

Summary
Describing, understanding, and modulating the function of the cell require elucidation of the structures of macromolecular assemblies. Here, we describe an integrative method for modeling heteromeric complexes using as a starting point disassembly pathways determined by native mass spectrometry (MS). In this method, the pathway data and other available information are encoded as a scoring function on the positions of the subunits of the complex. The method was assessed on its ability to reproduce the native contacts in five benchmark cases with simulated MS data and two cases with real MS data. To illustrate the power of our method, we purified the yeast initiation factor 3 (eIF3) complex and characterized it by native MS and chemical crosslinking MS. We established substoichiometric binding of eIF5 and derived a model for the five-subunit eIF3 complex, at domain level, consistent with its role as a scaffold for other initiation factors.
Graphical Abstract

Highlights
-
•
Integrative MS method allows topological characterization of heteromeric complexes
-
•
Intersubunit crosslinks increase the precision of the predicted topologies
-
•
A 3D model of eIF3:eIF5 complex was built using restraints from MS-based methods
-
•
Integrative modeling reveals two submodules within eIF3: eIF3b:i:g and eIF3a:c
Politis et al. develop a method for integrating diverse mass spectrometry-based data into topological models of protein complexes. The method was benchmarked on a number of known complexes and used to reveal the architecture of the eIF3 in complex with eIF5.
Introduction
Macromolecular assemblies perform many functions in the cell, such as RNA synthesis, translation, protein folding, and degradation (Lander et al., 2012b; Lasker et al., 2012; Robinson et al., 2007; Russel et al., 2012; Ward et al., 2013). Determining the structures of such assemblies is the primary goal of structural biology. However, structure determination of transient and heterogeneous assemblies by a single experimental method often fails rendering structural insights into critical biological machines intractable. Integrative structural biology approaches, which combine information from various techniques, can address this challenge, thus enabling structural insights for systems that are refractive to a single method (Alber et al., 2007, 2008; Heck, 2008; Lander et al., 2012b; Robinson et al., 2007; Russel et al., 2012; Sali et al., 2003; Ward et al., 2013).
MS approaches are characterized by general applicability and low sample consumption and can provide useful and diverse data for integrative modeling analysis. For example, data derived from nondenaturing or native MS can yield the composition and stoichiometry of protein complexes as well as identify stable subcomplexes using gas- or solution-phase disruption techniques (Hall et al., 2012; Hernández and Robinson, 2007; Zhou et al., 2008). Coupling MS with ion mobility (IM) can be used additionally to map the structure and dynamics of complexes by determining their orientationally averaged collision cross-section (CCS) (Baldwin et al., 2011; Hall et al., 2012; Politis et al., 2010; Stengel et al., 2010; Uetrecht et al., 2011; van Duijn et al., 2012). Moreover, improvements in crosslinking (CX) chemistry, underpinned by progress in MS, have led to a renaissance in crosslinking mass spectrometry (CX-MS) (Leitner et al., 2010). CX-MS can now be used to measure residue-specific distances by identifying proximate lysines (and other residue types) crosslinked by chemical reagents (Chu et al., 2010; Maiolica et al., 2007; Rappsilber et al., 2000; Walzthoeni et al., 2012, 2013; Young et al., 2000). These advances, including the development of tools to identify crosslinked peptides (Chen et al., 2010b; Götze et al., 2012; Herzog et al., 2012; Maiolica et al., 2007; Xu et al., 2010), have resulted in integrative structural models of challenging macromolecular assemblies (Alber et al., 2007; Benesch and Ruotolo, 2011; Chen et al., 2010b; Lasker et al., 2012; Schmidt et al., 2013; Zhou et al., 2014).
Further structural information can be attained from disruption of protein interactions in solution or in the gas phase, followed by MS detection of the stoichiometry of the resulting subcomplexes to reveal more detailed subunit interactions. Dissociation in the gas phase is most often achieved by colliding the multimeric complex ion with neutral gas molecules, such as argon (collision-induced dissociation [CID]) (Benesch et al., 2006; Hall et al., 2012; Jurchen and Williams, 2003). CID typically results in the loss of individual peripheral subunits; the identification of successively “stripped” oligomers is informative about intersubunit contacts (Taverner et al., 2008). Solution disruption can be achieved by the addition of organic solvent, manipulation of the pH, or increasing the ionic strength (Hall et al., 2012; Heck, 2008; Hernández and Robinson, 2007; Zhou et al., 2008). In either scenario, the resulting disassembly routes can be distinguished by charge states and define pathways of subcomplexes, starting from the intact complex (Zhou et al., 2008). These disassembly pathways can be used as restraints for building models of the (sub)complexes consistent with the input data.
Previously, we developed an algorithm for determining subunit composition of (sub)complexes identified by MS (summing masses for interaction topology) (Taverner et al., 2008). This strategy enabled us to generate protein interaction networks of transient assemblies that were refractive to atomic structure determination (Taverner et al., 2008; Zhou et al., 2008). Most notably, these complexes included the 19S proteasome (Politis et al., 2014; Sharon et al., 2006), the exosome (Taverner et al., 2008), and the human eukaryotic initiation factor (eIF3) (Pukala et al., 2009; Zhou et al., 2008). Here, we extend these preliminary studies by developing, testing, and applying an integrative method for modeling the 3D low-resolution structures of assemblies. In this method, subunit connectivity information from native MS and CX-MS experiments is encoded as connectivity restraints, which are included in a scoring function for building 3D topological models of protein assemblies. Monte Carlo sampling is then guided by the scoring function to generate models consistent with the data. This modeling approach is implemented in Integrative Modeling Platform (IMP) (http://integrativemodeling.org), an open source software suite for computing and characterizing models of macromolecular assemblies based on varied types of data (Russel et al., 2012). We assessed our method based on its ability to reproduce the native contacts of five benchmark cases with simulated MS data as well as two benchmark cases where MS data and structures are available in the literature (Lasker et al., 2012; Park et al., 2010; Politis et al., 2013).
We also purified and analyzed by nondenaturing MS and CX-MS the intact yeast eIF3 assembly in complex with eIF5. Yeast eIF3, a heteromeric complex of six subunits, plays an important role in the highly regulated translation-initiation process in eukaryotes. Despite significant interest, its molecular architecture remains elusive, presumably due to its dynamic nature and its role as a scaffold for many other initiation factors (Valásek et al., 2002). Our integrative method results in a 3D topological model of the yeast eIF3:eIF5 complex.
Results
Approach
We adapted the iterative four step process for the integrative structure determination of an investigated assembly (Figure 1) (Russel et al., 2012; Alber et al., 2007) as follows: (1) gather data, including generation of a list of subcomplexes by native MS and CX-MS experiments, (2) define system representation and convert data into spatial restraints that comprise a scoring function, (3) sample the configurational space guided by the scoring function to generate an ensemble of good-scoring models, and (4) analyze the ensemble of good-scoring models.
Figure 1.

Generation and Evaluation of Models Based on MS Experiments
Proteins are isolated using protein purification (ia), and their composition is identified by MS-based proteomics (ib). The stoichiometry of the constituent subunits and their subcomplexes formed along the MS/MS (ic) disruption in solution experiments (id) and crosslinking experiments (ie) are subsequently determined. Next, a representation scheme is chosen with one sphere per subunit and a scoring function is defined using MS-based and excluded volume restraints (ii). The scoring function then guides the search for the models consistent with the experiments (iii). Finally, models are subjected to analysis and interpretation based on the identified contacts and configurations within the ensemble of models that fit the data (iv).
Step 1. Gather Data
In the first step, proteins are affinity purified, thus enabling isolation of the intact complex for subsequent MS analysis. The composition of the complex is established using standard proteomics experiments (Yates et al., 2005), and the stoichiometry of the intact complex is determined from MS in the native state (Hernández and Robinson, 2007). A list of interacting subcomplexes is extracted by performing controlled dissociation of activated complexes in the gas phase or by disruption in solution (Hall et al., 2012; Hernández and Robinson, 2007). Although tandem MS with CID and disruption in solution are undergone in different phases, they often provide complementary information (Zhou et al., 2008). In some cases, we also gather CX-MS data, providing additional connectivity information at resolutions ranging from domain to subunit levels.
