

Abstract
Inhibition of bacterial nitric oxide synthase (bNOS) has the potential to improve the efficacy of antimicrobials used to treat infections by Gram-positive pathogens Staphylococcus aureus and Bacillus anthracis. However, inhibitor specificity toward bNOS over the mammalian NOS (mNOS) isoforms remains a challenge because of the near identical NOS active sites. One key structural difference between the NOS isoforms is the amino acid composition of the pterin cofactor binding site that is adjacent to the NOS active site. Previously, we demonstrated that a NOS inhibitor targeting both the active and pterin sites was potent and functioned as an antimicrobial (Holden, et al. , Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 1812724145412). Here we present additional crystal structures, binding analyses, and bacterial killing studies of inhibitors that target both the active and pterin sites of a bNOS and function as antimicrobials. Together, these data provide a framework for continued development of bNOS inhibitors, as each molecule represents an excellent chemical scaffold for the design of isoform selective bNOS inhibitors.
Introduction
Structure-based drug design has played a key role in the development and characterization of isoform selective mammalian nitric oxide synthase (mNOS) inhibitors.1,2 NOS, an enzyme responsible in mammals for the production of the essential signaling molecule NO, can also promote a variety of disease pathologies if production of NO is unregulated.3 While structure-based inhibitor design efforts have largely focused on the three mNOS isoforms,1,2 recent characterization of bNOS as an antibacterial target against Gram-positive pathogens Staphylococcus aureus(4,5) and Bacillus anthracis(6) has promoted interest in the development of potent bNOS inhibitors. Between several bacterial gene deletion experiments and biological assays it is clear that bNOS functions in Gram-positive pathogens to provide a protective barrier against a variety of antibiotics, oxidative stress, and host antimicrobial peptides.4−7 Recently, we demonstrated that the effectiveness of a common antimicrobial is significantly improved when B. subtilis, a nonpathogenic model organism for B. anthracis, is also treated with a NOS inhibitor8 originally designed to selectively inhibit mammalian neuronal NOS (nNOS).
One of the major issues in the design of bNOS inhibitors is its structural similarity to mNOS isoforms. Avoiding the inhibition of endothelial NOS (eNOS) and inducible NOS (iNOS) is important given the role these mammalian isoforms play in regulating blood pressure9 and in the immune host defense system,10 respectively. Direct comparison of the mammalian and bacterial NOS structures/sequences reveals several key differences that could be exploited for a bNOS inhibitor design effort. The first key difference is between the domain architecture of the NOS isoforms. Each mNOS is a multidomained protein composed of both a reductase and oxygenase domain whose activity is regulated by calmodulin. In sharp contrast, bNOS is only composed of an oxygenase domain and is not regulated by calmodulin. Because bNOS is not covalently linked to its redox partners like mNOS, bNOS must utilize redox partners for activity.11,12 A second key difference is amino acid variances between the NOS active sites. For example, both bNOS and endothelial NOS (eNOS) have an Asn residue that directly interacts with the l-Arg substrate while this residue is Asp in nNOS and inducible NOS (iNOS). The active site Asp/Asn difference provided the initial structural underpinning for the design of nNOS selective inhibitors.13 Despite this difference in electrostatics between bNOS and nNOS, inhibitors that target the Asn residue might be detrimental if they also inhibit the critical eNOS isoform. Additional active site differences in bNOS include His128 (mammalian equivalent is Ser) and Ile218 (mammalian equivalent is Val). The slightly bulkier Ile adjacent to the O2 binding site has been shown to decrease the NO release rates in bNOS.14 The last key difference between mNOS and bNOS is present at the pterin cofactor-binding site. Because bNOS lacks the N-terminal Zn2+ binding motif present in mNOS, the pterin binding site is more exposed in bNOS,14 resulting in weaker micromolar binding affinity in bNOS15 vs the stronger nanomolar affinity in mNOS.16 While the physiologically relevant bNOS cofactor that binds to the bNOS pterin site remains unknown,17 inhibitors that target this site are an attractive avenue for structure-based drug design. These differences were first exploited by a NOS inhibitor that demonstrated antimicrobial properties against B. subtilis.8
Because NOS inhibitors that bind both the active
and pterin binding sites could allow for further design of bNOS inhibitor
specificity, we report here on several NOS inhibitors (1–7) we identified by solving crystal structures
that target both the active and pterin binding sites of B. subtilis NOS (bsNOS). From initial analysis of
NOS inhibitors (1–4), we were able
to design and synthesize two additional inhibitors (5 and 6) that also targeted the active and pterin binding
sites. Further characterization of inhibition was carried out using
inhibitor binding assays, enzyme assays, molecular dynamics simulations,
and bacterial assays to provide a structural framework for the continued
design of isoform-selective bNOS inhibitors that function as antimicrobials.
Results and Discussion
Aminopyridine Inhibitor Binding
Although S. aureus and B. anthracis are the primary targets, we use nonvirulent B. subtilis for routine screening. We also use B. subtilis NOS (bsNOS) for crystallography because bsNOS crystals diffract to much higher resolution than S. aureus NOS (saNOS) although the structures are identical. The root-mean-square standard deviation of Cα atoms between bsNOS and saNOS is 0.55 Å with 32–33 residues being identical within 10 Å of the heme iron. Therefore, structural insights gained from bsNOS are directly applicable to saNOS and B. anthracis NOS. Inhibitors 1–3 were initially designed to target nNOS and the structures of nNOS–1,2,3 have previously been reported.18,19 The structures of bsNOS with inhibitors 1–6 revealed that each compound interacts with the active site Glu-243 and heme propionate D through a series of H-bonds between the aminopyridine functional groups (Figure 1A–D and Table 1). While 1 and 2 only differ in the amine substituent at the para-position of the aromatic ring linker, it is clear that linker composition between the aminopyridine groups dictates the orientation of the inhibitor and the rotameric position of Arg-247. For example, in 1, Arg-247 reorients to form a π–cation interaction with the aromatic ring of the linker. This alternative rotamer was also observed with 4. In sharp contrast, the linkers of 2 and 3 are parallel to the heme group, and Arg-247 is observed in its native position. In the case of 2, the parallel orientation (relative to the heme group) of the aromatic ring within the linker is likely a result of the H-bond formed between the linker’s primary amine and heme propionate A.
Figure 1.

Active site view of bsNOS–inhibitor bound crystal structures with the inhibitor colored yellow, heme colored salmon, and select active site residues colored white. Inhibitor–protein H-bond distances are represented as black lines. The 2FO – FC maps are shown for (A) 1 contoured at 1.8σ, (B) 2 contoured at 1.4σ, (C) 3 contoured at 1.2σ, (D) 4 contoured at 1.8σ, (E) 5 contoured at 2.0σ, and (F) 6 contoured at 1.3σ.
