

Abstract
Fluorine-18 is the most frequently used radioisotope in positron emission tomography (PET) radiopharmaceuticals in both clinical and preclinical research. Its physical and nuclear characteristics (97% β+ decay, 109.7 min half-life, 635 keV positron energy), along with high specific activity and ease of large scale production, make it an attractive nuclide for radiochemical labeling and molecular imaging. Versatile chemistry including nucleophilic and electrophilic substitutions allows direct or indirect introduction of 18F into molecules of interest. The significant increase in 18F radiotracers for PET imaging accentuates the need for simple and efficient 18F-labeling procedures. In this review, we will describe the current radiosynthesis routes and strategies for 18F labeling of small molecules and biomolecules.
1. Introduction
Positron emission tomography (PET) is a nuclear medicine imaging technology that provides moderate-resolution, sensitive images of the biodistribution of a radiotracer in vivo.1 The technique has sufficient speed of acquisition to allow determination of pharmacokinetics of radiotracer uptake and distribution. The combination of PET imaging with a validated radiopharmaceutical can allow the images to provide interpretation of a biological function. In addition to applications for diagnosis of diseases,1 PET imaging can provide important insights for both drug discovery and development and for potentially limiting side effects due to off-target binding.2 The most important component to the future utility of PET technology is the development of novel, specific, validated radiotracers for clinically relevant targets and methods for their efficient preparation.
PET is based on the administration of radiolabeled molecules with positron emitting nuclides such a 15O, 13N, 11C, and 18F, with relatively short half-lives of 2.037, 9.965, 20.39, and 109.8 min, respectively.2 Of the nuclides mentioned above, 18F has the most ideal half-life for labeling of radiopharmaceuticals (small organic molecules, peptides, aptamers, and proteins) and has a unique and diverse chemistry for introduction into various molecules. In the discipline of medicinal chemistry, fluorine is a favorable atom in drug development due to its physical properties including small van der Waals radius (1.47 Å), high electronegativity, and ability to form a strong bond with carbon (C–F energy bond of 112 kcal/mol), which in comparison to a carbon–hydrogen bond (C–H = 98 kcal/mol) is more thermally stable and oxidation resistant.3,4 Fluorine can act as a bioisostere with hydrogen (size and valence electrons) and oxygen (size and electronegativity).4 As a result of its significance in the pharmaceutical field, several selective fluorination reagents for nucleophilic (F–) and electrophilic (F+) incorporation have been developed and have become commercially available (Figure 1).5,6
Figure 1.

Commercial fluorinating reagents for nucleophilic and electrophilic substitutions.
In the field of radiochemistry, fluorine-18 also gained high interest due to its favorable nuclear and physical characteristics, including high positron decay ratio (97%), relatively short half-life (109.7 min), and low positron energy (maximum 0.635 MeV). The positron energy results in a short diffusion range (<2.4 mm) that favorably increases the resolution limits of the PET images.7 Because of these nuclear properties and the ability to synthesize fluorine-18 in large quantities, there have been many radiosynthetic methods developed for incorporation of this radionuclide into biologically important and interesting molecules ranging from drug-like molecules to antibodies and oligonucleotides. Because of the short half-life, emphasis has been placed on developing radiosynthetic schemes that introduce the radionuclide in a late stage of the synthetic pathway. Some of its distinctive radiochemistry will be further discussed in this review.
1.1. Fluorine-18 Production
Fluorine-18 is produced with a cyclotron primarily by proton (1H) irradiation of 18O, a stable naturally occurring isotope of oxygen. When the target is liquid H218O, an aqueous solution of 18F-fluoride ion is obtained; when the target is 18O2 gas, 18F–F2 gas is obtained. 18F–F2 is also prepared from deuteron-irradiation of Ne. The production method used is dependent on the desired subsequent chemical reactions; 18F-fluoride is produced for use as a nucleophile, while 18F-fluorine is produced for use in electrophilic methods.8−11 The key differences between these two chemical forms are the specific activity (SA = radioactivity/mol) of the produced 18F isotope. Nucleophilic 18F-fluoride is produced by the efficient nuclear reaction 18O(p,n)18F to give a high amount of radioactivity (>370 GBq/batch). Nucleophilic 18F-fluoride is produced with specific activity in the range of 102 GBq/μmol.9 Electrophilic 18F–F2 has much lower specific activity (100–600 MBq/μmol) because fluorine-19 gas must be added as a carrier to extract the 18F–F2 from the target.9 The SA becomes crucial when working on low capacity systems (i.e., ligand–receptor binding). The addition of a carrier (19F–F2) leads to increased mass of the final radiotracer, which may result in receptor saturation and reduction of PET signal from specific binding.12 High mass may also cause pharmacological effects.9 The high yield from cyclotron production along with higher SA that is crucial for PET imaging of receptor–ligand interaction dictates that most of the fluorine-18 reactions in nuclear medicine use nucleophilic 18F-fluoride.
2. Nucleophilic Fluorination
Although fluoride ion is a strong nucleophile, in aqueous solution it forms hydrogen bonds with the surrounding water molecules and becomes unreactive for nucleophilic substitution.13 To achieve nucleophilic fluorination, the 18F-fluoride must be substantially dehydrated by evaporation of the water and subsequent displacement reactions conducted in polar aprotic organic solvents. The solubility and nucleophilicity of fluoride ion in organic solvents is enhanced by the addition of a phase transfer catalyst (PTC) (such that the cryptand Kryptofix222 complexes potassium) or by the addition of bulky tetrabutylammonium cation. Radiofluorinations are typically conducted in the presence of poorly nucleophilic bases (typically carbonate or bicarbonate ions). Thus, the aqueous solution of 18F-fluoride obtained from the cyclotron target is treated with the desired salt (cation and, if necessary, a complexing agent, and desired anion) prior to evaporation of the water;6,13 water removal is assisted by azeotropic distillation of water using CH3CN.
Once the process of drying is complete, fluoride can be introduced by SN2 mechanism into aliphatic positions or via nucleophilic aromatic substitution (SNAr) into aromatic molecules. A wide range of precursors, leaving groups, and reaction conditions can be utilized for 18F-fluoride nucleophilic substitution (Figure 2). Dipolar aprotic solvents such as dimethyl sulfoxide (DMSO), N,N-dimethylformamide (DMF), dimethylacetamide and CH3CN are preferred solvents.6 On the other hand, there are several publications that describe nucleophilic substitution using the polar protic solvents t-butanol and t-amylalcohol13−16 and some in which the addition of a low percentage of water enhances the yields.17 Optimization of conditions for a particular radiochemical synthesis will require exploration of solvent, temperature, counterion, and concentration.
Figure 2.

Examples of 18F-aliphatic and aromatic nucleophilic substitution.
2.1. Aliphatic Nucleophilic Fluorination
Aliphatic nucleophilic fluorination involves the SN2 substitution of 18F-fluoride into precursors that contain a leaving group. Choosing the best leaving group is a critical step of the radiosynthetic design and should take into consideration the reactivity of the leaving group and the stability of the precursor to basic conditions of the fluorination reaction.18 The reactivity of leaving groups has been studied in great detail under various conditions. The order of leaving ability is Cl < Br < I < 4-methylbenzenesulfonate (tosylate) ∼ methanesulfonate (mesylate) < 4-nitrobenzenesulfonate (nosylate) < trifluoromethanesulfonate (triflate).2 The better the leaving group, the more likely a competing elimination reaction will occur under basic conditions.9 The position of the leaving group also has an effect on the efficiency of the substitution (primary benzylic > primary aliphatic ≫ secondary aliphatic).9
The typical anions added to the radiofluorination reaction result in basic conditions and often lead to undesired formation of byproducts resulting from decomposition of the base-sensitive precursors. To avoid the aforementioned side reactions, several parameters need to be optimized: (1) ratio of PTC-to-base-to-precursor, (2) selection of a less basic counteranion such as oxalate, bicarbonate, etc., (3) reaction temperature, and (4) the choice of leaving group. More reactive leaving groups are more sensitive to elimination side reaction, especially with increasing temperatures.9 With careful evaluation of these parameters, nucleophilic aliphatic radiofluorinations are usually efficient in terms of radiochemical yield (RCY), require moderate temperature (room temperature to 100 °C), and need relatively short reaction time (usually up to 15 min).19
Depending on the stability of the precursor, the presence of the various functional groups and the reactivity of the leaving group, desired products of aliphatic fluorinations can be obtained in one or two steps. Typically, the first step is the substitution of the leaving group by 18F and then, if necessary, removal of protecting groups or conversion of the labeled intermediate into the desired product (Figure 3).18,20 Many of the radiopharmaceuticals used for human subject PET imaging, for example, 18F-FDG, 18F-FLT, 18F-FMISO, 18F-choline, 18F-FES, 18F-fluoroacetate, 18F-fluoro-α-methyltyrosine, O-(2-18F-fluoroethyl)-l-tyrosine (18F-FET), and 18F-fluoro furanyl norprogesterone (18F-FFNP), are synthesized via SN2 displacement of an aliphatic leaving group by 18F-fluoride.16,21−32
Figure 3.

Synthetic routes of known radiotracers by 18F-aliphatic nucleophilic mechanism.15,16,31,141
2.2. Nucleophilic Aromatic Fluorination
18F nucleophilic aromatic substitution requires sufficient activation of the phenyl ring, which can be achieved by electron withdrawing group(s) (such as −NO2, −CN, −CF3, or carbonyl groups) in the ortho or para position to the leaving group. Aromatic nucleophilic substitution is conducted in a polar aprotic solvent and requires higher temperatures than aliphatic substitution (typically above 100 °C) (Figure 4). Aromatic exchange of 19F by 18F is feasible in the presence of carbonate and Kyrptofix222 or anhydrous tetrabutylammonium fluoride (TBAF) in DMSO.33,34 However, it will result in a lower specific activity radiotracer, which may preclude imaging application in low binding capacity systems.
Figure 4.

18F-Aromatic nucleophilic substitution on NO2 and +NMe3 leaving groups.40,90
The most common and efficient leaving groups for no-carrier-added nucleophilic aromatic substitutions are trimethylammonium salt and nitro group.35 Lower temperatures (100–110 °C) are normally used for an aromatic fluorination on trimethylammonium group compared with a nitro group (120–180 °C). Therefore, acetonitrile is often used as solvent in a closed reactor system for trimethylammonium displacement. Because of the higher temperatures required for substitution of nitro group, DMF or DMSO are used as solvent. The choice of activating group, leaving group, and protecting group(s) must be considered during synthetic design in order to facilitate conversion of the initial radiofluorinated intermediate into the final product.
A number of 18F-radiopharmaceuticals have been synthesized by aromatic nucleophilic substitution and used in clinical trials, including 6-18F-fluorodopamine, (−)-6-18F-fluoronorepinephrine, and 6-18F-altanserin.36−40 However, the requirement that the aromatic ring must be electron deficient in order to achieve labeling severely limits the scope of aromatic nucleophilic substitution. Because of this, a great deal of effort has been expended to find easier, more general, and more efficient methods to achieve aromatic substitution.
2.2.1. Fluorination of Heteroarenes
Heteroarenes containing a nitrogen are more electron deficient than the corresponding aromatic hydrocarbon and, thus, are amenable to direct substitution for 18F-fluoride without an additional activating group.41 In comparison to a monosubstituted benzene, the substituted pyridine has lower LUMO energy at ortho and para position than benzene, which allows direct 18F substitution with high radiochemical yields, using NO2, N+Me3, Br, I, or Cl as a leaving group at 2- and 4-positions (Figure 5). 18F-labeling on heteroarenes is conducted in the presence of potassium carbonate and Kyrptofix222 with DMSO or DMF as solvent at high temperature (120–150 °C).41 Labeling of heteroarene moieties has been applied for small organic molecules as well as prosthetic groups for labeling biomolecules.42−46
Figure 5.

