

Abstract
Immunoassays have been translated into microfluidic device formats, but significant challenges relating to upstream sample processing still limit their applications. Here, stimuli-responsive polymer–antibody conjugates are utilized in a microfluidic immunoassay to enable rapid biomarker purification and enrichment as well as sensitive detection. The conjugates were constructed by covalently grafting poly(N-isopropylacrylamide) (PNIPAAm), a thermally responsive polymer, to the lysine residues of anti-prostate specific antigen (PSA) Immunoglobulin G (IgG) using carbodiimide chemistry via the polymer end-carboxylate. The antibody-PNIPAAm (capture) conjugates and antibody-alkaline phosphatase (detection) conjugates formed sandwich immunocomplexes via PSA binding in 50% human plasma. The complexes were loaded into a recirculating poly(dimethylsiloxane) microreactor, equipped with micropumps and transverse flow features, for subsequent separation, enrichment, and quantification. The immunocomplexes were captured by heating the solution to 39 °C, mixed over the transverse features for 2 min, and washed with warm buffer. In one approach, the assay utilized immunocomplex solution that was contained in an 80 nL microreactor, which was loaded with solution at room temperature and subsequently heated to 39 °C. The assay took 25 min and resulted in 37 pM PSA limit of detection (LOD), which is comparable to a plate ELISA employing the same antibody pair. In another approach, the microreactor was preheated to 39 °C, and immunocomplex solution was flowed through the reactor, mixed, and washed. When the specimen volume was increased to 7.5 μL by repeating the capture process three times, the higher specimen volume led to immunocomplex enrichment within the microreactor. The resulting assay LOD was 0.5 pM, which is 2 orders of magnitude lower than the plate ELISA. Both approaches generate antigen specific signal over a clinically significant range. The sample processing capabilities and subsequent utility in a biomarker assay demonstrate the opportunity for stimuli-responsive polymer–protein conjugates in novel diagnostic technologies.
Introduction
Detection of disease specific protein biomarkers in human bodily fluids can provide both diagnostic and prognostic value.1 However, biomarkers are often very dilute in the complex milieu of human blood.2 Therefore, current clinical diagnostic strategies are limited to techniques that require expert-run and time-consuming specimen processing with large sample volumes to purify and enrich biomarkers prior to assay,3 which are not ideal for many diagnostic settings3a,4 (e.g., global health settings). Combining biomarker separation and enrichment steps within a rapid and simple assay will overcome these limitations and enhance the utility of diagnostic assays.5
Microfluidic devices have been utilized for various applications6 and also offered the promise of rapid and simple assays suitable for use in distributed diagnostic settings.7 For example, Delmarche et al. introduced a one-step point-of-care microfluidic immunoassay, an integrated device with on-card detection and captured antibodies (Abs) utilized specimen addition to trigger a sandwich immunoassay powered by capillary forces.8 In another example, Murphy et al. have described a fluorescence competitive immunoassay for three different antigens using micromosaic capture Abs that were covalently immobilized in a mosaic pattern on silicon nitride microfluidic networks.9 While these assays could detect 10–12 M antigen in short assay times (<45 min), their performance is still limited by fundamental issues—surface bound capture Abs are unable to fully interrogate the sample volume rapidly due to antigen transport limitations to and from the surface, and conformational/mobility restrictions of the surface immobilized Ab.10
We have been investigating the use of stimuli-responsive polymer-Ab reagent systems10a,11 with complementary microfluidic devices to overcome some of the basic challenges of the microfluidic immunoassay. These molecular reagent systems enable efficient and rapid antigen binding and in situ sandwich immunocomplex assembly with the secondary labeling reagent in a homogeneous solution such as plasma.12 The reagent system can facilitate biomarker/immunocomplex separation, purification, and enrichment13 in microchannels that exhibit hydrophobic14 sidewalls and provide short transport distances and large surface area to volume ratios.15 Additionally, fluidic controls such as valves,16 pumps,17 and active features18 can be integrated to improve the transport and capture of the stimuli-responsive polymer-Ab complexes on the channel surfaces.19 Here, poly(N-isopropylacrylamide) (PNIPAAm) has been conjugated to a prostate specific antigen (PSA) Ab, an Immunoglobulin G (IgG), for purifying and concentrating the PSA sandwich immunocomplexes in a previously described circular microreactor with transverse flow generators without device surface functionalization.17,18b,20 Instead of dialysis,21 the conjugates were purified via ion exchange (IE) chromatography. In order to assist assay development, the conjugate binding properties were characterized via a competitive ELISA. In addition to concentrating immunocomplexes on a PDMS surface, the immunoassay here employs alkaline phosphatase (AP) to generate a fluorescent signal and generates the assay signal directly on the assay device. The stimuli-responsive reagent system and corresponding microreactor can concentrate the PSA immunocomplexes directly in human plasma with a strong enhancement of the biomarker specific signal.
Results and Discussion
System Design
A rapid PSA immunoassay with biomarker enrichment capability was enabled by using an Ab-PNIPAAm conjugate in conjunction with a recirculating microreactor module (Scheme 1). The assay started from forming sandwich immunocomplexes by mixing Ab-PNIPAAm conjugate (capture reagent) and Ab-AP conjugate (detection reagent) in 50% human plasma, spiked with PSA (antigen). This binding in a homogeneous solution format has been shown to overcome mass transport limitations on protein–ligand binding at solid surfaces.10a,22 The immunocomplexes in the human plasma specimen were then isolated by utilizing the recirculating microreactor module above the PNIPAAm transition temperature via the interactions between the device surface and the collapsed polymer within the conjugate.23 Compared to our previous immunoassay work,21 the PSA immunoassay here is different in assay format, which isolates immunocomplexes on a PDMS surface, in the detection system, which employs AP to generate a fluorescent signal, and in the detection procedure, which detects the assay signal directly within the microfluidic device. We evaluated two immunoassay approaches, 25 °C starting temperature with finite (80 nL) specimen volume and 39 °C starting temperature with immunocomplex enrichment.
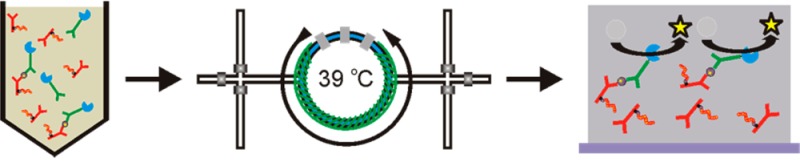
Scheme 1. Immunoassay Overview.
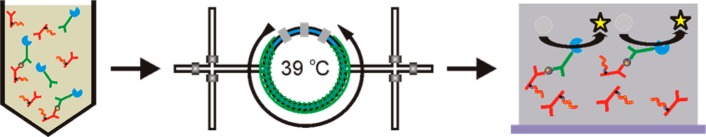
Immunocomplexes, composed of Ab-PNIPAAm conjugates, Ab-AP conjugates, and PSA, are formed in a 50% human plasma solution. Samples are loaded into the microfluidic device and surface is heated to 39 °C. Above 39 °C, immunocomplexes are separated from the human plasma solution by immobilization on the microfluidic device sidewalls through hydrophobic interactions. To enrich samples, the microfluidic device is washed to remove non-immobilized material and additional sample is injected with the temperature at 39 °C. Once sample is separated, enzyme substrate is loaded into the microfluidic device and turned over into fluorescent product by immobilized Ab-AP conjugates for detection.
PSA Antibody-PNIPAAm Conjugate Synthesis and Characterization
The conjugate was constructed using a protocol similar to that in our previous publication21 by end-grafting PNIPAAm chains to Ab’s lysine residues (Scheme 2). The PNIPAAm with an end-carboxylate was synthesized using thermally initiated reversible addition–fragmentation chain transfer (RAFT) polymerization. The gel permeation chromatography (GPC) results show the polymer exhibited Mn = 41.2 kDa with a polydispersity index (PDI) = 1.1. To facilitate the Ab conjugation, the PNIPAAm end-carboxylate was converted to tetrafluorophenyl (TFP) active ester.21 TFP was utilized to reducing hydrolysis rate for improving amine reactivity.24
Scheme 2. RAFT Polymerized PNIPAAm Was Functionalized with TFP to Create an Amine Reactive TFP-PNIPAAm Active Ester.
