

Abstract
Virus-based nanoparticles (VNPs) have been used for a wide range of applications, spanning basic materials science and translational medicine. Their propensity to self-assemble into precise structures that offer a three-dimensional scaffold for functionalization has led to their use as optical contrast agents and related biophotonics applications. A number of fluorescently labeled platforms have been developed and their utility in optical imaging demonstrated, yet their optical properties have not been investigated in detail. In this study, two VNPs of varying architectures were compared side-by-side to determine the impact of dye density, dye localization, conjugation chemistry, and microenvironment on the optical properties of the probes. Dyes were attached to icosahedral cowpea mosaic virus (CPMV) and rod-shaped tobacco mosaic virus (TMV) through a range of chemistries to target particular side chains displayed at specific locations around the virus. The fluorescence intensity and lifetime of the particles were determined, first using photochemical experiments on the benchtop, and second in imaging experiments using tissue culture experiments. The virus-based optical probes were found to be extraordinarily robust under ultrashort, pulsed laser light conditions with a significant amount of excitation energy, maintaining structural and chemical stability. The most effective fluorescence output was achieved through dye placement at optimized densities coupled to the exterior surface avoiding conjugated ring systems. Lifetime measurements indicate that fluorescence output depends not only on spacing the fluorophores, but also on dimer stacking and configurational changes leading to radiationless relaxation—and these processes are related to the conjugation chemistry and nanoparticle shape. For biological applications, the particles were also examined in tissue culture, from which it was found that the optical properties differed from those found on the benchtop due to effects from cellular processes and uptake kinetics. Data indicate that fluorescent cargos are released in the endolysosomal compartment of the cell targeted by the virus-based optical probes. These studies provide insight into the optical properties and fates of fluorescent proteinaceous imaging probes. The cellular release of cargo has implications not only for virus-based optical probes, but also for drug delivery and release systems.
Introduction
Fluorescent nanomaterials including nanostructures formed by viruses and protein cages have become versatile tools as photonic materials for a variety of applications, such as sensing,1,2 light harvesting,3 and optical imaging.4,5 Virus-based nanostructures are self-assembling systems that are highly symmetrical, dynamic, polyvalent, and monodisperse, rendering them one of the most advanced nanomaterials produced in nature. The proteinaceous capsid’s function is to protect the nucleic acid cargo; hence, their structures are extremely robust. Many structures have been solved to near-atomic resolution, allowing chemists, engineers, and physicists to tailor materials with atomic precision. For example, structure-based engineering allows the placement of metals with spatial control at the atomic level through genetic control, yielding unique plasmonic nanomaterials.6 The propensity to self-assemble into higher-order structures is another interesting feature; for example, hybrid virus-like nanoparticles encapsulating gold nanoparticles inside the virus shell were shown to crystallize into lattices exhibiting properties of plasmonic metamaterials.7 The structure of viral nanoparticles (VNPs) can be modified in several ways to allow for the loading of photonic and plasmonic materials within the internal cavity or conjugated to the exterior surfaces.8
Within the medical sector, virus-based probes combined with optical dyes are frequently used to study their biodistribution9−11 and to evaluate cellular internalization and localization.12,13 Fluorescently labeled virus-based materials can also be applied as tags for high-throughput flow cytometry applications.14 Last but not least, virus-based materials labeled with fluorophores have been demonstrated as excellent tools in optical and molecular imaging. For example, cowpea mosaic virus (CPMV), which naturally targets mammalian cells via interaction with surface expressed vimentin,15,16 has been applied to imaging of tumor neovasculature as well as the inflamed endothelium sites at which surface vimentin is upregulated.4,16,17 Other approaches include genetic or chemical incorporation of receptor-specific peptide ligands, which enables tissue-specific imaging in various preclinical animal models.5,18
It is clear that fluorescently labeled virus-based materials have become popular materials spanning a variety of applications in materials and medicine. While a variety of virus-based probes have been reported in the literature, detailed studies about their optical properties have not been reported. Therefore, we set out to determine the impact of spatial dye placement, dye density, conjugation chemistry, and microenvironment using an icosahedral platform, CPMV, measuring 30 nm in diameter, as well as the elongated and stiff rods formed by tobacco mosaic virus (TMV), measuring 300 × 18 nm. Distinct sets of fluorescently labeled CPMV and TMV nanoparticles were synthesized, and their optical properties were studied on the benchtop as well as in cellular environments. A combination of steady-state and time-resolved fluorescence measurements and flow cytometry and confocal microscopy was used. The overall goal of this study was to define some design rules for the construction of virus-based optical probes and to define their stability and fluorescence output in different settings. Understanding these properties is expected to help drive the development of fluorescent-based VNPs for applications in biophotonics and plasmonics.
Results and Discussion
Fluorescent Labeling of CPMV and TMV
The surface chemistry of CPMV and TMV is well understood. The 30-nm-sized CPMV displays 300 addressable lysine side chains per particle (Figure 1A), all of which are addressable using N-hydroxysuccinimide (NHS)-activated esters at large molar excess and overnight incubation.19,20 TMV forms a hollow tube measuring 300 × 18 nm with a 4-nm-wide interior channel. Its surface chemistry inside and out is well established. Native TMV contains solvent-exposed and addressable interior glutamic acids, Glu97 and Glu106 residues, which can be modified using carbodiimide coupling reactions. The exterior surface contains a solvent-exposed tyrosine side chain, Tyr139, which can be targeted and functionalized using diazonium coupling reactions.21 For our studies, we also considered a lysine mutant, TMVLys,22 which displays a genetically introduced lysine residue that replaces threonine at amino acid position 158. TMV consists of 2130 identical copies of its coat protein, which means that overall TMV displays 4260 solvent-exposed glutamic acids on the interior of the particle and 2130 addressable tyrosines on its exterior surface; the TMVLys mutants offers an additional 2130 surface-exposed lysine side chains on the exterior particle surface (Figure 1B).
Figure 1.

Schematic of CPMV and TMV modifications. (a,b) Structures of CPMV and TMV with amino acid residues available for modification shown on a single coat protein. (c) Labeling of CPMV with sulfo-Cy5 NHS ester. (d) Conjugation to interior glutamic acid residues through EDC coupling. (e) Exterior modification of tyrosine residues through diazonium coupling. (f) Conjugation of lysine mutant through NHS chemistry. (g) Click chemistry for dye attachment, with structure of sCy5 the same as shown in (c).
CPMV was labeled with cyanine dye sulfo-Cy5 using NHS-activated esters targeting surface lysines (Figure 1C). CPMV was incubated with NHS-sCy5 at various molar excesses yielding CPMV-sCy5 conjugates with varying densities of dye per particle. After completion of the reaction, the resulting CPMV-sCy5 conjugates were purified by dialysis using centrifugal filter units; the conjugates were characterized using a combination of UV–vis spectroscopy, native and denaturing gel electrophoresis, and transmission electron microscopy (TEM).
UV–vis spectroscopy was used to determine the degree of labeling. Absorbance was measured and the Beer–Lambert law and the fluorophore- and CPMV-specific extinction coefficients were used to determine the number of dyes per particle formulation. Molar excesses ranging from 1000 to 8000 dyes per particle were used to obtain formulations with 8, 20, 27, 40, 48, 55 sCy5 per CPMV particle (Supporting Information Figure S1). Native and denaturing gel electrophoresis confirmed covalent attachment of the dyes, and TEM imaging confirmed that the particles remained structurally sound after chemical modification (see below).