18F-Labeling on pyridine and quinoline derivatives.
2.2.2. Balz–Schiemann Reaction
When radiochemists began to explore radiofluorination of aromatic rings, they adapted literature procedures that had been used successfully for nonlabeled fluorine incorporation. One attempt to label aromatic rings with 18F– in the absence of SNAr activating groups and in a regiospecific manner was done using the Balz–Schiemann reaction, which involves the thermal decomposition/pyrolysis of aryl diazonium tetrafluoroborate salt in the presence of 18F-fluoride to give the corresponding 18F-fluoroarene (Figure 6).12,47 The Balz–Schiemann reaction proceeds through the formation of a carbocation intermediate, which may be trapped by any nucleophilic species and results in the formation of numerous products.9 The use of tetrafluoroborate as a counteranion theoretically limits the RCY to a maximum of 25% and decreases significantly the SA because of the exchange between 18F-fluoride and the four fluorine atoms of BF4–. Because of these limitations, the reaction has seen limited application in this field and, accordingly, was used sparsely in published research. Weinreich and co-workers described the labeling of 5-18F-fluoro-d/l-DOPA (an electron-rich arene) using the Balz–Schieman reaction of the corresponding diazonium tetrafluoroborate salt. This reaction involves six steps for the preparation of the precursor and two steps for the 18F-labeling; pyrolysis of the diazonium tetrafluoroborate at 120 °C in xylene, followed by hydrolysis and final purification on a chiral column to give the desired 5-18F-fluoro-l-DOPA. The labeling resulted in much lower RCY than the theoretical one (10%).48
Figure 6.

Balz–Schiemann reaction mechanism.
2.2.3. Wallach Reaction
Another approach rarely used for regiospecific labeling of electron-rich arene employs the Wallach reaction that involves acid catalyzed, thermal decomposition of aryl-triazenes in the presence of 18F-fluoride (Figure 7), leading to the corresponding fluoroarenes.12,49,50 A diazonium salt is believed to be an intermediate in the reaction mechanism. The reaction mechanism is also SN1 type, and the initially formed aryl cation can react with any available nucleophilic species, potentially yielding high amounts of side products and providing lower RCY in comparison to other labeling routes.9 Tewson et al. demonstrated the feasibility of the decomposition of aryl-piperidyl triazines in the presence of 18F-CsF to provide high specific activity aryl fluorides with RCY greater than 50%. The method was used for the no-carrier-added radiosynthesis of the neuroreceptor ligand, 18F-haloperidol.50
Figure 7.
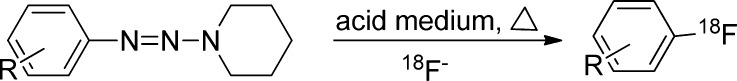
Wallach labeling reaction.
2.2.4. Diaryliodonium Salts
Because of the limitations of the Balz–Schiemann and Wallach reactions, new approaches have been sought. A much more efficient method to introduce an 18F into arenes has been accomplished using nucleophilic substitution of diaryliodonium salts.51−53 The regioselectivity of this fluorination reaction is guided by the electronic and steric features of the two aryl rings (Figures 8A,B).12,35,54 In the case of asymmetrical diaryliodonium salts, the nucleophilic substitution occurs on the more electron deficient aromatic component. In addition, the regioselectivity of the substitution is subject to an observed “ortho” effect. The ortho effect preferentially directs the 18F-fluoride substitution toward the aromatic ring which has substituent ortho to the iodonium moiety. Moreover, the RCYs increase when an additional ortho-substituent is introduced into the ring.9 The ortho effect is believed to be encouraged because, during the nucleophilic attack, an iodine-centered trigonal bipyramidal intermediate is formed and the sterically limiting ortho-substituted ring in the equatorial position favors the introduction of 18F into this moiety.54,55
Figure 8.

18F-Aromatic nucleophilic substitution on diaryliodonium salts. (A) 18F-Labeling on dihomoaryliodonium.54 (B) 18F-Labeling of aryl(2-thienyl)iodonium salts. (C) 18F-Labeling on 3-cyano-5-((2-(fluoromethyl)thiazol-4 yl)ethynyl)phenyl)(4-methoxyphenyl) iodonium salt.57
The degree of reactivity and the selectivity of radiofluorination of asymmetrical diaryliodonium salts was reported by Chun et al., which had shown that the selectivity for an ortho substituted product depends on the ortho substituents in the following order: 2,6-di-Me > 2,4,6-tri-Me > Br > Me > Et ∼ iPr ≫ H > OMe. The ortho effect is not purely dependent on the substituent bulk/steric influence but can be enhanced by the presence of one or more ortho hydrophobic groups (e.g., alkyl). The microenvironment created by these substituents is sufficiently lipophilic to support loose binding of 18F to the hypervalent iodine atom and then nucleophilic attack onto the adjacent lipophilic ortho-substituted ring. The electronic nature of the ortho substituents is also a factor in the product selectivity. The highly electron donating substituent ortho-OMe (an opposing substitutent) directs fluorination away from its ring, while the ortho-Br (a reinforcing substituent) enhances selection of its ring compared with ortho-Me.56
These reactions can be conducted in a microfluidic reactor in one single step.17 They typically required high temperature (140–200 °C) and the presence of base and Kryptofix222. In most cases, the 18F substitution is very efficient and results in high radiochemical yield (as determined by HPLC; isolated yields were not reported). The addition of a high concentration of a radical scavenger such as 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) significantly increased the labeling yield, perhaps by preventing the decomposition of the iodonium salt precursors before the fluorination reaction is complete (Figure 8C).57
The challenge of applying this methodology in radiochemistry is the design, synthesis, and purification of the required precursor. The entire molecule must be constructed with iodine in the ultimate location of the radioactive fluorine, and the molecule must either be substituted ortho to the desired site of fluorination or electron poor in relation to a thiophene or 4-methoxybenzene. This precursor structure must be compatible with the oxidative procedures for preparing the iodonium salt. Attempts to label more complicated small organic molecules with this method have resulted in much lower isolated RCY.57 Nevertheless, the method has promise for aromatic radiofluorination. Despite the increased scope provided to nucleophilic aromatic radiofluorinations by this method, it has, up to this point, seen limited applications to preparation of novel 18F-fluoro aromatic tracers for biological imaging.
3. Electrophilic Fluorination
As discussed under the heading “fluorine-18 production”, the production methods for electrophilic 18F–F2 require the addition of carrier 19F–F2 in order to extract the radioactivity from the cyclotron target. The maximum theoretical RCY of 18F–F2 is limited to 50%, because on every 18F atom there is a 19F atom as well. The produced 18F–F2 can be used as is or converted to less reactive and more selective fluorination agents such as acetylhypofluorite (18F-CH3COOF) for labeling.58,59 The more selective agents can be produced in the gas phase and carefully bubbled through the appropriate procurer solution.
One effort to improve the SA of 18F–F2 utilized a unique method for the conversion of 18F-fluoride into 18F-fluorine gas in an electrical discharge chamber.58,59 The 18F-fluoride was first converted to 18F-fluoromethane by nucleophilic substitution on methyl iodide.58,59 The conversion to 18F-fluoromethane was very facile (less than 6 min) and with high RCY (75%).58,59 The authors reported a SA of 5.5 TBq/μmol (148 Ci/μmol) for 18F-fluoride which decreased to 2.5 TBq/μmol (67.5 Ci/μmol) for 18F-fluoromethane. Then the CH318F was cooled and either purified by gas chromatography or used as produced and transferred to an electric discharge chamber containing the desired amount of carrier 19F-fluorine.58,59 The SA and yield of 18F–F2 were dependent on the amount of 18F-fluoromethane and carrier 19F–F2, along with the efficiency of the exchange reaction between the two.58,59 The authors reported a maximum exchange of 60%, as the amount of carrier 19F–F2 increased.58,59 The calculated SA of the final 18F-labeled radiopharmaceuticals, which was reported by the authors, was 15 GBq/μmol (0.4 Ci/μmol) at the end of synthesis.
Electrophilic 18F–F2 and its derivatives allow labeling of electron rich aromatic rings and alkenes, but because the regioselectivity is low, a mixture of fluoro isomers is obtained that presents challenging purification needs (Figure 9A).9,60,61 The regioselectivity of the 18F-fluorine can be increased by using organometallic precursors (Figure 9B); aryltrimethyltin is superior to arylmercury, aryltrimethylsilane, and aryltrimethylgermanium.9,62 As a direct outcome of improving the selectivity of 18F-fluorine, fewer byproducts form and higher RCY of the desired 18F product is realized.9,62
Figure 9.

(A) 18F2-Electrophilic substitution on 6-18F-fluoro-m-tyrosine.60 (B) Synthesis of 18F-FDOPA via 18F2-electrophilic substitution on aryltrimethyltin precursor.
One interesting class of mild electrophilic fluorination reagents are the N-fluoro compounds. Gouverneur’s group described the preparation and use of two such reagents: (1) 18F-N-fluorobenzenesulfonimide (18F-NFSi), a mild fluorination reagent used for the labeling of ethers, allylsilanes, and silyl enols,63 and (2) Selectfluor, prepared with high SA 18F–F2 via electrical discharge chamber as described above, for labeling a variety of small molecules (Figure 10).59,64 The availability of high specific activity 18F-Selectfluor as 18F-fluorination reagent may significantly advance electrophilic 18F-radiochemistry of electron rich arenes.59 Although utility of 18F-Selectfluor is very promising for those applications that can tolerate addition of some carrier 19F, the preparation of the electrical discharge chamber is not trivial. Consequently, the method has not been widely adopted in the field. Therefore, most of the clinically used radiopharmaceuticals, such as 18F-2-fluoro-l-tyrosine, 18F-6-fluoro-3,4-dihydroxy-l-phenylalanine (18F–F-DOPA), are prepared by electrophilic reaction using low SA 18F–F2.
Figure 10.

Labeling of fluorination reagent 18F-selectfluor using high specific activity 18F–F2.59
4. Transition Metal Mediated Radiofluorination
Transition metal mediated cross-coupling reactions that result in coupling of aromatic rings or the addition of nucleophiles to aromatic rings has been a significant area of research in organic chemistry. Applications of this methodology to fluorination have been reported recently.65,66 These reactions have a wide range of tolerable functional groups and, therefore, may increase the diversity of substrates that can be labeled with 18F-fluoride.5
Ritter and co-workers reported on the development and synthesis of an organopalladium-based fluorination reagent derived from fluoride and its application in labeling small aromatic molecules via late-stage fluorination (Figure 11A).65 Because of its high oxidative state (IV) in this type of complex, palladium functions as an oxidant and transfers the substrate to nucleophilic fluoride while being reduced to a lower oxidation state.65 The authors chose octahedral palladium(IV) in order to avoid undesired nucleophilic attack at the transition metal.65 Within the formed palladium–fluorine complex, the fluorine is partially negatively charged and the palladium is positively charged, forming a polar complex.65 Overall, this late-stage fluorination reaction requires two steps: capture of fluoride by the palladium complex and then its transfer via electrophilic fluorination to the appropriate aryl molecule.65 As in most fluorination reactions, nearly anhydrous conditions are required.
Figure 11.

