

Abstract
Fluorescence is essential for dynamic live cell imaging, and affinity reagents are required for quantification of endogenous proteins. Various fluorescent dyes can report on different aspects of biological trafficking, but must be independently conjugated to affinity reagents and characterized for specific biological readouts. Here we present the characterization of a new modular platform for small anti-EGFR affinity probes for studying rapid changes in receptor pools. A protein domain (FAP dL5**) that binds to malachite-green (MG) derivatives for fluorescence activation was expressed as a recombinant fusion to one or two copies of the compact EGFR binding affibody ZEGFR:1907. This is a recombinant and fluorogenic labeling reagent for native EGFR molecules. In vitro fluorescence assays demonstrated that the binding of these dyes to the FAP–affibody fusions produced thousand-fold fluorescence enhancements, with high binding affinity and fast association rates. Flow cytometry assays and fluorescence microscopy demonstrated that these probes label endogenous EGFR on A431 cells without disruption of EGFR function, and low nanomolar surface Kd values were observed with the double-ZEGFR:1907 constructs. The application of light-harvesting fluorogens (dyedrons) significantly improved the detected fluorescence signal. Altering the order of addition of the ligand, probe, and dyes allowed differentiation between surface and endocytotic pools of receptors to reveal the rapid dynamics of endocytic trafficking. Therefore, FAP/affibody coupling provides a new approach to construct compact and modular affinity probes that label endogenous proteins on living cells and can be used for studying rapid changes in receptor pools involved in trafficking.
Introduction
Labeling native proteins of interest for detection of receptor dynamics has been a major barrier for live cell imaging studies. Researchers have developed various ways to deliver fluorescence tags to proteins of interest, typically by fusions with fluorescent proteins or activatable tags (reviewed1). For dynamic studies of endogenous surface proteins, affinity probes or ligands attached to fluorescent dyes have been widely used. Antibodies can be directly coupled to dyes and characterized in vitro prior to use in studies ranging from single-cell to whole organisms.2−4 Due to the large sizes and potential multivalency, however, antibodies may affect the dynamics of targeted molecules. In addition, not all primary antibodies can be robustly conjugated to small molecules without loss of affinity or specificity.5 To overcome these potential limitations, alternative scaffolds have been developed as affinity tools such as immunoglobin-derived scFvs, diabodies, and RNA-based aptamers.6,7 Affibodies are the smallest alternate scaffold, only 58 amino acids, and are derived from the B-domain of staphylococcal protein A.8,9 The scaffold backbone was stabilized and the 13 residues of the binding surface were randomly mutated to create an affibody library for screening by phage display.10 Affibodies against many targets are available with high specificity and stability.11 These small probes can be produced recombinantly or chemically synthesized, providing a variety of strategies for probe production. When available, affibodies are potential antibody replacements for cell-based and animal studies.9,12−16
Affibody ZEGFR:1907 is one of the most developed probes, specifically binding the human epidermal growth factor receptor (EGFR) from the ErbB family. This family is one of the most well studied groups of receptor tyrosine kinases: ErbB1 (EGFR), ErB2, ErbB3, and ErbB4.17 These receptors homo- or heterodimerize to activate and transduce numerous cell signaling pathways involved in normal cell growth, migration, and resistance to apoptosis; and overexpression has been implicated in the development of many cancers.18 The abnormal expression levels of ErbB proteins or the mutations in ErbB proteins generally lead to more aggressive cancers and are also correlated with poor clinical outcomes.19,20 Several studies have suggested that downstream signaling is regulated by the trafficking of these receptors. Detecting the endogenous levels and understanding the trafficking dynamics of ErbB receptors will provide for a better understanding of signal transduction in ErbB overexpressing cell lines.
Several affibodies directly labeled with fluorescent modules have been reported. Fluorophore conjugated affibodies have been used to label cell surface EGFR for optical imaging, and this direct conjugation of dye to affibody requires additional processing and purification.13,21,22 Fluorescent proteins have also been used to label affibody as a recombinant probe for ErbB receptor targeting.11 Both dye-labeled and fluorescent protein-labeled affibodies lead to the problem of high background when applied to cells, requiring additional washing steps for high-specificity cell labeling. pH sensitive (acid-activated) dyes linked to the affibody have been used to report on internalization of the affibody–dye conjugate bound to EGFR,23 and requiring distinct dye conjugates to be synthesized and validated.
Fluorogen activating proteins (FAPs) are an emerging class of fluorescence-based molecular tags that have been used in a variety of trafficking assays due to the rapid noncovalent association and activation of a fluorogenic dye by the expressed protein tag.24,25 One FAP can exhibit distinct properties when combined with various dye derivatives. FAPs are derived from single chain variable fragment antibodies (scFv) that specifically recognize and activate fluorogenic dyes with high binding affinity and provide modularity in targeted labeling without the need for direct conjugation of dyes. Fluorogens are organic dyes that have low fluorescence signal when free in solution and show significantly enhanced fluorescence output upon binding to the FAP.26 These dye–FAP fluoromodules show several advantages as fluorescent tags in cell biological studies. FAPs are small expressible protein modules that have typically been cloned as a fusion to proteins of interest. Recent work has demonstrated the use as recombinant affinity tags for secondary detection of fluorescein27 or biotin modified proteins.28 The fast association and activation of fluorogen/FAP complexes shortens the time for labeling protocols. Since the fluorescence is dependent on the association of fluorogen to FAP, the fluoromodules allow order-of-addition and compartment selectivity to achieve subpopulation labeling for internalized receptors. A number of modifications to fluorogens enrich the functional properties of a single FAP for optical labeling, such as membrane permeability/exclusion,26 fluorescence brightness,29 and environmental sensitivity.25,29 Thus, fusion of an affibody to a FAP should provide a compact and modular affinity probe to instantaneously label pretargeted endogenous protein with simple and reliable labeling protocols using a variety of distinct fluorogen dyes. Several FAPs have been reported to activate fluorescence of malachite green (MG) and thiazole orange (TO) derivatives, as well as dimethylindole red (DIR), oxazole-thiazole-blue (OTB), and various derivatives of these dyes, resulting in a range of fluoromodules with excitation/emission properties at any desired laser wavelength and emission range, typically with affinities for fluorogens in the low nanomolar to picomolar range.26,30−32 The dL5** FAP is relatively small (24.2 kDa), and binds to MG with a low picomolar equilibrium dissociation constant.33 The extremely low unbound fluorescence background and the high fluorescent signal upon binding allow high contrast and no-wash signal detection.