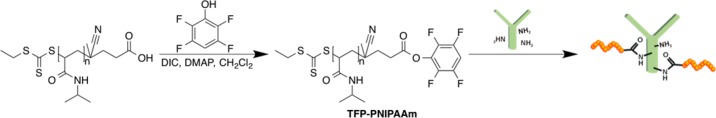
Functionalized polymer was subsequently conjugated to Ab to create an Ab-PNIPAAm conjugate. Conjugates were purified through a serial combination of thermal precipitation and ion exchange chromatography.
The conjugation was carried out by simply mixing TFP-PNIPAAm (in anhydrous DMSO) with PSA Ab.21 In addition to desalting and thermal precipitation, we employed IE chromatography for conjugate purification because PNIPAAm is neutral, and therefore facilitates the protein–polymer conjugate to be selectively captured and released from the IE resin. The purification process was monitored using a UV–vis spectrophotometer and gel electrophoresis (Figure 1). UV–vis spectrum of the conjugation reaction solution, RXN (Figure 1a, purple), exhibited absorbance peaks near 280 and 307 nm, which indicate the solution contains both Ab and PNIPAAm (trithiocarbonate CTA). After the conjugation, the solution was processed first with a desalting column to remove small molecule impurities, and then with thermal precipitation to separate native Ab (without polymer) in the solution by pelleting both Ab-PNIPAAm conjugates and excess PNIPAAm. UV–vis spectrum of the supernatant from thermal precipitation, Supernate (Figure 1a, green), exhibited a peak absorbance around 280 nm, which confirmed the solution contains mainly native Ab, IgG (Figure 1a, blue). The resuspended pellet, Pellet (Figure 1a, red), exhibited a broad absorbance around 307 nm. Figure 1c shows the resuspended pellet solution (Lane 2) with smear pattern and shifted toward higher molecular weight than native Ab, IgG (Lane 6), which confirmed the conjugation.
Figure 1.

Monitoring Ab-PNIPAAm conjugate purification process using UV–vis spectrophotometer and gel electrophoresis. (a) UV–vis spectra for the thermal precipitation process. The conjugation reaction solution, RXN (a, purple), exhibited absorbance near 280 and 307 nm, which indicates the solution contains both antibody and PNIPAAm (trithiocarbonate CTA). After the thermal precipitation, the spectrum of the supernatant, Supernate (a, green), exhibited a peak absorbance around 280 nm, which confirmed the solution contains mainly IgG (a, blue). The resuspended pellet, Pellet (a, red), exhibited a broad absorbance around 307 nm. (b) UV–vis spectra for the IE chromatography process. The spectrum of the purified conjugates, Conjugate (b, purple) exhibited an 280 nm absorbance peak with an PNIPAAm trithiocarbonate CTA shoulder, 307 nm, which confirms the IE chromatography successfully isolated Ab-PNIPAAm conjugates via dimethylaminopropyl resin and removed excess polymer by washing as the ion exchange (IE) effluent (red) and wash (green) consist mostly of PNIPAAm, as seen by strong 307 nm absorbance of trithiocarbonate CTA. (c) Gel electrophoresis results: Lane 1, ladder; Lane 2, resuspended pellet (from thermal precipitation); Lane 3, IE effluent (during loading); Lane 4, IE wash; Lane 5, released conjugate; Lane 6, IgG. The resuspended pellet solution (Lane 2) exhibit a smear pattern that shifted toward higher molecular weight than native IgG (Lane 6) confirms the conjugation. The results also confirm ion exchange chromatography successfully isolated Ab-PNIPAAm conjugates via dimethylaminopropyl resin because released conjugate (Lane 5) exhibits a smear pattern on the gel with molecular weight higher than the native IgG (Lane 6) and IE effluent (Lane 3) and wash (Lane 4) did not contain conjugates (PNIPAAm does not stain).
The pellet solution was further processed with IE chromatography to purify the Ab-PNIPAAm conjugates by removing excess PNIPAAm. Both IE effluent and wash (Figure 1c, Lane 3 and 4) did not contain conjugates but consisted mostly of PNIPAAm, as seen by strong 307 nm absorbance of trithiocarbonate CTA (Figure 1b, red and green), which suggests that dimethylaminopropyl resins successfully retained Ab-PNIPAAm conjugates. The released conjugates, Conjugate (Figure 1b, purple), exhibited a 280 nm absorbance peak with a PNIPAAm trithiocarbonate CTA shoulder, 307 nm, which confirms the conjugate contains both Ab and PNIPAAm. Figure 1c shows the released conjugate (Lane 5) also with a smear pattern with molecular weight higher than the native Ab, IgG (Lane 6), which confirms the IE chromatography successfully isolated Ab-PNIPAAm conjugates and removed excess polymer by washing. These results confirm the Ab-PNIPAAm conjugates were successfully purified.
In order to assist assay development, we evaluated the effect of polymer conjugation on Ab-antigen equilibrium binding via a competitive binding assay using 96-well PSA ELISA. In this assay, PSA (300 pM final concentration in PBS) was premixed with either native Ab, or Ab-PNIPAAm conjugates at 30, 300, 3000, or 30000 pM final concentration in PBS. After 15 min, these solutions were subjected to the 96-well ELISA protocol. The native Ab, Ab-PNIPAAm conjugate, and capture Ab in ELISA were the same monoclonal IgG that bind PSA at the identical epitope. Therefore, the 96-well ELISA was utilized to quantify unbound PSA at equilibrium in the solutions of native Abs (or Ab-PNIPAAm conjugates). As a control, PSA was incubated without native Ab (or Ab-PNIPAAm conjugate). The control allowed the quantification of signal for total PSA. Assuming total PSA equals the sum of unbound and bound PSA, the bound PSA was estimated by subtracting unbound PSA from total PSA.
Figure 2 shows bound PSA at equilibrium for various native Ab (or Ab-PNIPAAm conjugate) concentrations. The bound PSA increased with increasing concentration of native Ab (or Ab-PNIPAAm conjugate). Dashed lines are fittings via one site total binding model, which was also used to estimate KD values (GraphPad Prism). With our in-house ELISA, the native Ab (blue) KD is 1.192 nM, and the KD for Ab-PNIPAAm conjugates (red) is 1.599 nM. Compared to the native Ab, binding via Ab-PNIPAAm conjugates was lower at the same molar excess. However, our new stimuli-responsive reagent system enabled PSA binding similar to the native Ab by using more conjugates. Therefore, the downstream assay performance was not compromised.
Figure 2.
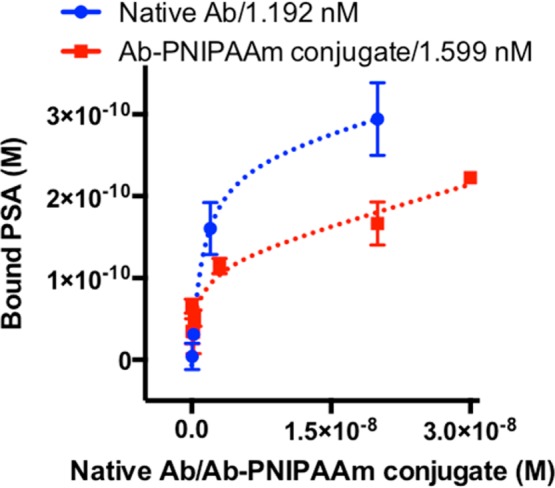
Equilibrium binding curves for native Ab (blue) and Ab-PNIPAAm conjugate (red). Error bars are standard deviations over three independent experiments. Dashed lines are fittings via one site total binding model, which was also used to estimate KD values (GraphPad Prism). The native Ab (blue) KD is 1.192 nM, and the KD for Ab-PNIPAAm conjugates (red) is 1.599 nM.