18F-Labeling by late-stage fluorination reaction. (A) 18F2-Electrophilic fluorination via organometallic palladium complex.65 (B) 18F-Fluoride reaction with nickel–aryl complex.66
Recently, the same group reported the syntheses of aryl and alkenyl fluorides from organometallic nickel complexes66 in a late-stage fluorination. This fluorination reaction could be conducted in a single synthetic step using aqueous fluoride, without the need of azeotropic dehydration of 18F-fluoride (Figure 11B). This late-stage fluorination method was applied to label small molecules, but its simplicity and the ability to use aqueous fluoride-18 (1% of the reaction volume is water) may portend future application for labeling more complex bioactive molecules for medical applications.
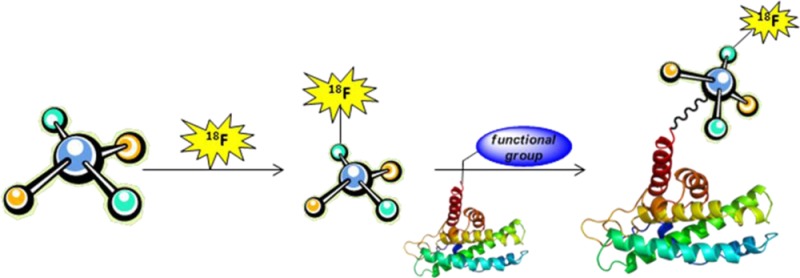
5. Radiolabeling of Biomolecules
Bioactive molecules, such as peptides, proteins, and oligonucleotides, are often used as molecular imaging agents because of their target specificity. Typically, these molecules do not have good stability to the common reaction conditions used in radiofluorination reactions, although there are exceptions. The radiolabeling of these molecules may be conducted by direct methods or indirect methods. Direct methods are those in which the fluoride is reacted directly with the molecule/biomolecule, which may have been previously modified to facilitate radiolabeling, and only subsequent purification is required to obtain the final product. Indirect methods require the prior radiosynthesis of a prosthetic group and subsequent bioconjugation to a molecular entity that has been modified for site specific reaction. In the sections that follow, we will discuss some direct and indirect methods that have been applied to radiolabeling biomolecules. This discussion will include preparation of unique prosthetic groups and methods to conjugate these prosthetic groups to the biomolecules.
5.1. Direct Labeling Methods for Biomolecules
A few successful attempts to introduce 18F directly into small peptides have been reported in the literature.67−72 We have reported on a one-step labeling strategy of peptide with 18F-fluoride by displacing an aromatic nitro group in an arene which is activated toward nucleophilic substitution by an ortho trifluoromethyl group. We applied this labeling method to cyclic RGD monomer and dimeric peptides. One downside of this labeling is the difficulty of separating the nitro-containing peptide precursors from the desired 18F-labeled products. Thus, the specific activity of the labeled peptide is related to the amount of precursor and radioactivity which are used.71 This method would not be expected to be generally applicable as the reaction conditions utilized high temperatures and basic conditions that may not be tolerated by most biomolecules.
Another example of such labeling, described by Hazari et al., was the synthesis of Si18F–dipropargyl glycerol scaffold, based on silicon-fluoride acceptor (Figure 12).73 The precursor for the labeling was prepared by conjugation of 4-(2,3-bis(prop-2-ynyloxy)propoxy)phenyl)-di-tert-butylsilane (SiH–dipropargyl glycerol) to 1-(2-azidoethyl)-4-(2-methoxyphenyl)piperazine through alkyne–azide Huisgen cycloaddition. 18F-labeling was conducted in the presence of K2CO3 and Kryptofix222 following azeotropic drying. The fluorination reaction was done in DMSO with 1% glacial acetic acid at 80 °C for 15 min.73 The desired labeled product was obtained in RCY of 50–60% following a 1 h synthesis time and applied for serotonin receptor PET imaging studies.
Figure 12.
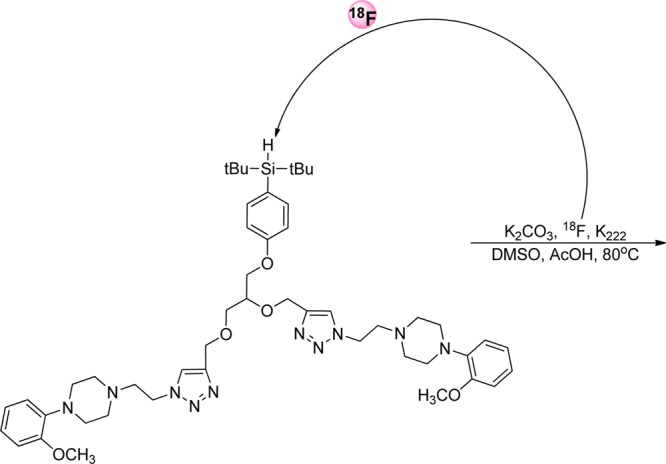
Direct 18F-labeling on the conjugated Si–dipropargyl glycerol prosthetic group.73
Perrin’s group has described the labeling of RGD peptides using 18F-aryltrifluoroborate.67−70 The labeling was done in the presence of potassium (mainly in its carbonate form) without the addition of PTC. The K18F solution was concentrated to near dryness. Thereafter, carrier 19F-fluoride was added to the mixture, followed by the peptide, which contains a boronate ester/borimidine and HCl acidic buffer at pH 2 (Figure 13).68−70 The acidification was required for the formation of three B–F bonds of an 18F-labeled aryltrifluoroborate anion.67−70 The efficiency of this methodology was confirmed by large-scale production of a 18F-aryltrifluoroborate-RGD peptide through direct 19F–18F exchange.70 However, the use of low pH may not be amenable for majority of biomolecules and may disrupt their biological activity.
Figure 13.
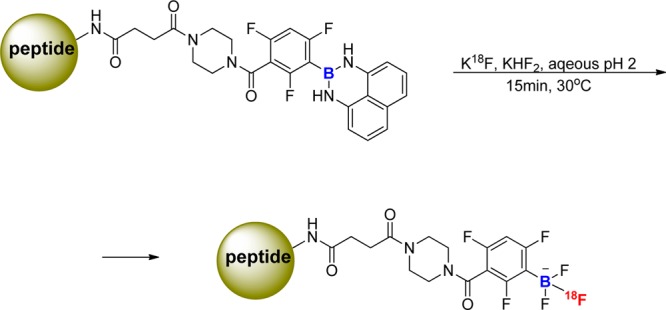
Labeling peptides using 18F-aryltrifluoroborate.68
In 2009, McBride et al. hypothesized that 18F, which was known to bind and form stable complexes with many metals, would form a stable NOTA Al–18F complex.74 The ensuing experiments resulted in successful chelation of Al18F into peptide conjugated with p-SCN-Bn-1,4,7-triazacyclononane-1,4,7-triacetic acid (NOTA) (Figure 14).74 The labeling was performed at pH 4 and required neither adding PTC nor azeotropic drying of water. The labeling is typically done at 100 °C for 15 min with a small amount of conjugated peptide and resulted in relatively high RCY.75 Although this is a promising methodology for relatively quick labeling of chelator-conjugated molecules, the conditions, such as excessive heating and low pH, may limit the scope of biomolecules that can be labeled by this approach. Since the initial report and subsequent work by the originators, this labeling methodology has been used by several groups for the labeling of small molecules, peptides, and proteins.76−87 Wan et al. reported recently on the first human study using a new lyophilized kit for the labeling of Al18F RGD dimer peptide.84
Figure 14.
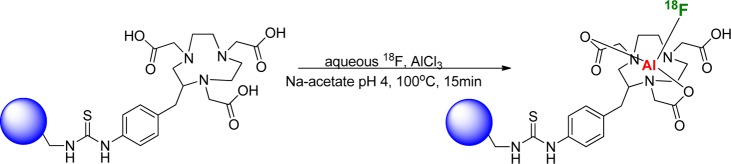
Labeling through Al18F methodology.74
5.2. Indirect Labeling Methods for Biomolecules
As discussed above, direct substitution methods usually require some nonphysiological conditions of pH or temperature. Most peptides, proteins, oligonucleotides, etc., do not tolerate such conditions and may undergo hydrolysis. Hence, labeling of biomolecules is often accomplished using a prosthetic group.2,88 The biomolecules are attached to the prosthetic groups mostly through amine- or thiol-reactive groups via acylation, alkylation, amidation, imidation, oxime, hydrazone formation,2 or using click chemistry (described below). The choice of prosthetic group is critical for radiotracer development, as they may adversely alter the physical and physiological characteristics of the labeled molecule.89 Methods for the site specific introduction of reactive thiols and other unique reactive functional groups must be utilized to develop novel, active, biological radiotracers.
18F-labeled prosthetic groups are prepared in a reaction employing from one to three synthetic steps and require subsequent purification processes to remove fluorination reagents (base and PTC), unreacted precursor, and other byproducts that can affect the conjugation with the biomolecule.90 Typically, the labeled biomolecule undergoes an additional purification process to obtain the desired product with high SA.88 The relatively short half-life of 18F is a challenge when designing a radiosynthesis that includes several synthetic and purification steps.
Despite the half-life limitations, numerous prosthetic groups have been developed for conjugation with biomolecules or small molecules using nucleophilic procedures described previously in this review (Figure 15).9,91−96 A great deal of work has been published using N-(hydroxysuccimidyl)-18F-fluorobenzoate (SFB) for coupling to amine functionalities on peptides and proteins and various 18F-fluorinated maleimide analogues for reaction with free thiols.90,97 Fluorinated N-hydroxysuccinimidyl esters and maleimides are typically synthesized in two or three labeling steps. Simplifications of their syntheses using one-pot reaction have been reported and appear to be routine in many laboratories.90,97−99 These prosthetic groups are of low volatility such that they can be concentrated without significant loss of radioactivity, thus, allowing scale up to clinical doses.
Figure 15.

