

Abstract
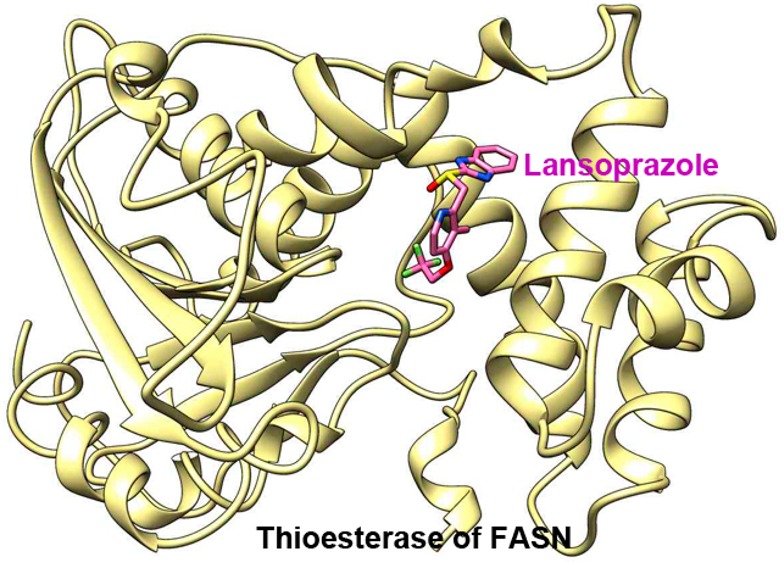
Fatty acid synthase (FASN), the enzyme responsible for de novo synthesis of free fatty acids, is up-regulated in many cancers. FASN is essential for cancer cell survival and contributes to drug resistance and poor prognosis. However, it is not expressed in most nonlipogenic normal tissues. Thus, FASN is a desirable target for drug discovery. Although different FASN inhibitors have been identified, none has successfully moved into clinical use. In this study, using in silico screening of an FDA-approved drug database, we identified proton pump inhibitors (PPIs) as effective inhibitors of the thioesterase activity of human FASN. Further investigation showed that PPIs inhibited proliferation and induced apoptosis of cancer cells. Supplementation of palmitate, the end product of FASN catalysis, rescued cancer cells from PPI-induced cell death. These findings provide new evidence for the mechanism by which this FDA-approved class of compounds may be acting on cancer cells.
Introduction
Human fatty acid synthase (FASN), consisting of 7-reaction domains, is the sole cytosolic enzyme responsible for synthesis of long-chain fatty acids, mainly 16-carbon palmitate.1−3 During palmitate synthesis, the growing fatty chain, tethered to the acyl carrier protein (ACP) domain, rotates between the other domains of FASN with addition of two carbons in each cycle.1−3 The thioesterase (TE) domain hydrolyzes the thioester bond between palmitate and ACP, releasing the free palmitate. FASN expression has been shown to play important roles in the formation, maintenance, and progression of many types of cancer4 and in the development of drug resistance.5−7 However, most nonlipogenic normal tissues do not express FASN. Thus, the development of an effective FASN inhibitor may have wide-reaching implications for many types of human cancers with high FASN expression. Unfortunately, despite past efforts, little progress has been made in finding a clinically useful FASN inhibitor.
Pancreatic cancers are the fourth leading cause of cancer-related deaths,8 and a majority of pancreatic cancer patients die within 6 months of diagnosis.9 FASN is overexpressed in pancreatic ductal adenocarcinomas and is positively associated with recurrence and negatively associated with overall survival.10 However, it is not expressed in normal pancreatic ductal epithelium.11 FASN has also been implicated in the increased resistance of pancreatic cancer cells to radiation and gemcitabine.6 Thus, targeting FASN may be an attractive approach for better treatment of pancreatic cancers and for eliminating drug resistance.
Recently, there has been great interest in repositioning FDA-approved drugs for treatment of human cancers.12 In this study, we searched for FDA-approved drugs that could potentially inhibit FASN using a crystal structure of FASN TE and performed virtual screening of a library of FDA-approved drugs targeting the active site of FASN TE, followed by a fluorogenic assay of top-scoring drugs using recombinant TE protein. We found that proton pump inhibitors (PPIs) effectively inhibited TE activity. PPIs are benzimidazole compounds13 that are FDA-approved therapeutics for treatment of a variety of acid-related diseases that plague the digestive system.14−16 Further examination showed that PPIs inhibited lipid synthesis, binding of a serine hydrolase probe to FASN, pancreatic cancer cell proliferation, and induced apoptosis of pancreatic cancer cells. Palmitate supplementation effectively rescued cancer cells from PPI-induced apoptosis. Thus, PPIs may exert anticancer activity in part by targeting and inhibiting the TE activity of human FASN, which is an important mechanistic consideration as PPIs are being repositioned for anticancer use.
Results
Identification of PPIs as FASN TE Inhibitors
To identify potential FASN TE inhibitors, we performed in silico screening of a library of 2417 FDA-approved drugs using DOCK programs and a crystal structure of FASN TE (PDB code 3TJM).17 The 200 top-scoring compounds were clustered based on their chemical structure, and 25 representative drugs from different clusters (Supporting Information Table S1) were selected for testing their ability to inhibit TE. For this purpose, we first purified recombinant FASN TE18,19 (Figure 1A) and adopted the fluorogenic assay using 4-methylumbelliferyl heptanoate (4-MUH) as a substrate, both as previously described.20−22 Figure 1B and Figure 1C show that the recombinant TE actively catalyzes hydrolysis of 4-MUH with a Km of 38.5 μM. Using this assay and purified TE, we tested the 25 top-scoring FDA-approved drugs with orlistat, a known inhibitor of FASN TE, as a positive control. As shown in Figure 2A, three drugs, 9, 16, and 18, reduced ≥40% of TE activity and, thus, were selected for further investigation. However, only 9 (pantoprazole) inhibited FASN TE activity in a dose-dependent manner (Figure 2B) with a Ki of 4.1 μM (Table 1). Drugs 16 (13-cis-retinoic acid) and 18 (sulcotidil) did not demonstrate the ability to inhibit FASN TE activity at lower concentration or in a dose-dependent manner (data not shown) and, consequently, were eliminated from further evaluation.