Table 1. Data Collection, Processing, and Refinement Statisticsa.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | N-ω-nitro-l-arginine (NOHA) | N-ω-hydroxy-l-arginine (L-NNA) | |
|---|---|---|---|---|---|---|---|---|---|
| a, b, c (Å) | 80.62, 95.0,1 62.77 | 80.52, 94.94, 63.49 | 80.97, 94.55, 125.12 | 80.38, 95.70, 63.07 | 79.70, 93.54, 62.30 | 79.85, 93.45, 62.48 | 80.42, 94.91, 62.86 | 80.93, 94.73, 62.03 | 80.55, 94.94, 62.13 |
| α, β, γ (deg) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| total observations | 230276 (11686) | 122521 (9449) | 358403 (14500) | 244251 (8510) | 226474 (10117) | 211092 (9519) | 344120 (12494) | 172015 | 212567 (9899) |
| unique observations | 56580 (2868) | 29389 (2258) | 63764 (4312) | 50162 (2562) | 34237 (2377) | 37512 (2303) | 69173 (3171) | 33624 | 39483 (2495) |
| resolution (Å) | 37.11–1.67 (1.73–1.67) | 44.14–2.09 (2.15–2.09) | 49.51–2.02 (2.07–2.02) | 52.66–1.74 (1.77–1.74) | 49.09–1.96 (2.01–1.96) | 49.21–1.90 (1.94–1.90) | 37.02–1.55 (1.58–1.55) | 37.65–1.98 (2.03–1.98) | 49.20–1.88 (1.92–1.88) |
| Rmerge | 0.037 (1.118) | 0.131 (0.882) | 0.053 (0.108) | 0.082 (0.396) | 0.144 (0.347) | 0.141 (0.346) | 0.039 (2.151) | 0.038 (1.303) | 0.073 (0.917) |
| RPIM | 0.032 (0.951) | 0.101 (0.690) | 0.031 (0.094) | 0.052 (0.357) | 0.082 (0.289) | 0.078 (0.268) | 0.029 (1.697) | 0.027 (0.950) | 0.045 (0.810) |
| CC1/2 | 0.999 (0.591) | 0.991 (0.674) | 0.999 (0.990) | 0.997 (0.867) | 0.995 (0.915) | 0.991 (0.868) | 0.999 (0.601) | 1.000 (0.657) | 0.998 (0.515) |
| I/σI | 17.74 (1.1) | 5.2 (1.1) | 21.6 (8.5) | 10.6 (2.3) | 7.4 (2.4) | 8.9 (3.0) | 15.0 (0.7) | 20.1 (1.0) | 18.7 (1.5) |
| completeness (%) | 99.79 (99.77) | 99.7 (99.8) | 99.7 (97.2) | 99.3 (94.8) | 99.9 (99.3) | 99.4 (95.7) | 98.3 (92.5) | 99.2 (97.1) | 99.9 (99.7) |
| multiplicity | 4.1 (4.1) | 4.2 (4.2) | 5.6 (3.4) | 4.9 (3.3) | 6.6 (4.3) | 5.6 (4.1) | 5.0 (3.9) | 5.1 (4.9) | 5.4 (4.0) |
| Wilson B-factor | 26.54 | 28.44 | 20.36 | 20.89 | 27.21 | 19.22 | 26.72 | 36.44 | 30.17 |
| Refinement | |||||||||
| resolution (Å) | 1.67 | 2.09 | 2.017 | 1.74 | 2.13 | 1.90 | 1.55 | 1.98 | 1.88 |
| no. reflections | 56494 | 29338 | 63603 | 49954 | 26698 | 36318 | 68910 | 26173 | 39297 |
| Rwork | 0.1648 | 0.1674 | 0.1718 | 0.1668 | 0.1525 | 0.1666 | 0.1782 | 0.1662 | 0.1724 |
| Rfree | 0.1884 | 0.2199 | 0.1987 | 0.1878 | 0.2014 | 0.2077 | 0.1998 | 0.2083 | 0.209 |
| no. of atoms | 3399 | 3280 | 6740 | 3318 | 3285 | 3357 | 3364 | 3206 | 3318 |
| macromolecules | 2951 | 2949 | 5873 | 2949 | 2944 | 2946 | 2941 | 2937 | 2940 |
| ligands | 106 | 85 | 167 | 87 | 100 | 89 | 114 | 100 | 106 |
| solvent | 342 | 246 | 700 | 282 | 241 | 322 | 309 | 169 | 272 |
| average B-factor | 31.9 | 40.5 | 28 | 28.3 | 34.6 | 28.9 | 42.9 | 48.4 | 45.1 |
| macromolecule | 30.8 | 40.1 | 26.8 | 27.9 | 34.2 | 27.9 | 42.3 | 48.5 | 44.8 |
| ligands | 30.8 | 41.9 | 30.5 | 23.6 | 32.6 | 30.3 | 40.7 | 46.7 | 44.1 |
| solvent | 41.1 | 44.6 | 37.5 | 34.5 | 39.9 | 37.4 | 49.5 | 47.3 | 48.5 |
| Ramachandran favored (%) | 98 | 96 | 97 | 97 | 97 | 98 | 97 | 98 | 98 |
| Ramachandran outliers (%) | 0 | 0.28 | 0.14 | 0 | 0 | 0 | 0 | 0 | 0 |
| rms deviations | |||||||||
| bond lengths (Å) | 0.007 | 0.008 | 0.004 | 0.007 | 0.007 | 0.007 | 0.006 | 0.008 | 0.007 |
| bond angles (deg) | 1.17 | 1.24 | 1.03 | 1.218 | 1.27 | 1.27 | 1.25 | 1.32 | 1.3 |
Values in parentheses are for the highest resolution shell.
Considering that a cyano substituent within the linker of 4 results in a π–cation interaction with Arg-247 and a H-bond between heme propionate D and the aminopyridine functional group, we anticipated that inhibitor binding to the pterin site could be further improved by addition of a secondary amino group to form an additional H-bond with heme propionate D and the cyano conjugated ring to maintain the π–cation interaction with Arg-247. These observations led to the synthesis of 5, whose linker unexpectedly adopted a parallel orientation to the heme group but maintained the π–cation interaction with Arg-247 (Figure 1E). To further improve inhibitor binding to the pterin site of bsNOS, we also synthesized 6; the design of 6 was based on the crystal structure of 2. The goal was to develop an inhibitor that formed a stable π–π stacking interaction with Trp-329 by replacing one of the aminopyridine groups of 2 with a pyrrolopyridine. The pyrrolopyridine should also be able to H-bond with heme propionate D. As observed with 2, compound 6 does indeed form a H-bond with the heme propionate and undergoes a π–π stacking interaction with Trp-329 as predicted from the modeling.
To further characterize inhibitor binding at the NOS active site, we measured the spectral binding constant, KS, for each inhibitor to both bsNOS and iNOS. On the basis of the measured KS for each inhibitor, binding to both bsNOS and iNOS was in the low μM range (Table 2). In addition, we also measured KS for bsNOS in the presence and absence of H4B. We anticipated the KS of inhibitors targeting the pterin site to increase in the presence of H4B, as inhibitor binding would be weakened owing to competition with the pterin molecule. However, in the case of inhibitors that tightly bind to the pterin site, for example 1, only a 2-fold difference in binding was observed (Table 2), which we consider to be a negligible difference because the KS of ligands L-NNA and NOHA, two ligands that do not bind to the pterin site, resulted in a 3.4- and 1.2-fold difference in binding, respectively. Another interpretation is that the binding affinity of 1 to the pterin site is significantly stronger than the binding affinity of H4B and that 1 is able to easily displace H4B. If this is true, for tight binders the KS should remain unchanged in the presence or absence of H4B. In contrast, for inhibitors that are transiently associated with the pterin site, as is observed with 5 and 6, it is not surprising that we also observe a negligible change in KS upon addition of H4B owing to the weak noncovalent interactions observed at the pterin site. Regardless, from the crystal structures (Figure 1), it is apparent that 1–6 may serve as reasonable templates for future structure-based drug design against bNOS, as all molecules do bind to the pterin-binding site.