PSA Immunoassay in the Microreactor
The PSA immunoassay utilized a microreactor to efficiently isolate sandwich immunocomplexes containing Ab-PNIPAAm conjugates within the recirculator via heating. We first evaluated the ability of our system to generate antigen specific signal over a clinically significant range by capturing the immunocomplexes from 80 nL of 50% human plasma spiked with 0, 150, 210, and 300 pM PSA.25 The human plasma dilution was meant to precondition specimens for minimizing variation across specimens and alleviating nonspecific protein interactions with capture surfaces.26 The immunoassay was carried out using the microfluidic immunoassay protocol illustrated in Scheme 3 and detailed in the Experimental Procedures section below. During the sample preparation step, the sandwich immunocomplexes were assembled in a homogeneous solution by mixing Ab-PNIPAAm conjugate (capture reagent) and Ab-AP conjugate (detection reagent) in 50% human plasma, spiked with PSA (antigen). After the microreactor was filled with immunocomplex solution and sealed (sample loading), the reactor was heated to 39 °C (heating). Heating above the transition temperature caused the PNIPAAm to phase transition from hydrophilic to hydrophobic, which leads to immocomplex precipitation and immobilization on the PDMS surface. Circulating the contained specimen (separation) brought precipitated complexes into contact with the PDMS surface, maximizing immunocomplex separation.20 After a buffer wash at 39 °C, 4-MUP substrate was loaded (substrate loading) into the device for generating fluorescent signal via the assay’s alkaline phosphatase (AP) detection conjugates. Additional circulation at 25 °C was employed during image acquisition to homogenize the captured immunocomplexes with the 4-MUP substrate while subsequently homogenizing the fluorescent product, methylumbelliferone (substrate development). Fluorescence images were acquired over 15 min and were analyzed using ImageJ to measure the fluorescent intensity in the microchannel.27 Normalized fluorescence intensity was calculated by first measuring the fluorescence intensity in the channel, which was then normalized to the native fluorescence of PDMS.
Scheme 3. Process for PSA Immunoassay with 80 nL Specimen Volume.
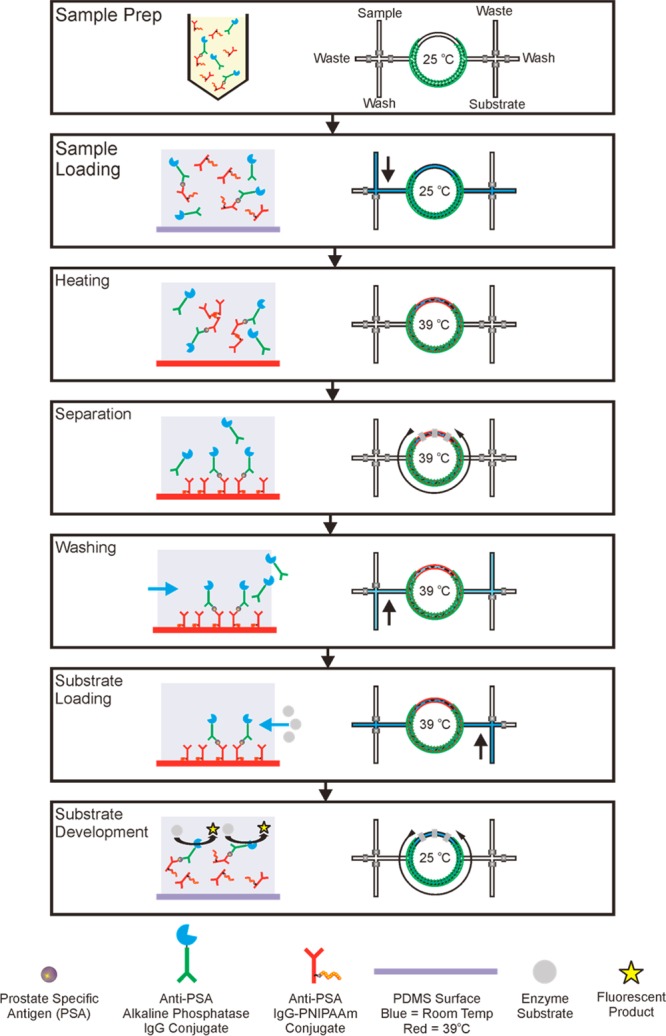
Sample Prep: form sandwich immunocomplexes in a homogeneous solution by mixing Ab-PNIPAAm conjugate (capture reagent) and Ab-AP conjugate (detection reagent) in 50% human plasma, spiked with PSA (antigen). Sample loading: fill microreactor with immunocomplex solution. Heating: raise the device temperature to 39 °C, which transitions sandwich immunocomplexes with Ab-PNIPAAm conjugates to be hydrophobic. Separation: capture immunocomplexes on the surface by circulating the contained specimens. Washing: remove excess detection reagent and other serum proteins via buffer wash. Substrate Loading: load 4-MUP substrate into the reactor. Substrate Development: lower the device temperature to 25 °C and activate the circulation to mix immunocomplexes with substrate for generating signal.
Figure 3 shows the normalized fluorescence intensity increased linearly with signal development time for each antigen concentration, indicating immunocomplexes were successfully immobilized on the microreactor surface. All assay evaluations show a strong linear correlation with R2 values between 0.96 and 0.99. The slope increased from 0.0099 to 0.0492 when the PSA concentration changed from 0 to 303 pM. At the end of the assay (15 min) specimens with spiked PSA show significantly higher fluorescence intensity than 0 pM specimen.
Figure 3.
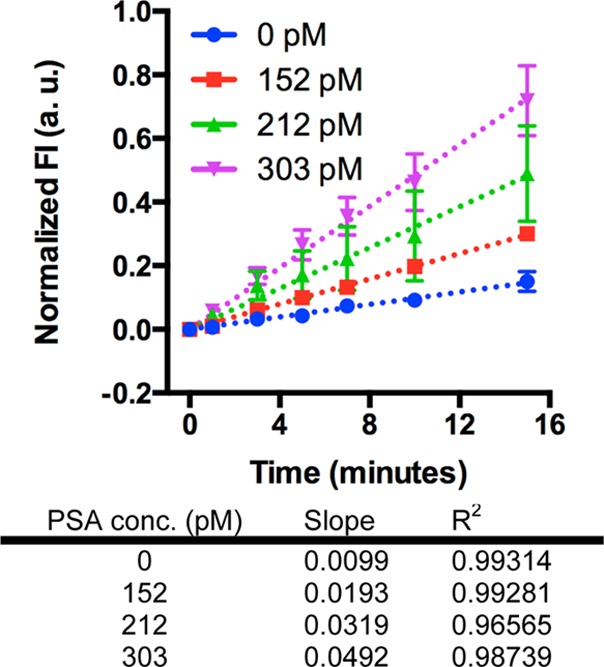
Results of immunoassays with 80 nL specimen volume. The fluorescence intensity increased with time for each antigen concentration measured. The slope increased with increasing antigen concentration as expected. Dashed lines are linear fits to the data and error bars are standard deviations over three independent experiments. Data points are for 0, 1, 3, 5, 7, 10, and 15 min only.
For a given PSA concentration, utilizing larger specimen volume provides higher numbers of PSA molecules, which results in more immunocomplexes. Therefore, efficiently concentrating immunocomplexes from larger specimen volume leads to higher assay signal (fluorescent intensity) and the resulting assay might exhibit a lower limit of detection (LOD), which can improve the assay sensitivity. Therefore, we applied the enrichment process to larger specimen volumes, 2.5 and 7.5 μL. We have previously demonstrated the analyte enrichment process via PNIPAAm conjugates using streptavidin as a model marker.20 Here the enrichment process was applied to the PSA sandwich immunocomplexes, which enable direct analyte detection in the microfluidic device via the detection reagent. The immunoassay with enrichment process is illustrated in Scheme 4, which is similar to the assay with finite volume. Instead of starting the assay with the device at 25 °C, this approach began with the device preheated to 39 °C prior to sample loading, which is above the conjugate transition temperature. Flowing immunocomplexes into a preheated device increased the volume of specimen exposed to immuocomplex capture conditions. After separation and washing, the new assay system can either proceed with substrate loading for detection or allow additional sample loadings, which can significantly increase the number of captured immunocomplexes to improve analyte detection.