Figure 1.

Determination of FASN TE kinetic parameters. (A) Expression and purification of recombinant FASN TE: CB, commassie blue staining; IB, Western blot. (B) Lineweaver–Burk plot analysis of recombinant TE using 4-MUH fluorogenic assay. (C) Kinetic analysis. The kinetic parameters of FASN TE were determined by plotting 4-MU product formed (pmol/min) vs the concentration of the 4-MUH substrate. The Km of the protein was determined using a one enzyme model with no weighing of the data.
Figure 2.
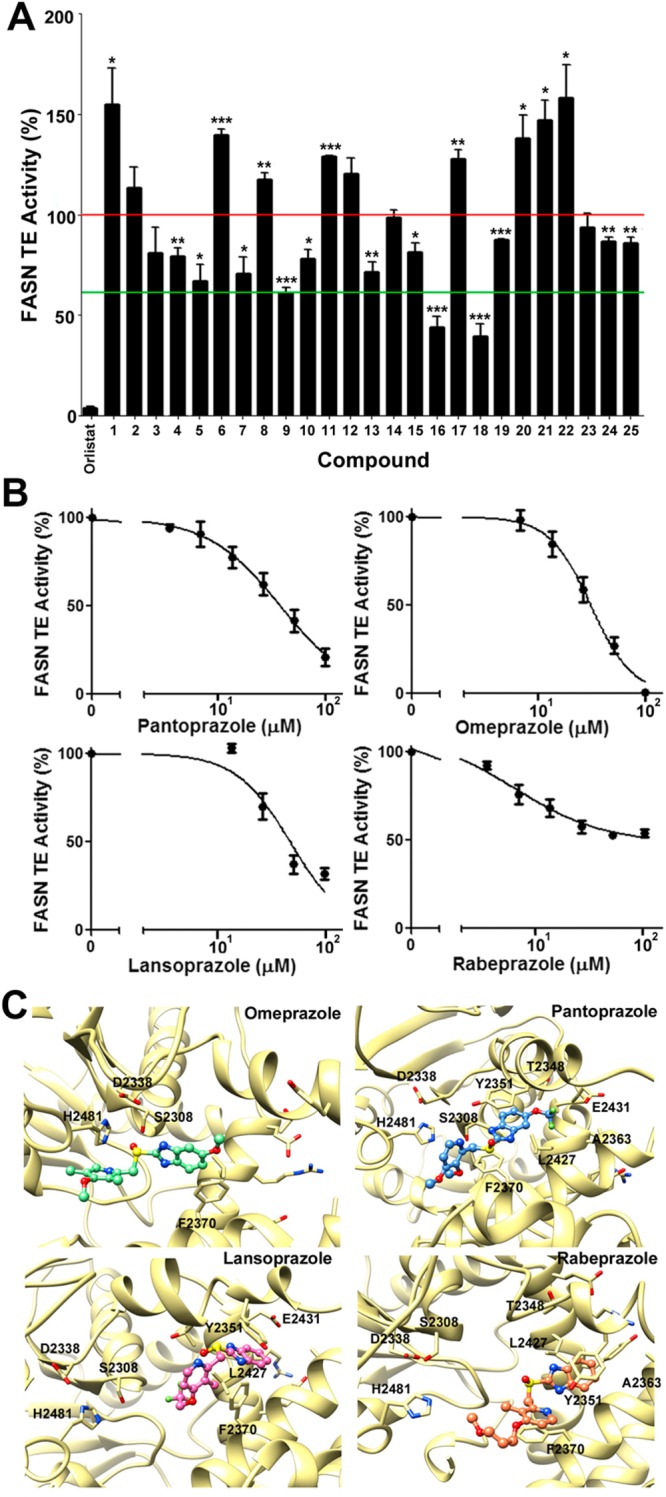
PPIs inhibit FASN TE activity. (A) Fluorogenic assay of 25 top-scoring FDA-approved drugs. The two horizontal lines indicate 100% and 60% TE activity (∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗, p < 0.001). (B) Dose-dependent inhibition of TE activity by PPIs. Each plot represents the average of three independent experiments. (C) Average simulated structures of PPIs bound to TE. TE is shown in gold ribbon. Omeprazole, pantoprazole lansoprazole, and rabeprazole are shown as ball and stick in green, blue, pink, and orange, respectively. In each panel, the catalytic triad residues and the residues predicted to interact with each PPI are labeled.
Table 1. Structures, IC50, Ki, and ΔGBind of PPIs.