Table 2. Imidazole Displacement Assay Used to Calculate the Spectral Binding Constants (KS) in the Absence (−) and Presence (+) of 50 μM H4B.
|
KS (μM) |
|||
|---|---|---|---|
| compound | bsNOS (−H4B) | bsNOS (+H4B) | iNOS (+H4B) |
| 1 | 0.91 ± 0.08 | 1.9 ± 0.1 | 2.9 ± 0.4 |
| 2 | 15 ± 3 | 47 ± 2 | 33 ± 8 |
| 3 | 16 ± 1 | 19 ± 1 | 19 ± 4 |
| 4 | 12 ± 1 | 19 ± 2 | 6.1 ± 0.6 |
| 5 | 39 ± 2 | 34 ± 4 | 16 ± 6 |
| 6 | 10 ± 2 | 12 ± 4 | 21 ± 8 |
| 7 | 13 ± 2 | 80 ± 19 | 93 ± 77 |
| N-ω-nitro-l-arginine (NOHA) | 1.32 ± 0.048 | 4.4 ± 0.3 | nd |
| N-ω-hydroxy-l-arginine (L-NNA) | 3.2 ± 0.511 | 1.9 ± 0.1 | nd |
Thiophenecarboximidamide Inhibitor Binding
Thiophenecarboximidamide inhibitors are expected to exhibit improved pharmacological properties over aminopyridine inhibitors but form similar complexes with nNOS,20 so we investigated the binding of 7 to bsNOS. Similar to 1–6, crystal structure analysis of 7 revealed that the inhibitor also bound at both the active and pterin sites (Figure 2A). Because 7 is an asymmetric inhibitor, with both ends of 7 being composed of a thiophenecarboximidamide, we wanted to investigate in more detail the noncovalent interactions that allow for the unique binding mode and orientation of 7 to bsNOS. From the crystal structures, we hypothesized that the inhibitor orientation observed in bsNOS-7 resulted from the 3.2 Å H-bond formed with His-128 and/or the hydrophobic contact shared between Ile-218 and one of the inhibitor aromatic rings. The observation of a 3.2 Å H-bond between 7 and His-128 (Figure 2A) was of particular interest because the corresponding residue in mNOS is a Ser. Similarly, it is feasible that the hydrophobic contact between Ile-218 and 7 would favor 7 to bind the aromatic ring lacking a polar substituent to form better nonpolar interactions with the larger Ile-218 side chain. However, site directed mutagenesis of bsNOS revealed that neither the H128S (Figure 2B) nor the I218V mutants (Figure 2C) resulted in an alternative bsNOS–7 binding mode. In fact, I218V-bsNOS resulted in a more stable H-bond, 2.7 vs 3.3 Å for WT, with His-128. Together, these data suggest that the binding mode of 7 results from the ability of 7 to bind in the pterin pocket in order for 7 to optimally interact with both the pterin and active sites, and this is likely observed in bsNOS because pterin binding is weak. The importance of 7 binding to the pterin site is evidenced by the 6-fold decrease in affinity in the presence of H4B (Table 2).
Figure 2.
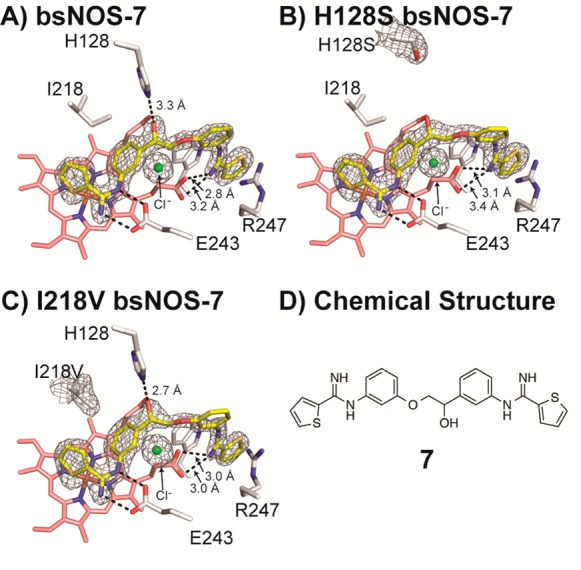
Neither His128 nor I218 provides binding specificity of 7 to bsNOS. (A) WT bsNOS–7 with 2FO– FC map contoured at 2.0σ reveals a 3.3 Å H-bond interaction between His128 and the hydroxyl group of 7. (B) H128S-bsNOS–7 crystal structure with 2FO – FC map contoured at 1.5σ. (C) I218V-bsNOS–7 crystal structure with 2FO – FC map contoured at 1.8σ. (D) 2D chemical structure of 7.
To further probe the binding mode of 7 to bsNOS and investigate the H-bond contribution of His-128 to the binding mode, we ran MD simulations for 25 ns. After allowing the system to equilibrate for 4.5 ns, the ligand was allowed to freely move. In both WT and H128S MD simulations 7 retains its interactions with the active site Glu-243 (Figure 3A) and is loosely bound to heme propionate D (Figure 3B), suggesting that the H-bond formed between 7 and Glu-243 is strong and the H-bond between 7 and heme propionate D is weak. In fact, the bond distance between 7 and heme propionate D exceeded 3.5 Å for 96.4% of the bsNOS MD trajectory. Moreover, comparison of the MD trajectories for bsNOS and H128S-bsNOS suggests that the H-bond between 7 and His128 is also transient and does not contribute toward the binding mode of 7. Distance measurements between the α-carbon of residue 128 and the hydroxyl group of 7 reveal His128 to function only by sterically restricting the movement of 7, as the atom distances range from 2.6 to 7.6 Å for bsNOS–7 and 4.7 to 10.7 Å for H128S-bsNOS–7 (Figure 3C). This is not surprising as His is a much bulkier residue then Ser.
Figure 3.
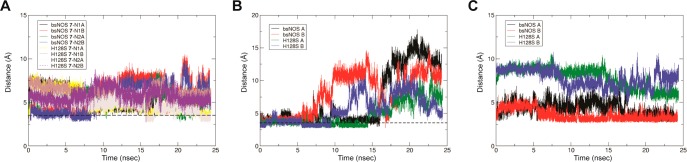
Molecular dynamics simulation of 7 bound to bsNOS and H128S-bsNOS reveals inhibitor is only tightly bound to active site Glu-243. (A) The distance measured between active site Glu-243 and the imine group of 7 bound to the active site is constant over time. The dotted line indicates 3.5 Å. (B) The distance measured between heme propionate D and the imine group bound to pterin site of 7 over time with the dotted line indicating a distance of 3.5 Å. (C) The distance measured between the hydroxyl group of 7 and α-carbon of residue 128.
Interestingly, in both WT and H128S MD simulations, the thiophenecarboximidamide headgroup of 7 initially bound at the pterin site was observed to rotate in and out of the pterin pocket. This rotation of the thiophenecarboximidamide rendered the pterin-binding site temporarily solvent exposed. More specifically, direct measurement of the bond distance between heme propionate D and the imine of 7 throughout the MD simulation revealed the distance between heme propionate D and the imine of 7 to be nonuniform within each chain of the bsNOS dimer (Figure 3B). Together, these data provide additional support that binding of 7 to the pterin site is relatively weak.
Inhibition–Activity Analysis
Both the aminopyridine and thiophenecarboximidamide based inhibitors bind and inhibit the three mNOS isoforms (Table 3). Considering 1–4 were originally designed to target nNOS, it is not surprising that 1–4 are good inhibitors of nNOS. Interestingly, a comparison of Ki’s for 2 and 6, with 6 having a bulkier pyrrolopyridine group then the aminopyridine present in 2, suggests the bulkier pyrrolopyridine group lowers the specificity toward the mNOS isoforms. Specifically, the potency of 6 toward nNOS decreased >4-fold as compared to the potency of 2 toward nNOS. Hence, the introduction of a bulkier group that also binds to the bNOS pterin site has the potential to lower inhibitor affinity to the mNOS isoforms.
Table 3. Ki Values of Inhibitors 1–7 with the Mammalian NOS Isoforms.
With the use of the recently described bBiDomain and YumC system for bNOS activity/inhibition measurements, we also evaluated the potency of each inhibitor at varying concentrations using a single time point approach.11 At all concentrations evaluated, 1 was the most potent, with 49.2 ± 1.2% nitrite detected at an inhibitor concentration of 300 μM. The increased potency of 1, compared to 2–7, is likely because 1 is able to form two separate 3.3 Å H-bonds with heme propionate D (Figure 1A), which accounts for an additional 1–3 kcal/mol in binding energy. Unlike 1, compounds 2–6 are unable to form a strong H-bond with the heme propionate D (Figure 1). On the basis of this limited data set, it is clear that formation of a strong H-bond with heme propionate D improves inhibitor potency for molecules that target the pterin site. Moreover, of the compounds evaluated here, it also is clear that inhibitor potency needs to be dramatically improved, as only one of these compounds lowered enzyme activity below 50% (Figure 4). The relatively weak potency observed might be the result of the inclusion of 50 μM H4B in the enzyme assay, as these inhibitors must compete with H4B. Moreover, because the physiological pterin group17 and its concentration within either S. aureus or B. anthracis is unknown, the actual in vivo potency may be substantially different from our in vitro results. Even so, the relative inhibitor potency of each inhibitor is a sufficient tool to guide inhibitor design. Therefore, improving the noncovalent protein–inhibitor interactions at the pterin-binding site will likely also improve inhibitor potency.