Additional data can be derived from a wide variety of other sources (Russel et al., 2012). Here, we also use information from crystal structures and homology models of subunits where available (Sali and Blundell, 1993) and standard packing densities for proteins when structural information is not accessible (Chen et al., 2010a).
Step 2. Define System Representation and Scoring Function
Each domain or subunit of the assembly is treated as a rigid collection of spheres at a resolution depending on the available information. A subunit or a domain of known atomic structure or with a reliable homology model is represented at atomic resolution, while a subunit or a domain for which no structure is available is represented as a single sphere with the radius derived from its mass, assuming average protein density (Hall et al., 2012); the latter representation assumes the globular shape of the subunit, although in principle other shapes can also be used.
The scoring function encodes the experimental data as a sum of individual restraints. It is used to guide the sampling of candidate models and to evaluate the generated models based on how well they fit the input data. Our scoring function is
| (Equation 1) |
where SMS and SCX-MS refer to connectivity restraints derived from native MS and CX-MS, respectively. These restraints enforce intersubunit contacts and proximities among the (sub)complexes in the disassembly pathway revealed by MS data. The SMS restraint enforces subcomplex connectivities as determined by native MS experiments, while the SCX-MS restraint is used to enforce pairwise domain or subunit connectivities (e.g., spheres are touching at least at one point). The distance proximity between amino acid residues, typically specified from CX-MS experiments, was not used in this study due to the lack of complete high-resolution structures (only parts of high-resolution structures were used in the case of the eIF3 complex). SEV is the excluded volume restraint that enforces a lower distance bound between domains and/or subunits (Supplemental Information available online) (Hall et al., 2012). It is noteworthy that all individual restraints from different techniques were simultaneously used in our search for topological models that match the data and therefore no weighting on the basis of the method utilized was performed.
Step 3. Sample Models that Fit the Data
With the scoring function in hand, we employ a Monte Carlo (MC) algorithm for sampling assembly configurations (Experimental Procedures and Supplemental Information). Typically, 50 MC steps are used for each model. MC sampling is followed by a conjugate gradients refinement of 250 steps (Russel et al., 2012). The resulting model is considered to fit the data and added to the ensemble that is passed to the next step for analysis if each restraint is satisfied. A connectivity restraint is considered to be satisfied if, for each required contact, there is a pair of spheres from the corresponding subunits within 5% of the sum of their radii of one another. To further enhance the robustness of our approach with respect to false positive interactions, we considered within the good solutions those models where one pairwise interaction disagreed with the data. An excluded volume restraint is satisfied if each sphere is disconnected from all other spheres within the assembly. As a guide, computing 50,000 refined models takes approximately 2 hr of CPU time (Intel Xeon E3 processor, 3GHz) for an assembly of five subunits.
Step 4. Analyze the Ensemble of Models that Fit the Data
We analyze the ensemble of generated models using intersubunit contact maps. The average contact map of the ensemble captures the precision with which the integrative modeling method defines intersubunit interactions. Contacts whose relative frequencies are near one are present in almost all structures that fit the data and are therefore highly probable, given the assumptions and data encoded in the modeling procedure. Contacts whose frequencies are near zero are correspondingly unlikely. If many contacts have intermediate frequencies, then the input information is insufficient and/or contradictory and the structure cannot be determined.
The contact maps were evaluated on one-to-one basis; for a given “native” contact between two subunits, we searched and subsequently assessed the same contact in the model structures. Thus, the contact frequency of a given pairwise interaction is equal to the fraction of the models with the specified native contact. When the native contact map is established, we compute the fraction of models that match the native contacts. For the models that do not match the native contacts, we know there are ambiguities in the interpretation of the input information.
Benchmark
Simulated Data
We tested our method initially on five artificial assemblies. These assemblies are heteromers of 4 to 13 subunits (Figures 2A and 2B; Figures S1, S2A, and S2B), two of which contain multiple copies of one or more subunits. For each of these benchmark cases, we generated artificial MS disassembly pathways (Figures S2C and S2D) based on the subunit sizes, the number of contacts with other subunits, and the relative subunit positions within the assembly (step 1 of Approach). To map the precision of the method as a function of the quantity of input data, we also varied the number of subcomplexes determined within the simulated disassembly pathways.
Figure 2.

Analysis of the Hypothetical, Heteromeric Complexes
(A and B) Simulated disassembly pathways were computed for two hypothetical heteromers: an ABCDE (A) and an AABBC complex (B). In the Inset, the native pairwise contact maps of both heteromeric complexes at subunit level are depicted.
(C) We plotted the fraction of models that match the native contacts for the AABBC and ABCDE hypothetical complexes.
(D) A contact frequency map is plotted for all intersubunit interactions within the studied heteromers.
We modeled the benchmark assemblies using one sphere per subunit. Overall, when subcomplexes and excluded volume restraints were used, we found only 7% to 12% of the ensemble matched the contacts of the hypothetical topological structure (Figure 2C and Table S1). However, more than 60% of the ensemble had all of the contacts of the hypothetical structure (Figure 2D). Knowledge of additional subcomplexes and pairwise interactions (Figure S2), which would usually be obtained from other complementary approaches, significantly increased precision (Tables S1A and S1B). For example, the fraction of models that match the native contacts for a hypothetical pentamer increased from 8% to 44% (Table S1A) when an additional eight crosslinks were added. We further assessed the ability of the restraints individually, and combined, to predict the native topology using the receiver operating characteristic (ROC) curves (Figure S3). The area under the ROC curve provides a measure of the information content of each restraint, where 0.5 indicates that the restraint cannot discriminate between correct and incorrect topologies (Hall et al., 2012). Consistent with the fraction of models that match the data, for both cases of hypothetical pentamers the area under ROC curve was markedly increased when information on subcomplexes coupled with knowledge of binary interactions (Figure S3). Finally, we clustered the ensemble of model structures (Hall et al., 2012). Based on hierarchical clustering of the low-resolution topological models generated, we identified one predominant cluster for the five subunit hypothetical complex (Figure S2E).
Experimental Data
We next applied our method to two well-characterized complexes for which a wealth of structural information is available: the seven-subunit clamp loader complex (γ3δδ′ψχ), which is involved in DNA replication, and the lid subcomplex of the 19S proteasome regulatory particle. For the 7-subunit clamp loader complex (full complex) from Escherichia coli, a 12-subcomplex dissasembly pathway had been identified previously from MS experiments (Park et al., 2010; Politis et al., 2010). An X-ray crystal structure is available for the minimal five-subunit clamp loader complex (γ3δδ′, Protein Data Bank [PDB] ID 1JR3). We performed calculations for both the minimal and full clamp loader complex, allowing us to compare our models with the established contacts determined crystallographically (Jeruzalmi et al., 2001).
Initially, we modeled the five-subunit clamp using one sphere per subunit. When using a seven-subcomplex disassembly pathway extracted from the literature (dashed disassembly pathway in Figure 3A) (Politis et al., 2013), 6% of the ensemble matched the native contacts. If we discard the order of the three identical gamma subunits in our data analysis, these numbers increase to 35.4% (Table S2). The ROC curves of the five-subunit clamp loader also showed that the ability of the restraints to predict the native topology significantly increases when subcomplex information is combined with pairwise interactions (Figure S3).
Figure 3.

Model of the Complexes Determined by Experimental Data
(A) Clamp loader complex: a disassembly pathway for the full clamp loader complex is constructed using MS/MS data. Subcomplexes within the dashed line represent the minimal five-subunit clamp loader for which an X-ray structure is available. A contact frequency map is plotted for all interactions within the seven-subunit subcomplex, allowing us to suggest a model of the previously unknown subcomplex from DNA polymerase III.