In this study, we present the functional validation of the FAP dL5**/ZEGFR:1907 fusion as a targeted probe that labels EGFR in cultured cells. The configuration of the fusion probes and the labeling scheme were optimized for detecting native receptor on the A431 cell surface in a no-wash assay format suitable for flow cytometry and fluorescence imaging. By applying different MG derivatives and order-of-addition experiments using this labeling protocol, we demonstrated enhanced brightness, receptor tracking, and surface selective labeling measurements. Modular labeling with distinct fluorogens allows for quantitative labeling of endocytosed and surface receptor pools. Compared to directly conjugated affibodies, these probes enable homogeneous assays where directly conjugated affibodies fail and provide a robust probe to assess changes in the localization of these native receptors.
Results
The results presented here demonstrate the functionality of an EGFR affibody affinity probe fused to a FAP that binds a far-red fluorogenic dye, MG, and various analogs of that fluorogen. The FAP and affibody retain their individual functions and allow for wash-free labeling of EGFR expressing cells for analysis by flow cytometry or fluorescent live-cell imaging.
Fusing the Affibody to FAP Preserves Fluorogen Activation Properties and EGFR Recognition
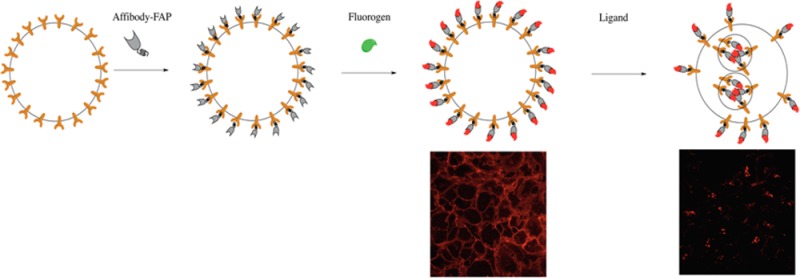
Fusion of proteins can sometimes perturb the function of the individual components. Especially with small proteins such as FAPs and affibodies, it is important to assess the function of each component of the fusion protein, ensuring preserved activity of both components, and to consider the impact of construct orientation (i.e., N-terminal FAP, C-terminal affibody or vice versa). We evaluated N and C-terminal fusions, as well as a single FAP flanked by two affibodies for their properties to bind and activate MG-fluorogens as well as their binding to EGFR on A431 cells.
The FAP was fused to the C-terminus (ZEGFR:1907-FAPdL5**, AF), to the N-terminus (FAPdL5**-ZEGFR:1907, FA) or between the dimeric affibody (ZEGFR:1907-FAPdL5**-ZEGFR:1907, AFA) (Scheme 1). The three fusion probes and the corresponding monomeric control proteins (ZEGFR:1907 as A and FAPdL5** as F) were cloned, expressed, and purified. In order to verify the functionality of the FAP with fusion to the affibody, the fluorescence properties of the probes were analyzed and compared with the control protein F. In the presence of MG-B-tau, a cell-excluded analog of the MG dye, the fluorescence scans of all three fusion probes AF, FA, and AFA featured a major and a minor excitation peak at 636 and 480 nm, and a single emission peak at 664 nm, which are consistent with the fluorescence spectra of F (Figure 1A). FA and AFA probes showed an enhanced fluorescence intensity compared to F and AF. Based on the measurement of fluorescence under equilibrium binding conditions, these three probes possessed subnanomolar dissociation constants, which were comparable to F alone (Figure 1B). A small, but significant decrease in association rate was observed in all FAP–affibody fusion proteins compared to F only. No detectable fluorescence activation and fluorogen binding by affibody alone was observed (Supporting Information Figure S1C). Hence, all fusion proteins preserve the fluorogen activating properties of unmodified FAP, and the affibody alone does not interfere with fluorogen activation.
Scheme 1. Representation of FAP/Affibody Fusion Constructs.

Figure 1.