Figure 4.
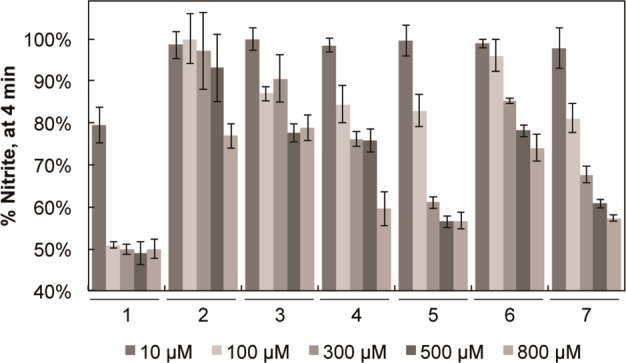
%Nitrite detected as a function of bBiDomain activity in the presence of NOS inhibitors at varying concentrations. On the basis of a single time point analysis, 1 is the most potent bNOS inhibitor. Error bars represent the mean ± the SEM for three separate experiments.
Bacterial Growth Inhibition
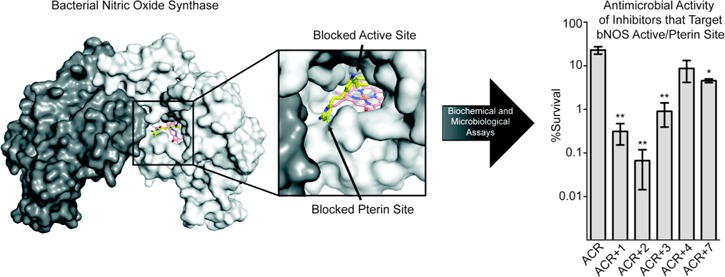
We previously showed that some NOS inhibitors developed for selective inhibition of nNOS worked synergistically with the antibiotic acriflavine (ACR)8 to inhibit the growth of B. subtilis. In the previous work, there was a good correlation between the binding affinity of a NOS inhibitor to bsNOS and its ability to work synergistically with the antibiotic ACR in bacterial killing. We thus would expect inhibitor 1 to be especially effective at bacterial growth inhibition, and this indeed is the case (Figure 5). Of the inhibitors tested, 1 is the most effective at working synergistically with ACR to block growth. With ACR alone, bacterial growth is 23% of control. In the presence of inhibitor 1 growth is 36% of control, but together with ACR the inhibition is close to 99%, clearly indicating a strong synergistic effect. While the correlation between being an effective bsNOS and a bacterial growth inhibitor are strong, 2 is an outlier. Inhibitor 2 does not bind as tightly to bsNOS nor is it as effective at blocking bsNOS activity, but it is, nevertheless, a good inhibitor of bacterial growth. Given that both 1 and 2 can inhibit bacterial growth on their own suggests that these compounds block bsNOS to render the bacteria more susceptible to ACR but may also influence additional non-bsNOS target(s).
Figure 5.

NOS inhibitors influence the % survival of B. subtilis. (A) B. subtilis treated with NOS inhibitors. Significance calculated using the Student’s t test between the measured CFU of B. subtilis treated with and without NOS inhibitors for each strain separately. (B) Bacterial survival of B. subtilis wt and Δnos decreases in the presence of 800 μM ACR and 500 μM NOS inhibitors. Significance calculated for each strain separately using the Student’s t test between the calculated % survival of ACR treated cells and % survival of ACR+NOS inhibitor treated cells. Error bars represent the mean ± the SEM of at least three replicates. Student’s t test gives ***p < 0.01, **p < 0.05, *p < 0.1.
Conclusion
Although the physiological pterin cofactor for bNOS remains unknown, NO production by bNOS requires the presence of a pterin group.21 Because of this strict pterin group requirement for activity and the significant differences in the pterin binding sites between mNOS and bNOS, inhibitors that target the pterin site may provide the key to unlocking the design and development of potent bNOS inhibitors. By taking a structure-based approach toward the identification of NOS inhibitors, we were able to identify several chemical scaffolds that inhibit bsNOS by binding to both the active and pterin sites. Several of these structures resulted in unexpected rotameric positions of active site residue Arg-247. These alternate rotameric positions will be important to consider for future structure-based drug design. Moreover, while the crystal structures and KS analysis provide definitive support for each molecule binding at the active site, MD simulations suggest that the binding of 7 is transient and, therefore, weak. A particularly important observation is that aminopyridine inhibitors bind better than the pyrrolopyridine inhibitor even though both bind in the pterin pocket, form stacking interactions with Trp329, and H-bonds with the heme propionate. One might have expected the pyrrolopyridine to bind more tightly given the larger surface area that should form more extensive interactions with Trp329. However, a key difference between the aminopyridine group and the pyrrolopyridines is that the aminopyridine has a higher pKa than the pyrrolopyridine and is likely to be at least partially protonated and positively charged at neutral pH thus giving stronger interactions with the heme propionate. This underscores the importance of strong electrostatic interactions with the heme propionate relative to π stacking interactions. Further development of potent bNOS inhibitors that work synergistically with antibiotics will thus require further modification of inhibitor scaffolds that take these lessons into account.
Experimental Procedures
Site Directed Mutagenesis
Active site mutations H128S and I218V were introduced using PfuTrubo (Agilent) on the previously reported bsNOS expression vector8 containing sERP mutations E24A/E25A/E316A.22
Expression and Purification
Isolation of recombinant B. subtilis NOS, bBiDomain, and YumC from Escherichia coli were followed as previously reported.11,14 The purification of mammalian NOS isoforms was also followed as previously reported.23−25
Crystallization and Structure Determination
bsNOS crystals were prepared, cryoprotected, and soaked with inhibitor as previously described for bsNOS–inhibitor complexes.8 To obtain inhibitor bound crystal structures of compounds 5, 6, and 7, H4B was removed from the cryosoak. X-ray data were collected at both the Stanford Synchotron Radiation Light (SSRL) Source and the Advanced Light Source (ALS). Data sets were indexed and integrated with either imosflm26 or XDS.27 Aimless was used to scale the data sets.28 Inhibitor restraints were constructed using PRODRG,29 and data sets were refined using PHENIX.30
Imidazole Displacement
The transition from an imidazole bound low-spin to a ligand bound high-spin state was measured as previously described8 using purified oxygenase domain of either bsNOS or iNOS. For trials that included H4B, H4B was added at 50 μM. The measured KS,apparent was used to calculate the KS(31) using the bsNOS imidazole KD of 384 μM and iNOS imidazole KD of 158 μM.32
Inhibition Assay using bBiDomain
NADPH dependent Bacillus subtilis NOS activity/inhibition was evaluated using bBiDomain and the flavodoxin reductase YumC, as previously described,11 with varying concentrations of inhibitor. Nitrite levels were measured as a function of NOS activity and calculated by the Griess reaction using a standard curve.