Scheme 4. Process for PSA Immunoassay with Enrichment Process.
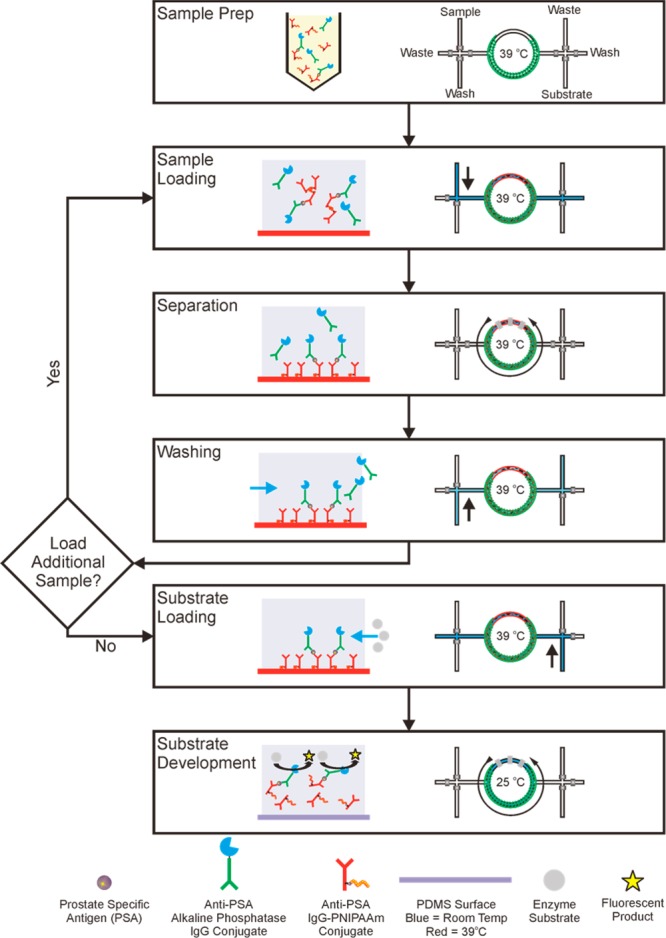
This process utilizes a device that has been preheated to 39 °C prior to sample loading to transition sandwich immunocomplexes with Ab-PNIPAAm conjugates to be hydrophobic during sample loading. Sample Prep: form sandwich immunocomplexes in a homogeneous solution by mixing Ab-PNIPAAm conjugate (capture reagent) and Ab-AP conjugate (detection reagent) in 50% human plasma, spiked with PSA (antigen). Sample loading: fill microreactor with immunocomplex solution. Separation: capture immunocomplexes on the microfluidic channel surface by circulating the contained specimens. Washing: remove other excess detection reagent and other serum proteins via buffer wash. Substrate Loading: load 4-MUP substrate into the reactor. Substrate Development: lower the device temperature to 25 °C and activate the circulation to mix immunocomplexes with substrate for generating signal.
These experiments utilized 50% human plasma spiked with 0, 30, and 150 pM PSA to mimic the clinical relevant range.25 One sample load consisted of flowing 2.5 μL spiked human plasma through the microreactor at 39 °C. Then, the aforementioned protocol was used to capture the immunocomplexes and generate fluorescent signal. The fluorescence images and their analysis are the same as the assay with finite volume.27 These experiments utilized both one (2.5 μL) and three sample (3 × 2.5 μL) loads. Figure 4 shows the normalized fluorescence intensity (using the same calculation as in Figure 3) increased linearly with substrate development time for each antigen concentration in both 2.5 and 7.5 μL assays, indicating that immunocomplexes were successfully immobilized on the PDMS surface. All assay evaluations show a strong linear correlation with R2 values between 0.95 and 0.98. When the PSA concentration changed from 0 to 152 pM, the slope increased from 0.0088 to 0.0585 for 2.5 μL assays and from 0.216 to 0.1386 for 7.5 μL assays. At the end of assay (15 min) specimens with spiked PSA show significantly higher fluorescence intensity than those with 0 pM specimen.
Figure 4.
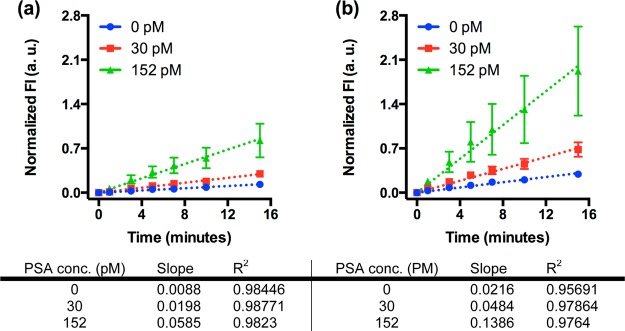
Results of immunoassays with larger specimen volume. The fluorescence intensity increased with time for each antigen concentration measured. Increasing the volume of sample loaded from (a) 2.5 μL to (b) 7.5 μL resulted in a modest increase in the rate of production for the null sample, while antigen containing sample increased significantly, thus decreasing the limit of detection. Dashed lines are linear fits to the data and error bars are standard deviations over three independent experiments. Data points are for 0, 1, 3, 5, 7, 10, and 15 min only.
Figure 5 shows the correlation between rate of fluorescence production and antigen concentration. The rate of fluorescence production is the calculated slope of normalized fluorescence intensity versus time from Figures 3 and 4. The rate of fluorescence production increased with antigen concentration, indicating immunocomplexes were successfully immobilized on the microreactor surface. For a given antigen concentration, the rate of fluorescence production increased with the number of specimen loads, indicating immunocomplexes were being enriched on the microfluidic device surface. All assay evaluations show a strong linear correlation with R2 values between 0.91 and 0.99. The slope of linear fit changed from 0.000 300 to 0.000 711 when the specimen volume increased from 2.5 to 7.5 μL. The signals for 0 pM PSA specimens were very similar for the assays using 80 nL and 2.5 μL, but was ∼2-fold higher for the assay using 7.5 μL, which might be caused by nonspecific immobilization of the detection reagent to the PDMS surface.28 For the assays with 150 pM PSA, the rate was 0.126 fluorescence units/min for 7.5 μL assay which was 6.4-fold higher than 80 nL and 2.3-fold higher than 2.5 μL evaluations. The increase in rate of fluorescence production for the 7.5 μL specimen was due to the increase in immunocomplexes present in the larger sample volume and due, presumably, to improved transport conditions in the channel. As immunocomplexes were immobilized on the channel wall, the channel cross section became occluded, reducing the distance from the channel centerline to the effective capture surface thus enabling an increase in the rate of immobilization at the expense of increased back pressure. Therefore, the assay signal was significantly higher.
Figure 5.
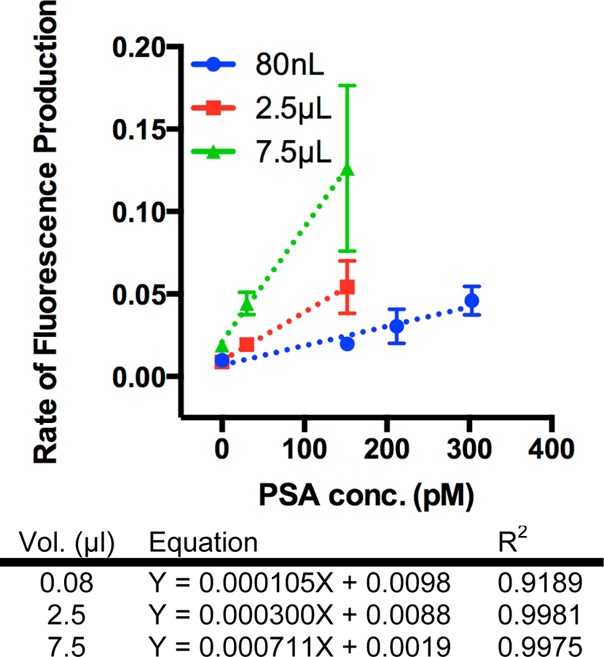
Rate of fluorescence production for immunoassays with finite volume (80 nL) and enrichment processes (2.5 and 7.5 μL). A linear relationship between rate of fluorescence production and antigen concentration was observed for all assays. In immunoassays with the enrichment process, increasing loaded volume from 2.5 to 7.5 μL showed a marked increase in rate of fluorescence production for a given antigen concentration, demonstrating enrichment on the microfluidic channel sidewalls. Dashed lines are linear fits to the data and error bars are standard deviations over three independent experiments.