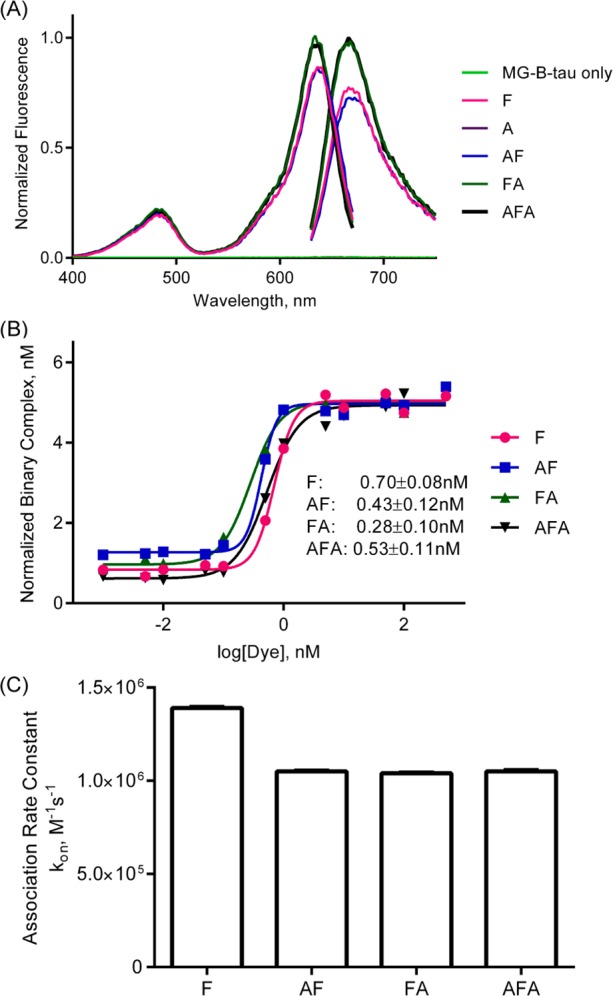
Characterization of recombinant probes binding to malachite green. (A) Fluorescence spectral properties of recombinant probes with malachite green. Excitation and emission scans of 1 μM MG-B-tau precomplexed with10 μM various probe constructs. (B) Fluorogen binding equilibrium analysis of recombinant probes. 5 nM samples of probes were assembled into fluorescence complexes as a function of MG-B-tau concentrations. The fluorescence intensity was corrected for fluorogen-only fluorescence background and then normalized to fluorescence of 1 nM FAP/MG complexes. (C) Analysis of association rate constants of recombinant probes. 4 nM samples of probes were mixed with 50 nM, 75 nM, 100 nM, 150 nM, or 200 nM of MG-B-tau, and the fluorescence intensity was monitored at 636/664 nm.
The labeling of cell surface EGFR by these fusion probes was tested with live-cell imaging. Three fusion probes AF, FA, and AFA all showed clear cell surface labeling, and control protein F failed to target to the cell surface (Figure 2D). By equilibrium binding fluorescence, the cell surface dissociation constant was estimated to be 122 ± 16 nM for AF, 101 ± 17 nM for FA, and 37 ± 6 nM for AFA (Figure 2A), which are slightly higher than the data from previous studies using fluorescent protein-conjugated ZEGFR:1907.11 The head-to-tail dimer configuration of affibody was previously reported to improve the affinity and avidity of EGFR binding.34 Probe AFA showed a 4-fold decrease in Kd values, which indicated that insertion of FAP had no obvious disruption on the function of the double affibody, and recapitulated the avidity-based binding enhancement previously reported. The AFA probe showed the lowest Kd, and therefore further experiments were performed with this version of the probe.
Figure 2.

Characterization of probes binding on A431 cell surface. (A) Dissociation constant analysis of probes on cell surface. 5 × 105/mL quantities of cells were incubated with probes for 1 h at 37 °C followed by 2 μM MG-B-tau for 5 min. Then cells were kept on ice for flow cytometry. The mean fluorescence intensity was corrected with background of cells incubating with F/MG and then normalized to mean fluorescence at 250 nM of probes. (B) Competition assay of nonfluorescent affibody A binding to the cell surface. Cells were labeled with 250 nM AFA or F and a serial dilution of A followed by 100 nM of MG-B-tau added prior to measurement. (C) Detection of receptor activation by Western blots. Starved cells were labeled with 250 nM probes followed by 100 nM of MG-B-tau and then cells were treated with 100 ng/mL EGF. Then cells were lysed for Western blot in order to detect phosphorylated EGFR and total EGFR. (D) Live-cell fluorescence microscopy of A431 cells labeled by various probes. Cells were labeled with 250 nM of probe and 100 nM of MG-B-tau prior to imaging or 100 nM of Cy5 conjugated affibody dimer. Scale bar 20 μm.
To validate that the conjugation of FAP preserves the EGFR binding specificity of affibody ZEGFR:1907, various concentrations of the control protein (A) were mixed with a fixed concentration of fusion probe (AFA) to label the cell surface EGFR of A431 cells. In these experiments, the nonfluorescent affibody protein (A) was used as a “cold” probe to compete with the FAP–affibody fusion protein for EGFR binding. Cell fluorescence was analyzed at equilibrium binding using a flow cytometry assay. The drop of fluorescence intensity was correlated to the increase of A concentration, verifying the specificity of EGFR binding by the AFA probe (Figure 2B). The titration with probe A revealed a nearly stoichiometric competition with the AFA protein, demonstrating that the specificity and affinity of the affibody are not compromised by fusion to the FAP. The binding of affibody–FAP fusion protein showed no interference with the EGF stimulation and no evidence of EGFR activation in the absence of EGF (Figure 2C).
Optimization of EGFR Labeling by FAP–Affibody Fusions
EGFR cell surface labeling was then optimized by varying labeling protocol and probe/dye concentrations. Three different labeling strategies for flow cytometry were explored. First, probes were precomplexed with excess fluorogen and the mixture was used to label cells. Second, probes were incubated with cells and then the unbound probes were washed off before adding fluorogen. The third approach is similar to the second one but without washing off unbound probes. All three approaches resulted in successful EGFR labeling based on fluorescence intensity from flow cytometry (data not shown). The first approach used the probe essentially as a fluorescent tag conjugated probe while the second approach resulted in lower fluorescence signal because of the relatively fast postwash dissociation of probes. Therefore, the third approach exhibited optimal labeling properties for rapid flow cytometric analysis. The same protocol was adopted for labeling cells during imaging. The labeling conditions were then optimized for maximal signal-to-background ratio with 250 nM of AFA and 100 nM of MG-B-tau. Under this condition, the probe could not be fully occupied by fluorogen, yet no further increase in signal at the cell surface was observed upon increased dye concentration, potentially implying a preference of fluorogen binding to EGFR-bound affibody. It is possible that the binding of two affibody domains to EGFR might position the two L5 domains in a complex-favoring state that improves the association of fluorogen.