Molecular Dynamics
Starting coordinates for molecular dynamics simulations were generated from crystal structures of both bsNOS–7 and H128S-bsNOS–7. Because bsNOS is present as a dimer in solution,33 a second subunit was generated from the crystal structure monomer using a C2 rotation axis. Hydrogens were added to the protein, 7, and solvent using psfgen of VMD 1.9.1.34 Bulk water was added to each system using the solvate command of VMD to generate a 25 Å cushion of solvent. As both structures were identical in overall size, the same unit cell box dimensions of (122 × 138 × 122) were used for each system. The final systems for bsNOS–7 and H128S-bsNOS–7 were composed of 194372 and 192503 atoms, respectively. Simulations were carried out on the Stampede supercomputer using NAMD 2.9.35 The CHARMM force fields employed to model the protein and heme were identical to those used in previous simulations.36,37 The CHARMM force field of 7 was generated using ParamChem (www.paramchem.org).38 The smooth partial Ewald mesh method was used in the calculation of Coulombic forces, while a Langevin thermostat was employed for the constant temperature simulations.39 Nosé–Hoover–Langevin piston was employed for pressure control.40 All simulations were run at 300 K and 1 atm with a time step of 1 fs. All bonds involving hydrogen atoms were held fixed using the SHAKE algorithm.41 Each structure was minimized for 1000 fs before the simulation was allowed to propagate. Positional restraints were placed on 7 during the first 4.5 ns of the simulation, allowing the protein to equilibrate before the inhibitor restraints were removed. Analyses of the simulations were carried out using locally developed analysis tools and VMD.34
Effect of Antimicrobial Induced Stress and NOS Inhibitors on B. subtilis
Isolation and characterization of B. subtilisΔnos strain were previously described.8 WT (American Type Culture Collection 23857) and ΔnosB. subtilis strains were grown separately to an OD600 ≈ 1.0 and diluted to OD600 = 0.6 in LB media. From the diluted cell stock, 80 μL was aliquoted into a sterile 1.5 mL Eppendorf tube and diluted to a final volume of 100 μL with M9 minimal media or a combination of either M9 minimal media, NOS inhibitor diluted in M9 minimal media, and/or acriflavine dissolved in M9 minimal media. Final concentrations of NOS inhibitor and acriflavine were 500 and 800 μM, respectively. Cells were treated for 30 min at 30 °C and then immediately serially diluted in M9 minimal media and plated on LB agar (supplemented with 0.5% glucose). Plates were incubated at 37 °C overnight. After overnight incubation, colony-forming units were counted and percent survival was calculated relative to the untreated cells.
Chemical Synthesis
Solvents were purified by passage through a solvent column composed of activated alumina and a supported copper redox catalyst. Reagents were purchased from Sigma-Aldrich and TCI America and were used without further purification. Normal phase flash column chromatography was performed using SuperFlash Si 50 prepacked silica cartridges and reverse phase flash column chromatography was performed using a SiliaSep C18 flash cartridge (25 g, 40–63 μm/230–400 mesh, pore size 60 Å) using 5–80% MeOH in water with an Agilent 971-FP flash purification system. 1H NMR and 13C NMR spectra were recorded in the indicated solvent on a Bruker Avance-III (500 and 126 MHz for 1H and 13C, respectively) spectrometer. MS was performed on a system consisting of an electrospray ionization (ESI) source in a Thermo Finnigan LCQ mass spectrometer. High resolution mass spectra were obtained using an Agilent 6210 LC-TOF spectrometer. The purities of the final compounds were evaluated on an Agilent 1260 analytical HPLC system using an Agilent Zorbax Eclipse XDB-C18 (4.6 mm × 50 mm, 5 μm) reverse phase column with UV absorbance and evaporative light scattering detection and were found to be ≥95%.
Compounds 1, 2, 3, and 7 were obtained from previous studies.18−20 To synthesize 4 (Scheme 1), Boc-protected 2-amino-4,6-dimethylpyridine (8) was treated with n-butyllithium and was mixed with 3,5-bis(bromomethyl)benzonitrile to give the corresponding intermediate (10). N-Methylation of 10 with 0.57 equiv of iodomethane and NaH followed by deprotection of the Boc protecting group gave target compound 4 in a moderate yield. The synthesis of 5 is shown in Scheme 2. Dibromophenethyl derivative 12 was prepared by coupling of 3,5-dibromobenzyl bromide with lithiated pyrrolyl-4,6-lutidine (11). This intermediate underwent microwave-assisted Rosenmund–von Braun reaction with CuCN to replace one of the bromides by CN (13). A Buchwald–Hartwig reaction of 13 with 2-pyridinylethylamine using Pd2(dba)3 and DavePhos gave the corresponding amine (14). The 2,5-dimethylpyrrole protecting group of 14 was removed with NH2OH·HCl using a microwave42 to generate 5. Compound 6 was obtained using the synthetic pathway shown in Scheme 3. Sonogashira coupling between 13 and 15(43,44) using Pd(PPh3)2Cl2 yielded 16. The produced alkyne of 16 underwent catalytic hydrogenation with Pd/C under hydrogen atmosphere. Microwave-assisted deprotection of the protecting groups on the aminopyridine ring with NH2OH·HCl and Raney nickel mediated hydrogenation of the CN group yielded 6.
Scheme 1.

Scheme 2.

Scheme 3.

Spectral Data
Di-tert-butyl (((5-Cyano-1,3-phenylene)bis(ethane-2,1-diyl))bis(4-methylpyridine-6,2-diyl))dicarbamate (10)
To a solution of tert-butyl (4,6-dimethylpyridin-2-yl)carbamate (8, 889 mg, 4.0 mmol) in THF (25 mL) was added n-BuLi (1.6 M solution in hexanes, 5.0 mL, 8.0 mmol). The reaction mixture was stirred for 30 min at 0 °C and was then cooled to −78 °C. To this solution, 3,5-bis(bromomethyl)benzonitrile (9, 578 mg, 2.0 mmol) in THF (2 mL) was added dropwise. After the mixture was stirred for an additional 10 min, it was quenched with H2O (50 mL). The organic layer was partitioned by addition of ethyl acetate (50 mL), dried with MgSO4, and concentrated in vacuo. The resulting brown oil was purified by flash chromatography (EtOAc/hexanes) to yield 10 (480 mg, 42%) as a pale-yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.64 (s, 2H), 7.50 (s, 2H), 7.29 (s, 2H), 7.19 (s, 1H), 6.57 (s, 2H), 2.96 (dd, J = 9.3, 5.9 Hz, 4H), 2.90–2.80 (m, 4H), 2.30 (s, 6H), 1.52 (s, 18H). 13C NMR (126 MHz, CDCl3) δ 158.25, 152.49, 151.52, 150.00, 143.03, 133.49, 129.65, 119.16, 118.81, 112.15, 110.34, 80.81, 38.99, 35.10, 28.27, 21.31. MS ESI [M + H]+ = 572.2.
3-(2-(6-Amino-4-methylpyridin-2-yl)ethyl)-5-(2-(4-methyl-6-(methylamino)pyridin-2-yl)ethyl)benzonitrile (4)
To a solution of Boc-protected aniline (400 mg, 0.70 mmol) in THF (2 mL), NaH (60% in oil, 17 mg, 0.4 mmol) was added at 0 °C. The reaction mixture was stirred for 15 min at the same temperature, and then methyl iodide (57 mg, 0.4 mmol) was added. The mixture was stirred for an additional 12 h at room temperature, and then quenched with H2O (10 mL). After addition of ethyl acetate (10 mL), the organic layer was partitioned, dried with MgSO4, and concentrated in vacuo. The resulting brown oil was mixed with 1 M HCl solution in MeOH, stirred for 5 h, and then concentrated in vacuo to give crude 4. The crude product was subjected to purification with a SiliaSep C18 flash cartridge (25g, 40–63 μm/230–400 mesh, pore size 60 Å) using 5–80% MeOH to give pure 4 (131 mg, 51%) as a colorless gel. 1H NMR (500 MHz, OD) δ 7.42–7.37 (m, 2H), 7.35 (s, 1H), 6.39 (s, 1H), 6.34 (s, 1H), 6.30 (s, 1H), 6.28 (s, 1H), 3.05–2.95 (m, 4H), 2.90 (s, 3H), 2.88–2.80 (m, 4H), 2.23 (s, 3H), 2.22 (s, 3H). 13C NMR (126 MHz, CD3OD) δ 160.36, 159.46, 157.59, 156.34, 153.29, 152.08, 144.59, 144.14, 134.97, 131.04, 130.88, 119.92, 114.82, 114.20, 113.14, 108.87, 106.37, 39.12, 38.68, 36.25, 36.18, 29.16, 21.32, 21.26. HRMS (ESI): calcd for C24H28N5 [M + H]+, 386.2339; found, 386.2333.