We next turned to TMV and its mutant TMVLys, which offer attractive platforms to evaluate how spatial dye placement, conjugation chemistry, and microenvironment would affect the fluorescence properties of the nanoparticle probes. The conjugation with sCy5 was achieved using a two-step protocol: first, alkyne ligation handles were introduced, and second, copper-catalyzed azide–alkyne cycloaddition (“click” chemistry) was carried out to introduce sCy5 azide. To decorate the interior surface, a terminal alkyne was incorporated into the interior channel of TMV by targeting glutamic acid residues, designated TMV-iAlk.21 To decorate the exterior TMV surface, tyrosine residues were targeted with the diazonium salt generated from 3-ethynylaniline to yield TMV-eAlk.23 Similarly, to target exterior lysine residues using the TMVLys mutant, an NHS-active ester was used to introduce alkynes to yield TMVLys-eAlk. The reaction schemes are shown in Figure 1D–G.
Alkyne labeling was carried out under forcing conditions, i.e., a large molar excess of alkyne to TMV and/or overnight incubation, to yield maximum conversion of the carboxylic acid, hydroxyphenyl ring, or amine functional groups into alkynes. In brief, 25 molar excess of propargylamine per coat protein was reacted using EDC coupling overnight to produce TMV-iAlk, 35 equiv of ethynylaniline diazonium salt was reacted for 30 min to yield TMV-eAlk, and 10 molar excess of NHS-alkyne was reacted overnight with TMVLys to form TMVLys-eAlk. To conjugate fluorophores to the TMV nanoparticles, TMV-iAlk, TMV-eAlk, and TMVLys-eAlk were incubated with sCy5 azide at various molar excesses yielding TMV-i-sCy5, TMV-e-sCy5, and TMVLys-e-sCy5 conjugates with varying densities of dye per particle. After completion of the reaction, the resulting TMV(Lys)-e/i-sCy5 conjugates were purified by ultracentrifugation; the conjugates were characterized using a combination of UV–vis spectroscopy, native and denaturing gel electrophoresis, and transmission electron microscopy (TEM) (see below).
As in the case of CPMV, UV–vis spectroscopy was used to determine the degree of labeling, with the minor adjustment of using TMV-specific extinction coefficients to determine the number of dyes per particle formulation. For TMV-i-sCy5, molar excesses from 0.2 to 6 dyes per coat protein (426 to 12 780 per particle) were used to obtain 68, 97, 222, 299, 402, 488, and 555 sCy5 per TMV particle; for TMV-e-sCy5, excesses from 0.3 to 6 dyes per coat protein resulted in 124, 164, 324, 396, 509, and 613 sCy5 per particle; and for TMVLys-sCy5, excesses ranging from 0.02 to 2 sCy5 per coat protein were used to obtain 81, 165, 234, 302, and 365 dyes per particle (Supporting Information Figure S2). Aggregation occurred for TMVLys above a molar excess of 2, with very little increase in labeling efficiency observed. Denaturing gel electrophoresis confirmed covalent attachment of the dyes and TEM imaging confirmed that the particles remained structurally sound after chemical modification (see below).
Fluorescence Properties of CPMV- and TMV-Dye Conjugates
To evaluate the fluorescence properties of CPMV and TMV-dye conjugates, steady-state and time-resolved fluorescence measurements were carried out (Figures 2 and 3).
Figure 2.
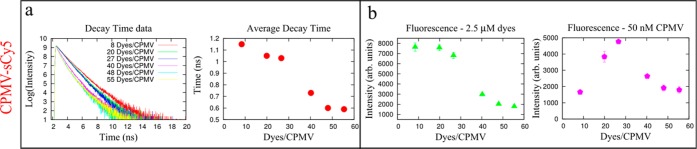
Lifetime and fluorescence characterization of CPMV-sCy5 formulations. (a) Fluorescence lifetime decay measurements as a function of dye number for CPMV-sCy5. (b) Fluorescence intensity measurements normalized for dye concentration (left) and protein concentration (right).
Figure 3.

Lifetime and fluorescence characterization of TMV-sCy5 for different formulations. (a, c, e) Lifetime decay measurements as a function of dye number for TMV-i-sCy5, TMV-e-sCy5, and TMVLys-sCy5, respectively. (b, d, f) Fluorescence intensity measurements normalized for dye concentration (left) and protein concentration (right) for TMV-i-sCy5, TMV-e-sCy5, and TMVLys-sCy5, respectively.
Here, time-resolved fluorescence spectroscopy is primarily employed to evaluate the lifetime of excitonic states and to differentiate between various processes leading to photoluminescence dimming. Short-lived excitonic states (picoseconds time scale) are mainly related to radiationless transitions between donors and acceptors located in close proximity (few nanometers), mostly resulting in fluorescence quenching. However, many molecular events such as rotational diffusion, resonance energy transfer, dimer trapping, and dynamic quenching occur on the same time scale as fluorescence decay. Thus, it is of vital importance to discriminate between the causes of the fluorescence quenching to design effective systems.
CPMV-sCy5 with dye loading between 8 and 55 dyes per CPMV were compared (data were normalized for dye or CPMV concentration, respectively). It was apparent that the sample with highest fluorescence intensity (FI) was not the sample with highest dye content: CPMV displaying 27 sCy5 dyes gave a fluorescence reading of 4768 cts, which is over 2.5 times the fluorescence intensity of 1795 cts for CPMV with 55 dyes at the same particle concentration. This indicates that maximum fluorescence intensity is achieved at sparse labeling with ∼30 dyes per particle; as the dye density increases, fluorescence quenching occurs, reducing the overall emission intensity. It is well-known that fluorescent molecules in close proximity (d < 10 nm) undergo interactions and couplings that may lead to resonant excitation energy transfer processes due to the partial overlap of their absorption and emission curves.24−26 This phenomenon is referred to as Förster resonant energy transfer (FRET) and involves a donor fluorophore in an excited electronic state, which may transfer its excitation energy to a nearby acceptor chromophore in a nonradiative way through long-range dipole–dipole interactions. FRET processes are associated with the observation of fluorescence quenching and reduction of emission decay times. It is also known that FRET strongly depends on the distance between donor and acceptor molecules and scales as R–6, where R is the molecular interdistance. Increasing the number of sCy5 dye molecules per CPMV results in a decrease of the distance between dye molecules and consequently leads to stronger dipolar coupling between them (Supporting Information Figure S3; the spacing between dye molecules was calculated based on the average distance between each dye’s nearest neighbor in multiple simulations where the dyes were randomly positioned on the available Lys side chains (the coordinates were determined from the structure of CPMV, which is available at viperdb.scripps.edu, file 1NY7).
The proximity of excitons is commonly considered to be responsible for fluorescence quenching. While the proximity does contribute to the overall optical properties of the CPMV-based probes, additional factors must be considered. In particular, the experimental evidence suggests that three radiation-less processes govern the disposition of the electronic excited state energy in this type of system: (i) energy transfer between dye molecules, (ii) trapping by dimers, and (iii) radiationless relaxation of the excited state. Dipolar coupling along with the intrinsic spectral overlap between absorption and emission bands create the conditions for an effective FRET. In the case of CPMV, fluorescence intensity measurements show an initial monotonic increase of emission as a function of the dye molecules for CPMV, and the maximum value is obtained for 27 dyes/CPMV. At this density, the average interdye separation is estimated to be about 8 nm and lifetime decay was measured to be longer than 1 ns, indicating the absolute absence of coupling between molecules. Above this level of loading the average separation distance between dye molecules become short enough to induce strong molecular coupling and to trigger FRET processes, causing a reduction of the emission. This was accompanied by a reduction of the fluorescence decay time by 43%, confirming that dye coupling and energy transfer are behind this effect (see Figure 2). However, this fluorescence lifetime reduction is not compatible with the formation of trapped dimers, which usually show much shorter decay time (<100 ps). This indicates that dimers undergo rapid configurational changes, which enhance the radiationless relaxation rates.