In comparison with Cy5 conjugated dimeric affibody, the FAP–affibody labeling showed surprising signal contrast and allowed direct imaging of cell-surface proteins without any washing (Figure 2D). Due to the intrinsic fluorescence of fluorophores, the labeling of cells with the Cy5 conjugated affibody showed a higher background signal if unbound probes were not washed off from cells.
Modularity of Labeling by FAP/EGFR Affibody
The structure of the FAP allows binding to a variety of modified fluorogens that are compatible with distinct biologically relevant responses. Based on the structural analysis, FAPdL5** binds to the thiphenylmethane core of the fluorogen, which allows the modification on the diethylene glycol linker without disruption of the FAP/fluorogen interaction.29,33 Several derivatives of MG have been reported with various membrane permeability and molecular brightness (Figure 3A).29 To demonstrate the capacity of labeling EGFR with different fluorogens, A431 cells were first labeled with AFA, and then different MG derivatives were added for fluorescence activation. The modifications on the linker showed no disruptive effects on the fluorescence intensity of labeled cells analyzed by both flow cytometry and live-cell imaging (Figure 3B and C). Hexa-Cy3-MG (HCM) is a new light-harvesting dyedron derivative of MG prepared by coupling six Cy3 molecules to one MG using azide–alkyne cycloaddition chemistry to a highly decorated lysine linker (Synthetic and characterization details in Supporting Information), enabling the energy transfer from multiple Cy3 moieties to significantly enhance the molecular brightness of the FAP/fluorogen complex (Supporting Information Figure S1A), while maintaining a fluorogenic activation ratio of ∼330-fold at 562 nm excitation. Labeling of EGFR on A431 cells by HCM magnified the fluorescence signal by about 6-fold comparing the Cy3 excitation channel to the MG excitation channel (Figure 3B and C), which strengthened the sensitivity of the EGFR detection. Despite the slightly elevated background signal in the Cy3 excitation channel, the signal-to-background ratio of cells labeled by AFA/HCM (about 6-fold) showed an improvement over cells labeled by other MG derivatives (about 5-fold) in the no-wash labeling format. These results showed the possibility of simply changing fluorogens for various experimental purposes.
Figure 3.

Modular capacity of probes for labeling EGFR on the cell surface. (A) Structures of the fluorogens used. The synthetic and analytical details were shown in Supporting Information or described previously.26 A431 cell labeled with FAP–affibody fusions and various malachite green derivatives were analyzed by flow cytometry (B) and live-cell fluorescence microscopy (C). 5 × 105/mL of cells were labeled with 250 nM of AFA or F followed by incubation with 100 nM of fluorogens for 5 min. Cells were then either analyzed by flow cytometry for mean fluorescence measurement or cell imaging. Scale bar 20 μm.
MG-B-tau is a fluorogen derivative excluded from the cell by the plasma membrane, enabling the tracking of receptor endocytosis from initially surface-bound AFA (Figure 4A). Cell surface EGFR was first labeled with probe/MG-B-tau and showed an expected cell surface pattern. After EGF stimulation, probe/MG-B-tau complex was carried along with EGFR undergoing endocytosis, which resulted in a loss of cell surface signal and an increase of punctate endocytic structures (Figure 4A and Supporting Information Movie S2). Altering the order of addition, when probe labeled cells were first stimulated by EGF and then subsequently stained with MG-B-tau, only cell surface signal was observed, indicating the residual cell-surface receptors not undergoing endocytosis (Figure 4B). A related decrease in fluorescent labeling was quantified by flow cytometry (Figure 4C), resulting in a significant (p < 0.0001) ∼50% reduction in probe-labeled AFA at the cell surface, when ligand addition preceded dye addition. By altering the order of addition, such probes can be used to compare trafficking changes of endogenous receptors in living cells due to rapid dye activation, in contrast to the typical differential permeabilization employed for antibody staining requiring long incubations.35,36
Figure 4.
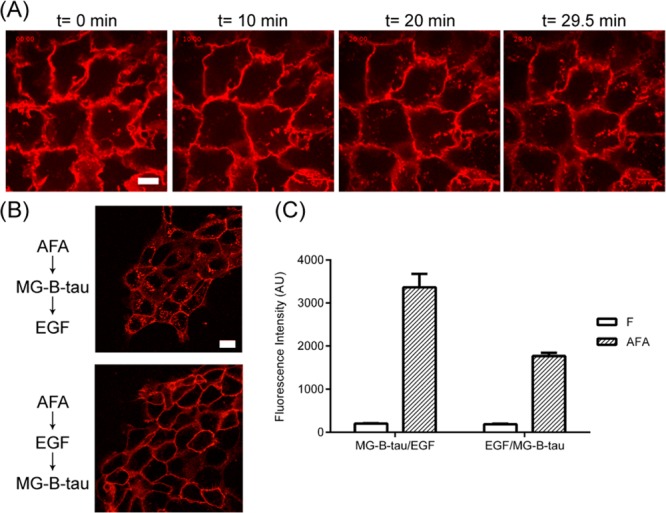
EGFR endocytosis tracking and subpopulation quantification using cell impermeable fluorogen (MG-B-tau). (A) Overnight starved cells were incubated with 250 nM of AFA followed by 100 nM MG-B-tau. Labeled cells were stimulated with 10 ng/mL EGF. The cell fluorescence was monitored over 30 min. (B) All cells are starved overnight and first labeled with AFA. The first group of cells were labeled with MG-B-tau and then stimulated with 100 ng/mL EGF for 15 min. The second group of cells were labeled with MG-B-tau after 15 min of EGF stimulation. Both groups of cells were analyzed by flow cytometry (B) and live-cell imaging (C). Scale bar 20 μm.