The data in Figure 5 were also utilized for estimating assays’ LOD, which is the antigen concentration for generating rate of fluorescence production = Rave + 3RstdevRave is the average rate of fluorescence production and Rstdev is the resulting standard deviation that were calculated using three independent 0 pM PSA samples. The calculated LOD for the microfluidic immunoassays are summarized in Table 1. For comparison purposes Table 1 also summarizes three FDA approved commercial PSA immunoassays, including the Elecsys test (Roche), Immuno-1 test (Bayer), and the Immulite test (Diagnostic Products Corporation). The values for sample volume and limit of detection were taken from product literature and relevant clinical evaluations.29
Table 1. Summary of Various Immunoassaysa.
| protocol | sample volume (μL) | limit of detection (pM) | limit of detection (molecules) | assay time (minutes) |
|---|---|---|---|---|
| immunoassay with finite volume | 0.08 | 37 | 1.8 × 106 | 25 |
| immunoassay with enrichment | 7.5 | 0.5 | 2.2 × 106 | 35 |
| 2.5 | 22 | 3.3 × 107 | 25 | |
| Plate ELISA | 100 | 43 | 1.3 × 109 | 270 |
| Elecsys (Roche) | 20 | 0.06 | 7.1 × 105 | 20–30 |
| Immuno-1 (Bayer) | 20 | 0.6 | 7.1 × 106 | 40 |
| Immulite (Diagnostic Products Corporation) | 50 | 0.09 | 2.7 × 106 | 95 |
The calculated LOD for the 80 nL immunoassay was 37 pM, which is comparable to plate based ELISA (43 pM) but the assay only took 25 min. The new assay system can enable highly efficient and rapid sandwich immunocomplex separation to generate antigen specific signal with an 80 nL specimen (ca. 2 × 106 PSA molecules), which is approximately 3 orders of magnitude less than plate ELISA. The calculated LOD for 2.5 μL assay was 22 pM, which was similar to the plate. However, the estimated LOD was 0.5 pM for 7.5 μL assay, which was a 2 orders of magnitude improvement from the plate ELISA. Compared to the FDA approved PSA immunoassays, our microfluidic immunoassay takes a similar amount of time but lower specimen volume. While our microfluidic immunoassay exhibits higher LOD in concentration, our assay system shows comparable LOD in number of antigen molecules.
The calculated LOD for the 80 nL immunoassay was 37 pM, which is comparable to plate based ELISA (43 pM) but the assay only took 25 min. The new assay system can generate antigen specific signal over clinical relevant concentrations with an 80 nL specimen (ca. 2 × 106 PSA molecules), which is approximately 3 orders of magnitude less than ELISA because the combination of Ab-PNIPAAm conjugates and microreacator enable highly efficient and rapid sandwich immunocomplex separation. The calculated LOD for 2.5 μL assay was 22 pM, which was similar to the plate. However, the estimated LOD was 0.5 pM for 7.5 μL assay, which was a 2 orders of magnitude improvement from the plate ELISA. 7.5 μL assays provide significantly more PSA molecules than 80 nL and 2.5 μL assays, which led to the antigen dependent increase in signal required to detect low antigen concentrations. Compared to FDA approved PSA immunoassays, our microfluidic immunoassay takes a similar amount of time to complete but consumes lower specimen volume. While our microfluidic immunoassay exhibits higher LOD in concentration, our assay system shows comparable LOD in number of antigen molecules because our assay system can efficiently capture immunocomplexes from a smaller specimen volume.
However, comparison with the commercialized PSA detection kits is very challenging because these kits are optimized with different antibody pairs and detection systems, including enzymes, substrates, and instruments. Antibody is the key reagent that dictates the immunoassay performance. Assay manufacturers would focus on developing antibodies with the best binding properties (e.g., kon, koff), which may be significantly better than the antibodies used in our assay. Additionally, these kits are utilized in conjunction with instruments that are optimized for their assays. Therefore, a direct comparison should not be based on few values.
Overall, the results indicate that the microfluidic immunoassay with finite specimen volume, heated from room temperature and captured, can be employed to measure antigen concentration over a finite range, while larger sample volumes flown into a preheated device can facilitate immunocomplex concentration and subsequent detection of dilute antigen concentrations.
Conclusion
We have developed a new microfluidic immunoassay system that can efficiently capture sandwich immunocomplexes from various volumes (80 nL to 7.5 μL) of 50% spiked human plasma via the combination of Ab-PNIPAAm conjugate and the microreactor. The microfluidic immunoassay takes ca. 30 min. The assay LOD with 80 nL specimen was comparable to the plate ELISA. When the specimen volume was increased to 7.5 μL, the assay system could concentrate immunocomplexes from the higher volume specimen and the resulting assay LOD was 2 orders of magnitude lower than plate ELISA. The low LOD can be attributed to solution phase formation of the immunocomplex, resulting in greater interrogation of the sample volume. Also, the high surface to volume ratio within the device, coupled with flow, increases the immunocomplex–surface interactions and immobilization. While the microdevice architecture described here is complex, these protocols could be completed using a less automated system. There are examples of handheld microfluidic devices30 that could find utility in this application, thus permitting point-of-care solutions with sub-pM limits of detection.
Experimental Procedures
Materials
All materials used will be listed in the corresponding procedure section. Unless stated, the materials are used without further processing or purification.
Polymer Synthesis
The trithiocarbonate chain transfer agent (CTA) ethyl cyanovaleric trithiocarbonate (ECT) was synthesized as previously described.31 2,2-Azobis(isobutyronitrile) (AIBN, Sigma-Aldrich) was used as an initiator. N-Isopropylacrylamide (NIPAAm) monomer (Sigma-Aldrich) was recrystallized from hexanes. NIPAAm was polymerized with ECT and AIBN as the initiator at 60 °C in dioxane under a N2 atmosphere for 16 h. The molar ratio of [monomer]/[CTA]/[initiator] was 450/1/0.1. The resulting polymer was purified by repeated cycles of precipitation in pentane (Sigma-Aldrich). The polymer was dried overnight in vacuo.
The resulting PNIPAAm was characterized using GPC performed on an Agilent 1200 series liquid chromatography system, equipped with TSKgel alpha 3000 and TSKgel alpha 4000 columns (TOSOH biosciences). The mobile phase was LiBr (0.01 M) in HPLC grade DMF at a flow rate of 1 mL min–1. MALS data were obtained on a miniDAWN TREOS (Wyatt Technologies Corp.) with 658 nm laser source, and three detectors at 45.8°, 90.0°, and 134.2°. The instrument calibration constant was 4.7460 × 10–5 V–1cm–1. Refractive index was measured using an Optilab Rex detector (Wyatt Technologies Corp.). The dn/dc value for the PNIPAAm was determined under the assumption of 100% mass recovery by injecting polymer samples at known concentrations into the RI detector postcolumn. The dn/dc value was then calculated using linear regression with the Astra 5.3.4.14 data analysis software package (Wyatt Technologies Corp.).
Antibody–PNIPAAm Conjugation
The PNIPAAm–antibody conjugation utilized PNIAAm with an active ester by converting the end-carboxylate (Scheme 2) to an amine-reactive tetrafluorophenol (TFP) ester. 1.5 g (30 μmol) PNIPAAm was mixed with 8 mg (48 μmol) TFP (Sigma-Aldrich) and 27 mg (219 μmol) N,N′-diisopropylcarbodiimide (DIC, Sigma-Aldrich) in 12 mL methylene chloride. 4-Dimethylaminopyridine (DMAP, Sigma-Aldrich) was added as a catalyst in a 1:10 molar ratio with PNIPAAm. The reaction proceeded overnight at room temperature in a nitrogen atmosphere and the polymer was purified by repeated cycles of precipitation in pentane. The polymer was dried overnight in vacuo and stored under vacuum at −20 °C.