2-(3,5-Dibromophenethyl)-6-(2,5-dimethyl-1H-pyrrol-1-yl)-4-methylpyridine (12)
To a solution of 11 (1.2 g, 6.0 mmol) in THF (25 mL) was added n-BuLi (1.6 M solution in hexanes, 3.75 mL, 6.0 mmol). The reaction mixture was stirred for 30 min at 0 °C and transferred to a solution of 3,5-dibromobenzyl bromide (1.64 g, 5.0 mmol) in THF (25 mL) at −78 °C via cannula. The reaction mixture was allowed to stir for an additional 20 min and then quenched with H2O (50 mL). After addition of ethyl acetate (50 mL), the organic layer was partitioned, dried with MgSO4, and concentrated by rotary evaporation. The residue was purified by flash chromatography (EtOAc/hexanes) to yield 12 (86%) as a pale-yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.51 (s, 1H), 7.26 (ss, 2H), 6.91 (s, 2H), 5.92 (s, 2H), 3.06 (q, J = 2.8 Hz, 4H), 2.40 (d, J = 1.6 Hz, 3H), 2.15 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 159.76, 151.73, 149.72, 145.43, 131.65, 130.43, 128.48, 122.77, 122.74, 120.42, 106.76, 39.02, 34.89, 21.01, 13.27. MS ESI [M + H]+ = 449. 3.
3-Bromo-5-(2-(6-(2,5-dimethyl-1H-pyrrol-1-yl)-4-methylpyridin-2-yl)ethyl)benzonitrile (13)
A mixture of 12 (448 mg, 1.0 mmol), CuCN (108 mg, 1.20 mmol), and DMF (4 mL) was heated at 220 °C for 20 min in the microwave cavity. The reaction mixture was then treated with dichloromethane (20 mL), filtered, and concentrated in vacuo. The residue was purified by flash chromatography to give the 13 (225 mg, 57%) as a pale-yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.63 (s, 1H), 7.57 (s, 1H), 7.41 (s, 1H), 6.93 (ss, 2H), 5.92 (s, 2H), 3.19–3.03 (m, 4H), 2.41 (s, 3H), 2.13 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 159.27, 151.82, 149.88, 145.04, 136.39, 132.31, 130.76, 128.44, 122.77, 122.75, 120.59, 117.45, 113.92, 106.81, 38.69, 34.58, 21.01, 13.26. MS ESI [M + H]+ = 394.5.
3-(2-(6-(2,5-Dimethyl-1H-pyrrol-1-yl)-4-methylpyridin-2-yl)ethyl)-5-((2-(pyridin-2-yl)ethyl)amino)benzonitrile (14)
A mixture of 3-bromobenzonitrile 13 (200 mg, 0.5 mmol), 2-(2-pyridinyl)ethylamine (73 mg, 0.60 mmol), Pd2(dba)3 (23 mg, 0.025 mmol), DavePhos (20 mg, 0.050 mmol), and NaOtBu (58 mg, 0.60 mmol) in THF (1.5 mL) and 1,4-dioxane (1.5 mL) was stirred at 80 °C for 12 h. The reaction mixture was then treated with diethyl ether (20 mL), filtered, and concentrated in vacuo. The residue was purified by flash chromatography (EtOAc/hexanes) to give 14 (157 mg, 72%) as a pale-yellow oil. 1H NMR (500 MHz, CDCl3) δ 8.62–8.55 (m, 1H), 7.65 (td, J = 7.6, 1.9 Hz, 1H), 7.22–7.16 (m, 2H), 6.93 (s, 1H), 6.88 (s, 1H), 6.77 (s, 1H), 6.67 (s, 1H), 6.64 (s, 1H), 5.91 (s, 2H), 4.65 (t, J = 5.6 Hz, 1H), 3.50 (q, J = 5.8 Hz, 2H), 3.15–3.03 (m, 4H), 3.02–2.93 (m, 2H), 2.39 (s, 3H), 2.20 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 160.25, 159.34, 151.66, 149.62, 149.37, 148.54, 143.70, 136.75, 128.45, 123.39, 122.65, 121.73, 120.69, 120.25, 119.66, 117.48, 112.99, 112.70, 106.72, 43.09, 39.17, 36.83, 35.43, 21.01, 13.25. MS ESI [M + H]+ = 436.5.
3-(2-(6-Amino-4-methylpyridin-2-yl)ethyl)-5-((2-(pyridin-2-yl)ethyl)amino)benzonitrile (5)
To a 5 mL microwave vial was added protected aminopyridine 14 (131 mg, 0.3 mmol), hydroxylamine HCl (105 mg, 1.5 mmol), EtOH (1.5 mL), and H2O (0.5 mL). After being sealed, the vial was shaken vigorously and then heated in the microwave irradiator for 30 min at 120 °C. The reaction mixture was allowed to cool to room temperature and then concentrated in vacuo and purified by flash column chromatography using a SiliaSep C18 flash cartridge (25 g, 40–63 μm/230–400 mesh, pore size 60 Å) with 5–80% MeOH in water to yield 5 (76%) as a pale-yellow gel. 1H NMR (500 MHz, CDCl3) δ 8.63–8.53 (m, 1H), 7.65 (td, J = 7.7, 1.8 Hz, 1H), 7.20 (dd, J = 7.6, 5.6 Hz, 2H), 6.81 (s, 1H), 6.67 (d, J = 1.4 Hz, 2H), 6.32 (s, 1H), 6.20 (s, 1H), 4.60 (t, J = 5.6 Hz, 1H), 3.51 (q, J = 6.2 Hz, 2H), 3.09 (t, J = 6.5 Hz, 2H), 2.91 (dd, J = 9.5, 5.7 Hz, 2H), 2.82 (dd, J = 9.5, 5.7 Hz, 2H), 2.20 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 159.38, 158.81, 158.22, 149.39, 149.27, 148.45, 144.12, 136.72, 123.39, 121.71, 120.89, 119.76, 117.54, 114.52, 112.88, 112.61, 106.68, 43.12, 39.34, 36.88, 35.68, 20.97. HRMS (ESI): calcd for C22H24N5 [M + H]+, 358.2026; found, 358.2024.
6-Ethynyl-1H-pyrrolo[2,3-b]pyridine (15)
Compound 15 was prepared as previously described,41,42 and the spectral data were in accordance with those previously reported.
3-((1H-Pyrrolo[2,3-b]pyridin-6-yl)ethynyl)-5-(2-(6-(2,5-dimethyl-1H-pyrrol-1-yl)-4-methylpyridin-2-yl)ethyl)benzonitrile (16)
A mixture of 13 (394 mg, 1.0 mmol), 15 (156 mg, 1.1 mmol), Pd(PPh3)2Cl2 (35 mg, 0.05 mmol), CuI (10 mg, 0.05 mmol), PPh3 (52 mg, 0.20 mmol), diethylamine (2 mL), and DMF (2 mL) was heated at 120 °C for 20 min in the microwave cavity. Then the reaction mixture was treated with diethyl ether (20 mL), filtered, and concentrated in vacuo. The residue was purified by flash chromatography (EtOAc/hexanes) to give 16 (405 mg, 89%) as a pale-yellow oil. 1H NMR (500 MHz, CDCl3) δ 11.54 (s, 1H), 8.00 (d, J = 8.0 Hz, 1H), 7.71 (t, J = 1.6 Hz, 1H), 7.68 (t, J = 1.7 Hz, 1H), 7.45 (dt, J = 3.4, 2.1 Hz, 2H), 7.39 (d, J = 8.0 Hz, 1H), 6.95 (d, J = 1.3 Hz, 1H), 6.92 (d, J = 1.3 Hz, 1H), 6.57 (dd, J = 3.5, 1.9 Hz, 1H), 5.93 (s, 2H), 3.17 (h, J = 2.6 Hz, 4H), 2.41 (s, 3H), 2.16 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 159.58, 151.80, 149.89, 148.47, 143.47, 136.14, 134.18, 132.66, 131.91, 129.07, 128.48, 127.35, 124.33, 122.78, 120.79, 120.56, 119.87, 118.16, 112.89, 106.82, 101.20, 91.73, 85.40, 38.75, 34.78, 21.04, 13.29. MS (ESI) e/z = 456.3 [M + H]+.