Overview of common 18F-labeled prosthetic groups.
Another prosthetic group with specificity for amino functionalities, 3-18F-fluoropropanesulfonyl chloride, was first reported by Kiesewetter and co-workers for labeling target molecules by forming sulfonamide derivatives.100 Loser et al. recently described the preparation of this same molecule using a two-step 18F-labeling procedure via the intermediate 3-18F-fluoropropyl thiocyanate (Figure 16).89 The resulting 3-18F-fluoropropanesulfonyl chloride was reacted with different primary and secondary aliphatic and aromatic amines at room temperature with or without various bases.89 RCY (calculated from radio-thin layer chromatography) were high (77–89%) for most of the reactions without the addition of base.89
Figure 16.
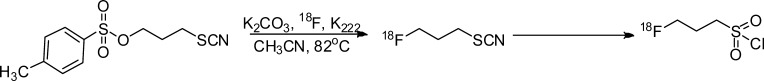
Radiosynthesis of 3-18F-fluoropropanesulfonyl chloride prosthetic group.89
Another interesting paper, recently published by Gao et al., described the fluorination of proteins and peptides via 4-fluorophenylboronic acid prosthetic group using an aqueous Suzuki–Miyaura coupling reaction with an optimized palladium ligand.101 This reaction requires three labeling steps, 18F nucleophilic fluorination on a diaryliodonium precursor, conversion to the 4-fluorophenylboronic acid prosthetic group, and palladium-catalyzed cross-coupling reaction with an appropriate small molecule, peptide, or larger biomolecule (Figure 17).101 The site specificity of the radiolabeling is achieved only by site specific introduction of an aryl iodide into the biomolecule by either chemical methods or genetic modification. Thus, the site of labeling can be selected so that the functional binding site of the protein is not impaired. The reported procedure produced the 4-18F-fluorophenylboronic acid in relatively low yield (5–10% corrected). The subsequent coupling yields with a functionalized peptide were on the order of 30%, but, unfortunately, coupling yields with larger proteins were quite low. Significant optimization will be required if this method is to become useful for labeling biomolecules.
Figure 17.
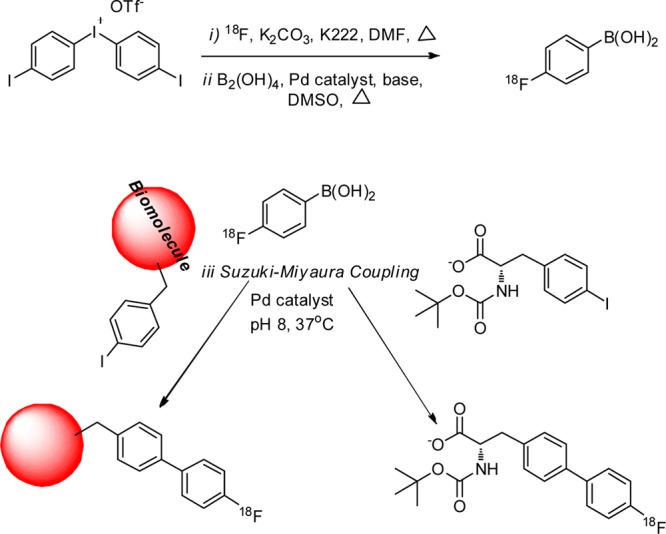
Suzuki–Miyaura coupling via 4-18F-fluorophenylboronic acid prosthetic group.101
5.3. Click Chemistry/Bioorthogonal Reactions
Bioorthogonal reactions are defined as chemical reactions that take place in living tissue but do not interact with the biological system and because of that, allow investigation of the various biological processes. In addition to being highly selective, the bioorthogonal reactions are typically rapid and can be conducted in biological media. Consequently, a number of bioorthogonal reaction strategies have been developed and used for incorporation of 18F-labeled prosthetic groups into drug-like and biomolecules (indirect labeling). To select a click chemistry approach, consideration must be given to the reaction rate, stoichiometry, concentration, and mass of biomolecule required for achieving good radiochemical yields and minimizing purification procedures. In this section, we will discuss the reactions that have been applied in 18F chemistry.
5.3.1. Copper(I)-Catalyzed Azide–Alkyne Cycloaddition
The Huisgen cycloaddition is a 1,3-dipolar cycloaddition of an azide and an acyclic alkyne to yield a 1,2,3-triazole. As originally described, excessive heat and prolonged reaction time are required to overcome the activation barrier of triazole formation from alkyne bond.102 Research led by K. Barry Sharpless first described the catalysis of the Huisgen cycloaddition by Cu(I) and created the name “click chemistry” to describe this type of reaction.103 Meldal104 and Sharpless103 groups exploited this reaction, described as copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) for the formation of various five-membered heterocycles (Figure 18). The scope of the reaction is quite large and, because alkynes and azides are typically inert in biological systems,105 qualifies as a bioorthogonal reaction.106 The orthogonal nature of cycloaddition reactions does not require protection of other functional groups and can be done in the presence of water and oxygen.107 Primary, secondary, and tertiary alkyl azides, aryl azides, and an azido sugar were reacted in the copper(I)-catalyzed cycloaddition with alkynes to give a variety of 1,4-substituted [1,2,3]-triazoles in peptide backbones or side chains as a mixture of two possible regioisomers.104,105,107−109 It is widely accepted that Cu(I) coordinates first to the terminal alkyne to form a copper(I) acetylide.103,104,110 Recently, Worrell et al. suggested a mechanism which involves two copper atoms within the cycloaddition step.106
Figure 18.
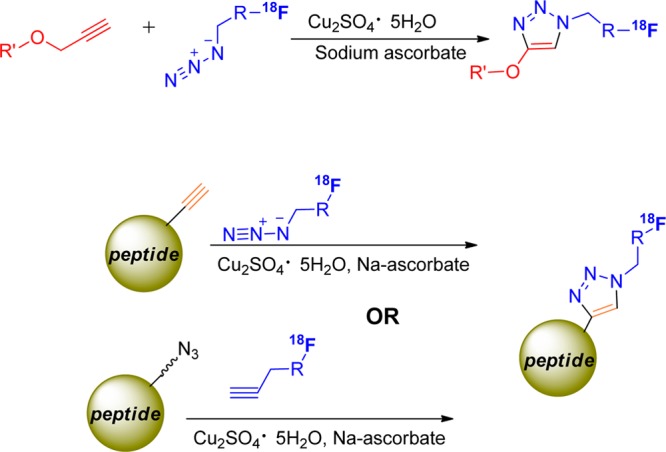
Examples of copper(I)-catalyzed azide–alkyne cycloaddition.
The high efficiency and selectivity of this reaction has been exploited for 18F labeling.105,110−112 A variety of 18F-labeled small molecules containing azides or alkynes have been prepared, and peptides and other biomolecules have been decorated with the corresponding reactive moiety. The cycloaddition reaction can be catalyzed with Cu(I) sources such as CuBr and CuI but is typically done with CuSO4, which becomes Cu(I) in situ in the presence of a reducing agent such as sodium ascorbate.
Ramenda et al. described the radiosynthesis of N-propargyl-N-methyl18F-fluorobenzenesulfonamide as a prosthetic group for Cu(I)-mediated [3 + 2] cycloaddition reactions. Its applicability was evaluated by conjugation to a peptide, human serum albumin (HSA) protein, and a RNA oligonucleotide.111,11318F labeling of peptide was evaluated using different amounts of peptide (0.1–0.4 mg) and in the presence of CuSO4 and sodium ascorbate in borate buffer. As more peptide was used, higher RCY was achieved.113 For HSA labeling, the use of CuSO4 and sodium ascorbate did not result in the desired labeled protein. The authors speculated that the sodium ascorbate induced partial or complete reduction of the disulfide bonds in the protein.113
The successful labeling of both HSA and RNA oligonucleotide was done using CuBr and oligotriazole tris[(1-benzyl-1H-1,2,3-triazol-4-yl)-methyl]-amine (TBTA), which is used to prevent the reoxidation of Cu(I) to Cu(II) (Figure 19).113 The stable Cu(I)–TBTA complex allows the cycloaddition reaction between azide-modified biomolecule and the 18F-arylalkynyl sulfonamide while avoiding generation of hydroxyl radicals and subsequent oxidative decomposition.113
Figure 19.

Synthesis of 18F-aryl sulfonamide as a building block for Cu(I)-mediated [3 + 2] cycloaddition reaction with RNA.113
5.3.2. Strain-Promoted Alkyne–Azide Cycloaddition
Strain-promoted alkyne–azide cycloaddition involves the reaction of azide with the bent triple bond of the cyclooctyne to give a triazole without the need for Cu(I) catalyst (Figure 20).102,114 In this reaction, the activation barrier is significantly reduced by the bent geometry of the triple bond of the eight-membered ring.102 Increased reactivity toward terminal azides can be achieved by modifying the cyclooctyne with electron withdrawing groups or conformationally restricted aromatic rings adjacent to the triple bond. More importantly, the elimination of toxic copper as a catalyst allows use of this alkyl–azide cycloaddition in living systems.110,114−117
Figure 20.
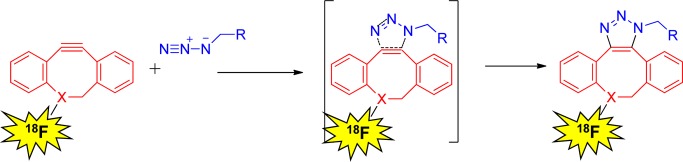
Strain-promoted alkyne–azide cycloaddition mechanism.
In recent years, several cyclooctynes and dibenzocyclooctynes have been used in strain-promoted reactions for 18F-labeling.114,118−122 As in the case of CuAAC reaction, two radioactive triazole regioisomers are formed and are detected by HPLC.114,123 These regioisomers might have different biological properties in vivo such as target binding affinity and pharmacokinetics. One example for such cycloaddition, reported by Carpenter et al., described the 1,3-dipolar cyloaddition of 18F-FB-azadibenzocyclooctyne (ADIBO) with alkyl azides (Figure 21). 18F-Fluoride was initially introduced into the cyclooctyne prosthetic group. Subsequent strain-promoted cyclization with small molecules was conducted. This reaction provided the desired product in high RCY (>70% yield of the cycloaddition reaction) in less than 1 h reaction time. In addition, the triazole product showed high stability in rat serum.123 The same group also applied this methodology for labeling peptides that had been site selectively functionalized with an azide. This cycloaddition reaction was conducted for 10 min at 35–45 °C, with a peptide substrate concentration of 1 mg (0.37 μmol) in 150 μL of ethanol and resulted in an isolated RCY of 12% based on the quantity of [18F]ADIBO (Figure 21).114 Sachin et al. described 18F labeling of ADIBO peptide precursors with an 18F-azide synthon under physiological conditions with quantitative RCY. One of the advances described in this work was the replacement of HPLC purification with a unique azide-containing scavenger resin that effectively removed unreacted ADIBO peptide precursor from the desired radiolabeled triazole product (Figure 22).122 This intriguing methodology can be easily transferred into a commercial kit, making it readily available for labeling biomolecules for clinical applications.
Figure 21.

1,3-Dipolar cycloaddition of 18FB-azadibenzocyclooctyne (ADIBO) with alkyl azide without Cu(I) catalyst.114,123
Figure 22.
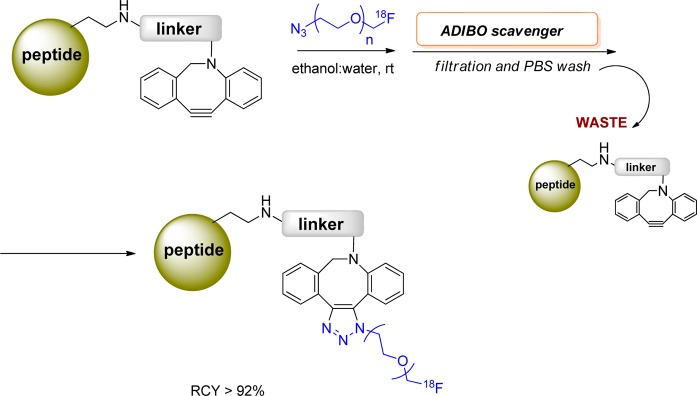
Strain-promoted alkyne–azide cycloaddition under nontoxic physiological conditions.122
5.3.3. Staudinger Ligation
Staudinger reaction occurs between azide and phosphine via an iminophosphorane, which is stable in organic solvents but is rapidly hydrolyzed in aqueous solution, to give the primary amine and phosphine oxide.124−126 Mechanistic studies revealed the potential to perform ligation reactions. An intramolecular electrophilic trap, such as an ester moiety, could capture the nucleophilic aza-ylide intermediate with the formation of a stable covalent adduct that upon rearrangement forms a stable amide bond; no hydrolysis of the aza-ylide is required.127 This method was first demonstrated in 2000 by Saxon and Bertozzi, in which coupling of azide and modified triarylphosphine yielded an amide bond.127
Although this method has proven to be very useful, the resulting amides contain a very lipophilic triphenylphosphonium oxide moiety as part of the product. As a result, the Bertozzi group and the Raines group independently developed the “traceless” Staudinger ligation for chemoselective synthesis in which the triphenylphosphonium oxide moiety is lost during the ligation reaction.128,129 The phosphine, stabilized by two phenyl groups, is linked to an acyl group via an ester or thioester. The intermediate aza-ylide attacks the carbonyl group, cleaving the ester or thioester bond. Subsequent hydrolysis gives an amide bond and releases the phosphine oxide moiety.128
For 18F-labeling of biomolecules, traceless Staudinger ligation does not require metal catalyst and allows a simple separation of the 18F-labeled amide from the phosphine oxide byproduct.130,131 It has been applied for indirect 18F-labeling via the synthesis of a prosthetic group followed by conjugation with phosphine derivatives and formation of amide bond.19,131−134 One example, published by Pretze et al., described the ligation between 18F-6-fluorohexanoyl-phosphane moiety and several azides such as benzyl azide, azidoacetic acid, and a 6-azido-galactose derivative (Figure 23).19 The Staudinger ligation has two major limitations. The first is the relatively slow kinetics that requires high concentration of the phosphine to obtain usable reaction rates for radiochemical synthesis. The second is the fact that the phosphine precursor is subject to oxidation that will demolish its reactivity.
Figure 23.