With regard to the fluorescence, similar trends were observed when comparing the TMV samples; the underlying photophysics however differed. Each sample reached a plateau at a specific dye-to-TMV ratio, and this was dependent on the spatial placement and independent of the chemistry processes. Specifically, for either of the externally labeled TMV samples, maximum FI was reached upon attachment of 165 sCy5 dyes to either tyrosine or lysine side chains. On the other hand, much higher dye loading was required to reach a maximal fluorescence intensity (FI) of 774 cts for TMV-i-sCy5 labeled with 402 sCy5 (Figure 3). When measuring the FI values of the exteriorly labeled particles with the same density of 165 dyes attached, we found that TMVLys-e-sCy5 reached an intensity of 1616 cts, which was 4.5 times higher compared to the FI value of 354 cts observed for TMV-e-sCy5. This evidence of a lower quantum yield for TMV-e-sCy5 can be related to the formation of dimers. In fact, it is important to note that lifetime measurements show very short-lived excitonic states (τ ≤ 100 ps), indicative of dimer trapping, above 300 dyes/TMV. It is possible that the planar shape of the tyrosine residue results in stacking of the sCy5 dyes on the high aspect ratio, flat TMV structure, therefore resulting in dimer trapping. In contrast, steady-state fluorescence data for TMV-i-sCy5 and TMVLys-e-sCy5 show higher values of FI with respect TMV-e-sCy5 (see Figure 3). Such evidence of the importance of conjugation chemistry is additionally corroborated by comparable decay times (τ ≥ 400 ps) for the systems showing higher FI.
It was interesting to note that stark differences were observed when comparing the various formulations. Normalized for particle concentration, the brightest CPMV sample (CPMV-sCy5 with 27 dyes) resulted in fluorescence intensities four times higher (FI ∼ 4768 cts) than the brightest TMV sample (TMVLys-e-sCy5, FI ∼ 1616 cts), despite both samples having comparable dye concentrations in the range of 0.2 to 0.3 mg/mL. For the brightest TMV samples, TMV-i-sCy5 had an intermediate FI about twice that of TMV-e-sCy5, while the brightest TMV sample across the board was TMVLys-e-sCy5 reaching a FI another factor of 2 times that of TMV-i-sCy5. These data indicate that not only spatial placement (inside versus outside), conjugation chemistry (see Figure 1), and dye density, but also nanoparticle shape and microenvironment dictate the fluorescence properties of (virus-based) nanoparticles. Time-resolved fluorescence measurements play a key role in gaining further insight in the processes behind the change of the radiative emission rate of the fluorophores. The measurements allow us to understand whether fluorescence quantum yield is affected by the chemical microenvironment or energy transport. At low values of dyes/TMV, electronic excited state energy transport occurs due to dipole–dipole interactions between the dye molecules, and the energy transport causes fluorescence depolarization effects while not affecting the fluorescence quantum yield.
The lowest FIs, accompanied by corresponding shortest fluorescence decay times (τ ≤ 100 ps), were observed when studying the TMV-e-sCy5 samples. This can be explained by the conjugation chemistry and the formation of dimers. In this case, the fluorophores were placed on the aromatic tyrosine residues via diazonium bonds, and electron delocalization in the conjugated ring systems induces a reduction of the spontaneous emission rate. It is generally more effective to conjugate dyes via nonaromatic systems to avoid quenching. On the other hand, the formation of dimers, as proven by the very short-lived states, represents a configuration with the fastest channel to release the excitation energy nonradiatively because of their extremely rapid conformational changes.
TMVLys-e-sCy5 reached higher FIs compared to those obtained for the TMV-i-sCy5; this may be explained by spatial localization: internal placement results in crowding of the dyes. The average distance of dyes in the TMV-i-sCy5 sample containing 402 dyes is 3 nm, compared to 8 nm for TMV-e-sCy5 and TMVLys-e-sCy5 with 165 dyes (see Supporting Information Figure S3). As 8 nm was also the interdye distance found for CPMV-sCy5 with 27 dyes, this is likely the optimal distance for sCy5 dyes spread along the exterior viral capsid wall. Indeed, the rate of energy transfer is inversely proportional to the sixth power of the distance between the donor and acceptor. Therefore, the efficiency of the transfer rapidly declines to zero at distances larger than the Förster radius. Förster radii have been experimentally determined for each specific donor–acceptor pair, and the majority of fluorophore pairs fall within the 5–10 nm range.27−29 Since the interior channel of TMV is only 4 nm wide, decreasing the dye labels to 165 dyes for TMV-i-sCy5 only increases the distance between dyes to about 3.6 nm. The balance between improving the fluorescence per dye through less dyes per particle to decrease the amount of quenching and increasing the fluorescence per particle by having more dye labels per particle is therefore altered for TMV-i-sCy5 compared to the other particles. Fluorescence lifetime data corroborates this explanation. In fact, the decay times of both TMVLys-e-sCy5 and TMV-e-sCy5 decrease linearly with the dye molecule number, whereas the decay times of TMV-i-sCy5 shows a saturation effect by reaching a plateau around 400 ps at a loading value of 222 dyes per particle.
Overall, these data indicate that design principles beyond interdye spacing must be considered to develop optical probes with maximized fluorescent output: interdye-spacing can be calculated based on the averaged distance between each dye’s nearest neighbors; dye-loading can be optimized through adjustment of the conjugation protocol (excess reagents used and incubation time). To avoid dimer trapping, dyes should be placed to avoid crowding and/or attachment via conjugated ring systems. To overcome losses due to configurational interactions leading to enhanced radiationless relaxation rates, one might consider alternative linker chemistries that may affix the fluorophore in stringent chemical positions.
These quantitative data were correlated with results from gel electrophoresis analysis of CPMV- and TMV-dye conjugates. Samples with highest FI were compared to samples with higher dye loading but lower FI. Specifically, CPMV and CPMV-sCy5 with 27 and 55 dyes per particle as well as TMVLys and TMVLys-e-sCy5 containing 165 and 365 dyes were analyzed by gel electrophoresis. First, CPMV samples were separated using native agarose gels (TMV cannot be analyzed under these experimental conditions, and therefore only denaturing gel analysis was performed). After electrophoretic separation of intact CPMV in native agarose gels, the particles were visualized under UV light as well as Maestro 2D fluorescence imaging system with a yellow (635 nm) long-pass filter and then stained with Coomassie blue followed by imaging under white light (Figure 4A). The mobility of CPMV in the native gel increases upon dye conjugation; the sCy5 dye is a negatively charged molecule, so as more dyes are conjugated to CPMV, the more negative the surface properties of CPMV (see also Supporting Information Figure S4). This enhances the mobility of CPMV-sCy5 toward the positively charged anode as a function of dye-loading. CPMV-sCy5 with 55 sCy5 molecules has the highest mobility compared to CPMV-sCy5 with 27 sCy5 dyes, and native CPMV has the slowest mobility. Analysis of the fluorescence signal intensity (Figure 4A, heat map) shows that CPMV-sCy5 with 27 sCy5 dyes has a higher fluorescence intensity compared to its counterpart displaying 55 dyes per particle, therefore further confirming the fluorescence lifetime and intensity measurements (see Figure 2).