(B) Proteasomal lid: interaction network of the proteasomal lid subunits as determined by previously published data including native MS (Sharon et al., 2006) and chemical crosslinking (Lasker et al., 2012). A disassembly pathway constructed using MS of the intact complex and MS/MS data. Six subcomplexes were identified. Contact frequency maps of the spheres making up the proteasomal subcomplex for the ensemble of best scoring models. A list of the most frequent interactions observed in the ensemble of models is shown. The proposed model structure of the eight-subunit proteasomal lid subcomplex assembled by its constituent subunits.
Next, we applied our method to the seven-subunit clamp loader complex. As before, we modeled it with one sphere per subunit. We used the 12-subcomplex disassembly pathway, incorporating data from the literature on the 5-subunit complex and additional MS data defining the ψχ heterodimer (Figure 3A) (Park et al., 2010; Politis et al., 2013). The average contact map contained contacts ranging from likely to unlikely. However, the most likely contacts are consistent with one another (Figure 3A). Clustering analysis based on low-resolution structures (Hall et al., 2012) showed a less prominent cluster in the case of the seven-subunit clamp loader complex than that of the five-subunit complex (Figures S4A and S4B).
Next, we applied our method to an eight-subunit subcomplex of the proteasomal lid Rpn3/5/6/7/8/9/11/Sem1 (Rpn12 has no data and was therefore excluded). We used a seven-subcomplex disassembly pathway reported in the literature (Sharon et al., 2006) and a limited number of pairwise interactions from CX-MS and other experiments published elsewhere (Fu et al., 2001; Lasker et al., 2012; Wollenberg and Swaffield, 2001) (Figure 3B and Table S3). Each subunit was represented by a set of overlapping spheres, one per domain with radii derived from their masses and protein density (Table 1) (Hall et al., 2012). The domains within the individual subunits were defined based on their homology models generated previously (Lasker et al., 2012). Overall, out of the 153 possible contacts, 13 contacts have high frequency (>60%) and 65 pairs low frequency (<10%), showing that the contacts were precisely defined by the input experimental information (Figure 3B). Hierarchical clustering analysis also confirmed a prominent cluster in the ensemble of models (Figure S4C). The high confidence contacts show good agreement with models suggested previously (Lander et al., 2012a; Lasker et al., 2012; Sharon et al., 2006).
Table 1.
Shape and Composition Information for Individual Components of the 19S Proteasomal Lid Assembly
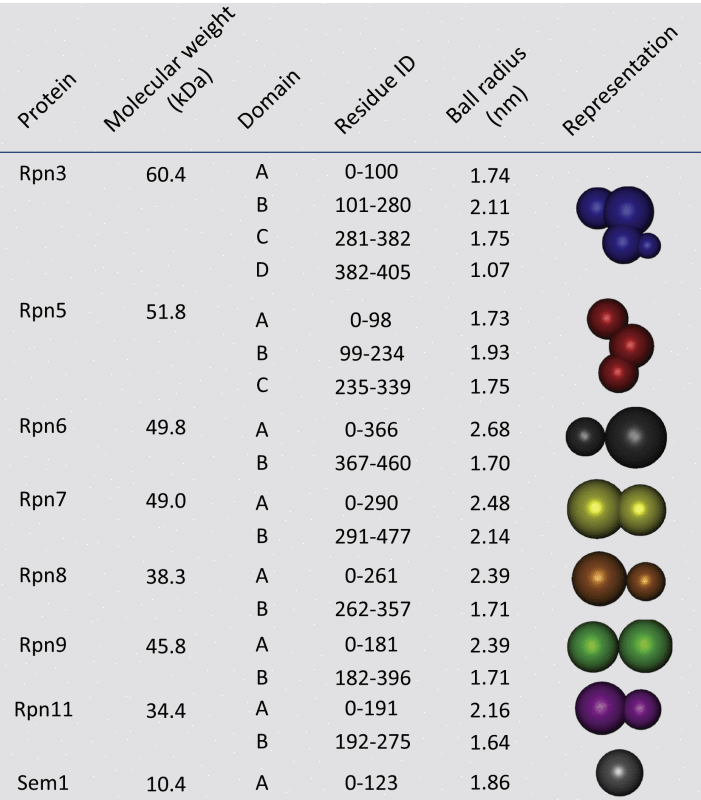 |
The proteins in the lid subassembly are decomposed and represented as spheres scaled by their corresponding mass. Each identified domain is represented as a sphere and the overall subunit as a rigid set of overlapping spheres. The positions of the spheres in a single subunit are computed from their homology models (Lasker et al., 2012).
The Architecture of the Yeast eIF3:eIF5 Complex
Finally, we applied our method to the yeast eIF3 complex. eIF3 is the largest of all initiation factors acting as a docking site for many eIFs that assemble onto the 40S ribosomal subunit (Valásek et al., 2002). eIF3 contains five core (eIF3a/Tif32, eIF3b/Prt1, eIF3c/Nip1, eIF3g/Tif35, eIF3i/Tif34) and one loosely associated, nonfunctional subunit (eIF3j/Her1) in yeast and 13 subunits in human (eIF3a–eIF3m) (Asano et al., 1998; Damoc et al., 2007). The intact eIF3 is refractive to crystallization, but our structural knowledge has been enhanced by two electron microscopy (EM) maps of the human complex, which revealed a five-lobed particle binding to the 40S ribosome on the opposite side of the 60S subunit (Siridechadilok et al., 2005; Spahn et al., 2001). A protein interaction network determined for the yeast eIF3, derived from a biochemical and structural analysis, yielded a model for a compact complex with a protruding eIF3a C-terminal domain (Khoshnevis et al., 2012). The yeast eIF3 is an excellent target for our method due to the lack of high-resolution structural information for intact eIF3, a consensus that human and yeast eIF3 complexes share a common core, and the conservation of its protein-protein interactions.
To probe the structure of the yeast eIF3:eIF5 complex, we purified the intact eIF3 complex from a Saccharomyces cerevisiae strain containing the tandem-affinity-purification (TAP)-tag at the C terminus of eIF3c (Nip1) by applying the TAP-tag strategy (Rigaut et al., 1999). To remove buffer components incompatible with nondenaturing MS, an additional size exclusion chromatography step was performed after TAP-tag purification. Omitting the second purification step leads to a relatively pure eIF3 complex that we used for CX experiments (Figure S5A). The eIF3 proteins as well as eIF5 (Tif5) were identified after in-gel digestion by liquid chromatography-tandem mass spectrometry (LC-MS/MS) (Table S4). For all subunits within the complex, a high number of spectra and peptide sequences were obtained, thus confirming the presence of the six anticipated protein subunits in the preparation (eIF3a–eIF3g and eIF5).
Next, we subjected the purified complex to native MS by employing a mass spectrometer modified for transmission of high-mass complexes (Sobott et al., 2002). The mass spectrum shows the presence of two protein complexes with masses of 364 and 410 kDa, corresponding to the intact eIF3 and eIF3:eIF5 complexes, respectively (Figure 4A and Table S5). The charge state distributions of these two species are characteristic of protein complexes that exist in solution (Hall and Robinson, 2012). Their subunit composition thus confirms substoichiometric binding of eIF5. The lower m/z region of the spectrum reveals different subcomplexes and single protein subunits (Figure 4B). The observed masses of subunits eIF3g (Tif35) and eIF3i (Tif34) as well as eIF5 (Tif5) agree well with their theoretical masses (Table S5). As expected for the tagged eIF3c (Nip1), we observed a mass increase corresponding to the remaining tag after TEV cleavage. Interestingly, eIF3b (Prt1) appeared to have a lower mass compared to its theoretical mass. Inspection of the DNA sequence of eIF3b (Prt1) revealed an alternative translation initiation site at Met40 (Figure S5B). The N-terminal sequence of eIF3b was not observed in LC-MS/MS analysis (Figure S5C), and the mass of the truncated form agrees well with that observed in our spectrum (Figures 4 and S5C; Table S5). The eIF3a (Tif32) subunit was not observed in isolation in any of our mass spectra; we therefore used its theoretical mass for calculation of theoretical masses of the intact eIF3 and eIF3-eIF5 complexes (Table S6). Having defined five of the six subunit masses, we were able to assign several stable subcomplexes formed in solution by comparing their observed masses with the expected ones (Figure 4 and Table S5). The predominant subcomplexes observed are g:i, b:i, b:g:i, and c:eIF5.