Staudinger ligation between 18F-6-fluorohexanoyl-phosphane and benzyl azide, azidoacetic acid, and a 6-azido-galactose.19
5.3.4. Tetrazine Ligation
Tetrazine ligation is a bioorthogonal cycloaddition based on inverse electron demand Diels–Alder reaction of s-tetrazine and trans-cyclooctene moieties.135 The inverse demand Diels–Alder reaction followed by a retro-Diels–Alder reaction with release of nitrogen yields either dihydropyradazines or pyradazines depending on the dienophile reactant (alkynes or alkenes).135,136 This reaction is characterized by fast reaction rate and, therefore, no need for excess amount of reactants or catalyst, which makes for a fast and efficient bioconjugation at low concentrations.135,136 It also tolerates a broad range of functionality and is not dependent on the solvent, i.e., can be performed in organic solvents, water, cell media, or cell lysate without reducing the high yield.135
Several publications described the 18F incorporation via tetrazine–trans-cyclooctene ligation (TTCO-ligation) by indirect (the use of prosthetic groups) or direct routes.136−140 Li et al. described the TTCO-ligation between 3,6-di(2-pyridyl)-s-tetrazine and 18F-trans-cyclooctene (18F-TCO) (Figure 24). The TTCO ligand was completely consumed within 10 s and resulted in high RCY (98%). The concentration of tetrazine was 21 μM and the 18F-TCO ranged from 0.2–2 μM. There was also a very small amount of side product (Figure 24), which is due to the slow rearrangement of the product with formation of regio- and stereoisomers.140 The feasibility of TTCO was further evaluated by conjugation of 18F-TCO with tetrazinyl-maleimide-conjugated to c(RGDyC) peptide and vascular endothelial growth factor (VEGF) protein, containing free cysteines (Figure 25). The click chemistry was done in DMSO and ethanol or DMSO and phosphate buffer to give the desired product with high RCY (quantitative yield for the RGD and 75% for VEGF, using 80–100 μM and 6 μM substrate concentrations, respectively).139 Unfortunately, the complementary reaction using an 18F-tetrazine was unsuccessful. Attempts to incorporate [18F]fluoride via aromatic and aliphatic nucleophilic substitutions into tetrazine precursors resulted in very low yields (<1%) because the tetrazine precursors proved unstable under basic fluorination conditions.140
Figure 24.