TFP-activated PNIPAAm (33 mg, 800 nmol) dissolved in anhydrous dimethyl sulfoxide (DMSO, Sigma-Aldrich) was added to 2 mg (1 mg mL–1 in pH 8.5 borate buffer) 5G6 monoclonal antibody (M86599M, Biodesign International). The reaction proceeded overnight at 4 °C. The antibody–PNIPAAm conjugates were purified via three sequential processes, desalting, thermal precipitation, and ion exchange. The reaction mixture was spun through a desalting column (89883, Thermo Scientific) primed with pH 7.4 PBS (P-5368, Sigma-Aldrich) to remove TFP and DMSO. Thermal precipitation, centrifugation at 45 °C, was utilized to remove nonconjugated Ab. The desalted reaction mixture was heated to 45 °C for 10 min, and then centrifuged at 10K rpm for 5 min at 45 °C. After the supernatant was collected (nonconjugated IgG) the pellet was resuspended in pH 7.4 PBS overnight at 4 °C. Thermal precipitation was repeated 2 additional times. Removal of nonconjugated IgG was determined by UV–vis spectroscopy and SDS-PAGE. Ion exchange was utilized to remove excess polymer. Three milliliters of anion ion-exchange resin (17–1287–10, GE Healthcare) was washed 5 times with 10 mL pH 8.5 Tris, followed by centrifugation at 1450 rpm. The resin was resuspended into 3 mL pH 8.5 Tris. One milliliter of resin was added to a spin column (732–6008, Bio-Rad) and spun at 1450 rpm for 5 min. 200 μL of the reaction mixture was added to the dry resin and allowed to mix overnight at 4 °C (conjugate binding step). The resin was then spun down and 200 μL pH 8.5 Tris was added and allowed to mix overnight at 4 °C (free PNIPAAm wash step). The resin was then spun down and 200 μL pH 8.5 Tris with 0.5 M NaCl was added and allowed to mix overnight at 4 °C (conjugate release step). Products of each step were collected and analyzed by UV–vis spectroscopy and SDS-PAGE.
Antibody–Alkaline Phosphatase Conjugation
Alkaline phosphatase (AP) conjugation kits (A-9002-001, Solulink) were used to conjugate AP to 200 μg of 5A6 monoclonal antibody (M86506M, Biodesign International) by following the manufacturer’s protocol. Briefly, primary amines of 5A6 IgG were functionalized with 6-hydrazino-nicotinic acid and purified by size exclusion chromatography. Functionalized IgG was subsequently conjugated to 4-formylbenzoate functionalized AP via aniline catalyzed bis-arylhydrazone formation.
Human Plasma
Pooled human plasma, with sodium citrate as an anticoagulant (IPLA-N-02, Innovative Research), was thawed and centrifuged at 3700 rpm for 30 min. Supernatant was filtered (6994–2504, Whatman) and stored at 4 °C for subsequent use.
PSA 96-Well Enzyme-Linked Immunosorbent Assay (ELISA)
To immobilize the captured antibody the NUNC Maxisorp 96-well plate was added with 100 μL 5G6 capture antibody (4 μg mL–1 pH 7.4 PBS), covered with plate film, and incubated overnight at 4 °C. The plate was then washed 3 times with PBS Tween (PBST, Fluka) using an automated plate washer (BioTek ELx50). The plate was added with 200 μL 2% bovine serum albumin (w/v in PBS), covered with plate film, and incubated for 2 h at room temperature. During incubation, PSA antigen was diluted to working concentrations (0, 2, 5, 10, and 25 ng mL–1) in a 50:50 PBS:human plasma solution. After incubation, each well was again washed 3 times with PBST. 100 μL antigen solutions were added to the plate and incubated for 1 h at room temperature. During incubation, the antibody–AP conjugate (5A6 epitope) was diluted to 50 ng mL–1 in PBST. After incubation, each well was again washed 3 times with PBST. 100 μL of the antibody–AP conjugate was added to each well, covered in plate film, and allowed to incubate for 1 h at room temperature. After incubation, each well was again washed 3 times with PBST. 100 μL 5 mM 4-methylumbelliferyl phosphate (4-MUP, Invitrogen) in pH 9.5 Tris was added to each well, covered with plate film, protected from light, and allowed to incubate at room temperature for 10 min. The plate film was removed and florescence (excitation: 360 nm, emission: 440 nm) was measured using a plate reader (Tecan).
Microfluidic Device Fabrication
Multilayer polydimethylsiloxane (PDMS) microfluidic devices were manufactured as described previously.20,23,32 Briefly, degassed 10:1 PDMS:cross-linker solution was cured on photoresist patterned silicon wafers for at least 2 h at 65 °C. The control layer master was bonded to a 40-μm-thick PDMS membrane using oxygen plasma bonding. This layer was then aligned and bonded to a fluid layer using methanol assisted oxygen plasma bonding. Devices were then allowed to sit overnight at 65 °C. Poly(ethylene glycol) acrylate (Sigma-Aldrich) was polymerized from the sidewalls of all input and output fluid channels. Prior to use, devices were aligned with an ITO heater and the control channels filled with nanopure water. Control channels were connected to positive pressure with valve channels regulated to 20 psi and mixing blades regulated to 5 psi. Valves and blades were actuated using custom LabView software. After use, devices were flushed with and stored in nanopure water at 4 °C.
Microfluidic Immunoassay
The procedure for the immunoassay on the microfluidic device is illustrated in Scheme 3. Prior to the assay on the device, the sandwich immunocomplex was formed in 50% human plasma outside of device. Using PBS as the diluent, PSA was diluted 10 000-fold to a working concentration of 10 nM and the antibody–AP conjugates were diluted 100-fold to 500 ng mL–1. Aliquots of diluted PSA (to achieve 150 pM, 210 pM, or 300 pM final concentration) in 50 μL human plasma were vortexed with 5.7 μL capture conjugate (30 nM). Five microliters of diluted detection conjugate was then added, followed by PBS to final volume as 100 μL. The solution was vortexed and allowed to mix at room temperature for 15 min.
Three microfluidic device inputs were filled with (1) immunocomplex in 50% plasma, (2) pH 9.5 Tris (50 mM Tris, 100 nM NaCl, 10 mM MgCl2) wash buffer, and (3) 5 mM 4-methylumbelliferyl phosphate (4-MUP, M-6491, Invitrogen) in pH 9.5 Tris, respectively. When the device was held at 25 °C, immunocomplexes were loaded into the recirculator at 5 μL min–1 for 30 s. After the recirculator was loaded and sealed the device temperature was raised to 39 °C and the solution in the recirculator were mixed for 2 min at 5 Hz. The recirculator was then washed with pH 9.5 Tris for 1 min at 1 μL min–1. For signal generation, MUP was loaded into the recirculator at 1 μL min–1 for 2 min. After the recirculator was sealed, the solution was mixed at 5 Hz and fluorescence images were acquired every 30 s (with the shutter closed between exposures) for 10 min and once again at 15 min. Post assay, the entire device was washed with pH 9.5 Tris as it cooled to room temperature. Finally, the device was heated to 70 °C and washed with a pH 9.5 Tris 10 mM EDTA solution for 15 min to inactivate any residual AP.
PSA Sandwich Immunocomplex Enrichment via Microfluidic Immuoassay
The assay with immunocomplex enrichment was carried out using the aforementioned protocol with minor modification (Scheme 4). Instead of starting the assay with a 25 °C device, the enrichment assay was carried out with a device that was maintained at 39 °C constantly. The immunocomplex solution was loaded into the preheated recirculator at 5 μL min–1 for 30 s (2.5 μL), followed by the mixing and washing processes. In order to enrich immunocomplexes within the recirculator, the load, wash, and mixing steps were repeated a total of three times to achieve a “7.5 μL” condition. The substrate load and turnover, device wash, and EDTA processes were all the same as the aforementioned protocol.