Figure 4.

Gel electrophoresis of particles used for cell studies. (a) Particles run on a 1.2% agarose gel visualized with fluorescence imaging (635 nm long-pass filter) and corresponding heat map, UV light, and after Coomassie staining: 1 = CPMV, 2 = CPMV-sCy5 with 27 dyes; 3 = CPMV-sCy5 with 55 dyes. (b) Particles on 4–12% SDS-PAGE gel run in MES buffer visualized with white light through a 635 nm long-pass filter, fluorescence imaging (635 nm long-pass filter) and corresponding heat map, and after Coomassie staining: 4 = TMV, 5 = TMVLys-sCy5 with 165 dyes, 6 = TMVLys-sCy5 with 365 dyes.
While native gels separate intact viral nanoparticles, in denaturing gels the coat proteins are separated. CPMV consists of 60 copies each of a small (24 kDa) and large (42 kDa) protein, and 2130 copies of a single 15 kDa protein form TMV particles. Both the small and large coat proteins of CPMV are detectable after electrophoretic separation and show the expected sizes; it is apparent that fluorophores were conjugated to both coat protein subunits. We hypothesize that the sCy5 dyes are located around a fivefold axis of CPMV near the interface of the small and large coat proteins where the most reactive lysines (Lys38 and Lys99) are located (see Figure 1A).19,20 The protein concentration between each sample was consistent, which is confirmed by comparable signal intensities in the Coomassie-stained gels. Analysis of the fluorescent signals is consistent with UV/vis quantification of dye loading, as CPMV-sCy5 with 55 sCy5 dyes displays a higher dye per protein density compared to CPMV-sCy5 with 27 sCy5 dyes. Since denatured proteins are imaged in these gels, any quenching effects are lost (Figure 4B). Similarly, fluorescence signal from denaturing gel electrophoresis of TMV samples also indicates a higher dye-to-protein ratio for TMV samples labeled with more dyes (a brighter signal is observed for TMVLys-e-sCy5 containing 365 dyes versus 165 dyes and TMVLys is not fluorescent, Figure 4B).
Stability of Fluorescently Labeled CPMV and TMV
We assessed the stability of CPMV and TMV conjugates, specifically addressing the question of whether CPMV and TMV-dye conjugates remain structurally sound and whether the dyes would remain covalently attached during fluorescence lifetime measurements. After lifetime measurements, the CPMV- and TMV-sCy5 conjugates were evaluated using TEM and native gel electrophoresis (Supporting Information Figure S4). The combination of methods provides insights into the structural integrity as measured by direct TEM imaging, and chemical stability as measured by detection and quantification of the covalent modifications. After completion of electrophoretic separation, CPMV bands were visualized under UV light. The electrophoretic mobility of the bands did not alter before and after measurements, indicating that the particles remain structurally sound and the fluorophores remain covalently attached; no free dye was detectable for any of the samples analyzed, therefore indicating that chemical stability is maintained. These data were in good agreement with TEM analysis of the samples, with the CPMV and TMV samples all found to be intact (Supporting Information Figure S4).
In addition, unlabeled CPMV samples were exposed to various pulse and power settings using various laser beams, both pulsed and continuous wave (CW), to test their stability under optical excitation. Different parameters, such as pulse duration, energy per pulse, repetition rate, wavelength, and average power, were selected to mimic various scenarios relevant to photonics and plasmonic applications (future direction); detailed procedures are described in the Supporting Information (see also Supporting Information Figures S5 and S6). In short, both CPMV and TMV were found to be very robust and stable; being able to endure significantly high excitation energy fluence for a high range of wavelengths makes them suitable candidates for biophotonic applications. Optical excitation by means of trains of ultrashort pulses (about 100 fs) represents a very strict physical requirement to understand radiative and nonradiative mechanisms between excitonic molecules. The virus particle stability has been investigated under severe optical excitation upon varying several physical parameters (wavelength, pulse duration, energy/pulse, and repetition rate) with the precise aim to evaluate stability and tolerances to be used for optical spectroscopies.
Fluorescently Labeled CPMV and TMV in Cell Imaging Applications
We then set out to determine whether the properties observed in the test tube would translate into cell imaging studies. We continued with both CPMV and TMV to additionally evaluate shape-dependent effects, specifically choosing to study TMVLys since it had superior fluorescent properties to internally and externally labeled wild-type particles. Fluorescence and lifetime measurements indicated that a CPMV formulation labeled at external lysine side chains with 27 sCy5 dyes is brighter (FI ∼ 4768 cts) compared to a CPMV sample labeled at external lysines with 55 sCy5 (FI ∼ 1795 cts). Similarly, TMV labeled with 165 sCy5 dyes on its genetically introduced, exterior lysine side chain was brighter (FI ∼ 1616 cts) compared to TMV samples labeled with 365 dyes conjugated using the same chemistry (FI ∼ 539 cts). To test the hypothesis that CPMV-sCy527 and TMVLys-e-sCy5165 would outperform their counterparts with higher dye loading (but reduced fluorescence intensity due to quenching), we performed quantitative flow cytometry studies using HeLa cells; these studies were complemented by confocal imaging studies (Figure 5).
Figure 5.

Cell interactions of CPMV-sCy5 and TMVLys-e-sCy5. (a) Flow cytometry studies with mean fluorescence intensity from CPMV antibody staining with Alexa Fluor 555 secondary (striped bars) and from sCy5 labels on the particles (solid bars). (b) Flow cytometry quantifying the percent of cells positive for TMV uptake determined from TMV antibody staining with Alexa Fluor 555 secondary (striped bars) and from sCy5 labels on the particles (solid bars), obtained from the indicated regions in the histograms to the left. (c,d) Confocal imaging of HeLa cells showing cellular uptake of CPMV-sCy5 and TMVLys-e-sCy5 after 3 h. Nuclei are shown in blue, endolysosomes stained with mouse anti-human Lamp-1 antibody and secondary Alexa Fluor 488 goat anti-mouse antibody are shown in green, and CPMV-sCy5 and TMVLys-e-sCy5 are shown in red. Colocalization is shown in yellow, with associated Manders’ coefficient as indicated. Scale bars = 20 μm.
In brief, CPMV or TMV-based sensors were incubated with HeLa cells at a concentration of 5 × 105 particles per cell; cells were collected at 30 min and 3 h post incubation with CPMV or TMV, fixed, permeabilized, and stained with anti-CPMV or anti-TMV antibodies and fluorescently labeled secondary antibodies. This allowed detection of the signals derived from the fluorophores conjugated to the plant virus-based nanoparticles as well as imaging of the nanoparticles through antibody staining.