Figure 4.

Mass Spectra of the Intact Yeast eIF3:eIF5 Complex and Its Stable Subcomplexes
(A) Mass spectrum showing the intact pentameric eIF3 (364.4 kDa) and eIF3:eIF5 (409.9 kDa) complexes was recorded.
(B) Stable subcomplexes were identified in the region of the low mass-to-charge ratio, showing single protein subunits (circles) and subcomplexes (triangles) of eIF3 and eIF5. The obtained subcomplexes included g:i, b:i, b:g:i, and c:eIF5.
To gain further insight into the protein-protein interactions within the eIF3-eIF5 complex, we performed chemical CX using the BS3 crosslinker. The proteins were crosslinked with a 1:1 mixture of nondeuterated (d0) and deuterated (d4) BS3. Crosslinked peptides were analyzed by LC-MS/MS after tryptic digestion (Experimental Procedures). The raw data were searched against a reduced database containing eIF3 protein subunits and eIF5. The search was performed using MassMatrix search engine, which utilizes a concatenated approach to identify protein-protein crosslinks (Xu et al., 2010). We performed two searches for each sample using both the light (d0) and the heavy (d4) BS3 crosslinkers. A decoy database search was also performed.
Using the MassMatrix search engine, we identified potential and decoy crosslink hits. The decoy hits were excluded from further analysis as they present false positives. In total, we obtained 2,062 potential crosslink hits. Manual validation of these crosslinks by their typical pairs of peaks in mass spectra (generated by the mixture of d0/d4-BS3) resulted in 1,440 crosslinked peptide spectra, corresponding to a false discovery rate of 30.2% for crosslinking analysis. Disregarding intraprotein and intrapeptide crosslinks, we identified 162 interprotein crosslinks (Table S6). To validate our crosslinking approach, we projected obtained crosslinks onto available crystal structures and measured Cα-Cα distances (Figure S6). All observed crosslinks are in reasonable range, confirming our crosslinking strategy. However, there are only few high-resolution structures available for eIF3/eIF5 and only five crosslinks could be projected onto the crystal structures. We therefore further strengthened our analysis by considering crosslinked peptides with at least four amino acid residues, detected in at least three spectra. As recommended for smaller protein complexes by previous studies (Trnka et al., 2014; Walzthoeni et al., 2012), we have manually inspected these MS/MS spectra for their quality. This filter yielded 18 confident protein-protein interaction assignments (Figure 5A).
Figure 5.

Model of the Yeast eIF3:eIF5 Complex
(A) Schematic representation of identified interprotein crosslinks of eIF3:eIF5 complex. The subunits are drawn proportional to their sequence length.
(B) A weighted interaction network, based on the frequency of the observed spectra, of the eIF3:eIF5 complex was built based on information from native MS and CX-MS.
(C) A disassembly pathway constructed based on native MS. Twelve subcomplexes, including eight dimers, were identified.
(D) A model of the eIF3-eIF5 complex is shown, assembled by integrative structure modeling based primarily on the native MS and CX-MS data, supplemented by excluded volume restraints. The dashed lines show an identified submodule of eIF3 complex (eIF3b:g:i).
For structural modeling, we represented the subunits using a total of 14 spheres, each representing a domain (Table 2). To increase the resolution of the final complex, we decomposed the individual eIF3 subunits into domains based on their sequence size and the location of the identified crosslinks (Figures 5A and 5B and Table 2). Our MS experiments yielded 9 unique interprotein contacts (Figure 5B) and 12 identified subcomplexes (Figure 5C). Overall, 21 interdomain contacts occurred often (>99%), while the rest exhibited either low (45 contacts with 5%–40% frequency) or nearly zero frequency (25 contacts) (Figure S7). The topological model derived from high-frequency contacts is shown in Figure 5D and Figure S7, suggesting previously unobserved interactions between eIF3 subunits as well as interactions between the eIF3 and eIF5 subunits, which are primarily formed via the N and C termini of eIF3c and eIF3a.
Table 2.
Shape and Composition Information for Individual Components of the Yeast eIF3:eIF5 Complex
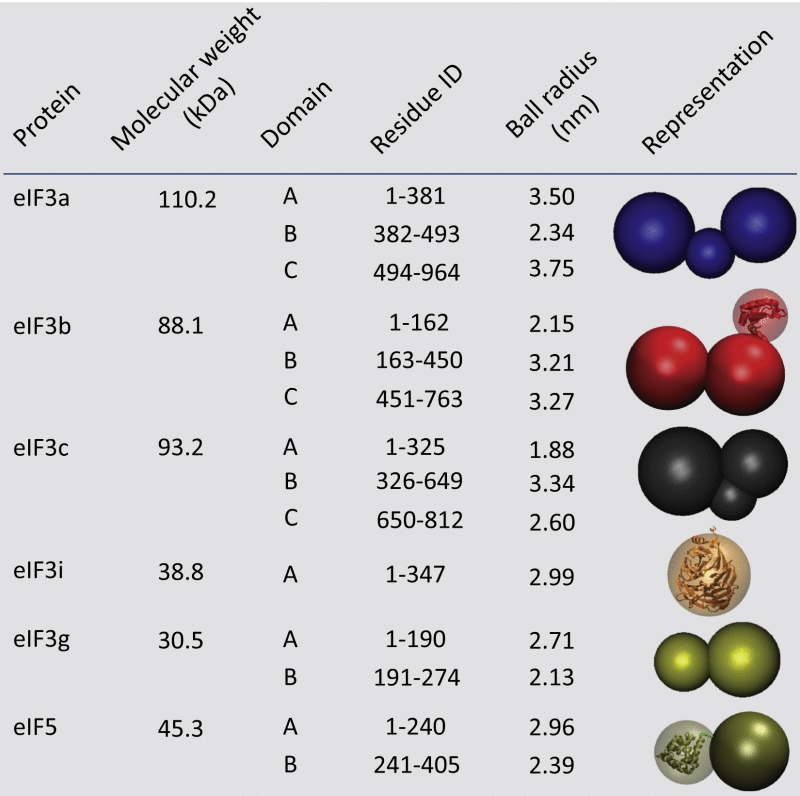 |
The subunits in the eIF3-eIF5 complex are decomposed and represented as spheres scaled using the corresponding mass. Each identified domain is represented as a sphere and the overall subunit as a set of overlapping spheres. Partial atomic coordinates were used to represent eIF3b (PDB ID 3NS6), eIF3i (PDB ID 3ZWL), and eIF5 (PDB ID 3FUL).
Our data suggest a high degree of connectivity consistent with a globular complex and at least one elongated module, namely eIF3b:g:i. (Figures 4 and 5). For this module, connectivity between eIF3b and eIF3g is mediated through eIF3i, as indicated by the pairwise interactions identified for eIF3b:i and eIF3i:g. Interestingly, no interaction between eIF3b and eIF3g was confirmed, consistent with the predicted topology (Figure 5D). Based on our results, it is interesting to speculate that two other submodules, namely eIF3a:c:eIF5 and eIF3a:b:c:eIF5, could be formed prior to the six-subunit complex. The six-subunit eIF3:eIF5 complex is formed mainly through an interaction involving the eIF3b and eIF5 subunits (Figure 5D). This model is consistent with an extensive binding interface between eIF3 and eIF5 that may allow recognition during discrete stages of the initiation factor cycle.