Acknowledgments
The authors would like to express their gratitude to the NIH for funding (EB000252). The authors would like to thank Dr. Albert Folch for the use of clean room facilities and equipment.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Anderson N. L. (2010) The clinical plasma proteome: a survey of clinical assays for proteins in plasma and serum. Clin. Chem. 56(2), 177–85. [DOI] [PubMed] [Google Scholar]
- a Anderson N. L.; Anderson N. G. (2002) The human plasma proteome: history, character, and diagnostic prospects. Mol. Cell. Proteomics 1(11), 845–67. [DOI] [PubMed] [Google Scholar]; b Thaxton C. S.; Elghanian R.; Thomas A. D.; Stoeva S. I.; Lee J. S.; Smith N. D.; Schaeffer A. J.; Klocker H.; Horninger W.; Bartsch G.; Mirkin C. A. (2009) Nanoparticle-based bio-barcode assay redefines ″undetectable″ PSA and biochemical recurrence after radical prostatectomy. Proc. Natl. Acad. Sci. U.S.A. 106(44), 18437–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lequin R. M. (2005) Enzyme immunoassay (EIA)/enzyme-linked immunosorbent assay (ELISA). Clin. Chem. 51(12), 2415–8. [DOI] [PubMed] [Google Scholar]; b Warsinke A. (2009) Point-of-care testing of proteins. Anal. Bioanal. Chem. 393(5), 1393–405. [DOI] [PubMed] [Google Scholar]
- a Blick K. E. (1999) Current trends in automation of immunoassays. J. Clin. Ligand Assay 22(1), 6–12. [Google Scholar]; b Diamandis E. P., and Christopoulos T. K. (1996) Immunoassay, p xxxii, Academic Press, San Diego. [Google Scholar]
- a Stern E.; Vacic A.; Rajan N. K.; Criscione J. M.; Park J.; Ilic B. R.; Mooney D. J.; Reed M. A.; Fahmy T. M. (2010) Label-free biomarker detection from whole blood. Nat. Nanotechnol. 5(2), 138–42. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wang Y. C.; Han J. Y. (2008) Pre-binding dynamic range and sensitivity enhancement for immuno-sensors using nanofluidic preconcentrator. Lab Chip 8(3), 392–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Chatterjee D.; Ytterberg A. J.; Son S. U.; Loo J. A.; Garrell R. L. (2010) Integration of protein processing steps on a droplet microfluidics platform for MALDI-MS analysis. Anal. Chem. 82(5), 2095–101. [DOI] [PubMed] [Google Scholar]; b Hunter R. A.; Privett B. J.; Henley W. H.; Breed E. R.; Liang Z.; Mittal R.; Yoseph B. P.; McDunn J. E.; Burd E. M.; Coopersmith C. M.; Ramsey J. M.; Schoenfisch M. H. (2013) Microfluidic amperometric sensor for analysis of nitric oxide in whole blood. Anal. Chem. 85(12), 6066–72. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Mohan R.; Mukherjee A.; Sevgen S. E.; Sanpitakseree C.; Lee J.; Schroeder C. M.; Kenis P. J. (2013) A multiplexed microfluidic platform for rapid antibiotic susceptibility testing. Biosens. Bioelectron. 49, 118–25. [DOI] [PubMed] [Google Scholar]
- Weigl B.; Domingo G.; Labarre P.; Gerlach J. (2008) Towards non- and minimally instrumented, microfluidics-based diagnostic devices. Lab Chip 8(12), 1999–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gervais L.; Delamarche E. (2009) Toward one-step point-of-care immunodiagnostics using capillary-driven microfluidics and PDMS substrates. Lab Chip 9(23), 3330–7. [DOI] [PubMed] [Google Scholar]
- Murphy B. M.; He X.; Dandy D.; Henry C. S. (2008) Competitive immunoassays for simultaneous detection of metabolites and proteins using micromosaic patterning. Anal. Chem. 80(2), 444–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Auditore-Hargreaves K.; Houghton R. L.; Monji N.; Priest J. H.; Hoffman A. S.; Nowinski R. C. (1987) Phase-separation immunoassays. Clin. Chem. 33(9), 1509–16. [PubMed] [Google Scholar]; b Hage D. S. (1998) Survey of recent advances in analytical applications of immunoaffinity chromatography. J. Chromatogr., B 715(1), 3–28. [DOI] [PubMed] [Google Scholar]; c Peoples M. C.; Karnes H. T. (2008) Microfluidic immunoaffinity separations for bioanalysis. J. Chromatogr., B 866(1–2), 14–25. [DOI] [PubMed] [Google Scholar]; d Sebra R. P.; Masters K. S.; Bowman C. N.; Anseth K. S. (2005) Surface grafted antibodies: controlled architecture permits enhanced antigen detection. Langmuir 21(24), 10907–11. [DOI] [PubMed] [Google Scholar]; e Sebra R. P.; Masters K. S.; Cheung C. Y.; Bowman C. N.; Anseth K. S. (2006) Detection of antigens in biologically complex fluids with photografted whole antibodies. Anal. Chem. 78(9), 3144–51. [DOI] [PubMed] [Google Scholar]
- a Fong R. B.; Ding Z. L.; Hoffman A. S.; Stayton P. S. (2002) Affinity separation using an Fv antibody fragment-″smart″ polymer conjugate. Biotechnol. Bioeng. 79(3), 271–276. [DOI] [PubMed] [Google Scholar]; b Hoffman A. S.; Stayton P. S. (2007) Conjugates of stimuli-responsive polymers and proteins. Prog. Polym. Sci. 32(8–9), 922–932. [Google Scholar]; c Kulkarni S.; Schilli C.; Grin B.; Muller A. H.; Hoffman A. S.; Stayton P. S. (2006) Controlling the aggregation of conjugates of streptavidin with smart block copolymers prepared via the RAFT copolymerization technique. Biomacromolecules 7(10), 2736–41. [DOI] [PubMed] [Google Scholar]; d Sugiura S.; Imano W.; Takagi T.; Sakai K.; Kanamori T. (2009) Thermoresponsive protein adsorption of poly(N-isopropylacrylamide)-modified streptavidin on polydimethylsiloxane microchannel surfaces. Biosens. Bioelectron. 24(5), 1135–40. [DOI] [PubMed] [Google Scholar]
- Blake R. C. 2nd; Pavlov A. R.; Blake D. A. (1999) Automated kinetic exclusion assays to quantify protein binding interactions in homogeneous solution. Anal. Biochem. 272(2), 123–34. [DOI] [PubMed] [Google Scholar]
- a Lai J. J.; Hoffman J. M.; Ebara M.; Hoffman A. S.; Estournes C.; Wattiaux A.; Stayton P. S. (2007) Dual magnetic-/temperature-responsive nanoparticles for microfluidic separations and assays. Langmuir 23(13), 7385–91. [DOI] [PubMed] [Google Scholar]; b Lai J. J.; Nelson K. E.; Nash M. A.; Hoffman A. S.; Yager P.; Stayton P. S. (2009) Dynamic bioprocessing and microfluidic transport control with smart magnetic nanoparticles in laminar-flow devices. Lab Chip 9(14), 1997–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y. N.; Whitesides G. M. (1998) Soft lithography. Annu. Rev. Mater. Sci. 28, 153–184. [Google Scholar]
- Squires T. M.; Quake S. R. (2005) Microfluidics: Fluid physics at the nL scale. Rev. Mod. Phys. 77(3), 977–1026. [Google Scholar]
- Studer V.; Hang G.; Pandolfi A.; Ortiz M.; Anderson W. F.; Quake S. R. (2004) Scaling properties of a low-actuation pressure microfluidic valve. J. Appl. Phys. 95(1), 393–398. [Google Scholar]
- Chou H. P.; Unger M. A.; Quake S. R.; Microfabricated A. (2001) Rotary Pump. Biomed. Microdev. 3(4), 323–330. [Google Scholar]
- a Hsu C. H.; Folch A., Spatio-temporally-complex concentration profiles using a tunable chaotic micromixer. Appl. Phys. Lett. 2006, 89 (14);. [Google Scholar]; b Stroock A. D.; Dertinger S. K.; Ajdari A.; Mezic I.; Stone H. A.; Whitesides G. M. (2002) Chaotic mixer for microchannels. Science 295(5555), 647–51. [DOI] [PubMed] [Google Scholar]