In the case of CPMV, antibody staining indicated that the more dyes are conjugated, the more efficiently CPMV nanoparticles are being internalized by the cells, and this effect is more profound at longer incubation times (Figure 5A). It should be noted that flow cytometry does not differentiate between bound and internalized nanoparticles; however, cell uptake was confirmed by confocal microscopy (Figure 5C). Increased cell uptake with increasing numbers of dyes was also observed when studying the TMV formulations (Figure 5B). Increased cell uptake as a function of dye-loading could be explained by sCy5 dye–cell membrane interactions. It may also be possible that altered surface charges contribute to the cell uptake efficiency; however, conjugation of the sCy5 dye contributes to an increased negative charge of the plant virus-based nanoparticles (e.g., see Figure 4); first, the sCy5 itself is negatively charged, and second, conjugation to the surface lysine residues reduces the positive charge contributions. Cell membranes are negatively charged, and therefore positively charged materials interact more strongly with cells.30−32 We hypothesize that the planar, hydrophobic structure of the sCy5 dye interacts directly with the lipid bilayer of the cell membrane to promote cell binding, which then may trigger endocytosis of the nanoparticles. We had previously observed that when potato virus X (PVX) nanoparticles are modified with PEG of a molecular weight of 2000 Da and Alexa Fluor 647, cell interactions are enhanced compared to non-PEGylated PVX. In contrast, cell interactions are reduced when PVX is labeled with the same PEG coating but displays Oregon Green 488 (instead of the Alexa Fluor 647 dye),33 supporting the hypothesis that the fluorophore may play a role in mediating cell interactions and uptake.
Next, cell uptake (as determined based on antibody staining) was compared with the signal strength obtained when cells were imaged based on the conjugated sCy5 dyes: the more dyes that are conjugated the brighter the signals obtained from the cells (Figure 5A,B). The CPMV-sCy555 formulation had a fluorescence signal approximately 1.5 times as strong as that of the CPMV-sCy527 formulation (1.5 at 30 min and 1.6 at 2 h). On the other hand, cell uptake (based on the antibody staining) of the CPMV-sCy555 formulation was even higher, with mean antibody fluorescence 2.3 times as strong at that of the CPMV-sCy527 formulation. Taking the ratio (2.3:1.5), we find that the CPMV-sCy527 formulation indeed is still brighter, with a 1.5-fold higher fluorescence intensity per particle. Since the test tube fluorescence intensity of CPMV-sCy527 was about 2.6-fold stronger compared to that of CPMV-sCy555, this indicates that the somewhat unexpected signal increase for the CPMV-sCy555 formulation can only be in part explained by the increased cell uptake. Other factors may contribute to the fluorescence signal enhancement. Confocal microscopy imaging and colocalization studies indicate that CPMV samples are taken up by cells via endocytosis and colocalize with the endolysosome (Lamp-1 marker, Mander’s colocalization coefficient 0.83, Figure 5C). The endolysosome is a low pH compartment with high protease and hydrolase activity, which may at least in part lead to the dissociation or degradation of the capsid proteins within the 3 h time frame; these structural changes may relax the conformation and packing of the dyes, therefore overcoming quenching and enhancing the fluorescence properties.
In the case of TMV, cell uptake was generally less efficient and only apparent after a 3 h incubation period using TMV displaying 365 sCy5 dyes per particle (Figure 6B). Cell uptake and colocalization with the endolysosome was also confirmed (Figure 6D). The shape of the nanocarrier may explain the reduced cell uptake: the high aspect ratio and stiff nature of the TMV rod (300 × 18 nm) may reduce nonspecific cell interactions and cell uptake kinetics.34−37 It should be noted that targeting ligands were not applied in this study; therefore, cell uptake depends on nonspecific cell interactions, at least in the case of TMV.
Figure 6.

Chemical stability of TMVLys-e-A488 in lysosomal extract over time. (a) Representative SDS-PAGE results; data shown are obtained after 1 day incubation of TMVLys-e-A488 in lysosomal extract. After electrophoretic separation, gels were imaged first under UV light, then stained with Coomassie staining and photographed under white light. M = SeeBluePlus2 protein size standard, the molecular weights (in kDa) are indicated on the left. 1 = BSA protein in KP pH 7.0 (negative control, BSA protein and multimers are detected on the gel), 2 = BSA in lysosomal extract (positive control demonstrating protein degradation as expected), 3 = TMVLys-e-A488 in pH 7.0, 4 = TMVLys-e-A488 in KP pH 5 (acidity has no effect on the chemical stability), 5 = TMVLys-e-A488 in lysosomal extract; release of the fluorophores are apparent. (b) Plots of fluorescence to coat protein intensity (normalized to control lane 3) as measured by lane analysis tool using ImageJ software. TMVLys-e-A488 in KP buffer at pH 7 and pH 5 remain stable, with the A488 stably attached over time; A488 release is detected for TMVLys-e-A488 exposed to lysosomal extracts.
Manchester et al. reported that, despite being a plant pathogen, CPMV binds to and is internalized by mammalian cells through surface-expressed vimentin, a protein that is overexpressed on tumor endothelial cells;15,16 we demonstrated that this interaction can be exploited to target CPMV-based nanoparticles to cancer cells, including HeLa cells expressing surface vimentin.38 Such receptor-specific interactions have not been described for TMV. Therefore, differences in surface chemistry and shape of the nanocarriers may explain their distinct cell uptake behavior. Even though TMV cell uptake is negligible, low level fluorescence signals are detectable and consistent with findings reported for CPMV; the more dye conjugated to TMV, the more efficient its cell uptake properties and the brighter the fluorescence obtained from the conjugated sCy5 dye.
Cell studies pinpoint distinct fluorescent properties of VNPs in cellular environments that do not match the properties determined on the benchtop. These observations indicate structural changes or metabolic degradation of the carrier after cell entry. To gain an initial understanding of this phenomenon, we set out to determine whether chemical stability of the virus-based optical probes was maintained within the endolysosomal compartment. A fluorescently labeled TMVLys-e-A488 sample (conjugated with Alexa Fluor 488) was prepared to enable visualization of fluorescent protein bands and free dye under UV light after electrophoretic separation. TMVLys-e-A488 was incubated in a lysosomal extract obtained from liver tissue of Balb/C mice; then, its integrity over time was examined. Indeed, the data indicate release of the fluorescent cargo from the coat proteins; the coat proteins, however, appeared to remain intact even after several days of exposure (as measured by their molecular weight; Figure 6). Cargo release was also evaluated by exposure of the TMVLys-e-A488 sample to low pH. pH-triggered release was not observed, therefore indicating that enzymatic activity may cause the cleavage of the fluorescent cargo. In ongoing studies, we are elucidating the underlying mechanism to determine the time course and enzymes involved. The release of the fluorophores after cell entry results in reduced quenching and hence increased fluorescence output within the cell, which is consistent with the observations made in the cell imaging study (see Figure 5).
In summary, cell studies indicate distinct behavior of the CPMV and TMV carriers; CPMV exhibits favorable cell uptake properties. Data indicate that dye-labeling influences cell interactions; the more dyes that were conjugated, the stronger the cell interactions. Fluorescent dyes are frequently used in preclinical evaluation of nanocarriers for imaging or drug delivery; therefore it is important to carefully evaluate and test each formulation to understand the nonspecific (and potentially nondesired) contributions from organic fluorophores on cell-specificity and uptake rates. While fluorescence and lifetime measurements of CPMV and TMV with less dye exhibit enhanced fluorescence properties, this may not translate into cell imaging studies in vitro; altered cell uptake properties and chemical degradation of the optical within the endolysosome results in brighter signals from particles with increased dye loading.