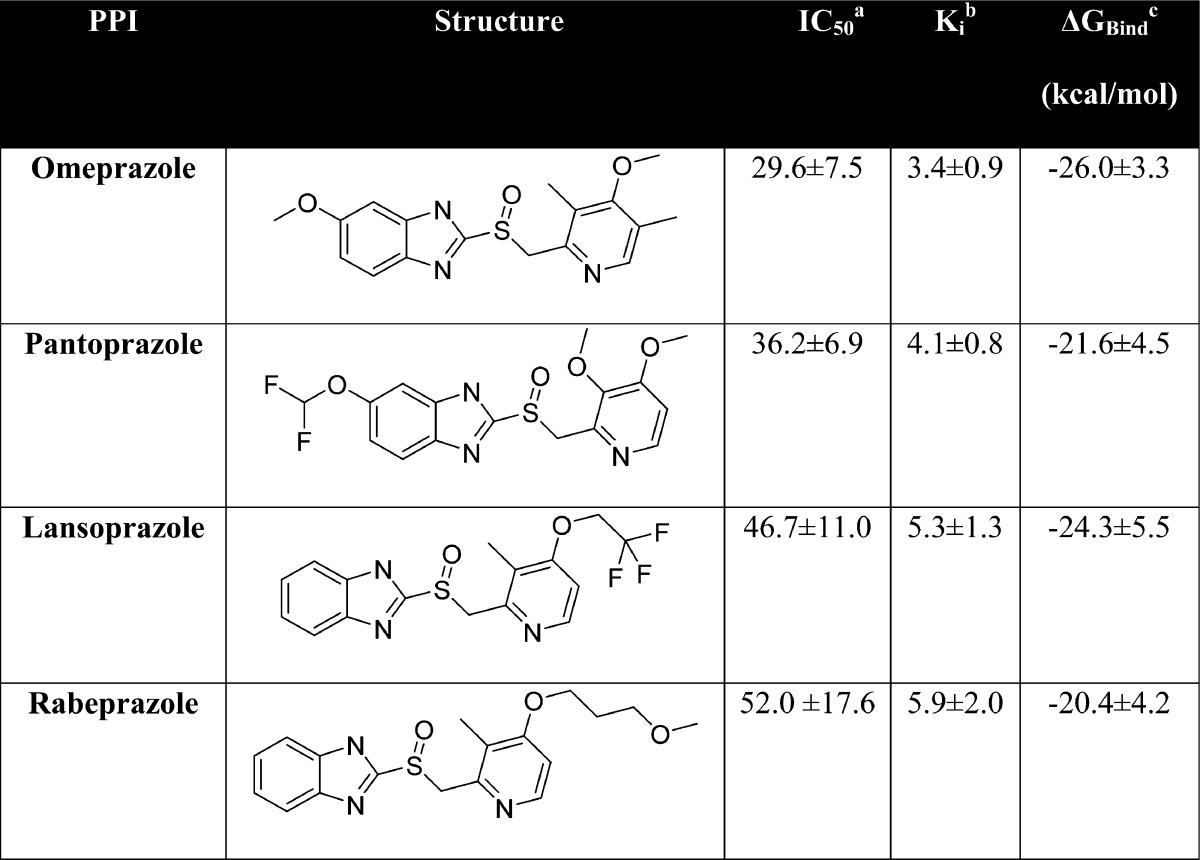
IC50 is the concentration of PPIs required to inhibit 50% of the recombinant TE activity, as measured using the 4-MUH fluorogenic assay.
Ki, the inhibition constant, was calculated from the IC50 using the Cheng–Prusoff equation.37
ΔGBind, the binding free energy, was calculated by Poisson–Boltzmann surface analysis (PBSA), where ΔGbind = Gcomplex – GTE – GPPI and G = Gsolute + Gsolvent.
We next sought to determine if any drugs from the cluster containing pantoprazole could also potentially inhibit FASN TE activity. Interestingly, the remaining drugs in this cluster of the 200 top-scoring compounds were other PPIs including omeprazole, lansoprazole, and rabeprazole. As shown in Figure 2B and Table 1, each of these PPIs similarly inhibited TE in a dose-dependent manner with Ki values of 3.4–5.9 μM with an activity ranking of omeprazole > pantoprazole > lansoprazole > rabeprazole. These findings suggest that increasing the size of either the 2-pyridylmethyl or the benzimidazole group of the compounds may slightly decrease the activity of PPI in inhibiting TE activity.
Binding Modes of PPIs
To predict a possible binding mode for each PPI within FASN TE, we used the well-established AMBER 12 suite of programs to perform molecular dynamics (MD) simulations of each PPI docked in the active site of FASN TE and calculated the binding free energy (ΔGbind) using Poisson–Boltzmann surface area (PBSA) analyses. Table 1 shows that the ΔGbind is favorable and that omeprazole has the highest while rabeprazole has the lowest ΔGbind, similar to the ranking of their experimental Ki values.
Next, the simulated average structure of each PPI within FASN TE was examined in detail. As shown in Figure 2C, omeprazole, with the most favorable ΔGbind and Ki, shows potential for the formation of a strong hydrogen bond between the active site serine residue (Ser2308) of the catalytic triad of TE and the sulfoxide moiety of omeprazole, which may prevent Ser2308 from nucleophilically attacking a substrate with an ester moiety. Interestingly, pantoprazole, lansoprazole, and rabeprazole are not predicted to have apparent interaction with any of the catalytic triad residues, Asp2338-His2481-Ser2308. However, the hydrophobic benzamidazole moiety of these PPIs may interact with residues of the “specificity channel”, which is predicted to accommodate the growing carbon chain during fatty acid synthesis,19 and thus, these PPIs may block access of the fatty acid chain to the channel. Residues in the channel that interact with PPIs include Thr2348, Tyr2351, Ala2363, Phe2370, Leu2427, and Glu2431 for pantoprazole; Tyr2351, Phe2370, Leu2427, and Glu2431 for lansoprazole; and Thr2348, Ala2363, Leu2427, Tyr2351, and Phe2370 for rabeprazole. Omeprazole also interacts with the channel residue Phe2370. These potential interactions provide rationale as to the mechanism by which PPIs inhibit TE activity. However, experimental structure analysis is clearly needed to validate the predicted binding mode of PPIs and specific residues of interaction within FASN TE.