- Liu J.; Williams B. A.; Gwirtz R. M.; Wold B. J.; Quake S. (2006) Enhanced signals and fast nucleic acid hybridization by microfluidic chaotic mixing. Angew. Chem. 45(22), 3618–23. [DOI] [PubMed] [Google Scholar]
- Hoffman J. M.; Ebara M.; Lai J. J.; Hoffman A. S.; Folch A.; Stayton P. S. (2010) A helical flow, circular microreactor for separating and enriching ″smart″ polymer-antibody capture reagents. Lab Chip 10(22), 3130–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golden A. L.; Battrell C. F.; Pennell S.; Hoffman A. S.; Lai J. J.; Stayton P. S. (2010) Simple fluidic system for purifying and concentrating diagnostic biomarkers using stimuli-responsive antibody conjugates and membranes. Bioconjugate Chem. 21(10), 1820–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Schuck P. (1996) Kinetics of ligand binding to receptor immobilized in a polymer matrix, as detected with an evanescent wave biosensor. I. A computer simulation of the influence of mass transport. Biophys. J. 70(3), 1230–49. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Schuck P.; Minton A. P. (1996) Kinetic analysis of biosensor data: elementary tests for self-consistency. Trends Biochem. Sci. 21(12), 458–60. [DOI] [PubMed] [Google Scholar]; c Schuck P.; Minton A. P. (1996) Analysis of mass transport-limited binding kinetics in evanescent wave biosensors. Anal. Biochem. 240(2), 262–72. [DOI] [PubMed] [Google Scholar]
- Ebara M.; Hoffman J. M.; Stayton P. S.; Hoffman A. S. (2007) Surface modification of microfluidic channels by UV-mediated graft polymerization of non-fouling and ’smart’ polymers. Radiat. Phys. Chem. 76(8–9), 1409–1413. [Google Scholar]
- a Dordi B.; Schonherr H.; Vancso G. J. (2003) Reactivity in the confinement of self-assembled monolayers: Chain length effects on the hydrolysis of N-hydroxysuccinimide ester disulfides on gold. Langmuir 19(14), 5780–5786. [Google Scholar]; b Tournier E. J. M.; Wallach J.; Blond P. (1998) Sulfosuccinimidyl 4-(N-maleimidomethyl)-1-cyclohexane carboxylate as a bifunctional immobilization agent. Optimization of the coupling conditions. Anal. Chim. Acta 361(1–2), 33–44. [Google Scholar]
- a Avery K. N. L.; Blazeby J. M.; Lane J. A.; Neal D. E.; Hamdy F. C.; Donovan J. L. (2008) Decision-making about PSA testing and prostate biopsies: A qualitative study embedded in a primary care randomised trial. Eur. Urol. 53(6), 1186–1193. [DOI] [PubMed] [Google Scholar]; b Chou R.; Croswell J. M.; Dana T.; Bougatsos C.; Blazina I.; Fu R.; Gleitsmann K.; Koenig H. C.; Lam C.; Maltz A.; Rugge J. B.; Lin K. (2011) Screening for prostate cancer: a review of the evidence for the U.S. Preventive Services Task Force. Ann. Int. Med. 155(11), 762–71. [DOI] [PubMed] [Google Scholar]; c Partin A. W.; Criley S. R.; Subong E. N. P.; Zincke H.; Walsh P. C.; Oesterling J. E. (1996) Standard versus age-specific prostate specific antigen reference ranges among men with clinically localized prostate cancer: A pathological analysis. J. Urol. 155(4), 1336–1339. [PubMed] [Google Scholar]; d Recker F.; Kwiatkowski M. K.; Huber A.; Stamm B.; Lehmann K.; Tscholl R. (2001) Prospective detection of clinically relevant prostate cancer in the prostate specific antigen range 1 to 3 ng./ml. combined with free-to-total ratio 20% or less: the Aarau experience. J. Urol. 166(3), 851–5. [PubMed] [Google Scholar]; e Schroder F. H.; Hugosson J.; Roobol M. J.; Tammela T. L. J.; Ciatto S.; Nelen V.; Kwiatkowski M.; Lujan M.; Lilja H.; Zappa M.; Denis L. J.; Recker F.; Paez A.; Maattanen L.; Bangma C. H.; Aus G.; Carlsson S.; Villers A.; Rebillard X.; van der Kwast T.; Kujala P. M.; Blijenberg B. G.; Stenman U. H.; Huber A.; Taari K.; Hakama M.; Moss S. M.; de Koning H. J.; Auvinen A.; Investigators E. (2012) Prostate-cancer mortality at 11 years of follow-up. New Engl J. Med. 366(11), 981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Tate J.; Ward G. (2004) Interferences in immunoassay. Clin. Biochem. Rev. (Ultimo, Aust.) 25(2), 105–20. [PMC free article] [PubMed] [Google Scholar]; b Rosenberg-Hasson Y.; Hansmann L.; Liedtke M.; Herschmann I.; Maecker H. T. (2014) Effects of serum and plasma matrices on multiplex immunoassays. Immunol. Res. 58(2–3), 224–33. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Dodig S. (2009) Interferences in quantitative immunochemical methods. Biochem. Medica 19(1), 50–62. [Google Scholar]
- Thompson J. C.; Mazoh J. A.; Hochberg A.; Tseng S. Y.; Seago J. L. (1991) Enzyme-amplified rate conductimetric immunoassay. Anal. Biochem. 194(2), 295–301. [DOI] [PubMed] [Google Scholar]
- Lionello A.; Josserand J.; Jensen H.; Girault H. H. (2005) Dynamic protein adsorption in microchannels by ″stop-flow″ and continuous flow. Lab Chip 5(10), 1096–103. [DOI] [PubMed] [Google Scholar]
- a Forest J. C.; Masse J.; Lane A. (1998) Evaluation of the analytical performance of the Boehringer Mannheim Elecsys 2010 immunoanalyzer. Clin. Biochem. 31(2), 81–8. [DOI] [PubMed] [Google Scholar]; b Morris D. L.; Dillon P. W.; Very D. L.; Ng P.; Kish L.; Goldblatt J. L.; Bruzek D. J.; Chan D. W.; Ahmed M. S.; Witek D.; Fritsche H. A.; Smith C.; Schwartz D.; Schwartz M. K.; Noteboom J. L.; Vessella R. L.; Yeung K. K.; Allard W. J. (1998) Bayer Immuno 1 PSA Assay: an automated, ultrasensitive method to quantitate total PSA in serum. J. Clin. Lab. Anal. 12(1), 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ferguson R. A.; Yu H.; Kalyvas M.; Zammit S.; Diamandis E. P. (1996) Ultrasensitive detection of prostate-specific antigen by a time-resolved immunofluorometric assay and the Immulite immunochemiluminescent third-generation assay: potential applications in prostate and breast cancers. Clin. Chem. 42(5), 675–84. [PubMed] [Google Scholar]
- a Pugia M. J.; Blankenstein G.; Peters R. P.; Profitt J. A.; Kadel K.; Willms T.; Sommer R.; Kuo H. H.; Schulman L. S. (2005) Microfluidic tool box as technology platform for hand-held diagnostics. Clin. Chem. 51(10), 1923–1932. [DOI] [PubMed] [Google Scholar]; b Huang C. J.; Chen Y. H.; Wang C. H.; Chou T. C.; Lee G. B. (2007) Integrated microfluidic systems for automatic glucose sensing and insulin injection. Sens. Actuators, B 122(2), 461–468. [Google Scholar]
- Henry S. M.; Convertine A. J.; Benoit D. S.; Hoffman A. S.; Stayton P. S. (2009) End-functionalized polymers and junction-functionalized diblock copolymers via RAFT chain extension with maleimido monomers. Bioconjugate Chem. 20(6), 1122–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebara M.; Hoffman J. M.; Hoffman A. S.; Stayton P. S. (2006) Switchable surface traps for injectable bead-based chromatography in PDMS microfluidic channels. Lab Chip 6(7), 843–8. [DOI] [PubMed] [Google Scholar]