3-(2-(1H-Pyrrolo[2,3-b]pyridin-6-yl)ethyl)-5-(2-(6-amino-4-methylpyridin-2-yl)ethyl)benzonitrile (17)
After stirring 16 (400 mg, 0.88 mmol) and Pd/C (10%, 200 mg) in EtOH (10 mL) for 30 min at ambient temperature under a hydrogen atmosphere, the reaction mixture was filtered through Celite and concentrated in vacuo. The residue was mixed with hydroxylamine HCl (308 mg, 4.4 mmol), EtOH (1.5 mL), and H2O (0.5 mL) in a microwave vial. The vial was capped, shaken vigorously, and then heated in the microwave irradiator for 30 min at 120 °C. After cooling the vial to room temperature, the reaction mixture was concentrated in vacuo and purified using a SiliaSep C18 flash cartridge (25g, 40–63 μm/230–400 mesh, pore size 60 Å) with 5–80% MeOH to give pure 17 (180 mg, 57%) as a pale-brown gel. 1H NMR (500 MHz, CD3OD) δ 7.89 (d, J = 8.0 Hz, 1H), 7.39 (s, 1H), 7.35–7.31 (m, 2H), 7.27 (s, 1H), 6.91 (d, J = 8.0 Hz, 1H), 6.46 (d, J = 3.5 Hz, 1H), 6.28 (s, 1H), 6.21 (s, 1H), 3.10 (td, J = 5.1, 4.0, 2.6 Hz, 4H), 2.92 (dd, J = 9.1, 6.7 Hz, 2H), 2.72 (dd, J = 9.1, 6.7 Hz, 2H), 2.17 (s, 3H). 13C NMR (126 MHz, CD3OD) δ 160.67, 158.71, 154.78, 151.29, 149.18, 144.64, 144.53, 135.00, 130.88, 130.83, 130.58, 126.30, 120.33, 120.00, 116.62, 114.81, 113.01, 108.12, 101.30, 40.35, 39.87, 37.17, 36.50, 21.01. HRMS (ESI): calcd for C24H24N5 [M + H]+, 382.2026; found, 358.2027.
6-(3-(2-(1H-Pyrrolo[2,3-b]pyridin-6-yl)ethyl)-5-(aminomethyl)phenethyl)-4-methylpyridin-2-amine (6)
A solution of 17 (100 mg, 0.28 mmol) in MeOH (10 mL) was stirred with Raney-Ni (50% in water, 0.2 mL) for 1 h at ambient temperature under a hydrogen atmosphere. The reaction mixture was filtered through Celite, concentrated in vacuo, and purified using prep-HPLC to yield 6 (63 mg, 62%). 1H NMR (500 MHz, CD3OD) δ 8.31 (s, 2H), 7.74 (d, J = 7.9 Hz, 1H), 7.18 (d, J = 3.5 Hz, 1H), 7.05 (s, 1H), 7.02 (s, 1H), 6.93 (s, 1H), 6.80 (d, J = 7.9 Hz, 1H), 6.77–6.67 (m, 1H), 6.35 (s, 1H), 6.31 (d, J = 3.5 Hz, 1H), 6.27 (s, 1H), 3.92 (s, 2H), 2.98 (dd, J = 8.7, 5.6 Hz, 2H), 2.92 (dd, J = 8.6, 5.6 Hz, 2H), 2.81 (t, J = 7.6 Hz, 2H), 2.72 (t, J = 7.4 Hz, 2H), 2.13 (s, 3H). 13C NMR (126 MHz, CD3OD) δ 157.84, 156.30, 155.35, 149.22, 144.46, 142.96, 134.90, 130.80, 130.53, 128.24, 127.83, 126.42, 120.45, 116.61, 115.67, 114.68, 110.01, 101.45, 44.42, 40.77, 37.78, 37.05, 36.31, 21.73. HRMS (ESI): calcd for C24H28N5 [M + H]+, 386.2339; found, 386.2338.
Acknowledgments
This work was supported by National Institute of Health grants GM57353 (to T.L.P.), GM49725 (to R.B.S.), and NIH predoctoral training grant T32 GM108561 (S.A.H.). Simulations were carried out on Stampede through an Xsede grant to S.A.H., J.H., and T.L.P. (TG-MCB130001). This work involves research carried out at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences.
Glossary
Abbreviations Used
- NOS
nitric oxide synthase
- bNOS
bacterial nitric oxide synthase
- mNOS
mammalian nitric oxide synthase
- nNOS
neuronal nitric oxide synthase
- eNOS
endothelial nitric oxide synthase
- iNOS
inducible nitric oxide synthase
- bsNOS
Bacillus subtilis nitric oxide synthase
- ACR
acriflavine
- H4B
tetrahydrobiopterin
- KS
spectral binding constant
- L-NNA
N-ω-nitro-l-arginine
- L-NOHA
N-ω-hydroxy-l-arginine
- MD
molecular dynamics
Accession Codes
Atomic coordinates and structure factors for compounds 1–7 complexed to wild type bsNOS and 7 complexed the H128S and I218V have been deposited in the Protein Data Bank with the ID codes 4D3J, 4D3I, 4D3K, 4D3M, 4D3N, 4D3O, 4D3T, 4D3U, and 4D3V, respectively.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Poulos T. L.; Li H. Structural basis for isoform-selective inhibition in nitric oxide synthase. Acc. Chem. Res. 2013, 46, 390–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman R. B. Design of selective neuronal nitric oxide synthase inhibitors for the prevention and treatment of neurodegenerative diseases. Acc. Chem. Res. 2009, 42, 439–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallance P.; Leiper J. Blocking NO synthesis: how, where and why?. Nature Rev. Drug Discovery 2002, 1, 939–950. [DOI] [PubMed] [Google Scholar]
- van Sorge N. M.; Beasley F. C.; Gusarov I.; Gonzalez D. J.; von Kockritz-Blickwede M.; Anik S.; Borkowski A. W.; Dorrestein P. C.; Nudler E.; Nizet V. Methicillin-resistant Staphylococcus aureus bacterial nitric-oxide synthase affects antibiotic sensitivity and skin abscess development. J. Biol. Chem. 2013, 288, 6417–6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusarov I.; Shatalin K.; Starodubtseva M.; Nudler E. Endogenous nitric oxide protects bacteria against a wide spectrum of antibiotics. Science 2009, 325, 1380–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shatalin K.; Gusarov I.; Avetissova E.; Shatalina Y.; McQuade L. E.; Lippard S. J.; Nudler E. Bacillus anthracis-derived nitric oxide is essential for pathogen virulence and survival in macrophages. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 1009–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusarov I.; Nudler E. NO-mediated cytoprotection: instant adaptation to oxidative stress in bacteria. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 13855–13860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden J. K.; Li H.; Jing Q.; Kang S.; Richo J.; Silverman R. B.; Poulos T. L. Structural and biological studies on bacterial nitric oxide synthase inhibitors. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 18127–18131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K.; Shimamura K.; Sekiguchi F.; Sunano S. Effects of NG-nitro-l-arginine on the blood pressure of spontaneously hypertensive rats with different degrees of hypertension. Clin. Exp. Hypertens. 2001, 23, 533–544. [DOI] [PubMed] [Google Scholar]
- Bogdan C. Nitric oxide and the immune response. Nature Immunol. 2001, 2, 907–916. [DOI] [PubMed] [Google Scholar]
- Holden J. K.; Lim N.; Poulos T. L. Identification of Redox Partners and Development of a Novel Chimeric Bacterial Nitric Oxide Synthase for Structure–Activity Analyses. J. Biol. Chem. 2014, 289, 29437–29445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z. Q.; Lawson R. J.; Buddha M. R.; Wei C. C.; Crane B. R.; Munro A. W.; Stuehr D. J. Bacterial flavodoxins support nitric oxide production by Bacillus subtilis nitric oxide synthase. J. Biol. Chem. 2007, 282, 2196–2202. [DOI] [PubMed] [Google Scholar]