Conclusions
This study validated the fusion of the FAP/fluorogen module with an affibody as a promising tool for targeted labeling. This affinity probe preserves the capacity of the FAP to enhance fluorescence upon fluorogen binding, along with the specificity and sensitivity of EGFR labeling by affibody. The fast kinetics of FAP/fluorogen association accelerates the fluorescence detection and avoids the long incubation of the secondary antibody in conventional immunostaining protocols. A surprisingly high signal-to-background labeling protocol was developed, allowing specific labeling without any washing steps, in contrast to conventional fluorescent affibody reagents. The compatibility of FAPs with modified fluorogens provides more flexibility for targeted labeling to serve different experimental objectives, using a common affinity probe. The cell impermeable MG derivative associated with the probe can track EGFR through endocytosis, and revealed changes in surface abundance upon stimulation with EGF. This flow cytometric analysis of surface receptor abundance pre- and poststimulation may provide a high-throughput approach to evaluate drugs or cellular components that alter the receptor endocytosis or surface abundance.
The use of modular fluorogen substitution with a common affinity reagent may enable a wide range of applications. The targeting of FAP/affibody probe with dyedrons dramatically elevated the fluorescence signal, and provided a secondary excitation channel, which may facilitate quantitative analysis of low abundance proteins or pulse-chase labeling to tag differentially trafficked receptor subsets.24 Because the fluorogen can be linked to various cargo, while retaining sub-nanomolar Kd values, it may be possible to directly target radionuclides and nanomaterials for PET/SPECT imaging or MRI/photoacoustic imaging.37−40 FAPs with separable spectroscopic properties can be used to tag affibodies against different targets to achieve multicolor labeling.26,32 These properties make affibody–FAP fusions a potentially versatile tool in cell biology studies and promise improved, nontoxic clinical tools to detect cells with overexpressed receptors as guidance for further surgical procedures.41
Materials and Methods
DNA Construction
The Escherichia coli (E. coli) bacterial strain MACH1-T1 (Invitrogen) was used as the host for cloning. The pET21a vector was modified to include an N-terminal 10XHis and GST followed by an HRV3C protease cleavage site. Multiple cloning sites were introduced after the HRV3C protease site in the order of HindIII, NheI, BamHI, SpeI, KpnI, and XhoI from 5′ to 3′. Fragments of FAPdL5** and affibody ZEGFR:1907 were amplified from pPNL629 and pJET1.2, respectively. In construct ZEGFR:1907, affibody was inserted into the modified pET21a vector using HindIII and XhoI sites. FAP was inserted between NheI and BamHI sites to make construct FAPdL5:**. In construct FAPdL5**-ZEGFR:1907, FAP was introduced into the vector between HindIII and NheI sites; affibody was inserted between KpnI and XhoI sites. In constructs ZEGFR:1907-FAPdL5**, FAP and affibody were introduced using KpnI, XhoI, HindIII, and NheI sites. For construct ZEGFR:1907-FAPdL5**-ZEGFR:1907, affibody was introduced into ZEGFR:1907-FAPdL5** construct through HindIII and BamHI sites.
Protein Expression and Purification
Expression of recombinant proteins was carried out in the E. coli strain Rosetta-gami 2 (DE3) (Novagen). The plasmids were transformed into competent cells and fresh colonies were grown in 5 mL overnights with 12.5 μg/mL tetracycline, 34 μg/mL chloramphenicol, and 50 μg/mL ampicillin. The 5 mL cultures were then added to 500 mL LB+GB (10 g/L tryptone, 5 g/L yeast extract, 4 g/L NaCl with 100 mM phosphate pH 7.2 and supplemented with 20 mM succinic acid, 0.4% glycerol) to an OD of 0.8 at 37 °C, the temperature was dropped to 22 °C for 1 h and then cultures were induced with 500 μM IPTG and supplemented with 0.4% glucose for 18 h growth at 22 °C. Cells were pelleted and washed once with cold PBS before freezing at −20 °C. The pellets were resuspended in 3 mL of wash buffer A (50 mM Tric-Cl pH 7.5, 750 mM NaCl, 0.1% Triton X-100, 0.02% Tween-20, 50 mM imidazole) and sonicated with 10, 15 s pulses prior to dilution with 15 mL of wash buffer A. This lysate was centrifuged 30 min at 20 000g and the supernatant was incubated with Ni-NTA agarose beads (Thermo Fisher) for 2 h at 4 °C with rocking. After binding, beads were washed with 10 mL of wash buffer A and then put on a column and washed with wash buffer 150 (same as wash buffer A but with 150 mM NaCl). His tagged HRV 3C protease was used to cleave the FAP–affibody away from His-GST at 4 °C overnight and protease was then removed by incubating with additional Ni-NTA beads at 4 °C for 2 h. Protein released by the proteolytic digestion was collected as flowthrough and was then purified on Superdex 75 Gel Filtration Colume (GE Healthcare) by fast protein liquid chromatography (BioLogic DuoFlow, Biorad). Endotoxin was removed from the purified protein by endotoxin removal resin (Thermo Fisher). Purity was evaluated using SDS-PAGE and protein was quantified using a DU730 UV/vis spectrophotometer based on the absorbance at 280 nm (Beckman Coulter Inc.).