Tetrazine–trans-cyclooctene ligation between 3,6-di(2-pyridyl)-s-tetrazine and 18F-trans-cyclooctene.140
Figure 25.
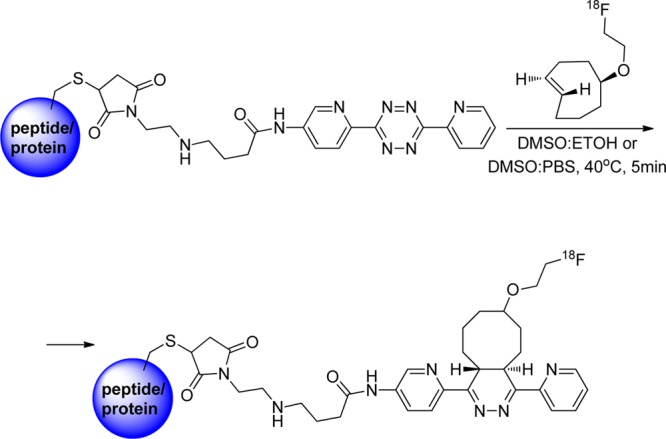
Tetrazine–trans-cyclooctene ligation of 18F-trans-cyclooctenes with biomolecules.139
This method seems to be the most promising of the click methods in terms of high yield and very fast reaction rates. A number of the chemical entities for the click chemistry pairs have been made commercially available. However, the precursor for 18F-TCO is not yet commercially available. The TCO precursor is prepared by a photochemical isomerization with kinetic trapping;135 both procedures are rarely practiced in radiochemistry laboratories. This precursor will need to be a commercial product in order for the method to be thoroughly studied. Questions remain as to the volatility of 18F-TCO, which will determine the ability to concentrate the radioactive component for synthesis of sufficient material for clinical use.
6. Conclusions
Fluorine-18 labeling chemistry has developed tremendously over the last decades. Because of higher specific activity, most syntheses use no-carrier-added nucleophilic fluoride-18 rather than electrophilic fluorine. Some of the 18F-labeling strategies require time-consuming and challenging radiosynthesis, and, thus, are not clinically viable.
Numerous labeling methods to incorporate 18F-fluoride into aliphatic or aromatic substrates have been developed, attempting to achieve easier and more time-efficient radiosyntheses, with high specific activity and high yield of desired products. Traditional nucleophilic aromatic radiofluorinations require electronically deficient rings; however, newer methods using aryl iodonium salts or palladium and nickel complexes have provided efficient radiofluorination of electron rich arenes. These approaches expand the scope of radiofluorination substrates and place the challenge on the ability to synthesize the required precursor molecules.
The need for easy and rapid labeling techniques of biomolecules, using biocompatible conditions, is still not fully answered. Typically, biomolecules are functionalized with a reactive functional group, (i.e., amine, azide, alkyne, thiol, metal chelator) and radiolabeled through reaction with the appropriate prosthetic group or radiometal. The use of click chemical reactions, including strain-promoted alkyne–azide cycloaddition and tetrazine ligation, for rapid labeling of biomolecules has been heavily exploited. The development of the method for conjugation of 18F-AlF with NOTA has allowed 18F-fluoride to be utilized in chelation labeling for small and large biomolecules. In addition, the development of purification resins (such as the azide resin) for separation of the undesired precursors may further promote the use of click chemistry in clinical translation of novel 18F-labeled radiotracers.
This review highlights the great arsenal of useful radiofluorination strategies available to prepare the next generation of 18F radiotracers for PET that will provide patients with more sensitive diagnosis, faster and more accurate evaluation of therapeutics, and ultimately improved outcomes.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Hargreaves R. J.; Rabiner E. A. (2014) Translational PET imaging research. Neurobiol. Dis. 61, 32–38. [DOI] [PubMed] [Google Scholar]
- Jacobson O.; Chen X. (2010) PET designated fluoride-18 production and chemistry. Curr. Top. Med. Chem. 10, 1048–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park B. K.; Kitteringham N. R. (1994) Effects of fluorine substitution on drug metabolism: pharmacological and toxicological implications. Drug Metab. Rev. 26, 605–643. [DOI] [PubMed] [Google Scholar]
- Barnes-Seeman D.; Beck J.; Springer C. (2014) Fluorinated compounds in medicinal chemistry: recent applications, synthetic advances and matched-pair analyses. Curr. Top. Med. Chem. 14, 855–864. [DOI] [PubMed] [Google Scholar]
- Hollingworth C.; Gouverneur V. (2012) Transition metal catalysis and nucleophilic fluorination. Chem. Commun. (Cambridge, U. K.) 48, 2929–2942. [DOI] [PubMed] [Google Scholar]
- Furuya T.; Kuttruff C. A.; Ritter T. (2008) Carbon–fluorine bond formation. Curr. Opin. Drug Discovery Dev. 11, 803–819. [PubMed] [Google Scholar]
- Pretze M.; Grosse-Gehling P.; Mamat C. (2011) Cross-coupling reactions as valuable tool for the preparation of PET radiotracers. Molecules 16, 1129–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casella V.; Ido T.; Wolf A. P.; Fowler J. S.; MacGregor R. R.; Ruth T. J. (1980) Anhydrous F-18 labeled elemental fluorine for radiopharmaceutical preparation. J. Nucl. Med. 21, 750–7. [PubMed] [Google Scholar]
- Coenen H. H. (2007) Fluorine-18 labeling methods: features and possibilities of basic reactions. Ernst Schering Res. Found. Workshop 15–50. [DOI] [PubMed] [Google Scholar]
- Nickles R. J.; Daube M. E.; Ruth T. J. (1984) An [18O]O2 Target for the Production of [18F]F2. Int. J. Appl. Radiat. Isot. 35, 117–122. [Google Scholar]
- Palmer A. J.; Clark J. C.; Goulding R. W. (1977) The preparation of fluorine-18 labelled radiopharmaceuticals. Int. J. Appl. Radiat. Isot. 28, 53–65. [DOI] [PubMed] [Google Scholar]
- Cai L.; Lu S.; Pike V. W. (2008) Chemistry with [18F]Fluoride Ion. Eur. J. Org. Chem. 2853–2873. [Google Scholar]
- Kim D. W.; Jeong H. J.; Lim S. T.; Sohn M. H.; Katzenellenbogen J. A.; Chi D. Y. (2008) Facile nucleophilic fluorination reactions using tert-alcohols as a reaction medium: significantly enhanced reactivity of alkali metal fluorides and improved selectivity. J. Org. Chem. 73, 957–962. [DOI] [PubMed] [Google Scholar]
- Kim D. W.; Ahn D. S.; Oh Y. H.; Lee S.; Kil H. S.; Oh S. J.; Lee S. J.; Kim J. S.; Ryu J. S.; Moon D. H.; Chi D. Y. (2006) A new class of SN2 reactions catalyzed by protic solvents: facile fluorination for isotopic labeling of diagnostic molecules. J. Am. Chem. Soc. 128, 16394–16397. [DOI] [PubMed] [Google Scholar]
- Kim D. W.; Choe Y. S.; Chi D. Y. (2003) A new nucleophilic fluorine-18 labeling method for aliphatic mesylates: reaction in ionic liquids shows tolerance for water. Nucl. Med. Biol. 30, 345–350. [DOI] [PubMed] [Google Scholar]
- Rodnick M. E.; Brooks A. F.; Hockley B. G.; Henderson B. D.; Scott P. J. (2013) A fully-automated one-pot synthesis of [18F]fluoromethylcholine with reduced dimethylaminoethanol contamination via [18F]fluoromethyl tosylate. Appl. Radiat. Isot. 78, 26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun J. H.; Pike V. W. (2013) Single-step syntheses of no-carrier-added functionalized [18F]fluoroarenes as labeling synthons from diaryliodonium salts. Org. Biomol. Chem. 11, 6300–6306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M. R.; Suzuki K. (2007) [18F]Fluoroalkyl agents: synthesis, reactivity and application for development of PET ligands in molecular imaging. Curr. Top. Med. Chem. 7, 1817–1828. [DOI] [PubMed] [Google Scholar]
- Pretze M.; Wuest F.; Peppel T.; Köckerling M.; Mamat C. (2010) The traceless Staudinger ligation with fluorine-18: a novel and versatile labeling technique for the synthesis of PET-radiotracers. Tetrahedron Lett. 51, 6410–6414. [Google Scholar]
- Zhou D.; Chu W.; Chen D. L.; Wang Q.; Reichert D. E.; Rothfuss J.; D’Avignon A.; Welch M. J.; Mach R. H. (2009) [18F]- and [11C]-Labeled N-benzyl-isatin sulfonamide analogues as PET tracers for apoptosis: synthesis, radiolabeling mechanism, and in vivo imaging study of apoptosis in Fas-treated mice using [11C]WC-98. Org. Biomol. Chem. 7, 1337–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallabhajosula S. (2007) (18)F-Labeled positron emission tomographic radiopharmaceuticals in oncology: an overview of radiochemistry and mechanisms of tumor localization. Semin. Nucl. Med. 37, 400–419. [DOI] [PubMed] [Google Scholar]
- Gaeta C. M.; Vercher-Conejero J. L.; Sher A. C.; Kohan A.; Rubbert C.; Avril N. (2013) Recurrent and metastatic breast cancer PET, PET/CT, PET/MRI: FDG and new biomarkers. Q. J. Nucl. Med. Mol. Imaging 57, 352–366. [PubMed] [Google Scholar]
- van Kruchten M.; Glaudemans A. W.; de Vries E. F.; Beets-Tan R. G.; Schroder C. P.; Dierckx R. A.; de Vries E. G.; Hospers G. A. (2012) PET imaging of estrogen receptors as a diagnostic tool for breast cancer patients presenting with a clinical dilemma. J. Nucl. Med. 53, 182–90. [DOI] [PubMed] [Google Scholar]
- Dehdashti F.; Laforest R.; Gao F.; Aft R. L.; Dence C. S.; Zhou D.; Shoghi K. I.; Siegel B. A.; Katzenellenbogen J. A.; Welch M. J. (2012) Assessment of progesterone receptors in breast carcinoma by PET with 21–18F-fluoro-16alpha,17alpha-[(R)-(1′-alpha-furylmethylidene)dioxy]-19-norpregn-4-ene-3,20-dione. J. Nucl. Med. 53, 363–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoh C. K.; Burris H. A. III; Bendell J. C.; Tarazi J.; Rosbrook B.; Kim S.; Infante J. R.; Reid T. R. (2014) Intermittent dosing of axitinib combined with chemotherapy is supported by (18)FLT-PET in gastrointestinal tumours. Br. J. Cancer 110, 875–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thureau S.; Chaumet-Riffaud P.; Modzelewski R.; Fernandez P.; Tessonnier L.; Vervueren L.; Cachin F.; Berriolo-Riedinger A.; Olivier P.; Kolesnikov-Gauthier H.; Blagosklonov O.; Bridji B.; Devillers A.; Collombier L.; Courbon F.; Gremillet E.; Houzard C.; Caignon J. M.; Roux J.; Aide N.; Brenot-Rossi I.; Doyeux K.; Dubray B.; Vera P. (2013) Interobserver agreement of qualitative analysis and tumor delineation of 18F-fluoromisonidazole and 3′-deoxy-3′-18F-fluorothymidine PET images in lung cancer. J. Nucl. Med. 54, 1543–1550. [DOI] [PubMed] [Google Scholar]
- Segard T.; Robins P. D.; Yusoff I. F.; Ee H.; Morandeau L.; Campbell E. M.; Francis R. J. (2013) Detection of hypoxia with 18F-fluoromisonidazole (18F-FMISO) PET/CT in suspected or proven pancreatic cancer. Clin. Nucl. Med. 38, 1–6. [DOI] [PubMed] [Google Scholar]
- Panebianco V.; Sciarra A.; Lisi D.; Galati F.; Buonocore V.; Catalano C.; Gentile V.; Laghi A.; Passariello R. (2012) Prostate cancer: 1HMRS-DCEMR at 3T versus [(18)F]choline PET/CT in the detection of local prostate cancer recurrence in men with biochemical progression after radical retropubic prostatectomy (RRP). Eur. J. Radiol. 81, 700–8. [DOI] [PubMed] [Google Scholar]
- Gulyas B.; Halldin C. (2012) New PET radiopharmaceuticals beyond FDG for brain tumor imaging. Q. J. Nucl. Med. Mol. Imaging 56, 173–90. [PubMed] [Google Scholar]
- Heinzel A.; Muller D.; Langen K. J.; Blaum M.; Verburg F. A.; Mottaghy F. M.; Galldiks N. (2013) The use of O-(2–18F-fluoroethyl)-l-tyrosine PET for treatment management of bevacizumab and irinotecan in patients with recurrent high-grade glioma: a cost-effectiveness analysis. J. Nucl. Med. 54, 1217–1222. [DOI] [PubMed] [Google Scholar]
- Grierson J. R.; Shields A. F. (2000) Radiosynthesis of 3′-deoxy-3′-[(18)F]fluorothymidine: [(18)F]FLT for imaging of cellular proliferation in vivo. Nucl. Med. Biol. 27, 143–156. [DOI] [PubMed] [Google Scholar]
- Yoo J.; Dence C. S.; Sharp T. L.; Katzenellenbogen J. A.; Welch M. J. (2005) Synthesis of an estrogen receptor beta-selective radioligand: 5-[18F]fluoro-(2R,3S)-2,3-bis(4-hydroxyphenyl)pentanenitrile and comparison of in vivo distribution with 16alpha-[18F]fluoro-17beta-estradiol. J. Med. Chem. 48, 6366–6378. [DOI] [PubMed] [Google Scholar]
- Sun H.; DiMagno S. G. (2006) Room-temperature nucleophilic aromatic fluorination: experimental and theoretical studies. Angew. Chem., Int. Ed. Engl. 45, 2720–5. [DOI] [PubMed] [Google Scholar]
- Blom E.; Karimi F.; Langstrom B. (2009) [18F]/19F exchange in fluorine containing compounds for potential use in 18F-labelling strategies. J. Labelled Compd. Radiopharm. 52, 504–511. [Google Scholar]
- Tredwell M.; Gouverneur V. (2012) 18F labeling of arenes. Angew. Chem., Int. Ed. Engl. 51, 11426–11437. [DOI] [PubMed] [Google Scholar]
- Timmers H. J.; Eisenhofer G.; Carrasquillo J. A.; Chen C. C.; Whatley M.; Ling A.; Adams K. T.; Pacak K. (2009) Use of 6-[18F]-fluorodopamine positron emission tomography (PET) as first-line investigation for the diagnosis and localization of non-metastatic and metastatic phaeochromocytoma (PHEO). Clin. Endocrinol. (Oxford, U. K.) 71, 11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y. S.; Fowler J. S.; Gatley S. J.; Dewey S. L.; Wolf A. P.; Schlyer D. J. (1991) Synthesis of high specific activity 6-[18F]fluorodopamine for positron emission tomography studies of sympathetic nervous tissue. J. Med. Chem. 34, 861–863. [DOI] [PubMed] [Google Scholar]