PPIs Inhibit Cancer Cell Proliferation by Inducing Apoptosis
To determine the utility of PPIs in inhibiting cancer cell proliferation, we performed colony formation assay of BxPC-3 pancreatic cancer cells in the presence of PPIs along with orlistat as a control. The survival of BxPC-3 cells was dose-dependently inhibited by all four PPIs (Figure 3A). The relative potency of PPIs is lansoprazole > rabeprazole > omeprazole > pantoprazole with IC50 values ranging from 6.7 to 18.5 μM. We also tested lansoprazole against another pancreatic cancer cell line, PANC-1 and showed a dose-dependent inhibition with an IC50 of 58.6 μM (Figure 3B). As a comparison, we also examined the potency of orlistat in both cell lines and found that the IC50 of orlistat is 8.5 μM for BxPC-3 and 68 μM for PANC-1 cells, slightly less potent than lansoprazole. To determine if PPIs possibly induce apoptosis, we performed ELISA to quantitate the amount of cytoplasmic histone-associated DNA-fragments using a cell death detection ELISA kit (Roche) and Western blot analysis of cleaved poly(ADP-ribose) polymerase 1 (PARP-1). As shown in Figure 3C,D, lansoprazole dose-dependently caused formation of DNA fragments and cleaved PARP-1, indicating that lansoprazole treatment causes apoptosis in a dose-dependent manner.
Figure 3.

PPIs inhibit survival and induce apoptosis. (A). Effect of PPIs on survival of BxPC-3 cells as determined using colony formation assay. Orlistat was used as a control. Each plot represents the average of three independent experiments. (B) Effect of lansoprazole and orlistat on survial of PANC-1 cells as determined using colony formation assay. (C) Lansoprazole induction of apoptosis. Apoptosis was measured by quantifying the amount of cytoplasmic histone-associated DNA-fragments in PANC-1 cells following lansoprazole treatment. (D) Lansoprazole-induced PARP-1 cleavage: cPARP, cleaved PARP-1. Actin was used as a loading control.
Lansoprazole Does Not Affect Intracellular pH
PPIs are known to irreversibly inhibit H+/K+ ATPases23−25 and may cause cancer cell death by affecting pH homeostasis. To test this possibility, we examined the intracellular pH of BxPC-3 cells treated in the absence or presence of different concentrations of lansoprazole. As shown in Figure 4A, lansoprazole treatment had no significant effect on intracellular pH. We then tested extracellular pH following lansoprazole treatment and also found no change in extracellular pH (data not shown). Thus, PPI-induced cancer cell death may not be due to changes in pH homeostasis.
Figure 4.

Effect of lansoprazole on intracellular pH and FASN. (A) Intracellular pH. Intracellular pH was measured in BxPC-3 cells using the pHrodo red intracellular pH dye following lansoprazole treatment. Each data point represents the average of three independent experiments. (B) Lipid synthesis. Inhibition of [14C]acetate incorporation into lipids in the presence of different concentrations of lansoprazole or orlistat was quantified in PANC-1 and/or BxPC-3 cells. The plots shown are representatives of three independent experiments. (C, D) Dose-dependent lansoprazole inhibition of FP serine hydrolase probe labeling (C) and expression (D) of FASN. Arrowhead indicates FP probe-labeled FASN. Asterisks indicate FP probe-labeled other serine hydrolases. Actin was used as a loading control for FASN.
Lansoprazole Inhibits Cellular FASN Activity
To investigate if lansoprazole inhibits cellular FASN, we performed a FASN activity assay by determining lipid synthesis in the presence of lansoprazole in live cells. As shown in Figure 4B, lansoprazole inhibited lipid synthesis dose-dependently in both PANC-1 and BxPC-3 cells with IC50 values of ∼93 and ∼124 μM, respectively. The known inhibitor of FASN TE, orlistat, also inhibited FASN activity in PANC-1 cells with an IC50 of ∼203 μM (Figure 4B). It is noteworthy that the IC50 of lansoprazole and orlistat required to inhibit lipid synthesis is higher than that for inhibiting cell survival (Figure 3). This discrepancy may be due to the difference in treatment duration used for the two different assays. While cells were treated by lansoprazole or orlistat for 10–14 days for the colony formation survival assay, the treatment was only 4 h for the lipid synthesis assays.
To ensure that lansoprazole inhibits FASN by binding to the active site of cellular FASN, we performed a probe binding displacement experiment using the ActivX desthiobiotin-fluorophosphonate (FP) serine hydrolase probe, which can covalently bind to the Ser residue in the catalytic triad of TE.18 For this purpose, PANC-1 cell lysate was incubated with the FP probe in the presence and absence of lansoprazole and subjected to Western blot analysis probed with streptavidin-conjugated HRP. As shown in Figure 4A, lansoprazole inhibited labeling of FASN by the FP probe in a dose-dependent manner, suggesting that lansoprazole inhibits FASN by directly interacting with the TE active site. However, lansoprazole treatment had no effect on the binding of the FP probe to other Ser hydrolases, suggesting that lansoprazole selectively inhibits FASN TE. Lansoprazole also had no effect on the total FASN level. Together, these results indicate that lansoprazole inhibits FASN TE, which is not likely due to an unknown artificial effect but possibly due, at least in part, to the direct binding to and inhibition of the TE active site.