Conclusions
Using CPMV and TMV as scaffolds, we synthesized a set of fluorescent-labeled virus-based nanoparticles. Our data show that density, spatial placement, conjugation chemistry, and microenvironment affect the optical properties of the probes. The brightest probes were obtained using CPMV with sparse dye labeling (CPMV-sCy5 with 27 dyes); its fluorescence intensity was about 3× higher compared to the brightest TMV sample (TMVLys-e-sCy5, FI ∼ 1616), even though both samples contain comparable dye concentrations with dye distances estimated at ∼8 nm. This dye interdistance is the theoretical minimum to avoid coupling and FRET that are responsible for photoluminescence quenching effects. The differences between the probes may be explained by differences in the microenvironment; it is possible that aromatic amino acids in proximity to Lys158 on the TMV scaffold interfere with emission. It is interesting to note that others have investigated the fluorescence properties of dye-labeled CPMV and reported that CPMV labeled with 70 dyes of A488 or 120 dyes of A555 did not show any apparent quenching,4 therefore indicating that the nature of the fluorophore is another variable to consider. Conjugation chemistry matters; our data confirm that coupling of fluorophores to aromatic tyrosine residues via diazonium coupling results in probes the least bright, which is consistent with electron delocalization in the conjugated ring systems resulting in quenching. Last, placement of dyes on the interior capsid surface is less efficient because of significant coupling of the densely located fluorophores, and this is consistent using TMV (this study) as well as CPMV.39
Strikingly, stability investigations show that viruses (CPMV and TMV) can be exposed to pulsed laser light characterized by a significant amount of excitation energy fluence while remaining intact. Ultrashort pulses with different wavelengths and intensity, typically used for laser spectroscopy studies, leave the structure of the virus unmodified because of reduced absorption coefficient. The overall low absorbance is responsible for moderate increase of the local temperature, therefore avoiding effects of thermal denaturation of viruses.
Finally, cell studies indicate distinct behavior of the CPMV and TMV carriers displaying dyes at various ratios. CPMV exhibits favorable cell uptake properties compared to TMV, and this could be explained by a combination of nanoparticle shape and molecular recognition. The spherical shape of CPMV may enhance its cell uptake properties compared to the stiff, elongated rod; further, CPMV targets surface-exposed vimentin on HeLa cells, therefore mediating receptor-specific internalization.15,16 Our data also demonstrate that dye-labeling influences cell interactions; the more dyes that were conjugated, the stronger the cell interactions, and this may be a result of the dye interacting with the cell membranes. Because various fluorescent dyes are utilized in preclinical imaging of targeted nanoparticles, it is important to use proper controls to delineate and differentiate between effects from the fluorophore versus targeting ligands. Furthermore, the brightest particles on the bench may not necessarily result in the brightest candidates in cells; our data indicate that the changing environment may affect the structural and hence the optical properties of the fluorescent probes. We provide insights into the fate of the optical probes in cells. Our data indicate that enzymatic cleavage alters the fluorescence of the optical probes in the cellular environment. Therefore, benchtop testing of optical probes does not necessarily reflect their optical properties in vivo.
In summary, the rules for designing the brightest probes are to conjugate the fluorophores to the exterior surface targeting nonaromatic residues and yielding a dye density with a dye distance of at least 8–10 nm; further, one should consider targeting various locations on the capsid surface to minimize effects from the microenvironment resulting in quenching; the latter could be achieved with VNPs through genetic design and insertion of lysine side chains at various specified locations. Furthermore, while the probes may exhibit extraordinary stability on the benchtop, the cellular machinery, including proteases and hydrolases, may contribute to structural changes, which impact the optical properties in cells. This not only has implications for applications of protein-based probes in optical imaging, but also provides insights for their development as drug delivery vehicles with a built-in cargo release strategy.
Experimental Section
Materials
Sulfo-Cy5 (sCy5) NHS ester and azide were purchased from Lumiprobe (Hallandale Beach, FL). Propargylamine was supplied from Sigma-Aldrich (St. Louis, MO), n-hydroxybenzotriazole (HOBt) from Chem-Impex International (Wood Dale, IL), ethyldimethylpropylcarbodiimide (EDC) from Pierce Biotechnology (Rockford, IL), propargyl-NHS ester from Click Chemistry Tools (Scottsdale, AZ), and 3-ethynylaniline and dimethyl sulfoxide (DMSO) from Fisher. HeLa cells were supplied from ATCC (Manassas, VA). Cell culture reagents minimal essential medium (MEM), fetal bovine serum (FBS), l-glutamine, and penicillin–streptomycin (pen-strep), as well as secondary goat anti-mouse Alexa Fluor 488 antibody were purchased from Life Technologies (Grand Island, NY). Mouse anti-human LAMP-1 came from Biolegend (San Diego, CA).
Propagation and Purification of CPMV and TMV/TMVLys
CPMV was propagated using Vigna unguiculata (black eyed peas) plants, and wild-type TMV and TMVLys mutants22 were propagated using Nicotiana benthamiana plants (a tobacco species). CPMV40 and TMV/TMVLys41 particles were purified from infected leaves using established procedures yielding 100 mgs of CPMV or TMV/TMVLys per 100 g infected leaf material.
Bioconjugation
For CPMV, reactions were carried out with 1000 to 8000 mol equiv of sCy5 NHS ester per particle at a final concentration of 2 mg/mL CPMV in 0.1 M potassium phosphate buffer (pH 7.0) with 10% (v/v) DMSO. The resultant CPMV-sCy5 was purified using centrifugal filter units with a 10 kDa molecular weight cutoff (Millipore). TMV and TMVLys click reactions were all two-step reactions: alkyne handles were first added to the particles, then sCy5 azide was attached through Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC). For TMV-iAlk, 25 molar excess of propargylamine per coat protein was reacted using EDC coupling with 45 molar excess of both EDC and HOBt at a final concentration of 2 mg/mL TMV in HEPES buffer. For TMV-eAlk, 35 equiv of 3-ethynylaniline diazonium salt formed by mixing 400 μL of 0.3 M p-toluenesulfonic acid monohydrate with 75 μL of 0.68 M 3-ethynylaniline and 25 μL of 3.0 M sodium nitrite for an hour on ice protected from light was added to TMV at a final concentration of 2 mg/mL TMV in borate buffer for 30 min on ice protected from light. For TMVLys-eAlk, the same reaction conditions as for CPMV were used, except 10 equiv of propargyl-NHS ester per coat protein were added. TMV-iAlk, TMV-eAlk, and TMVLys-eAlk were then mixed with sCy5 azide at molar excesses ranging from 0.2 to 6 dyes per coat protein for TMV-iAlk, 0.3 to 6 for TMV-eAlk, and 0.02 to 2 per coat protein for TMVLys-eAlk at final concentrations of 1 mg/mL TMV in 10 mM potassium phosphate buffer (pH 7.4) for 30 min on ice with 2 mM aminoguanidine, 2 mM ascorbic acid sodium salt, and 1 mM copper sulfate. The reaction was then stopped with 2.5 mM EDTA. TMVLys-e-A488 used for stability studies through exposure to lysosomal extracts was synthesized via an overnight reaction using TMVLys and Alexa Fluor488 (A488) succinimidyl ester at a molar excess of 10 A488 per coat protein in 0.1 M potassium phosphate buffer containing 10% DMSO by volume. All reactions were purified through ultracentrifugation purification. Yields after purification for each reaction step are 80–90% (as measured based on protein concentration).
UV–vis Spectroscopy
To determine the dye attachment density, the concentrations of the particles and dyes were determined using UV–vis spectroscopy. The particle-specific extinction coefficient at 260 nm is 8.1 mg–1 mL cm–1 for CPMV and 3 mg–1 mL cm–1 for TMV, while the extinction coefficient of sCy5 at 646 nm is 271 000 M–1 cm–1 and the extinction coefficient of AF488 at 495 nm is 73 000 M–1 cm–1. It should be noted that both TMV and CPMV are RNA viruses; the encapsulated RNA molecules result in increased absorbance at 260 nm (versus 280 nm absorbance derived from the protein shell).