- Volkow N. D.; Fowler J. S.; Ding Y. S. (1996) Cardiotoxic properties of cocaine: studies with positron emission tomography. NIDA Res. Monogr 163, 159–174. [PubMed] [Google Scholar]
- Quednow B. B.; Treyer V.; Hasler F.; Dorig N.; Wyss M. T.; Burger C.; Rentsch K. M.; Westera G.; Schubiger P. A.; Buck A.; Vollenweider F. X. (2012) Assessment of serotonin release capacity in the human brain using dexfenfluramine challenge and [18F]altanserin positron emission tomography. Neuroimage 59, 3922–3932. [DOI] [PubMed] [Google Scholar]
- Hamacher K.; Coenen H. H. (2006) No-carrier-added nucleophilic 18F-labelling in an electrochemical cell exemplified by the routine production of [18F]altanserin. Appl. Radiat. Isot. 64, 989–94. [DOI] [PubMed] [Google Scholar]
- Dolle F. (2005) Fluorine-18-labelled fluoropyridines: advances in radiopharmaceutical design. Curr. Pharm. Des 11, 3221–3235. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Hall A. W.; Horti A. G. (2004) Efficient synthesis of 6-chloro-3-((2-(S)-azetidinyl)methoxy)-5-((E)-2-(2-[18F]fluoropyridin-4-yl)vinyl)pyridine ([18F]NIDA 52289), a very high affnity radioligand for nicotinic acetylcholine receptors. J. Labelled Compd. Radiopharm. 47, 385–392. [Google Scholar]
- Liu H.; Liu S.; Miao Z.; Deng Z.; Shen B.; Hong X.; Cheng Z. (2013) Development of 18F-labeled picolinamide probes for PET imaging of malignant melanoma. J. Med. Chem. 56, 895–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karramkam M.; Hinnen F.; Berrehouma M.; Hlavacek C.; Vaufrey F.; Halldin C.; McCarron J. A.; Pike V. W.; Dolle F. (2003) Synthesis of a [6-pyridinyl-18F]-labelled fluoro derivative of WAY-100635 as a candidate radioligand for brain 5-HT1A receptor imaging with PET. Bioorg. Med. Chem. 11, 2769–2782. [DOI] [PubMed] [Google Scholar]
- Liang S. H.; Collier T. L.; Rotstein B. H.; Lewis R.; Steck M.; Vasdev N. (2013) Rapid microfluidic flow hydrogenation for reduction or deprotection of 18F-labeled compounds. Chem. Commun. (Cambridge, U. K.) 49, 8755–8757. [DOI] [PubMed] [Google Scholar]
- Kuhnast B.; de Bruin B.; Hinnen F.; Tavitian B.; Dolle F. (2004) Design and synthesis of a new [18F]fluoropyridine-based haloacetamide reagent for the labeling of oligonucleotides: 2-bromo-N-[3-(2-[18F]fluoropyridin-3-yloxy)propyl]acetamide. Bioconjugate Chem. 15, 617–627. [DOI] [PubMed] [Google Scholar]
- Balz G.; Schiemann G. (1927) Über aromatische Fluorverbindungen, I.: Ein neues Verfahren zu ihrer Darstellung. Chem. Ber. 60, 1186–1190. [Google Scholar]
- Argentini M.; Wiese C.; Weinreich R. (1994) Syntheses of 5-fluoro-d/l-dopa and [18F]5-fluoro-l-dopa. J. Fluorine Chem. 68, 141–144. [Google Scholar]
- Wallach O. (1886) Über dasVerhalten einiger Diazo- und Diazoamidoverbindungen. Justus Liebigs Ann. Chem. 235, 242–255. [Google Scholar]
- Tewson T. J.; Raichle M. E.; Welch M. J. (1980) Preliminary studies with [18F]haloperidol: a radioligand for in vivo studies of the dopamine receptors. Brain Res. 192, 291–295. [DOI] [PubMed] [Google Scholar]
- Pike V. W.; Aigbirhio F. I. (1995) Reactions of cyclotron-produced [18F]fluoride with diaryliodonium salts? A novel single-step route to no-carrier-added [18]fluoroarenes. J. Chem. Soc., Chem. Commun. 2215–2216. [Google Scholar]
- Basuli F.; Wu H.; Griffiths G. L. (2011) Syntheses of meta-[F]Fluorobenzaldehyde and meta-[F]Fluorobenzylbromide from Phenyl(3-formylphenyl) Iodonium Salt Precursors. J. Labelled Compd. Radiopharm. 54, 224–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B. C.; Lee K. C.; Lee H.; Mach R. H.; Katzenellenbogen J. A. (2007) Strategies for the labeling of halogen-substituted peroxisome proliferator-activated receptor gamma ligands: potential positron emission tomography and single photon emission computed tomography imaging agents. Bioconjugate Chem. 18, 514–523. [DOI] [PubMed] [Google Scholar]
- Ross T. L.; Ermert J.; Hocke C.; Coenen H. H. (2007) Nucleophilic 18F-fluorination of heteroaromatic iodonium salts with no-carrier-added [18F]fluoride. J. Am. Chem. Soc. 129, 8018–8025. [DOI] [PubMed] [Google Scholar]
- Hostetler E. D.; Jonson S. D.; Welch M. J.; Katzenellenbogen J. A. (1999) Synthesis of 2-[(18)F]Fluoroestradiol, a Potential Diagnostic Imaging Agent for Breast Cancer: Strategies to Achieve Nucleophilic Substitution of an Electron-Rich Aromatic Ring with [(18)F]F(−). J. Org. Chem. 64, 178–185. [DOI] [PubMed] [Google Scholar]
- Chun J. H.; Lu S.; Lee Y. S.; Pike V. W. (2010) Fast and high-yield microreactor syntheses of ortho-substituted [(18)F]fluoroarenes from reactions of [(18)F]fluoride ion with diaryliodonium salts. J. Org. Chem. 75, 3332–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telu S.; Chun J. H.; Simeon F. G.; Lu S.; Pike V. W. (2011) Syntheses of mGluR5 PET radioligands through the radiofluorination of diaryliodonium tosylates. Org. Biomol. Chem. 9, 6629–6638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman J.; Solin O. (1997) Fluorine-18-labeled fluorine gas for synthesis of tracer molecules. Nucl. Med. Biol. 24, 677–683. [DOI] [PubMed] [Google Scholar]
- Teare H.; Robins E. G.; Kirjavainen A.; Forsback S.; Sandford G.; Solin O.; Luthra S. K.; Gouverneur V. (2010) Radiosynthesis and evaluation of [18F]Selectfluor bis(triflate). Angew. Chem., Int. Ed. Engl. 49, 6821–6824. [DOI] [PubMed] [Google Scholar]
- Murali D.; Flores L. G.; Roberts A. D.; Nickles R. J.; DeJesus O. T. (2003) Aromatic l-amino acid decarboxylase (AAAD) inhibitors as carcinoid tumor-imaging agents: synthesis of 18F-labeled alpha-fluoromethyl-6-fluoro-m-tyrosine (FM-6-FmT). Appl. Radiat. Isot. 59, 237–243. [DOI] [PubMed] [Google Scholar]
- Furuya T.; Klein J. E.; Ritter T. (2010) C–F Bond Formation for the Synthesis of Aryl Fluorides. Synthesis 2010, 1804–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coenen H. H.; Moerlein S. M. (1987) Regiospecific Aromatic Fluorodemetallation of group IVb Metalloarenes Using Elemental Fluorine or Acetyl Hypofluorite. J. Fluorine Chem. 36, 63–75. [Google Scholar]
- Teare H.; Robins E. G.; Arstad E.; Luthra S. K.; Gouverneur V. (2007) Synthesis and reactivity of [18F]-N-fluorobenzenesulfonimide. Chem. Commun. (Cambridge, U. K.) 2330–2332. [DOI] [PubMed] [Google Scholar]
- Stenhagen I. S.; Kirjavainen A. K.; Forsback S. J.; Jorgensen C. G.; Robins E. G.; Luthra S. K.; Solin O.; Gouverneur V. (2013) [18F]Fluorination of an arylboronic ester using [18F]selectfluor bis(triflate): application to 6-[18F]fluoro-l-DOPA. Chem. Commun. (Cambridge, U. K.) 49, 1386–1388. [DOI] [PubMed] [Google Scholar]
- Lee E.; Kamlet A. S.; Powers D. C.; Neumann C. N.; Boursalian G. B.; Furuya T.; Choi D. C.; Hooker J. M.; Ritter T. (2011) A fluoride-derived electrophilic late-stage fluorination reagent for PET imaging. Science 334, 639–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E.; Hooker J. M.; Ritter T. (2012) Nickel-mediated oxidative fluorination for PET with aqueous [18F] fluoride. J. Am. Chem. Soc. 134, 17456–17458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting R.; Harwig C.; auf dem Keller U.; McCormick S.; Austin P.; Overall C. M.; Adam M. J.; Ruth T. J.; Perrin D. M. (2008) Toward [18F]-labeled aryltrifluoroborate radiotracers: in vivo positron emission tomography imaging of stable aryltrifluoroborate clearance in mice. J. Am. Chem. Soc. 130, 12045–12055. [DOI] [PubMed] [Google Scholar]
- Li Y.; Liu Z.; Lozada J.; Wong M. Q.; Lin K. S.; Yapp D.; Perrin D. M. (2013) Single step 18F-labeling of dimeric cycloRGD for functional PET imaging of tumors in mice. Nucl. Med. Biol. 40, 959–966. [DOI] [PubMed] [Google Scholar]
- Li Y.; Liu Z.; Harwig C. W.; Pourghiasian M.; Lau J.; Lin K. S.; Schaffer P.; Benard F.; Perrin D. M. (2013) (18)F-click labeling of a bombesin antagonist with an alkyne-(18)F-ArBF(3) (−): in vivo PET imaging of tumors expressing the GRP-receptor. Am. J. Nucl. Med. Mol. Imaging 3, 57–70. [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Li Y.; Lozada J.; Wong M. Q.; Greene J.; Lin K. S.; Yapp D.; Perrin D. M. (2013) Kit-like 18F-labeling of RGD-19F-arytrifluroborate in high yield and at extraordinarily high specific activity with preliminary in vivo tumor imaging. Nucl. Med. Biol. 40, 841–849. [DOI] [PubMed] [Google Scholar]
- Jacobson O.; Zhu L.; Ma Y.; Weiss I. D.; Sun X.; Niu G.; Kiesewetter D. O.; Chen X. (2011) Rapid and simple one-step F-18 labeling of peptides. Bioconjugate Chem. 22, 422–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becaud J.; Mu L.; Karramkam M.; Schubiger P. A.; Ametamey S. M.; Graham K.; Stellfeld T.; Lehmann L.; Borkowski S.; Berndorff D.; Dinkelborg L.; Srinivasan A.; Smits R.; Koksch B. (2009) Direct one-step 18F-labeling of peptides via nucleophilic aromatic substitution. Bioconjugate Chem. 20, 2254–2261. [DOI] [PubMed] [Google Scholar]
- Hazari P. P.; Schulz J.; Vimont D.; Chadha N.; Allard M.; Szlosek-Pinaud M.; Fouquet E.; Mishra A. K. (2014) A new SiF-dipropargyl glycerol scaffold as a versatile prosthetic group to design dimeric radioligands: synthesis of the [(18) F]BMPPSiF tracer to image serotonin receptors. ChemMedChem 9, 337–349. [DOI] [PubMed] [Google Scholar]
- McBride W. J.; Sharkey R. M.; Karacay H.; D’Souza C. A.; Rossi E. A.; Laverman P.; Chang C. H.; Boerman O. C.; Goldenberg D. M. (2009) A novel method of 18F radiolabeling for PET. J. Nucl. Med. 50, 991–998. [DOI] [PubMed] [Google Scholar]
- Laverman P.; McBride W. J.; Sharkey R. M.; Goldenberg D. M.; Boerman O. C. (2014) Al(18) F labeling of peptides and proteins. J. Labelled Compd. Radiopharm. 57, 219–223. [DOI] [PubMed] [Google Scholar]
- McBride W. J.; D’Souza C. A.; Sharkey R. M.; Karacay H.; Rossi E. A.; Chang C. H.; Goldenberg D. M. (2010) Improved 18F labeling of peptides with a fluoride–aluminum–chelate complex. Bioconjugate Chem. 21, 1331–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laverman P.; McBride W. J.; Sharkey R. M.; Eek A.; Joosten L.; Oyen W. J.; Goldenberg D. M.; Boerman O. C. (2010) A novel facile method of labeling octreotide with (18)F-fluorine. J. Nucl. Med. 51, 454–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laverman P.; D’Souza C. A.; Eek A.; McBride W. J.; Sharkey R. M.; Oyen W. J.; Goldenberg D. M.; Boerman O. C. (2011) Optimized labeling of NOTA-conjugated octreotide with F-18. Tumor Biol. 33, 427–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride W. J.; D’Souza C. A.; Sharkey R. M.; Goldenberg D. M. (2011) The radiolabeling of proteins by the [18F]AlF method. Appl. Radiat. Isot. 70, 200–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang L.; Li W.; Guo N.; Ma Y.; Zhu L.; Kiesewetter D. O.; Shen B.; Niu G.; Chen X. (2011) Comparison study of [18F]FAl-NOTA-PRGD2, [18F]FPPRGD2, and [68Ga]Ga-NOTA-PRGD2 for PET imaging of U87MG tumors in mice. Bioconjugate Chem. 22, 2415–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.; Yue X.; Lang L.; Kiesewetter D. O.; Li F.; Zhu Z.; Niu G.; Chen X. (2014) Longitudinal PET imaging of muscular inflammation using 18F-DPA-714 and 18F-Alfatide II and differentiation with tumors. Theranostics 4, 546–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipowska M.; Klenc J.; Shetty D.; Nye J. A.; Shim H.; Taylor A. T. (2014) Al18F-NODA-butyric acid: biological evaluation of a new PET renal radiotracer. Nucl. Med. Biol. 41, 248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan W.; Guo N.; Pan D.; Yu C.; Weng Y.; Luo S.; Ding H.; Xu Y.; Wang L.; Lang L.; Xie Q.; Yang M.; Chen X. (2013) First experience of 18F-alfatide in lung cancer patients using a new lyophilized kit for rapid radiofluorination. J. Nucl. Med. 54, 691–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiesewetter D. O.; Guo N.; Guo J.; Gao H.; Zhu L.; Ma Y.; Niu G.; Chen X. (2012) Evaluation of an [(18)F]AlF-NOTA Analog of Exendin-4 for Imaging of GLP-1 Receptor in Insulinoma. Theranostics 2, 999–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y.; Pan D.; Zhu C.; Xu Q.; Wang L.; Chen F.; Yang R.; Luo S.; Yang M.; Yan Y. (2014) Pilot Study of a Novel F-labeled FSHR Probe for Tumor Imaging. Mol. Imaging Biol. 16, 578–585. [DOI] [PubMed] [Google Scholar]
- Hoigebazar L.; Jeong J. M.; Lee J. Y.; Shetty D.; Yang B. Y.; Lee Y. S.; Lee D. S.; Chung J. K.; Lee M. C. (2012) Syntheses of 2-nitroimidazole derivatives conjugated with 1,4,7-triazacyclononane-N,N′-diacetic acid labeled with F-18 using an aluminum complex method for hypoxia imaging. J. Med. Chem. 55, 3155–3162. [DOI] [PubMed] [Google Scholar]