Palmitate Supplementation Rescues Cells from Lansoprazole Cytotoxicity
To further investigate the inhibition of FASN by lansoprazole, we tested if palmitate, the end product of FASN catalysis, can rescue cells from lansoprazole-induced apoptosis. First, we tested if palmitate supplementation alone affects cell survival. As shown in Figure 5A, supplementation with 3.75 μM palimitate had no significant effect on BxPC-3 cell survival. It also did not reduce FASN expression via potential feedback effect (Figure 5B). However, supplementation with 3.75 μM palmitate significantly increased cellular resistance to lansoprazole (Figure 5C) and reduced lansoprazole-induced apoptosis (Figure 5D). Thus, lansoprazole likely causes cell death by inhibiting FASN and production of palmitate, which can be rescued with palmitate supplementation.
Figure 5.

Palmitate supplementation rescues lansoprazole inhibition. (A) Effect of palmitate on cell growth compared to DMSO control, as measured by MTT assays (n = 3, p = 0.19). (B) Western blot analysis of palmitate effect on FASN expression. Actin was used as a loading control. (C) Effect of palmitate on lansoprazole cytotoxicity as measured by MTT assay (n = 3; ∗∗∗, p < 0.001). (D) Effect of palmitate on lansoprazole-induced apoptosis (n = 3; ∗∗∗, p < 0.001).
Lansoprazole Is More Effective in Cells with Higher FASN Activity
The data in Figure 3 show that BxPC-3 cells are ∼9-fold more sensitive than PANC-1 cells to lansoprazole treatment. To examine the underlining cause for the difference, we first examined FASN expression and FASN activity in these cells. As shown in Figure 6A, PANC-1 cells have a higher FASN expression level than BxPC-3 cells but with less FASN activity. Thus, FASN protein level does not directly correlate with FASN activity and endogenous FASN in PANC-1 cells may be less effective in synthesizing lipids (see discussion below). The above finding also indicates that cells with higher FASN activity may be more sensitive to lansoprazole treatment in the survival assay (see also discussion below).
Figure 6.

Differential effects of lansoprazole on cells with varying FASN activity. (A) FASN protein level and activity in PANC-1 and BxPC-3 cells. (n = 3; ∗, p < 0.05). (B) FASN protein level and activity in PANC-1/FASN and control PANC-1/Vec cells (n = 3). (C) Lansoprazole cytotoxicity on PANC-1/Vec and PANC-1/FASN cells as determined using colony formation assay. (n = 3; ∗, p < 0.05; ∗∗, p < 0.01).
To test this possibility, we took advantage of a previously established stable PANC-1/FASN cell line with overexpression of ectopic wild-type FASN (Figure 6B). We have shown previously that PANC-1/FASN cells have higher FASN activity than the vector-transfected PANC-1/Vec cells due to ectopic expression of the wild type FASN6 (see also Figure 6B). We performed a colony formation survival assay for both PANC-1/FASN and PANC-1/Vec cells in the presence of lansoprazole. As shown in Figure 6C, PANC-1/FASN cells with higher FASN activity are significantly more sensitive to lansoprazole than the control PANC-1/Vec cells. Thus, we conclude that cells with higher FASN activity are likely more sensitive to lansoprazole inhibition of survival.
Discussion and Conclusion
Recently, there has been considerable interest in repositioning FDA-approved drugs for cancer treatments. The results of the current study are the first to demonstrate that PPIs directly bind to the active site and inhibit FASN TE and, thus, provide a fundamental basis for repositioning PPIs as anticancer therapeutics. Considering that long-term and high-dose PPI treatment has been shown to be well tolerated in patients with few side effects,26,27 repositioning PPIs as anticancer drugs will unlikely have added toxicity.
Use of pantoprazole alone has been shown to induce apoptosis of gastric cancer cells both in vitro and in vivo28 and pretreatment with PPIs sensitized cancer cells to chemotherapeutic agents cisplatin, 5-FU, and vinblastine in vitro and cisplatin in vivo.29 Clinical trials are also being performed to evaluate the use of PPIs in combination with chemotherapeutic drugs for cancer treatment. For example, an ongoing phase I trial is investigating the use of pantoprazole in combination with doxorubicin in advanced cancer patients with solid tumors.30 However, it remains to be determined whether the effect of PPIs in suppressing tumor growth and chemosensitization in vivo and in clinical trials is due to inhibition of FASN. The fact that FASN plays an important role in cancer cell survival and in drug resistance4 and that PPIs inhibit FASN as shown in this study is consistent with the observations of both in vitro and in vivo studies. Furthermore, our finding that the effect of lansoprazole is reversed upon palmitate supplementation indicates that it may be important to restrict high fat diets in future clinical use of PPIs to increase PPI efficacy during chemotherapy.
Our finding that PPIs may be more effective in cells that have higher FASN activity is very important for designing future personalized treatments. Although it is unknown why cells with higher FASN activity are more sensitive to PPIs and orlistat inhibition of survival, it is possible that cells such as BxPC-3 may require, or are “addicted” to, higher FASN activity for survival and, thus, are more sensitive to FASN inhibition. It is also noteworthy that FASN activity in PANC-1 cells is lower than that in BxPC-3 cells, albeit PANC-1 cells have a higher FASN protein level. Although acetyl-CoA carboxylase is the known rate-limiting enzyme for lipid synthesis,31 we found that ectopic expression of FASN in PANC-1 cells was able to further increase lipid synthesis. Thus, the endogenous FASN in PANC-1 cells may be less effective in producing lipids possibly due to potential mutations or post-translational modifications. Further studies are clearly needed to determine if the endogenous FASN in PANC-1 cells has any defective mutations or is post-translationally modified, which may reduce FASN activity.