Transmission Electron Microscopy (TEM)
Carbon-coated copper TEM grids (Electron Microscopy Sciences) were placed over 20 μL drops of particles diluted to 0.1 mg/mL with deionized water. The particles were allowed to adsorb for 5 min, and then the grid was briefly rinsed with deionized water followed by negative staining with 2% (w/v) uranyl acetate for 1 min. Samples were observed using a Zeiss Libra 200FE transmission electron microscope operated at 200 kV.
Gel Electrophoresis
Native particles were analyzed by 1.2% (w/v) agarose gel electrophoresis (1 h at 100 V) in 1× TBE running buffer with ethidium bromide. Individual coat proteins were analyzed by denaturing 4–12% NuPAGE (Invitrogen) polyacrylamide gel electrophoresis (50 min at 200 V) in 1× MOPS running buffer. The gels were stained with Coomassie for protein content. Images of the gels were taken using an AlphaImager imaging system (Biosciences) for UV and white light images and a Maestro 2D fluorescence imaging system with yellow excitation and emission filters for fluorescent images.
Fluorescence Measurements
50 μL of dye-labeled CPMV and TMV diluted in 0.1 M potassium phosphate buffer were added in triplicate to a black 384-well plate at concentrations of 2.5 μM sCy5, 50 nM CPMV, or 5 nM TMV. Fluorescence intensity was measured using a Tecan Infinite 200 plate reader with excitation/emission wavelengths of 600 and 665 nm.
Lifetime Measurements
For lifetime measurements, a well-established and advanced technique based on ultrafast time correlated single-photon counting spectroscopy (TCSPC) was used. Samples were excited by means of an ultrafast pulsed light source at 4 MHz, with a pulse duration of 140 fs, at a wavelength of 360 nm. The exciting light was produced by a Ti:sapphire laser (model Chamaleon by Coherent) coupled to a second harmonic generator (SHG) module and to a pulse picker. This arrangement allowed synchronization of the exciting pulses with the acquisition card of a multipronged spectrofluorometer (Edinburgh) equipped with a last generation MCP-PMT (microchannel plate photomultiplier) for detecting fluorescence light and measuring fluorescence lifetime (time resolution ∼5 ps). The decay time data were fitted with multiexponential functions f(t) = A1 × exp(−t/τ1) + A2 × exp(−t/τ2) + ···, while the average decay times were calculated by means of a weighted average τ = (A1 × τ1 + A2 × τ2 + ···)/(A1 + A2 + ···).
Flow Cytometry
HeLa cells were grown in MEM supplemented with 10% (v/v) FBS, 1% (v/v) l-glutamine, and 1% (v/v) pen-strep at 37 °C in 5% CO2. The cells were seeded at a density of 5 000 000 cells/200 μL MEM/well onto an untreated 96-well v-bottom plate. Particles were added in triplicate at a concentration of 500 000 CPMV or TMV per cell and incubated for 3 h. Free particles were then removed by washing the cells twice through centrifugation at 500 × g for 4 min, gently removing the supernatant, and resuspending the cells in FACS buffer (0.1 mL of 0.5 M EDTA, 0.5 mL FBS, 1.25 mL of 1 M HEPES, pH 7.0 in 50 mL Ca2+/Mg2+-free PBS). The cells were fixed in 2% (v/v) paraformaldehyde in FACS buffer for 10 min at room temperature, then washed twice more before analysis on a BD LSR II flow cytometer (10 000 events per sample). Data were analyzed using FlowJo software (Tree Star).
Confocal Microscopy
HeLa cells were seeded at a density of 25 000 cells/250 μL MEM/well onto glass coverslips placed in an untreated 24-well plate. They were allowed to grow for 24 h before the media was replaced with 200 μL of fresh MEM containing 2 500 000 particles per cell (4.089 μg TMV or 0.581 μg CPMV). The cells were incubated at 37 °C in 5% CO2 for 3 h, then washed with DPBS and fixed using 4% (v/v) paraformaldehyde and 0.3% (v/v) glutaraldehyde in DPBS for 5 min. The cells were then permeabilized with 0.2% (v/v) Triton X-100 in DPBS for 2 min and blocked with 5% (v/v) goat serum (GS) in DPBS for 45 min. Endolysosomes were stained using mouse anti-human LAMP-1 at a 1:200 dilution in 5% GS for an hour. The cells were incubated with secondary goat anti-mouse Alexa Fluor 488 at a 1:500 dilution in 5% GS for another hour. After each step, the coverslips were washed with DPBS 3×. The coverslips were then mounted onto glass slides using Fluoroshield with DAPI (Sigma) and sealed using nail polish. Confocal images were obtained using Olympus FluoView FV1000 LSCM. The results were processed and analyzed using ImageJ 1.44o (http://imagej.nih.gov/ij).
Stability Studies in Lysosomal Extracts
All animal studies were carried out using IACUC-approved procedures. Female Balb/c mice were starved overnight and euthanized using carbon dioxide inhalation. Livers were removed and stored at −80 °C until ready for use. Lysosomes were extracted using the Lysosome Isolation Kit (Sigma-Aldrich) and the presence of lysosomal enzymes was determined using the Acid Phosphatase Assay Kit (Sigma-Aldrich). Following lysosomal extraction and characterization, samples was frozen at −80 °C until ready for use. BSA was used as the internal standard to determine the enzymatic activity of the extracts, because we found that BSA is easily degraded in the lysosomal extract (Figure 5). TMVLys-e-A488 samples were incubated in KP buffer pH 7 or pH 5 or in lysosomal extracts: TMVLys-e-A488 was added to the extract at a concentration of 1 mg/mL and hydrochloric acid was used to adjust the pH to 5. Samples were incubated at 37 °C under gentle agitation, and time course studies were conducted. Aliquots of 15 μL were taken at specific times and characterized using SDS gel electrophoresis. Gels were analyzed using ImageJ 1.44o (http://imagej.nih.gov/ij).
Acknowledgments
This work was funded by a grant from the National Science Foundation, CMMI NM 1333651 (to N.F.S.). A.M.W. was supported by NIH T32 HL105338 Training Grant. A.D.L. acknowledges support from Italian Project PRIN 2012, Protocol No. 2012JHFYMC. G.S. acknowledges support of the Ohio Third Frontier Project, “Research Cluster on Surfaces in Advanced Materials” (RC-SAM). A.E.C. was supported in part by NIH grants T32 GM007250 and TL1 TR000441. Prof. Christina Wege, Dr. Sabine Eiben, Dr. Fania Geiger, and team (University of Stuttgart) are thanked for providing the TMV lysine mutant. Case Western Reserve University Farm is thanked for help with scaled-up plant propagation.