Discussion
We have described an integrative modeling method for computing 3D models of heteromeric protein complexes using structural information derived from MS-based experiments. The main information used was the stoichiometry of intact complex and subcomplexes in the gas phase. In addition, other MS-based structural information, such as in-solution disruption, affinity purification, and chemical CX-MS, was also used to map subunit interactions. The method is implemented using IMP, an open source computer package for building models of macromolecular assemblies by integrating varied data. As a result, a wide variety of other types of data, such as electron microscopy density maps, can be added to the method. However, there are no EM density maps available for the yeast eIF3-eIF5 protein complex studied here. So far, only one study analyzed this complex by electron microscopy, yielding 2D class averages that are not of sufficient resolution for integration into modeling approaches (Khoshnevis et al., 2012).
The method was assessed by showing that it can reproduce native interactions from a benchmark set of five hypothetical assemblies and two real assemblies with known structures. Overall, 28%–56% of the benchmark complex ensembles exhibit >90% contact similarity to the corresponding native structures. However, the models were not necessarily of high precision when only subcomplexes from MS data were used (e.g., only 6% of the five-subunit clamp loader complex matched the native contacts). Nevertheless, as there are 13 unique sets of contacts that satisfy the data (due to the repeated subunit), the precision of the model ensemble is commensurate with that of the data. As a result, achieving higher precision in this case would require more data.
The precision with which the contacts can be determined depends on the quantity, accuracy, and lack of ambiguity of our input data. We first assessed the precision of our method as a function of the input information from the MS experiments and the size of the assemblies. In our benchmark, predictions for which >7% of the models match the native contacts were made for up to five subunits, when only information from the native MS experiments was considered. When information on pairwise interactions was added, predictions were made for up to seven subunits and reached precision values of up to 44% (Tables S1A and S1B). For (sub)complexes involving a higher number of subunits, precise predictions can only be made when information from MS experiments is complemented with other data (e.g., CX-MS, homology modeling, affinity purification MS).
The method was then applied to the yeast eIF3-eIF5 complex whose structure has not yet been solved. Based on the connectivity implied by the native MS and chemical CX data, we suggest a set of interprotein contacts for the core of the eIF3:eIF5 complex. We compared our results to the previously published studies on human (Valásek et al., 2002; Zhou et al., 2008) and yeast eIF3 complexes (Khoshnevis et al., 2012). Although some “core” pairwise interactions between the eIF3 subunits are retained (g:i, a:b, a:c, b:i), we also predicted several previously unobserved protein-protein interactions. In particular, the eIF3a interacts with the eIF3i subunit, while eIF3b and eIF3c interact through their C-terminal domains. In addition, contrary to previously published data for the human complex (Valásek et al., 2002), eIF5 was found to be located centrally, in the groove formed by eIFa, eIFb, and eIFc subunits. The subunit eIF5 integrated into the eIF3:eIF5 complex more closely than anticipated, consistent with the role of eIF3 as a scaffold for binding additional initiation factors during multifactor complex formation.
Significance
We have developed and implemented an integrative modeling method for topological elucidations of heteromeric complexes. These complexes usually elude structural determination by single experimental methods, thus impeding our understanding of their function. We established the method by reconstructing the topologies in a benchmark of known heteromeric complexes using restraints from MS experiments. We further integrated structural information derived from various MS experiments into our modeling strategy to propose a high-confidence model of eIF3-eIF5 complex, a frustrating target for conventional structural biology approaches. Although the protocol presented here has allowed a limited flexibility in the topological predictions, it is restricted to capturing coexisting topologies with large structural differences or highly flexible assemblies. This can be obtained by combining structural data from MS connectivities with flexibility information derived from IM-MS, comparative crosslinking (Schmidt and Robinson, 2014; Schmidt et al., 2013) and HDX-MS methods.
The benchmark scripts that we used to validate the method are available online (see Experimental Procedures). In addition, the main restraint used is available as part of IMP (the IMP::core::MSConnectivityRestraint). As a result, other members of the MS community can readily use our modeling protocol to analyze MS data and integrate it with other data available for complexes of interest. We anticipate that the resulting structural models will contribute to the understanding of the function and evolution of macromolecular complexes as well as providing a starting point for higher resolution structural studies.
Experimental Procedures
MS Connectivity Restraint
To build topological models of complexes consistent with experimental data, we designed and implemented a connectivity restraint as described elsewhere (Alber et al., 2008) for the disassembly pathway data. The connectivity restraint requires as input a disassembly pathway built from the connectivity information derived from nondenaturing MS-based experiments. Pairwise interactions between subunits or domains can also be added in these pathways. Each node in the pathway represents a subcomplex identified in the experiments. Protein subunits or domains are represented as single spheres with radii derived from subunit CCS or, where unavailable, from factoring subunit masses with spherical density (Hall et al., 2012). In disassembly pathways, the parent is the intact complex, while the child subcomplexes must be consistent with their parents in the pathway, both in terms of makeup (when there are multiple, indistinguishable subunits) and their connectivity. The restraint evaluates how well the spheres (e.g., subunits or domains) are connected and together with other available restraints are summed to give a scoring function, which is used to guide the search for “good” topological models by Monte Carlo sampling.
Purification of eIF3
The eIF3 complex was purified employing the TAP-Tag strategy (Rigaut et al., 1999) using a yeast strain (Saccharomyces cerevisiae) expressing the TAP-tag at the C terminus of subunit eIF3c. Briefly, cells were grown under standard conditions until they reached an optical density of 3–5 and were lysed in 10 mM Tris-HCl (pH 8.0), 150 mM NaCl, and 0.1% (v/v) NP40 using a Bead Beater (BioSpecProducts) with glass beads (0.5 mm; Soda Lime). The supernatant was obtained by centrifugation and IgG beads were added. After incubation at 4°C for ∼2 hr, the supernatant was removed and the IgG beads were washed with 10 mM Tris-HCl (pH 8.0), 150 mM NaCl, 0.1% (v/v) NP40, 0.5 mM EDTA, and 1 mM DTT. The beads were resuspended in the same buffer and TEV protease was added. After incubation for 2 hr at room temperature, the supernatant was collected and incubated with calmodulin beads in 10 mM Tris-HCl (pH 8.0), 10 mM β-mercaptoethanol, 150 mM NaCl, 1 mM MgCl, 1 mM imidazole, 2 mM CaCl, and 0.1% (v/v) NP40. After overnight incubation at 4°C, the complex was eluted with 10 mM Tris-HCl (pH 8.0), 10 mM β-mercaptoethanol, 150 mM NaCl, 1 mM MgCl, 1 mM imidazole, 2 mM EGTA, and 0.1% (v/v) NP40. For MS analysis of the intact complexes, an additional gel filtration step was performed to exchange the buffer for 200 mM ammonium acetate (pH 7.0). Gel filtration was performed at a flow rate of 0.5 ml/min on a Superdex 200 column.
Mass Spectrometry of Intact eIF3
Approximately 10 μM of the purified complex was analyzed in 200 mM ammonium acetate (pH 7.0). Spectra were acquired on a Q-ToF II mass spectrometer (Waters) modified for high mass transmission (Sobott et al., 2002) and proteins were electrosprayed using gold coated glass capillaries prepared in-house (Hernández and Robinson, 2007). Typical MS conditions were capillary voltage 1.6–1.7 kV, cone voltage 50–100 V, extractor 5 V, collision voltage 20–80 V, and backing pressure ∼1.0 × 10−2 mbar. Spectra were calibrated externally using Cesium iodide (100 mg/ml). MS spectra were processed and analyzed using Masslynx version 4 (Waters).