Experimental Procedures
In Silico Screening
The in silico screening was performed as we previously described.32 Briefly, the high resolution FASN TE crystal structure with a polyunsaturated fatty acid adduct17 was obtained from the RCSB Protein Data Bank (code 3TJM) with missing residues modeled using ModLoop33 and was prepared for in silico screening using the DOCK suite of programs. A library containing 2417 FDA approved ligands was downloaded from the ZINC database34−36 and was used for rigid docking with 500 orientations, which were scored using the grid program within DOCK. The 200 top-scoring compounds were then subjected to AMBER analysis and clustering using the clustering tool Library MCS. Compounds with unfavorable AMBER scores were eliminated. The 200 top-scoring compounds in each cluster were visually examined using the Chimera visualization program for the selection of representative compounds for further testing using a fluorogenic assay. Drugs containing lactone or lactam moieties, moieties that could be nucleophically attacked by the Ser residue in the active site of TE to form a covalent bond (such as an epoxide ring), and long-chain hydrocarbon moieties were given special preference. The final 25 drugs were selected, which met all above criteria (Supporting Information Table S1).
Purity Statement
All compounds tested were purchased from reputable sources (Sigma-Aldrich, Toronto Research Chemicals, Cayman Chemical), with purity of ≥95%, as determined by standard analytical methods. The certificate of analysis for the final selected compounds including pantoprazole (Toronto Research Chemicals), lansoprazole, omeprazole, and rabeprazole (Sigma) as well as NMR and MS spectra confirming their identity are shown in Supporting Information purity file.
Fluorogenic TE Activity Assay
The fluorogenic TE activity assay was performed as previously described.22 Briefly, each assay was performed in opaque black, flat-bottom 96-well plates (Corning), with each well containing 500 nM purified TE in buffer A (100 mM Tris-HCl, 50 mM NaCl, 0.05% Brij35, pH 7.5). For PPI inhibition, recombinant TE was preincubated with PPIs at 37 °C for 30 min. The hydrolysis reaction was started by addition of 300 μM 4-MUH (Sigma) and incubation at 37 °C for 1 h. Fluorescence due to liberated 4-MU was measured at 355/460 nm. The Ki value for each inhibitor candidate was calculated using the Cheng and Prusoff equation.37
MD Simulation and Estimation of ΔGbind
The binding free energies (ΔGbind) of PPIs within FASN TE were calculated as we previously described38 using the final docked pose of each PPI in the active site of TE. Briefly, to refine the position of the initially docked structure within the FASN TE active site, energy minimization was performed, followed by 10 ns production MD simulations. Finally, 50 snapshots were extracted from the final 5 ns of the production trajectories, and the binding free energy was calculated using the MM-PBSA method.39 All simulations and subsequent calculations were performed using the AMBER 12 molecular dynamics package.
Colony Formation Survival and Apoptosis Assays
These assays were performed as previously described.5,40,41 Briefly, cells were seeded in six-well plates (100 cells/well for PANC-1 and 200 cells/well for BxPC-3) and cultured for 24 h before addition of PPIs or DMSO vehicle. The cells were continuously cultured in the presence of PPIs or DMSO for 10–14 days followed by staining with crystal violet and counting manually.
For the apoptosis assay, BxPC-3 cells were seeded in 12-well plates (18 000 cells/well) and cultured for 24 h before treatment with lansoprazole or DMSO control for 72 h. The cells were then harvested and subjected to analysis using the cell death detection ELISA assay kit (Roche) according to the manufacturer’s instructions. For detection of cleaved PARP, BxPC-3 cells were seeded in six-well plates (200 000 cells/well) and cultured for 24 h before treatment with lansoprazole or DMSO control for 24 h. Cells were then collected and subjected to Western blot analysis of cleaved PARP using an antibody specific to the cleaved PARP (Cell Signaling).
Determination of pH
To measure intracellular pH, BxPC-3 cells were seeded in 96-well plates (2000 cells/well) and cultured for 24 h, followed by treatment with lansoprazole or DMSO control for 72 h. The cells were then incubated with the pHrodo red intracellular pH dye (Molecular Probes), and the fluorescence was measured directly in the 96-well plates according to the manufacturer’s instructions. The fluorescence values were then converted to pH using the intracellular pH calibration kit (Molecular Probes) and a standard curve of fluorescence vs pH, created according to the manufacturer’s instructions. For extracellular pH, BxPC-3 cells in six-well plates (55 000 cells/well) were treated with 100 μM lansoprazole or DMSO control for 72 h. The culture medium was collected, cleared of cell debris by centrifugation, and used for pH determination using a pH meter (Fisher Scientific).
Fatty Acid Synthase Activity Assay
Fatty acid synthase activity was determined using [14C]acetate incorporation assay as previously described.18 Briefly, cells were seeded in 12-well plates (100 000 cells/well), cultured for 24 h, and incubated for 2 h at 37 °C in the presence of 1 μCi/mL [14C]acetate (PerkinElmer). Lipids were then extracted using the Folch extraction method,42 dried, resuspended in CHCl3, and radioactivity was determined using a scintillation counter. For inhibition of FASN, the cells were treated with PPIs for 4 h prior to incubation with [14C]acetate and analysis.
Serine hydrolase probe displacement assay
The serine hydrolase probe displacement assay was performed as previously described.18 Briefly, lysate of PANC-1 cells was pretreated with varying concentrations of lansoprazole or DMSO control for 30 min at room temperature, followed by treatment with 5 μM ActivX desthiobiotin-fluorophosphonate (FP) serine hydrolase probe (Thermo) for 30 min at room temperature. Reactions were stopped by addition of SDS–PAGE loading buffer and boiling for 10 min, followed by Western blot analysis probed with streptavidin-conjugated HRP and ECL for visualization.