Supporting Information Available
Additional UV–vis spectra, interdye distance calculations, and TEM images of fluorescently labeled particles. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Dedication
This article is dedicated to the memory of Daniela Pucci, friend and collaborator, who died on April 24th, 2014.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Sapsford K. E.; Soto C. M.; Blum A. S.; Chatterji A.; Lin T.; Johnson J. E.; Ligler F. S.; Ratna B. R. (2006) Biosens. Bioelectron. 21, 1668–1673. [DOI] [PubMed] [Google Scholar]
- Soto C. M.; Blum A. S.; Vora G. J.; Lebedev N.; Meador C. E.; Won A. P.; Chatterji A.; Johnson J. E.; Ratna B. R. (2006) J. Am. Chem. Soc. 128, 5184–5189. [DOI] [PubMed] [Google Scholar]
- Miller R. A.; Presley A. D.; Francis M. B. (2007) J. Am. Chem. Soc. 129, 3104–3109. [DOI] [PubMed] [Google Scholar]
- Lewis J. D.; Destito G.; Zijlstra A.; Gonzalez M. J.; Quigley J. P.; Manchester M.; Stuhlmann H. (2006) Nat. Med. 12, 354–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz N. F.; Ablack A. L.; Hickey J. L.; Ablack J.; Manocha B.; Mymryk J. S.; Luyt L. G.; Lewis J. D. (2011) Small 7, 1664–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana J.; Dressick W. J.; Phelps J.; Johnson J. E.; Rendell R. W.; Sampson T.; Ratna B. R.; Soto C. M. (2014) Small 10, 3058–3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J.; DuFort C.; Daniel M. C.; Murali A.; Chen C.; Gopinath K.; Stein B.; De M.; Rotello V. M.; Holzenburg A.; Kao C. C.; Dragnea B. (2007) Proc. Natl. Acad. Sci. U. S. A. 104, 1354–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokorski J. K.; Steinmetz N. F. (2011) Mol. Pharmaceutics 8, 29–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruckman M. A.; Randolph L. N.; VanMeter A.; Hern S.; Shoffstall A. J.; Taurog R. E.; Steinmetz N. F. (2014) Virology 449, 163–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rae C. S.; Khor I. W.; Wang Q.; Destito G.; Gonzalez M. J.; Singh P.; Thomas D. M.; Estrada M. N.; Powell E.; Finn M. G.; Manchester M. (2005) Virology 343, 224–235. [DOI] [PubMed] [Google Scholar]
- Shukla S.; Wen A. M.; Ayat N. R.; Commandeur U.; Gopalkrishnan R.; Broome A. M.; Lozada K. W.; Keri R. A.; Steinmetz N. F. (2014) Nanomedicine (Lond) 9, 221–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aljabali A. A.; Shukla S.; Lomonossoff G. P.; Steinmetz N. F.; Evans D. J. (2013) Mol. Pharmaceutics 10, 3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildiz I.; Lee K. L.; Chen K.; Shukla S.; Steinmetz N. F. (2013) J. Controlled Release 172, 568–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson K. L.; Liu J. L. (2012) Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 4, 511–524. [DOI] [PubMed] [Google Scholar]
- Koudelka K. J.; Rae C. S.; Gonzalez M. J.; Manchester M. (2007) J. Virol. 81, 1632–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koudelka K. J.; Destito G.; Plummer E. M.; Trauger S. A.; Siuzdak G.; Manchester M. (2009) PLoS Pathog. 5, e1000417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plummer E. M.; Thomas D.; Destito G.; Shriver L. P.; Manchester M. (2012) Nanomedicine (London) 7, 877–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruckman M. A.; Jiang K.; Simpson E. J.; Randolph L. N.; Luyt L. G.; Yu X.; Steinmetz N. F. (2014) Nano Lett. 14, 1551–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterji A.; Ochoa W.; Paine M.; Ratna B. R.; Johnson J. E.; Lin T. (2004) Chem. Biol. 11, 855–863. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Kaltgrad E.; Lin T.; Johnson J. E.; Finn M. G. (2002) Chem. Biol. 9, 805–811. [DOI] [PubMed] [Google Scholar]
- Schlick T. L.; Ding Z.; Kovacs E. W.; Francis M. B. (2005) J. Am. Chem. Soc. 127, 3718–3723. [DOI] [PubMed] [Google Scholar]
- Geiger F. C.; Eber F. J.; Eiben S.; Mueller A.; Jeske H.; Spatz J. P.; Wege C. (2013) Nanoscale 5, 3808–3816. [DOI] [PubMed] [Google Scholar]
- Bruckman M. A.; Kaur G.; Lee L. A.; Xie F.; Sepulveda J.; Breitenkamp R.; Zhang X.; Joralemon M.; Russell T. P.; Emrick T.; Wang Q. (2008) ChemBioChem 9, 519–523. [DOI] [PubMed] [Google Scholar]
- Schlosser M.; Lochbrunner S. (2006) J. Phys. Chem. B 110, 6001–6009. [DOI] [PubMed] [Google Scholar]
- Schwartz D. E.; Gong P.; Shepard K. L. (2008) Biosens. Bioelectron. 24, 383–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soujon D.; Becker K.; Rogach A. L.; Feldmann J.; Weller H.; Talapin D. V.; Lupton J. M. (2007) J. Phys. Chem. C 111, 11511–11515. [Google Scholar]
- Clapp A. R.; Medintz I. L.; Mauro J. M.; Fisher B. R.; Bawendi M. G.; Mattoussi H. (2003) J. Am. Chem. Soc. 126, 301–310. [DOI] [PubMed] [Google Scholar]
- Strat D.; Dolp F.; von Einem B.; Steinmetz C.; von Arnim C. A.; Rueck A. (2011) J. Biomed. Opt. 16, 026002. [DOI] [PubMed] [Google Scholar]
- Yefimova S. L.; Tkacheva T. N.; Kurilchenko I. Y.; Sorokin A. V.; Malyukin Y. V. (2013) J. Appl. Spectrosc. 79, 914–921. [Google Scholar]
- Arnida; Janát-Amsbury M. M.; Ray A.; Peterson C. M.; Ghandehari H. (2011) Eur. J. Pharm. Biopharm. 77, 417–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnida; Malugin A.; Ghandehari H. (2010) J. Appl. Toxicol. 30, 212–217. [DOI] [PubMed] [Google Scholar]
- Gratton S. E. A.; Ropp P. A.; Pohlhaus P. D.; Luft J. C.; Madden V. J.; Napier M. E.; DeSimone J. M. (2008) Proc. Natl. Acad. Sci. U. S. A. 105, 11613–11618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz N. F.; Mertens M. E.; Taurog R. E.; Johnson J. E.; Commandeur U.; Fischer R.; Manchester M. (2010) Nano Lett. 10, 305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chithrani B. D.; Chan W. C. W. (2007) Nano Lett. 7, 1542–1550. [DOI] [PubMed] [Google Scholar]
- Chithrani B. D.; Ghazani A. A.; Chan W. C. W. (2006) Nano Lett. 6, 662–668. [DOI] [PubMed] [Google Scholar]
- Nan A.; Bai X.; Son S. J.; Lee S. B.; Ghandehari H. (2008) Nano Lett. 8, 2150–2154. [DOI] [PubMed] [Google Scholar]
- Schaeublin N. M.; Braydich-Stolle L. K.; Maurer E. I.; Park K.; MacCuspie R. I.; Afrooz A. R.; Vaia R. A.; Saleh N. B.; Hussain S. M. (2012) Langmuir 28, 3248–3258. [DOI] [PubMed] [Google Scholar]
- Steinmetz N. F.; Cho C. F.; Ablack A.; Lewis J. D.; Manchester M. (2011) Nanomedicine (London) 6, 351–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen A. M.; Shukla S.; Saxena P.; Aljabali A. A.; Yildiz I.; Dey S.; Mealy J. E.; Yang A. C.; Evans D. J.; Lomonossoff G. P.; Steinmetz N. F. (2012) Biomacromolecules 13, 3990–4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho C. F.; Shukla S.; Simpson E. J.; Steinmetz N. F.; Luyt L. G.; Lewis J. D. (2014) Methods Mol. Biol. 1108, 211–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruckman M. A.; Steinmetz N. F. (2014) Methods Mol. Biol. 1108, 173–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.