Protein Identification by LC-MS/MS and Database Search
Proteins were digested with Trypsin in-gel after separation by SDS-PAGE (Shevchenko et al., 1996). For LC-MS/MS analysis, tryptic peptides were separated by nanoflow reversed-phase liquid chromatography using a DionexUltiMate 3000 RSLC nano System (Thermo Scientific; mobile phase A, 0.1% [v/v] formic acid [FA]; mobile phase B, 80% [v/v] ACN/0.1% [v/v] FA) coupled to an LTQ-Orbitrap XL hybrid mass spectrometer (Thermo Scientific). The peptides were loaded onto a trap column (HPLC column Acclaim PepMap 100, C18, 100 μm inner diameter, particle size 5 μm; Thermo Scientific) and separated with a flow rate of 300 nL/min on an analytical C18 capillary column (50 cm, HPLC column Acclaim PepMap 100, C18, 75 μm inner diameter, particle size 3 μm; Thermo Scientific), with a gradient of 5%–80% (v/v) mobile phase B over 74 min. Peptides were directly eluted into the mass spectrometer.
Typical mass spectrometric conditions were spray voltage of 1.8 kV; capillary temperature of 180°C; and normalized collision energy of 35% at an activation of q = 0.25 and an activation time of 30 ms. The LTQ-Orbitrap XL was operated in the data-dependent mode. Survey full-scan MS spectra were acquired in the orbitrap (m/z 300–2,000) with a resolution of 30,000 at m/z 400 and an automatic gain control (AGC) target at 106. The five most intense ions were selected for CID MS/MS fragmentation in the linear ion trap at an AGC target of 30,000. Detection in the linear ion trap of previously selected ions was dynamically excluded for 30 s. Singly charged ions as well as ions with unrecognized charge state were also excluded. Internal calibration of the orbitrap was performed using the lock mass option (lock mass: m/z 445.120025) (Olsen et al., 2005).
Raw data were searched against the National Center for Biotechnology Information nonredundant database with taxonomy filter Saccharomyces cerevisiae (baker’s yeast) (2011-11-21, 16245521 sequences) using the Mascot search engine v2.3.02 (Matrix Science). The mass accuracy filter was 10 ppm for precursor ions and 0.5 Da for MS/MS fragment ions. Peptides were defined to be tryptic with maximally two missed cleavage sites. Carbamidomethylation of cysteines and oxidation of methionine residues were allowed as variable modifications.
Chemical Crosslinking and Identification of Crosslinked Peptides
Before crosslinking, the purification buffer was exchanged for PBS using Vivaspin concentrators (MWCO 50 kDa; Sartorius). A 1:1 mixture of 2.5 mM deuterated (d4) and nondeuterated (d0) BS3 (Thermo Scientific) were added to the purified eIF3:eIF5 complex. The reaction mixture was incubated for 30 min at 26°C and 450 rpm in a thermomixer. The crosslinked proteins were separated by SDS-PAGE followed by in-gel digestion (Shevchenko et al., 1996) or were precipitated with ethanol and digested in solution with RapiGest SF Surfactant (Waters). The peptides obtained after in-solution digestion were redissolved in 20% (v/v) ACN and 4% (v/v) FA and were separated by cation exchange chromatography using SCX stage tips (Thermo Scientific) according to the manufacturer’s protocol. Peptides were eluted with different concentrations of ammonium acetate (25–500 mM) and dried in a vacuum centrifuge. The mixture of crosslinked and noncrosslinked peptides was analyzed by nanoLC-MS/MS (see above).
Potential crosslinks were identified by using the MassMatrix Database Search Engine (Xu et al., 2010). Raw data were converted to mzXML files using MM_File_Conversion_Tool (www.massmatrix.net). mzXML files were searched against a reduced database containing eIF3/eIF5 protein subunits and a reversed database. Search parameters were as follows: peptides were defined as tryptic with a maximum of two missed cleavage sites. Carbamidomethylation of cysteines and oxidation of methionine residues were allowed as variable modifications. The mass accuracy filter was 10 ppm for precursor ions and 0.8 Da for fragment ions. Minimum pp and pp2 values were 5.0 and minimum pptag was 1.3. The maximum number of crosslinks per peptide was 1. All searches were performed twice, including the deuterated and the nondeuterated crosslinker, respectively. Potential crosslinks were validated manually by checking the presence of the corresponding peak pair in the MS spectra generated by the d4/d0-BS3 mixture, and high-confidence interprotein crosslinks were identified by the quality of their MS/MS spectrum.
Data, Software, and Results
The implementation of our integrative protocol is based on the open source IMP software package (http://integrativemodeling.org). The input data files, IMP modeling scripts, and output models for the seven benchmark cases and the eIF3:eIF5 complex are available at https://github.com/apolitis/eif3 and https://github.com/apolitis/ms_benchmark.
Author Contributions
A.P. and C.S. conceived the study. A.P., C.S., A.S., and C.V.R. designed the research. A.P. and C.S. performed the research and analysed the data. E.T., K.L., and D.R. contributed in the development and implementation of the computer code. A.M.S. and Y.G. performed part of native MS experiments and contributed reagents. A.P., C.S., D.R., A.S., and C.V.R. wrote the paper.
Acknowledgments
The authors would like to thank Idlir Liko, Matteo Degiacomi (University of Oxford), and Zoe Hall (University of Cambridge) as well as Ben Webb (UCSF) for many useful discussions and suggestions. This work was funded by the PROSPECTS (Proteomics Specification in Space and Time Grant HEALTH-F4-2008-201648) within the European Union 7th Framework Program (A.P., C.V.R.), the BBSRC (BB/I02626X/1) (A.P.), the Wellcome Trust (008150/B/09/Z) (C.V.R., C.S., and Y.G.), and the NIH/NIGMS R01 grant GM083960 (A.S.). C.V.R. is a Royal Society Professor.
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/).
Contributor Information
Andrej Sali, Email: sali@salilab.org.
Carol V. Robinson, Email: carol.robinson@chem.ox.ac.uk.
Supplemental Information
The two crosslinked proteins, the sequences of the crosslinked peptides, the crosslinked amino acid residues, and the number of identified MSMS spectra are given for every crosslink. Identified crosslinks are grouped. Intraprotein and intrapeptide crosslinks as well as interprotein crosslinks (red) are listed. Crosslinks with peptides of at least 4 amino acids that showed high quality spectra are highlighted in grey (18 protein-protein interactions). XLs that could not uniquely be assigned are not listed.