Protein Conjugation
Double affibody was cloned with an additional C-terminal cysteine. Purified protein in PBS was first treated with 10-fold molar excess TCEP (Tris(2-Carboxyethyl)phosphine) for 30 min and then was incubated with 5-fold molar excess Cy5-maleimide at 4 °C overnight. Excess dye was removed by dialysis. The purity of conjugated protein was analyzed by analytical FPLC.
Fluorescence Analysis
Fluorescence was measured using a Tecan M1000 plate reader (Tecan Group Ltd.). For fluorescence activation and emission scans, 10 μM of probes were complexed with 1 μM of fluorogen overnight at 4 °C with agitation. To measure dissociation constants, 5 nM of probes were complexed with various concentrations of fluorogen at 4 °C overnight with agitation. The fluorescence intensity was measured at the excitation of 636 nm and emission of 664 nm, and then the results were analyzed by nonlinear regression fit of one-site ligand depletion model in GraphPad Prism 6. Association rate was measured by adding fluorogen with various concentrations to constant probe. The fluorescence intensity at 636 nm excitation and 664 nm emission was monitored. Then, the data were analyzed by nonlinear regression fit of association kinetics with multiple concentrations in Graph Pad Prism 6.
Cell Culture
A431 cells (ATCC) were cultured in DMEM (Thermo Fisher) supplemented with 10% fetal bovine serum (FisherBrand). Cells were passaged after washing in PBS by incubation with PBS supplemented with 8 mM EDTA for 15 min at 37 °C. Then, the released cells were washed twice with PBS supplemented with 0.5% BSA and resuspended in PBS with 0.5% BSA to the desired cell density.
Flow Cytometry Analysis
Cell suspensions with a density of 5 × 105 cells/mL were incubated with probes at 37 °C for 1 h. After 5 min incubations with fluorogen, cell suspensions were kept on ice prior to analysis by flow cytometry (BD FACSVantage SE flow cytomoter). The data were collected and analyzed by a BD FACSDiva workstation. To measure cell surface dissociation constants, cells were incubated with various concentrations of each probe and then stained with 2 μM of fluorogen. To optimize the fluorogen concentration, cells were labeled with 250 nM of double affibody and then stained with various concentrations of fluorogen. In the competition assay, cells were incubated with 250 nM FAP–double affibody fusion and various concentration of cold affibody followed by 100 nM of fluorogen. To compare different fluorogens, 250 nM of probe and 100 nM of fluorogen was used to label cells.
Fluorescence Microscopy
Cells were seeded onto 35 mm glass bottom dishes (MatTek, Ashland, MA) 1 day prior to imaging. Cells were washed with PBS and maintained in OPTI-MEM (Invitrogen) supplemented with 5% fetal bovine serum (Invitrogen). Proteins were incubated with cells at 37 °C for 1 h and 100 nM of fluorogen was added before live cell imaging. Cell microscopy was conducted on LSM-510_Meta_Duoscan Inverted Confocal Microscope (Zeiss).
Western Blot
Cells were starved in serum-free medium overnight and then cells were treated with 250 nM probes for 1 h, 100 nM MG-B-tau for 5 min, and 100 ng/mL EGF for 10 min. Thereafter, cells were lyzed in TGH buffer buffer (1% Triton X-100, 10% glycerol, 50 mM NaCl, 50 mM Hepes, pH 7.3, 5 mM EDTA) with a protease inhibitor cocktail and phosphatase inhibitor cocktail. Cell lysates were concentrated through acetone precipitation and then loaded on 12% SDS-PAGE gels. After transfer, the membrane was blocked with 5% BSA for 1 h and then probed for rabbit antibody against phosphorylated EGFR and total EGFR, followed by HRP-conjugated antirabbit antibody (Cell signaling).
Acknowledgments
This work was supported by NIH TCNP grant U54GM103529 (Y.W., C.T., B.S., J.F., M.B.), and in part by the NIH R01GM086237 (B.S., M.B.) and the Max Planck Society (D.J.A.J., S.O.).
Glossary
Abbreviations
- EGFR
epidermal growth factor receptor
- FAP
fluorogen activating protein
- MG
malachite green derivative fluorogen
- A
affibody ZEGFR:1907
- F
FAP dL5**
- AF
ZEGFR:1907-FAPdL5**
- FA
FAPdL5**-ZEGFR:1907
- AFA
ZEGFR:1907-FAPdL5**-ZEGFR:1907
Supporting Information Available
Detailed synthesis and characterization of HCM and additional spectroscopic, biochemical and imaging validation data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
Josef D. Franke, Creighton University, Department of Biology, Omaha, Nebraska, U.S.A.
Author Present Address
Stephan Ort, Edwards Lifesciences Services GmbH, München, Germany.