IC50 and Statistical Calculations
All IC50 values and statistical calculations were performed using Prism5 (GraphPad). IC50 values were calculated using the log(inhibitor) vs normalized response regression equation. All statistics were calculated using a two-tailed Student’s t test.
Acknowledgments
The authors thank Dr. David Jones at Indiana University School of Medicine for expertise and assistance in calculating FASN TE enzymatic parameters and the IU Big Red supercomputer for CPU time. This work was supported in part by an NIH/NCI NRSA Grant 5F31 CA165603-03 (V.E.F.) and a DOD Predoctoral Fellowship W81XWH-10-1-0057 (X.W.).
Glossary
Abbreviations Used
- 4-MUH
4-methylumbelliferyl heptanoate
- ACP
acyl carrier protein
- FASN
fatty acid synthase
- FP
fluorophosphonate
- PBSA
Poisson–Boltzmann surface area
- PPI
proton pump inhibitor
- TE
thioesterase
Supporting Information Available
Purity of compounds, Table S1 of top-scoring FDA-approved drugs, and molecular formula strings in csv format. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Wakil S. J.; Stoops J. K.; Joshi V. C. Fatty acid synthesis and its regulation. Annu. Rev. Biochem. 1983, 52, 537–579. [DOI] [PubMed] [Google Scholar]
- Wakil S. J. Fatty acid synthase, a proficient multifunctional enzyme. Biochemistry 1989, 28, 4523–4530. [DOI] [PubMed] [Google Scholar]
- Smith S. The animal fatty acid synthase: one gene, one polypeptide, seven enzymes. FASEB J. 1994, 8, 1248–1259. [PubMed] [Google Scholar]
- Liu H.; Liu J. Y.; Wu X.; Zhang J. T. Biochemistry, molecular biology, and pharmacology of fatty acid synthase, an emerging therapeutic target and diagnosis/prognosis marker. Int. J. Biochem. Mol. Biol. 2010, 1, 69–89. [PMC free article] [PubMed] [Google Scholar]
- Liu H.; Liu Y.; Zhang J. T. A new mechanism of drug resistance in breast cancer cells: fatty acid synthase overexpression-mediated palmitate overproduction. Mol. Cancer Ther. 2008, 7, 263–270. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Liu H.; Li Z.; Zhao Z.; Yip-Schneider M.; Fan Q.; Schmidt C. M.; Chiorean E. G.; Xie J.; Cheng L.; Chen J. H.; Zhang J. T. Role of fatty acid synthase in gemcitabine and radiation resistance of pancreatic cancers. Int. J. Biochem. Mol. Biol. 2011, 2, 89–98. [PMC free article] [PubMed] [Google Scholar]
- Kao Y. C.; Lee S. W.; Lin L. C.; Chen L. T.; Hsing C. H.; Hsu H. P.; Huang H. Y.; Shiue Y. L.; Chen T. J.; Li C. F. Fatty acid synthase overexpression confers an independent prognosticator and associates with radiation resistance in nasopharyngeal carcinoma. Tumour Biol. 2013, 34, 759–768. [DOI] [PubMed] [Google Scholar]
- Greenlee R. T.; Murray T.; Bolden S.; Wingo P. A. Cancer statistics, 2000. Ca—Cancer J. Clin. 2000, 50, 7–33. [DOI] [PubMed] [Google Scholar]
- Janes R. H. Jr.; Niederhuber J. E.; Chmiel J. S.; Winchester D. P.; Ocwieja K. C.; Karnell J. H.; Clive R. E.; Menck H. R. National patterns of care for pancreatic cancer. Results of a survey by the Commission on Cancer. Ann. Surg. 1996, 223, 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alo P. L.; Amini M.; Piro F.; Pizzuti L.; Sebastiani V.; Botti C.; Murari R.; Zotti G.; Di Tondo U. Immunohistochemical expression and prognostic significance of fatty acid synthase in pancreatic carcinoma. Anticancer Res. 2007, 27, 2523–2527. [PubMed] [Google Scholar]
- Walter K.; Hong S. M.; Nyhan S.; Canto M.; Fedarko N.; Klein A.; Griffith M.; Omura N.; Medghalchi S.; Kuhajdal F.; Goggins M. Serum fatty acid synthase as a marker of pancreatic neoplasia. Cancer Epidemiol., Biomarkers Prev. 2009, 18, 2380–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim J. S.; Liu J. O. Recent advances in drug repositioning for the discovery of new anticancer drugs. Int. J. Biol. Sci. 2014, 10, 654–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellenius E.; Berglindh T.; Sachs G.; Olbe L.; Elander B.; Sjostrand S. E.; Wallmark B. Substituted benzimidazoles inhibit gastric acid secretion by blocking (H+ + K+)ATPase. Nature 1981, 290, 159–161. [DOI] [PubMed] [Google Scholar]
- Holt S. Proton-pump inhibition for acid-related disease. South. Med. J. 1991, 84, 1078–1087. [DOI] [PubMed] [Google Scholar]
- Stedman C. A.; Barclay M. L. Review article: Comparison of the pharmacokinetics, acid suppression and efficacy of proton pump inhibitors. Aliment. Pharmacol. Ther. 2000, 14, 963–978. [DOI] [PubMed] [Google Scholar]
- Der G. An overview of proton pump inhibitors. Gastroenterol. Nurs. 2003, 26, 182–190. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Chakravarty B.; Zheng F.; Gu Z.; Wu H.; Mao J.; Wakil S. J.; Quiocho F. A. Crystal structure of FAS thioesterase domain with polyunsaturated fatty acyl adduct and inhibition by dihomo-gamma-linolenic acid. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 15757–15762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kridel S. J.; Axelrod F.; Rozenkrantz N.; Smith J. W. Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity. Cancer Res. 2004, 64, 2070–2075. [DOI] [PubMed] [Google Scholar]