- Wester H. J.; Schottelius M. (2007) Fluorine-18 labeling of peptides and proteins. Ernst Schering Res. Found. Workshop 79–111. [DOI] [PubMed] [Google Scholar]
- Loser R.; Fischer S.; Hiller A.; Kockerling M.; Funke U.; Maisonial A.; Brust P.; Steinbach J. (2013) Use of 3-[(18)F]fluoropropanesulfonyl chloride as a prosthetic agent for the radiolabelling of amines: investigation of precursor molecules, labelling conditions and enzymatic stability of the corresponding sulfonamides. Beilstein J. Org. Chem. 9, 1002–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiesewetter D. O.; Jacobson O.; Lang L.; Chen X. (2011) Automated radiochemical synthesis of [18F]FBEM: a thiol reactive synthon for radiofluorination of peptides and proteins. Appl. Radiat. Isot. 69, 410–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olberg D. E.; Hjelstuen O. K. (2010) Labeling strategies of peptides with (1)(8)F for positron emission tomography. Curr. Top. Med. Chem. 10, 1669–1679. [DOI] [PubMed] [Google Scholar]
- Fani M.; Maecke H. R.; Okarvi S. M. (2012) Radiolabeled peptides: valuable tools for the detection and treatment of cancer. Theranostics 2, 481–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z.; Kandeel F. (2010) 18F-labeled proteins. Curr. Pharm. Biotechnol. 11, 572–580. [DOI] [PubMed] [Google Scholar]
- Carberry P.; Lieberman B. P.; Ploessl K.; Choi S. R.; Haase D. N.; Kung H. F. (2011) New F-18 prosthetic group via oxime coupling. Bioconjugate Chem. 22, 642–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dialer L. O.; Selivanova S. V.; Muller C. J.; Muller A.; Stellfeld T.; Graham K.; Dinkelborg L. M.; Kramer S. D.; Schibli R.; Reiher M.; Ametamey S. M. (2013) Studies toward the development of new silicon-containing building blocks for the direct (18)F-labeling of peptides. J. Med. Chem. 56, 7552–7563. [DOI] [PubMed] [Google Scholar]
- Koslowsky I.; Mercer J.; Wuest F. (2010) Synthesis and application of 4-[(18)F]fluorobenzylamine: a versatile building block for the preparation of PET radiotracers. Org. Biomol. Chem. 8, 4730–4735. [DOI] [PubMed] [Google Scholar]
- Vaidyanathan G.; Zalutsky M. R. (2006) Synthesis of N-succinimidyl 4-[18F]fluorobenzoate, an agent for labeling proteins and peptides with 18F. Nature Protoc. 1, 1655–1661. [DOI] [PubMed] [Google Scholar]
- Toyokuni T.; Walsh J. C.; Dominguez A.; Phelps M. E.; Barrio J. R.; Gambhir S. S.; Satyamurthy N. (2003) Synthesis of a new heterobifunctional linker, N-[4-(aminooxy)butyl]maleimide, for facile access to a thiol-reactive 18F-labeling agent. Bioconjugate Chem. 14, 1253–1259. [DOI] [PubMed] [Google Scholar]
- Yue X.; Yan X.; Wu C.; Niu G.; Ma Y.; Jacobson O.; Shen B.; Kiesewetter D. O.; Chen X. (2014) One-pot two-step radiosynthesis of a new (18)F-labeled thiol reactive prosthetic group and its conjugate for insulinoma imaging. Mol. Pharmaceutics 11, 3875–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.; Lang L.; Ma Y.; Kiesewetter D. O. (2008) [18F]Fluoropropylsulfonyl chloride: a new reagent for radiolabeling primary and secondary amines for PET imaging. J. Labelled Compd. Radiopharm. 51, 23–27. [Google Scholar]
- Gao Z.; Gouverneur V.; Davis B. G. (2013) Enhanced aqueous Suzuki–Miyaura coupling allows site-specific polypeptide 18F-labeling. J. Am. Chem. Soc. 135, 13612–13615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewett J. C.; Bertozzi C. R. (2010) Cu-free click cycloaddition reactions in chemical biology. Chem. Soc. Rev. 39, 1272–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostovtsev V. V.; Green L. G.; Fokin V. V.; Sharpless K. B. (2002) A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem., Int. Ed. Engl. 41, 2596–2599. [DOI] [PubMed] [Google Scholar]
- Tornoe C. W.; Christensen C.; Meldal M. (2002) Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 67, 3057–3064. [DOI] [PubMed] [Google Scholar]
- Jia L.; Cheng Z.; Shi L.; Li J.; Wang C.; Jiang D.; Zhou W.; Meng H.; Qi Y.; Cheng D.; Zhang L. (2013) Fluorine-18 labeling by click chemistry: multiple probes in one pot. Appl. Radiat. Isot. 75, 64–70. [DOI] [PubMed] [Google Scholar]
- Worrell B. T.; Malik J. A.; Fokin V. V. (2013) Direct evidence of a dinuclear copper intermediate in Cu(I)-catalyzed azide–alkyne cycloadditions. Science 340, 457–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thonon D.; Kech C.; Paris J.; Lemaire C.; Luxen A. (2009) New strategy for the preparation of clickable peptides and labeling with 1-(azidomethyl)-4-[(18)F]-fluorobenzene for PET. Bioconjugate Chem. 20, 817–823. [DOI] [PubMed] [Google Scholar]
- Gill H. S.; Marik J. (2011) Preparation of 18F-labeled peptides using the copper(I)-catalyzed azide–alkyne 1,3-dipolar cycloaddition. Nature Protoc 6, 1718–1725. [DOI] [PubMed] [Google Scholar]
- Amblard F.; Cho J. H.; Schinazi R. F. (2009) Cu(I)-catalyzed Huisgen azide–alkyne 1,3-dipolar cycloaddition reaction in nucleoside, nucleotide, and oligonucleotide chemistry. Chem. Rev. 109, 4207–4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pretze M.; Pietzsch D.; Mamat C. (2013) Recent trends in bioorthogonal click-radiolabeling reactions using fluorine-18. Molecules 18, 8618–8665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser M.; Robins E. G. (2009) ‘Click labelling’ in PET radiochemistry. J. Labelled Compd. Radiopharm. 52, 407–414. [Google Scholar]
- Pretze M.; Mamat C. (2013) Automated preparation of [18F]AFP and [18F]BFP: two novel bifunctional 18F-labeling building blocks for Huisgen-click. J. Fluorine Chem. 150, 25–35. [Google Scholar]
- Ramenda T.; Steinbach J.; Wuest F. (2013) 4-[18F]Fluoro-N-methyl-N-(propyl-2-yn-1-yl)benzenesulfonamide ([18F]F-SA): a versatile building block for labeling of peptides, proteins and oligonucleotides with fluorine-18 via Cu(I)-mediated click chemistry. Amino Acids 44, 1167–1180. [DOI] [PubMed] [Google Scholar]
- Hausner S. H.; Carpenter R. D.; Bauer N.; Sutcliffe J. L. (2013) Evaluation of an integrin alphavbeta6-specific peptide labeled with [18F]fluorine by copper-free, strain-promoted click chemistry. Nucl. Med. Biol. 40, 233–239. [DOI] [PubMed] [Google Scholar]
- Baskin J. M.; Prescher J. A.; Laughlin S. T.; Agard N. J.; Chang P. V.; Miller I. A.; Lo A.; Codelli J. A.; Bertozzi C. R. (2007) Copper-free click chemistry for dynamic in vivo imaging. Proc. Natl. Acad. Sci. U. S. A. 104, 16793–16797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewett J. C.; Sletten E. M.; Bertozzi C. R. (2010) Rapid Cu-free click chemistry with readily synthesized biarylazacyclooctynones. J. Am. Chem. Soc. 132, 3688–3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agard N. J.; Prescher J. A.; Bertozzi C. R. (2004) A strain-promoted [3 + 2] azide–alkyne cycloaddition for covalent modification of biomolecules in living systems. J. Am. Chem. Soc. 126, 15046–15047. [DOI] [PubMed] [Google Scholar]
- Chen K.; Wang X.; Lin W. Y.; Shen C. K.-F.; Yap L.-P.; Hughes L. D.; Conti P. S. (2012) Strain-Promoted Catalyst-Free Click Chemistry for Rapid Construction of 64Cu-Labeled PET Imaging Probes. ACS Med. Chem. Lett. 3, 1019–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvet V.; Wuest M.; Wuest F. (2011) Copper-free click chemistry with the short-lived positron emitter fluorine-18. Org. Biomol. Chem. 9, 7393–9. [DOI] [PubMed] [Google Scholar]
- Campbell-Verduyn L. S.; Mirfeizi L.; Schoonen A. K.; Dierckx R. A.; Elsinga P. H.; Feringa B. L. (2011) Strain-promoted copper-free “click” chemistry for 18F radiolabeling of bombesin. Angew. Chem., Int. Ed. Engl. 50, 11117–111120. [DOI] [PubMed] [Google Scholar]
- Evans H. L.; Slade R. L.; Carroll L.; Smith G.; Nguyen Q. D.; Iddon L.; Kamaly N.; Stockmann H.; Leeper F. J.; Aboagye E. O.; Spivey A. C. (2012) Copper-free click—a promising tool for pre-targeted PET imaging. Chem. Commun. (Cambridge, U. K.) 48, 991–993. [DOI] [PubMed] [Google Scholar]
- Sachin K.; Jadhav V. H.; Kim E. M.; Kim H. L.; Lee S. B.; Jeong H. J.; Lim S. T.; Sohn M. H.; Kim D. W. (2012) F-18 labeling protocol of peptides based on chemically orthogonal strain-promoted cycloaddition under physiologically friendly reaction conditions. Bioconjugate Chem. 23, 1680–1686. [DOI] [PubMed] [Google Scholar]
- Carpenter R. D.; Hausner S. H.; Sutcliffe J. L. (2011) Copper-Free Click for PET: Rapid 1,3-Dipolar Cycloadditions with a Fluorine-18 Cyclooctyne. ACS Med. Chem. Lett. 2, 885–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn M.; Breinbauer R. (2004) The Staudinger ligation—a gift to chemical biology. Angew. Chem., Int. Ed. Engl. 43, 3106–3116. [DOI] [PubMed] [Google Scholar]
- Leffler J. E.; Temple R. D. (1967) The Staudinger Reaction between Triarylphosphines and Azides. A Study of the Mechanism. J. Am. Chem. Soc. 89, 5235–5246. [Google Scholar]
- Nepomniaschiy N.; Grimminger V.; Cohen A.; DiGiovanni S.; Lashuel H. A.; Brik A. (2008) Switch peptide via Staudinger reaction. Org. Lett. 10, 5243–5246. [DOI] [PubMed] [Google Scholar]
- Saxon E.; Bertozzi C. R. (2000) Cell surface engineering by a modified Staudinger reaction. Science 287, 2007–2010. [DOI] [PubMed] [Google Scholar]
- Saxon E.; Armstrong J. I.; Bertozzi C. R. (2000) A “traceless” Staudinger ligation for the chemoselective synthesis of amide bonds. Org. Lett. 2, 2141–2143. [DOI] [PubMed] [Google Scholar]
- Nilsson B. L.; Kiessling L. L.; Raines R. T. (2000) Staudinger ligation: a peptide from a thioester and azide. Org. Lett. 2, 1939–1941. [DOI] [PubMed] [Google Scholar]
- Lin F. L.; Hoyt H. M.; van Halbeek H.; Bergman R. G.; Bertozzi C. R. (2005) Mechanistic investigation of the staudinger ligation. J. Am. Chem. Soc. 127, 2686–2695. [DOI] [PubMed] [Google Scholar]
- Carroll L.; Boldon S.; Bejot R.; Moore J. E.; Declerck J.; Gouverneur V. (2011) The traceless Staudinger ligation for indirect 18F-radiolabelling. Org. Biomol. Chem. 9, 136–140. [DOI] [PubMed] [Google Scholar]
- Gaeta A.; Woodcraft J.; Plant S.; Goggi J.; Jones P.; Battle M.; Trigg W.; Luthra S. K.; Glaser M. (2010) Use of 2-[(18)F]fluoroethylazide for the Staudinger ligation—preparation and characterisation of GABA(A) receptor binding 4-quinolones. Bioorg. Med. Chem. Lett. 20, 4649–4652. [DOI] [PubMed] [Google Scholar]
- Mamat C.; Franke M.; Peppel T.; Kockerling M.; Steinbach J. (2011) Synthesis, structure determination, and (radio-)fluorination of novelfunctionalized phosphanes suitable for the traceless Staudinger ligation. Tetrahedron 67, 4521–4529. [Google Scholar]
- Liu L.; Hong Z. Y.; Wong C. H. (2006) Convergent glycopeptide synthesis by traceless Staudinger ligation and enzymatic coupling. ChemBioChem 7, 429–432. [DOI] [PubMed] [Google Scholar]
- Blackman M. L.; Royzen M.; Fox J. M. (2008) Tetrazine ligation: fast bioconjugation based on inverse-electron-demand Diels–Alder reactivity. J. Am. Chem. Soc. 130, 13518–13519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraj N. K.; Weissleder R. (2011) Biomedical applications of tetrazine cycloadditions. Acc. Chem. Res. 44, 816–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossin R.; Verkerk P. R.; van den Bosch S. M.; Vulders R. C.; Verel I.; Lub J.; Robillard M. S. (2010) In vivo chemistry for pretargeted tumor imaging in live mice. Angew. Chem., Int. Ed. Engl. 49, 3375–3378. [DOI] [PubMed] [Google Scholar]
- Devaraj N. K.; Upadhyay R.; Haun J. B.; Hilderbrand S. A.; Weissleder R. (2009) Fast and sensitive pretargeted labeling of cancer cells through a tetrazine/trans-cyclooctene cycloaddition. Angew. Chem., Int. Ed. Engl. 48, 7013–7016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.; Hassink M.; Selvaraj R.; Yap L. P.; Park R.; Wang H.; Chen X.; Fox J. M.; Li Z.; Conti P. S. (2013) Efficient 18F labeling of cysteine-containing peptides and proteins using tetrazine–trans-cyclooctene ligation. Mol. Imaging 12, 121–128. [PMC free article] [PubMed] [Google Scholar]
- Li Z.; Cai H.; Hassink M.; Blackman M. L.; Brown R. C.; Conti P. S.; Fox J. M. (2010) Tetrazine–trans-cyclooctene ligation for the rapid construction of 18F labeled probes. Chem. Commun. (Cambridge, U. K.) 46, 8043–8045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori T.; Kasamatsu S.; Mosdzianowski C.; Welch M. J.; Yonekura Y.; Fujibayashi Y. (2006) Automatic synthesis of 16 alpha-[(18)F]fluoro-17beta-estradiol using a cassette-type [(18)F]fluorodeoxyglucose synthesizer. Nucl. Med. Biol. 33, 281–286. [DOI] [PubMed] [Google Scholar]