- Flinspach M. L.; Li H.; Jamal J.; Yang W.; Huang H.; Hah J. M.; Gomez-Vidal J. A.; Litzinger E. A.; Silverman R. B.; Poulos T. L. Structural basis for dipeptide amide isoform-selective inhibition of neuronal nitric oxide synthase. Nature Struct. Mol. Biol. 2004, 11, 54–59. [DOI] [PubMed] [Google Scholar]
- Pant K.; Bilwes A. M.; Adak S.; Stuehr D. J.; Crane B. R. Structure of a nitric oxide synthase heme protein from Bacillus subtilis. Biochemistry 2002, 41, 11071–11079. [DOI] [PubMed] [Google Scholar]
- Crane B. R.; Sudhamsu J.; Patel B. A. Bacterial nitric oxide synthases. Annu. Rev. Biochem. 2010, 79, 445–470. [DOI] [PubMed] [Google Scholar]
- Tejero J.; Stuehr D. Tetrahydrobiopterin in nitric oxide synthase. IUBMB Life 2013, 65, 358–365. [DOI] [PubMed] [Google Scholar]
- Sudhamsu J.; Crane B. R. Bacterial nitric oxide synthases: what are they good for?. Trends Microbiol. 2009, 17, 212–218. [DOI] [PubMed] [Google Scholar]
- Delker S. L.; Xue F.; Li H.; Jamal J.; Silverman R. B.; Poulos T. L. Role of zinc in isoform-selective inhibitor binding to neuronal nitric oxide synthase. Biochemistry 2010, 49, 10803–10810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.; Li H.; Martasek P.; Roman L. J.; Poulos T. L.; Silverman R. B. Structure-guided design of selective inhibitors of neuronal nitric oxide synthase. J. Med. Chem. 2013, 56, 3024–3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.; Li H.; Yang S.; Chreifi G.; Martasek P.; Roman L. J.; Meyskens F. L.; Poulos T. L.; Silverman R. B. Potent and selective double-headed thiophene-2-carboximidamide inhibitors of neuronal nitric oxide synthase for the treatment of melanoma. J. Med. Chem. 2014, 57, 686–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adak S.; Aulak K. S.; Stuehr D. J. Direct evidence for nitric oxide production by a nitric-oxide synthase-like protein from Bacillus subtilis. J. Biol. Chem. 2002, 277, 16167–16171. [DOI] [PubMed] [Google Scholar]
- Goldschmidt L.; Cooper D. R.; Derewenda Z. S.; Eisenberg D. Toward rational protein crystallization: A Web server for the design of crystallizable protein variants. Protein Sci. 2007, 16, 1569–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Raman C. S.; Glaser C. B.; Blasko E.; Young T. A.; Parkinson J. F.; Whitlow M.; Poulos T. L. Crystal structures of zinc-free and -bound heme domain of human inducible nitric oxide synthase. Implications for dimer stability and comparison with endothelial nitric oxide synthase. J. Biol. Chem. 1999, 274, 21276–21284. [DOI] [PubMed] [Google Scholar]
- Gerber N. C.; Ortiz de Montellano P. R. Neuronal nitric oxide synthase. Expression in Escherichia coli, irreversible inhibition by phenyldiazene, and active site topology. J. Biol. Chem. 1995, 270, 17791–17796. [DOI] [PubMed] [Google Scholar]
- Martasek P.; Liu Q.; Liu J.; Roman L. J.; Gross S. S.; Sessa W. C.; Masters B. S. Characterization of bovine endothelial nitric oxide synthase expressed in E. coli. Biochem. Biophys. Res. Commun. 1996, 219, 359–365. [DOI] [PubMed] [Google Scholar]
- Leslie A. G. W.; Powell H. R.. Processing diffraction data with MOSFLM. Evolving Methods for Macromolecular Crystallography, NATO Science Series; Springer Netherlands: Dordrecht, The Netherlands, 2007; Vol. 245, pp 41–51 [Google Scholar]
- Kabsch W. Xds. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. Scaling and assessment of data quality. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2006, 62, 72–82. [DOI] [PubMed] [Google Scholar]
- Schuttelkopf A. W.; van Aalten D. M. PRODRG: a tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 1355–1363. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Mustyakimov M.; Afonine P. V.; Langan P. Generalized X-ray and neutron crystallographic analysis: more accurate and complete structures for biological macromolecules. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2009, 65, 567–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman L. J.; Sheta E. A.; Martasek P.; Gross S. S.; Liu Q.; Masters B. S. High-level expression of functional rat neuronal nitric oxide synthase in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 8428–8432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z. Q.; Wei C. C.; Sharma M.; Pant K.; Crane B. R.; Stuehr D. J. A conserved Val to Ile switch near the heme pocket of animal and bacterial nitric oxide synthases helps determine their distinct catalytic profiles. J. Biol. Chem. 2004, 279, 19018–19025. [DOI] [PubMed] [Google Scholar]
- Pant K.; Crane B. R. Structure of a loose dimer: an intermediate in nitric oxide synthase assembly. J. Mol. Biol. 2005, 352, 932–940. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: visual molecular dynamics. J. Mol. Graphics 1996, 1433–3827–38. [DOI] [PubMed] [Google Scholar]
- Kale L.; Skeel R.; Bhandarkar M.; Brunner R.; Gursoy A.; Krawetz N.; Phillips J.; Shinozaki A.; Varadarajan K.; Schulten K. NAMD2: Greater scalability for parallel molecular dynamics. J. Comput. Phys. 1999, 151, 283–312. [Google Scholar]
- Mackerell A. D.; Feig M.; Brooks C. L. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004, 25, 1400–1415. [DOI] [PubMed] [Google Scholar]
- Madrona Y.; Hollingsworth S. A.; Khan B.; Poulos T. L. P450cin active site water: implications for substrate binding and solvent accessibility. Biochemistry 2013, 52, 5039–5050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; Hatcher E.; Acharya C.; Kundu S.; Zhong S.; Shim J.; Darian E.; Guvench O.; Lopes P.; Vorobyov I.; MacKerell A. D. CHARMM General Force Field: A Force Field for Drug-Like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darden T.; York D.; Pedersen L. Particle Mesh Ewald—an N.Log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar]
- Martyna G. J.; Tobias D. J.; Klein M. L. Constant-Pressure Molecular-Dynamics Algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar]
- Miyamoto S.; Kollman P. A. Settle—An Analytical Version of the Shake and Rattle Algorithm for Rigid Water Models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar]
- Walia A.; Kang S.; Silverman R. B. Microwave-Assisted Protection of Primary Amines as 2,5-Dimethylpyrroles and Their Orthogonal Deprotection. J. Organomet. Chem. 2013, 78, 10931–10937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minakata S.; Itoh S.; Komatsu M.; Ohshiro Y. Functionalization of 1H-Pyrrolo[2,3-b]pyridine. Bull. Chem. Soc. Jpn. 1992, 65, 2992–2997. [Google Scholar]
- Minakata S.; Komatsu M.; Ohshiro Y. Regioselective Functionalization of 1H-Pyrrolo[2,3-b]Pyridine Via Its N-Oxide. Synthesis 1992, 661–663. [Google Scholar]