References
- Alber F., Dokudovskaya S., Veenhoff L.M., Zhang W., Kipper J., Devos D., Suprapto A., Karni-Schmidt O., Williams R., Chait B.T. Determining the architectures of macromolecular assemblies. Nature. 2007;450:683–694. doi: 10.1038/nature06404. [DOI] [PubMed] [Google Scholar]
- Alber F., Förster F., Korkin D., Topf M., Sali A. Integrating diverse data for structure determination of macromolecular assemblies. Annu. Rev. Biochem. 2008;77:443–477. doi: 10.1146/annurev.biochem.77.060407.135530. [DOI] [PubMed] [Google Scholar]
- Asano K., Phan L., Anderson J., Hinnebusch A.G. Complex formation by all five homologues of mammalian translation initiation factor 3 subunits from yeast Saccharomyces cerevisiae. J. Biol. Chem. 1998;273:18573–18585. doi: 10.1074/jbc.273.29.18573. [DOI] [PubMed] [Google Scholar]
- Baldwin A.J., Lioe H., Hilton G.R., Baker L.A., Rubinstein J.L., Kay L.E., Benesch J.L. The polydispersity of alphaB-crystallin is rationalized by an interconverting polyhedral architecture. Structure (London, England: 1993) 2011;19:1855–1863. doi: 10.1016/j.str.2011.09.015. [DOI] [PubMed] [Google Scholar]
- Benesch J.L., Ruotolo B.T. Mass spectrometry: come of age for structural and dynamical biology. Curr. Opin. Struct. Biol. 2011;21:641–649. doi: 10.1016/j.sbi.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benesch J.L., Aquilina J.A., Ruotolo B.T., Sobott F., Robinson C.V. Tandem mass spectrometry reveals the quaternary organization of macromolecular assemblies. Chem. Biol. 2006;13:597–605. doi: 10.1016/j.chembiol.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Chen J., Feige M.J., Franzmann T.M., Bepperling A., Buchner J. Regions outside the alpha-crystallin domain of the small heat shock protein Hsp26 are required for its dimerization. J. Mol. Biol. 2010;398:122–131. doi: 10.1016/j.jmb.2010.02.022. [DOI] [PubMed] [Google Scholar]
- Chen Z.A., Jawhari A., Fischer L., Buchen C., Tahir S., Kamenski T., Rasmussen M., Lariviere L., Bukowski-Wills J.C., Nilges M. Architecture of the RNA polymerase II-TFIIF complex revealed by cross-linking and mass spectrometry. EMBO J. 2010;29:717–726. doi: 10.1038/emboj.2009.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu F., Baker P.R., Burlingame A.L., Chalkley R.J. Finding chimeras: a bioinformatics strategy for identification of cross-linked peptides. Mol. Cell. Proteomics. 2010;9:25–31. doi: 10.1074/mcp.M800555-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damoc E., Fraser C.S., Zhou M., Videler H., Mayeur G.L., Hershey J.W., Doudna J.A., Robinson C.V., Leary J.A. Structural characterization of the human eukaryotic initiation factor 3 protein complex by mass spectrometry. Mol. Cell. Proteomics. 2007;6:1135–1146. doi: 10.1074/mcp.M600399-MCP200. [DOI] [PubMed] [Google Scholar]
- Fu H., Reis N., Lee Y., Glickman M.H., Vierstra R.D. Subunit interaction maps for the regulatory particle of the 26S proteasome and the COP9 signalosome. EMBO J. 2001;20:7096–7107. doi: 10.1093/emboj/20.24.7096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götze M., Pettelkau J., Schaks S., Bosse K., Ihling C.H., Krauth F., Fritzsche R., Kühn U., Sinz A. StavroX—a software for analyzing crosslinked products in protein interaction studies. J. Am. Soc. Mass Spectrom. 2012;23:76–87. doi: 10.1007/s13361-011-0261-2. [DOI] [PubMed] [Google Scholar]
- Hall Z., Robinson C.V. Do charge state signatures guarantee protein conformations? J. Am. Soc. Mass Spectrom. 2012;23:1161–1168. doi: 10.1007/s13361-012-0393-z. [DOI] [PubMed] [Google Scholar]
- Hall Z., Politis A., Robinson C.V. Structural modeling of heteromeric protein complexes from disassembly pathways and ion mobility-mass spectrometry. Structure (London, England: 1993) 2012;20:1596–1609. doi: 10.1016/j.str.2012.07.001. [DOI] [PubMed] [Google Scholar]
- Heck A.J. Native mass spectrometry: a bridge between interactomics and structural biology. Nat. Methods. 2008;5:927–933. doi: 10.1038/nmeth.1265. [DOI] [PubMed] [Google Scholar]
- Hernández H., Robinson C.V. Determining the stoichiometry and interactions of macromolecular assemblies from mass spectrometry. Nat. Protoc. 2007;2:715–726. doi: 10.1038/nprot.2007.73. [DOI] [PubMed] [Google Scholar]
- Herzog F., Kahraman A., Boehringer D., Mak R., Bracher A., Walzthoeni T., Leitner A., Beck M., Hartl F.U., Ban N. Structural probing of a protein phosphatase 2A network by chemical cross-linking and mass spectrometry. Science. 2012;337:1348–1352. doi: 10.1126/science.1221483. [DOI] [PubMed] [Google Scholar]
- Jeruzalmi D., O’Donnell M., Kuriyan J. Crystal structure of the processivity clamp loader gamma (gamma) complex of E. coli DNA polymerase III. Cell. 2001;106:429–441. doi: 10.1016/s0092-8674(01)00463-9. [DOI] [PubMed] [Google Scholar]
- Jurchen J.C., Williams E.R. Origin of asymmetric charge partitioning in the dissociation of gas-phase protein homodimers. J. Am. Chem. Soc. 2003;125:2817–2826. doi: 10.1021/ja0211508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoshnevis S., Hauer F., Milón P., Stark H., Ficner R. Novel insights into the architecture and protein interaction network of yeast eIF3. RNA. 2012;18:2306–2319. doi: 10.1261/rna.032532.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander G.C., Estrin E., Matyskiela M.E., Bashore C., Nogales E., Martin A. Complete subunit architecture of the proteasome regulatory particle. Nature. 2012;482:186–191. doi: 10.1038/nature10774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander G.C., Saibil H.R., Nogales E. Go hybrid: EM, crystallography, and beyond. Curr. Opin. Struct. Biol. 2012;22:627–635. doi: 10.1016/j.sbi.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasker K., Förster F., Bohn S., Walzthoeni T., Villa E., Unverdorben P., Beck F., Aebersold R., Sali A., Baumeister W. Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proc. Natl. Acad. Sci. USA. 2012;109:1380–1387. doi: 10.1073/pnas.1120559109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitner A., Walzthoeni T., Kahraman A., Herzog F., Rinner O., Beck M., Aebersold R. Probing native protein structures by chemical cross-linking, mass spectrometry, and bioinformatics. Mol. Cell. Proteomics. 2010;9:1634–1649. doi: 10.1074/mcp.R000001-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiolica A., Cittaro D., Borsotti D., Sennels L., Ciferri C., Tarricone C., Musacchio A., Rappsilber J. Structural analysis of multiprotein complexes by cross-linking, mass spectrometry, and database searching. Mol. Cell. Proteomics. 2007;6:2200–2211. doi: 10.1074/mcp.M700274-MCP200. [DOI] [PubMed] [Google Scholar]
- Olsen J.V., de Godoy L.M., Li G., Macek B., Mortensen P., Pesch R., Makarov A., Lange O., Horning S., Mann M. Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol. Cell. Proteomics. 2005;4:2010–2021. doi: 10.1074/mcp.T500030-MCP200. [DOI] [PubMed] [Google Scholar]
- Park A.Y., Jergic S., Politis A., Ruotolo B.T., Hirshberg D., Jessop L.L., Beck J.L., Barsky D., O’Donnell M., Dixon N.E. A single subunit directs the assembly of the Escherichia coli DNA sliding clamp loader. Structure (London, England: 1993) 2010;18:285–292. doi: 10.1016/j.str.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politis A., Park A.Y., Hyung S.J., Barsky D., Ruotolo B.T., Robinson C.V. Integrating ion mobility mass spectrometry with molecular modelling to determine the architecture of multiprotein complexes. PLoS ONE. 2010;5:e12080. doi: 10.1371/journal.pone.0012080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politis A., Park A.Y., Hall Z., Ruotolo B.T., Robinson C.V. Integrative modelling coupled with ion mobility mass spectrometry reveals structural features of the clamp loader in complex with single-stranded DNA binding protein. J. Mol. Biol. 2013;425:4790–4801. doi: 10.1016/j.jmb.2013.04.006. [DOI] [PubMed] [Google Scholar]