The authors declare the following competing financial interest(s): Marcel Bruchez is a founder and Chief Technology Officer of Sharp Edge Labs, a company focused on developing FAP tags for drug discovery.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Saurabh S.; Bruchez M. P. (2014) Chapter 15 Targeting Dyes for Biology, in Cell Membrane Nanodomains: From Biochemistry to Nanoscopy, pp 341–360, CRC Press. [Google Scholar]
- Nelson A. L.; Dhimolea E.; Reichert J. M. (2010) Development trends for human monoclonal antibody therapeutics. Nat. Rev. Drug Discovery 9, 767–774. [DOI] [PubMed] [Google Scholar]
- Zhao H.; Cui K.; Muschenborn A.; Wong S. T. (2008) Progress of engineered antibody-targeted molecular imaging for solid tumors (Review). Mol. Med. Rep. 1, 131–134. [PubMed] [Google Scholar]
- Bai M.; Bornhop D. J. (2012) Recent advances in receptor-targeted fluorescent probes for in vivo cancer imaging. Curr. Med. Chem. 19, 4742–4758. [DOI] [PubMed] [Google Scholar]
- Peluso P.; Wilson D. S.; Do D.; Tran H.; Venkatasubbaiah M.; Quincy D.; Heidecker B.; Poindexter K.; Tolani N.; Phelan M.; Witte K.; Jung L. S.; Wagner P.; Nock S. (2003) Optimizing antibody immobilization strategies for the construction of protein microarrays. Anal. Biochem. 312, 113–124. [DOI] [PubMed] [Google Scholar]
- Hosse R. J.; Rothe A.; Power B. E. (2006) A new generation of protein display scaffolds for molecular recognition. Protein Sci. 15, 14–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binz H. K.; Amstutz P.; Pluckthun A. (2005) Engineering novel binding proteins from nonimmunoglobulin domains. Nat. Biotechnol. 23, 1257–1268. [DOI] [PubMed] [Google Scholar]
- Nord K.; Nilsson J.; Nilsson B.; Uhlen M.; Nygren P. A. (1995) A combinatorial library of an alpha-helical bacterial receptor domain. Protein Eng. 8, 601–608. [DOI] [PubMed] [Google Scholar]
- Lofblom J.; Feldwisch J.; Tolmachev V.; Carlsson J.; Stahl S.; Frejd F. Y. (2010) Affibody molecules: engineered proteins for therapeutic, diagnostic and biotechnological applications. FEBS Lett. 584, 2670–2680. [DOI] [PubMed] [Google Scholar]
- Friedman M.; Nordberg E.; Hoiden-Guthenberg I.; Brismar H.; Adams G. P.; Nilsson F. Y.; Carlsson J.; Stahl S. (2007) Phage display selection of Affibody molecules with specific binding to the extracellular domain of the epidermal growth factor receptor. Protein Eng. Des. Sel. 20, 189–199. [DOI] [PubMed] [Google Scholar]
- Lyakhov I.; Zielinski R.; Kuban M.; Kramer-Marek G.; Fisher R.; Chertov O.; Bindu L.; Capala J. (2010) HER2- and EGFR-specific affiprobes: novel recombinant optical probes for cell imaging. ChemBioChem 11, 345–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson F. Y.; Tolmachev V. (2007) Affibody molecules: new protein domains for molecular imaging and targeted tumor therapy. Curr. Opin. Drug Discovery Dev. 10, 167–175. [PubMed] [Google Scholar]
- Lee S. B.; Hassan M.; Fisher R.; Chertov O.; Chernomordik V.; Kramer-Marek G.; Gandjbakhche A.; Capala J. (2008) Affibody molecules for in vivo characterization of HER2-positive tumors by near-infrared imaging. Clin. Cancer Res. 14, 3840–3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronqvist N.; Malm M.; Gostring L.; Gunneriusson E.; Nilsson M.; Hoiden Guthenberg I.; Gedda L.; Frejd F. Y.; Stahl S.; Lofblom J. (2011) Combining phage and staphylococcal surface display for generation of ErbB3-specific Affibody molecules. Protein Eng. Des. Sel. 24, 385–396. [DOI] [PubMed] [Google Scholar]
- Gunneriusson E.; Nord K.; Uhlen M.; Nygren P. (1999) Affinity maturation of a Taq DNA polymerase specific affibody by helix shuffling. Protein Eng. 12, 873–878. [DOI] [PubMed] [Google Scholar]
- Lindborg M.; Cortez E.; Hoiden-Guthenberg I.; Gunneriusson E.; von Hage E.; Syud F.; Morrison M.; Abrahmsen L.; Herne N.; Pietras K.; Frejd F. Y. (2011) Engineered high-affinity affibody molecules targeting platelet-derived growth factor receptor beta in vivo. J. Mol. Biol. 407, 298–315. [DOI] [PubMed] [Google Scholar]
- Yarden Y.; Sliwkowski M. X. (2001) Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2, 127–137. [DOI] [PubMed] [Google Scholar]
- Blume-Jensen P.; Hunter T. (2001) Oncogenic kinase signalling. Nature 411, 355–365. [DOI] [PubMed] [Google Scholar]
- Slamon D. J.; Clark G. M.; Wong S. G.; Levin W. J.; Ullrich A.; McGuire W. L. (1987) Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235, 177–182. [DOI] [PubMed] [Google Scholar]
- Marmor M. D.; Skaria K. B.; Yarden Y. (2004) Signal transduction and oncogenesis by ErbB/HER receptors. Int. J. Radiat. Oncol. Biol. Phys. 58, 903–913. [DOI] [PubMed] [Google Scholar]
- Miao Z.; Ren G.; Liu H.; Jiang L.; Cheng Z. (2010) Cy5.5-labeled Affibody molecule for near-infrared fluorescent optical imaging of epidermal growth factor receptor positive tumors. J. Biomed. Opt. 15, 036007. [DOI] [PubMed] [Google Scholar]
- Gong H.; Kovar J.; Little G.; Chen H.; Olive D. M. (2010) In vivo imaging of xenograft tumors using an epidermal growth factor receptor-specific affibody molecule labeled with a near-infrared fluorophore. Neoplasia 12, 139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gostring L.; Chew M. T.; Orlova A.; Hoiden-Guthenberg I.; Wennborg A.; Carlsson J.; Frejd F. Y. (2010) Quantification of internalization of EGFR-binding Affibody molecules: Methodological aspects. Int. J. Oncol. 36, 757–763. [DOI] [PubMed] [Google Scholar]