- Pemble C. W. 4th; Johnson L. C.; Kridel S. J.; Lowther W. T. Crystal structure of the thioesterase domain of human fatty acid synthase inhibited by Orlistat. Nat. Struct. Mol. Biol. 2007, 14, 704–709. [DOI] [PubMed] [Google Scholar]
- Jacks T. J.; Kircher H. W. Fluorometric assay for the hydrolytic activity of lipase using fatty acyl esters of 4-methylumbelliferone. Anal. Biochem. 1967, 21, 279–285. [DOI] [PubMed] [Google Scholar]
- Purohit V. C.; Richardson R. D.; Smith J. W.; Romo D. Practical, catalytic, asymmetric synthesis of beta-lactones via a sequential ketene dimerization/hydrogenation process: inhibitors of the thioesterase domain of fatty acid synthase. J. Org. Chem. 2006, 71, 4549–4558. [DOI] [PubMed] [Google Scholar]
- Richardson R. D.; Smith J. W. Novel antagonists of the thioesterase domain of human fatty acid synthase. Mol. Cancer Ther. 2007, 6, 2120–2126. [DOI] [PubMed] [Google Scholar]
- Sachs G.; Shin J. M.; Besancon M.; Prinz C. The continuing development of gastric acid pump inhibitors. Aliment. Pharmacol. Ther. 1993, 7Suppl. 14–12(discussion 29–31). [DOI] [PubMed] [Google Scholar]
- Sachs G.; Shin J. M.; Briving C.; Wallmark B.; Hersey S. The pharmacology of the gastric acid pump: the H+,K+ ATPase. Annu. Rev. Pharmacol. Toxicol. 1995, 35, 277–305. [DOI] [PubMed] [Google Scholar]
- Sachs G.; Shin J. M.; Howden C. W. Review article: The clinical pharmacology of proton pump inhibitors. Aliment. Pharmacol. Ther. 2006, 23Suppl. 22–8. [DOI] [PubMed] [Google Scholar]
- Thomson A. B.; Sauve M. D.; Kassam N.; Kamitakahara H. Safety of the long-term use of proton pump inhibitors. World J. Gastroenterol. 2010, 16, 2323–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y. X.; Metz D. C. Safety of proton pump inhibitor exposure. Gastroenterology 2010, 139, 1115–1127. [DOI] [PubMed] [Google Scholar]
- Yeo M.; Kim D. K.; Kim Y. B.; Oh T. Y.; Lee J. E.; Cho S. W.; Kim H. C.; Hahm K. B. Selective induction of apoptosis with proton pump inhibitor in gastric cancer cells. Clin. Cancer Res. 2004, 10, 8687–8696. [DOI] [PubMed] [Google Scholar]
- Luciani F.; Spada M.; De Milito A.; Molinari A.; Rivoltini L.; Montinaro A.; Marra M.; Lugini L.; Logozzi M.; Lozupone F.; Federici C.; Iessi E.; Parmiani G.; Arancia G.; Belardelli F.; Fais S. Effect of proton pump inhibitor pretreatment on resistance of solid tumors to cytotoxic drugs. J. Natl. Cancer Inst. 2004, 96, 1702–1713. [DOI] [PubMed] [Google Scholar]
- Study evaluating pantoprazole with doxorubicin for advanced cancer patients with extension cohort of patients with solid tumors. NLM Identifier: NCT01163903. http://clinicaltrials.gov/ct2/show/NCT01163903 (accessed Jan 29, 2014).
- Tong L. Acetyl-coenzyme A carboxylase: crucial metabolic enzyme and attractive target for drug discovery. Cell. Mol. Life Sci. 2005, 62, 1784–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W.; Dong Z.; Wang F.; Peng H.; Liu J. Y.; Zhang J. T. A small molecule compound targeting STAT3 DNA-binding domain inhibits cancer cell proliferation, migration, and invasion. ACS Chem. Biol. 2014, 9, 1188–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiser A.; Sali A. ModLoop: automated modeling of loops in protein structures. Bioinformatics 2003, 19, 2500–2501. [DOI] [PubMed] [Google Scholar]
- Irwin J. J.; Shoichet B. K. ZINC—a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin J.; Shoichet B. The ZINC database as a new research tool for ligand discovery. Abstr. Pap.—Am. Chem. Soc. 2005, 230, U1009. [Google Scholar]
- Irwin J. J.; Sterling T.; Mysinger M. M.; Bolstad E. S.; Coleman R. G. ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y.; Prusoff W. H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- Liu J. Y.; Li Z.; Li H.; Zhang J. T. Critical residue that promotes protein dimerization: a story of partially exposed Phe25 in 14-3-3sigma. J. Chem. Inf. Model. 2011, 51, 2612–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rastelli G.; Del Rio A.; Degliesposti G.; Sgobba M. Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. J. Comput. Chem. 2010, 31, 797–810. [DOI] [PubMed] [Google Scholar]
- Li Z.; Dong Z.; Myer D.; Yip-Schneider M.; Liu J.; Cui P.; Schmidt C. M.; Zhang J. T. Role of 14-3-3sigma in poor prognosis and in radiation and drug resistance of human pancreatic cancers. BMC Cancer 2010, 10, 598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.; Wu X.; Dong Z.; Luo Z.; Zhao Z.; Xu Y.; Zhang J. T. Fatty acid synthase causes drug resistance by inhibiting TNF-alpha and ceramide production. J. Lipid Res. 2013, 54, 776–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folch J.; Lees M.; Sloane Stanley G. H. A simple method for the isolation and purification of total lipids from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.