aqueous Solution and Crystalline Salts by Using K-and L3-Edge XANES Spectroscopy. Chem.—Eur. J. 2010, 162684–692. [DOI] [PubMed] [Google Scholar]
- Taylor D. M. The Bioinorganic Chemistry of Actinides in Blood. J. Alloys Compd. 1998, 271, 6–10. [Google Scholar]
- Odoh S. O.; Bylaska E. J.; de Jong W. A. Coordination and Hydrolysis of Plutonium Ions in Aqueous Solution Using Car–Parrinello Molecular Dynamics Free Energy Simulations. J. Phys. Chem. A 2013, 1174712256–12267. [DOI] [PubMed] [Google Scholar]
- Wilson R. E.; Skanthakumar S.; Burns P. C.; Soderholm L. Structure of the homoleptic thorium (IV) aqua ion [Th (H2O) 10] Br4. Angew. Chem. 2007, 119428189–8191. [DOI] [PubMed] [Google Scholar]
- Sono M.; Roach M. P.; Coulter E. D.; Dawson J. H. Heme-containing oxygenases. Chem. Rev. 1996, 9672841–2888. [DOI] [PubMed] [Google Scholar]
- Kirm M.; Krupa J.; Makhov V.; Negodin E.; Zimmerer G.; Gesland J. 6d5f and 5f2 configurations of U4+ doped into LiYF4 and YF3 crystals. J. Lumin. 2003, 104185–92. [Google Scholar]
- Frick R. J.; Pribil A. B.; Hofer T. S.; Randolf B. R.; Bhattacharjee A.; Rode B. M. Structure and Dynamics of the U4+ Ion in Aqueous Solution: An ab Initio Quantum Mechanical Charge Field Molecular Dynamics Study. Inorg. Chem. 2009, 4893993–4002. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: visual molecular dynamics. J. Mol. Graphics 1996, 14133–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.