- Fisher G. W.; Fuhrman M. H.; Adler S. A.; Szent-Gyorgyi C.; Waggoner A. S.; Jarvik J. W. (2014) Self-checking cell-based assays for GPCR desensitization and resensitization. J. Biomol. Screen. 19, 1220–1226. [DOI] [PubMed] [Google Scholar]
- Grover A.; Schmidt B. F.; Salter R. D.; Watkins S. C.; Waggoner A. S.; Bruchez M. P. (2012) Genetically encoded pH sensor for tracking surface proteins through endocytosis. Angew. Chem., Int. Ed. Engl. 51, 4838–4842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szent-Gyorgyi C.; Schmidt B. F.; Creeger Y.; Fisher G. W.; Zakel K. L.; Adler S.; Fitzpatrick J. A.; Woolford C. A.; Yan Q.; Vasilev K. V.; Berget P. B.; Bruchez M. P.; Jarvik J. W.; Waggoner A. (2008) Fluorogen-activating single-chain antibodies for imaging cell surface proteins. Nat. Biotechnol. 26, 235–240. [DOI] [PubMed] [Google Scholar]
- Saunders M. J.; Block E.; Sorkin A.; Waggoner A. S.; Bruchez M. P. (2014) A bifunctional converter: fluorescein quenching scFv/fluorogen activating protein for photostability and improved signal to noise in fluorescence experiments. Bioconjugate Chem. 25(8), 1556–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo E.; Jarvik J. (2014) Fluorogen-activating scFv biosensors target surface markers on live cells via streptavidin or single-chain avidin. Mol. Biotechnol. 56, 585–590. [DOI] [PubMed] [Google Scholar]
- Szent-Gyorgyi C.; Schmidt B. F.; Fitzpatrick J. A.; Bruchez M. P. (2010) Fluorogenic dendrons with multiple donor chromophores as bright genetically targeted and activated probes. J. Am. Chem. Soc. 132, 11103–11109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozhalici-Unal H.; Pow C. L.; Marks S. A.; Jesper L. D.; Silva G. L.; Shank N. I.; Jones E. W.; Burnette J. M. 3rd; Berget P. B.; Armitage B. A. (2008) A rainbow of fluoromodules: a promiscuous scFv protein binds to and activates a diverse set of fluorogenic cyanine dyes. J. Am. Chem. Soc. 130, 12620–12621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senutovitch N.; Stanfield R. L.; Bhattacharyya S.; Rule G. S.; Wilson I. A.; Armitage B. A.; Waggoner A. S.; Berget P. B. (2012) A variable light domain fluorogen activating protein homodimerizes to activate dimethylindole red. Biochemistry 51, 2471–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotti K. J.; Silva G. L.; Creeger Y.; Robertson K. L.; Waggoner A. S.; Berget P. B.; Armitage B. A. (2011) Blue fluorescent dye-protein complexes based on fluorogenic cyanine dyes and single chain antibody fragments. Org. Biomol. Chem. 9, 1012–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szent-Gyorgyi C.; Stanfield R. L.; Andreko S.; Dempsey A.; Ahmed M.; Capek S.; Waggoner A. S.; Wilson I. A.; Bruchez M. P. (2013) Malachite green mediates homodimerization of antibody VL domains to form a fluorescent ternary complex with singular symmetric interfaces. J. Mol. Biol. 425, 4595–4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolmachev V.; Friedman M.; Sandstrom M.; Eriksson T. L.; Rosik D.; Hodik M.; Stahl S.; Frejd F. Y.; Orlova A. (2009) Affibody molecules for epidermal growth factor receptor targeting in vivo: aspects of dimerization and labeling chemistry. J. Nucl. Med. 50, 274–283. [DOI] [PubMed] [Google Scholar]
- Haglund K.; Sigismund S.; Polo S.; Szymkiewicz I.; Di Fiore P. P.; Dikic I. (2003) Multiple monoubiquitination of RTKs is sufficient for their endocytosis and degradation. Nat. Cell Biol. 5, 461–466. [DOI] [PubMed] [Google Scholar]
- Mosesson Y.; Shtiegman K.; Katz M.; Zwang Y.; Vereb G.; Szollosi J.; Yarden Y. (2003) Endocytosis of receptor tyrosine kinases is driven by monoubiquitylation, not polyubiquitylation. J. Biol. Chem. 278, 21323–6. [DOI] [PubMed] [Google Scholar]
- Saurabh S.; Beck L. E.; Maji S.; Baty C. J.; Wang Y.; Yan Q.; Watkins S. C.; Bruchez M. P. (2014) Multiplexed modular genetic targeting of quantum dots. ACS Nano 8, 11138–11146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.; Xie J.; Chen X. (2010) Peptide-based probes for targeted molecular imaging. Biochemistry 49, 1364–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z.; De Jesus O. P.; Namavari M.; De A.; Levi J.; Webster J. M.; Zhang R.; Lee B.; Syud F. A.; Gambhir S. S. (2008) Small-animal PET imaging of human epidermal growth factor receptor type 2 expression with site-specific 18F-labeled protein scaffold molecules. J. Nucl. Med. 49, 804–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altai M.; Strand J.; Rosik D.; Selvaraju R. K.; Eriksson Karlstrom A.; Orlova A.; Tolmachev V. (2013) Influence of nuclides and chelators on imaging using affibody molecules: comparative evaluation of recombinant affibody molecules site-specifically labeled with (6)(8)Ga and (1)(1)(1)In via maleimido derivatives of DOTA and NODAGA. Bioconjugate Chem. 24, 1102–1109. [DOI] [PubMed] [Google Scholar]
- Kantelhardt S. R.; Caarls W.; de Vries A. H.; Hagen G. M.; Jovin T. M.; Schulz-Schaeffer W.; Rohde V.; Giese A.; Arndt-Jovin D. J. (2010) Specific visualization of glioma cells in living low-grade tumor tissue. PLoS One 5, e11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.