- Politis A., Stengel F., Hall Z., Hernández H., Leitner A., Walzthoeni T., Robinson C.V., Aebersold R. A mass spectrometry-based hybrid method for structural modeling of protein complexes. Nat. Methods. 2014;11:403–406. doi: 10.1038/nmeth.2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pukala T.L., Ruotolo B.T., Zhou M., Politis A., Stefanescu R., Leary J.A., Robinson C.V. Subunit architecture of multiprotein assemblies determined using restraints from gas-phase measurements. Structure (London, England: 1993) 2009;17:1235–1243. doi: 10.1016/j.str.2009.07.013. [DOI] [PubMed] [Google Scholar]
- Rappsilber J., Siniossoglou S., Hurt E.C., Mann M. A generic strategy to analyze the spatial organization of multi-protein complexes by cross-linking and mass spectrometry. Anal. Chem. 2000;72:267–275. doi: 10.1021/ac991081o. [DOI] [PubMed] [Google Scholar]
- Rigaut G., Shevchenko A., Rutz B., Wilm M., Mann M., Séraphin B. A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 1999;17:1030–1032. doi: 10.1038/13732. [DOI] [PubMed] [Google Scholar]
- Robinson C.V., Sali A., Baumeister W. The molecular sociology of the cell. Nature. 2007;450:973–982. doi: 10.1038/nature06523. [DOI] [PubMed] [Google Scholar]
- Russel D., Lasker K., Webb B., Velázquez-Muriel J., Tjioe E., Schneidman-Duhovny D., Peterson B., Sali A. Putting the pieces together: integrative modeling platform software for structure determination of macromolecular assemblies. PLoS Biol. 2012;10:e1001244. doi: 10.1371/journal.pbio.1001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A., Blundell T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Sali A., Glaeser R., Earnest T., Baumeister W. From words to literature in structural proteomics. Nature. 2003;422:216–225. doi: 10.1038/nature01513. [DOI] [PubMed] [Google Scholar]
- Schmidt C., Robinson C.V. A comparative cross-linking strategy to probe conformational changes in protein complexes. Nat. Protoc. 2014;9:2224–2236. doi: 10.1038/nprot.2014.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt C., Zhou M., Marriott H., Morgner N., Politis A., Robinson C.V. Comparative cross-linking and mass spectrometry of an intact F-type ATPase suggest a role for phosphorylation. Nat. Commun. 2013;4:1985. doi: 10.1038/ncomms2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon M., Taverner T., Ambroggio X.I., Deshaies R.J., Robinson C.V. Structural organization of the 19S proteasome lid: insights from MS of intact complexes. PLoS Biol. 2006;4:e267. doi: 10.1371/journal.pbio.0040267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A., Wilm M., Vorm O., Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- Siridechadilok B., Fraser C.S., Hall R.J., Doudna J.A., Nogales E. Structural roles for human translation factor eIF3 in initiation of protein synthesis. Science. 2005;310:1513–1515. doi: 10.1126/science.1118977. [DOI] [PubMed] [Google Scholar]
- Sobott F., Hernández H., McCammon M.G., Tito M.A., Robinson C.V. A tandem mass spectrometer for improved transmission and analysis of large macromolecular assemblies. Anal. Chem. 2002;74:1402–1407. doi: 10.1021/ac0110552. [DOI] [PubMed] [Google Scholar]
- Spahn C.M., Kieft J.S., Grassucci R.A., Penczek P.A., Zhou K., Doudna J.A., Frank J. Hepatitis C virus IRES RNA-induced changes in the conformation of the 40s ribosomal subunit. Science. 2001;291:1959–1962. doi: 10.1126/science.1058409. [DOI] [PubMed] [Google Scholar]
- Stengel F., Baldwin A.J., Painter A.J., Jaya N., Basha E., Kay L.E., Vierling E., Robinson C.V., Benesch J.L. Quaternary dynamics and plasticity underlie small heat shock protein chaperone function. Proc. Natl. Acad. Sci. USA. 2010;107:2007–2012. doi: 10.1073/pnas.0910126107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverner T., Hernández H., Sharon M., Ruotolo B.T., Matak-Vinković D., Devos D., Russell R.B., Robinson C.V. Subunit architecture of intact protein complexes from mass spectrometry and homology modeling. Acc. Chem. Res. 2008;41:617–627. doi: 10.1021/ar700218q. [DOI] [PubMed] [Google Scholar]
- Trnka M.J., Baker P.R., Robinson P.J., Burlingame A.L., Chalkley R.J. Matching cross-linked peptide spectra: only as good as the worse identification. Mol. Cell. Proteomics. 2014;13:420–434. doi: 10.1074/mcp.M113.034009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetrecht C., Barbu I.M., Shoemaker G.K., van Duijn E., Heck A.J. Interrogating viral capsid assembly with ion mobility-mass spectrometry. Nat. Chem. 2011;3:126–132. doi: 10.1038/nchem.947. [DOI] [PubMed] [Google Scholar]
- Valásek L., Nielsen K.H., Hinnebusch A.G. Direct eIF2-eIF3 contact in the multifactor complex is important for translation initiation in vivo. EMBO J. 2002;21:5886–5898. doi: 10.1093/emboj/cdf563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Duijn E., Barbu I.M., Barendregt A., Jore M.M., Wiedenheft B., Lundgren M., Westra E.R., Brouns S.J., Doudna J.A., van der Oost J. Native tandem and ion mobility mass spectrometry highlight structural and modular similarities in clustered-regularly-interspaced shot-palindromic-repeats (CRISPR)-associated protein complexes from Escherichia coli and Pseudomonas aeruginosa. Mol. Cell. Proteomics. 2012;11:1430–1441. doi: 10.1074/mcp.M112.020263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walzthoeni T., Claassen M., Leitner A., Herzog F., Bohn S., Förster F., Beck M., Aebersold R. False discovery rate estimation for cross-linked peptides identified by mass spectrometry. Nat. Methods. 2012;9:901–903. doi: 10.1038/nmeth.2103. [DOI] [PubMed] [Google Scholar]
- Walzthoeni T., Leitner A., Stengel F., Aebersold R. Mass spectrometry supported determination of protein complex structure. Curr. Opin. Struct. Biol. 2013;23:252–260. doi: 10.1016/j.sbi.2013.02.008. [DOI] [PubMed] [Google Scholar]
- Ward A.B., Sali A., Wilson I.A. Biochemistry. Integrative structural biology. Science. 2013;339:913–915. doi: 10.1126/science.1228565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollenberg K., Swaffield J.C. Evolution of proteasomal ATPases. Mol. Biol. Evol. 2001;18:962–974. doi: 10.1093/oxfordjournals.molbev.a003897. [DOI] [PubMed] [Google Scholar]
- Xu H., Hsu P.H., Zhang L., Tsai M.D., Freitas M.A. Database search algorithm for identification of intact cross-links in proteins and peptides using tandem mass spectrometry. J. Proteome Res. 2010;9:3384–3393. doi: 10.1021/pr100369y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates J.R., 3rd, Gilchrist A., Howell K.E., Bergeron J.J. Proteomics of organelles and large cellular structures. Nat. Rev. Mol. Cell Biol. 2005;6:702–714. doi: 10.1038/nrm1711. [DOI] [PubMed] [Google Scholar]
- Young M.M., Tang N., Hempel J.C., Oshiro C.M., Taylor E.W., Kuntz I.D., Gibson B.W., Dollinger G. High throughput protein fold identification by using experimental constraints derived from intramolecular cross-links and mass spectrometry. Proc. Natl. Acad. Sci. USA. 2000;97:5802–5806. doi: 10.1073/pnas.090099097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M., Sandercock A.M., Fraser C.S., Ridlova G., Stephens E., Schenauer M.R., Yokoi-Fong T., Barsky D., Leary J.A., Hershey J.W. Mass spectrometry reveals modularity and a complete subunit interaction map of the eukaryotic translation factor eIF3. Proc. Natl. Acad. Sci. USA. 2008;105:18139–18144. doi: 10.1073/pnas.0801313105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M., Politis A., Davies R., Liko I., Wu K.-J., Stewart A.G., Stock D., Robinson C.V. Ion mobility-mass spectrometry of a rotary ATPase reveals ATP-induced reduction in conformational flexibility. Nat. Chem. 2014;6:208–215. doi: 10.1038/nchem.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The two crosslinked proteins, the sequences of the crosslinked peptides, the crosslinked amino acid residues, and the number of identified MSMS spectra are given for every crosslink. Identified crosslinks are grouped. Intraprotein and intrapeptide crosslinks as well as interprotein crosslinks (red) are listed. Crosslinks with peptides of at least 4 amino acids that showed high quality spectra are highlighted in grey (18 protein-protein interactions). XLs that could not uniquely be assigned are not listed.