

Supplemental digital content is available in the text.
KEYWORDS/ABBREVIATIONS: Acute peritonitis, OTII, proliferation, regulatory T cell, T-cell receptor, AP—acute peritonitis, CFSE—carboxyfluorescein succinimidyl ester, CLP—cecal ligation and puncture, CASP—colon ascendens stent peritonitis, CTLA-4—Cytotoxic T-Lymphocyte Antigen 4, Foxp3—Forkhead box P3, gMFI—geometric mean of fluorescence intensity, MHCII—major histocompatibility complex class II, OVA—ovalbumin, Treg—regulatory T cell, TCR—T-cell receptor, Teff—T effector cell, WT—wild type
ABSTRACT
During sepsis, CD4+ T cells express activation markers within the first 24 h. In the present study, the mechanisms of T-cell activation and its consequences were addressed in an acute peritonitis model in mice. The response of CD4+ T cells to sepsis induction was compared between OTII mice, characterized by ovalbumin-specific T-cell receptor–transgenic T cells, and C57BL/6 controls (wild type [WT] mice). Because ovalbumin was absent during peritonitis, the OTII CD4+ T cells could not be activated by canonical antigen recognition. In both OTII and WT control mice, CD4+ T effector cells and CD4+ Foxp3+ regulatory T cells (Tregs) expressed the activation marker CD69 early after sepsis onset. However, full activation with upregulation of CD25 and proliferation took place only in the presence of the antigen. Besides this, the fraction of Tregs was lower in OTII than that in WT mice. Sepsis mortality was increased in OTII mice. Our data show that, in sepsis, partial activation of CD4+ T cells is induced by a T-cell receptor–independent pathway, whereas full stimulation and proliferation require a specific antigen. Antigen-dependent T-cell effector functions as well as Treg activity may contribute to sepsis survival.
INTRODUCTION
Sepsis remains a leading cause of death in intensive care units worldwide (1), despite extensive research and improvements in intensive care therapy. Although lethality has decreased across the last years to currently 30% to 55%, depending on severity (2, 3), sepsis incidence is increasing because of the aging of the population (3). The Surviving Sepsis Campaign estimates that 18 million cases occur annually, with costs of approximately US$ 22,000 per patient (1, 3).
It was first assumed that an adaptive immune response would need at least 2 days to establish itself (4)—too late to influence the hyperacute disease course in sepsis. Interest in the adaptive immune response was raised when it became apparent that, first, T cells show signs of activation within a few hours of sepsis onset in both humans and mice (2, 5–7) and, second, mainly lymphocytes undergo apoptosis within the first 24 h of the septic insult.
The mechanisms of T-cell activation in sepsis are insufficiently understood. There are at least two possibilities. First, T cells can be activated via their master switch, the T-cell receptor (TCR). This requires canonical recognition of an antigenic peptide in the context of the major histocompatibility complex (MHC) on antigen-presenting cells. Specific antigen recognition results in activation of approximately 1 out of 100,000 T cells, with subsequent differentiation and proliferation, leading to a highly specific adaptive immune response. Second, cytokines such as interleukin 12 (IL-12), IL-18, or type I interferons (IFNs), typically released by activated innate immune cells, have antigen-independent stimulating effects on T cells, which has been termed the “innate pathway” of T-cell activation (8). Both processes could occur during sepsis because the systemic dissemination of the endogenous microbiota floods the immune system with a wealth of both antigens and innate stimuli.
Regarding T-cell function in sepsis, different observations have been reported. As indicated by the expression of Ki-67 in humans, T cells proliferate in response to activation early during sepsis (5). Later in the disease course, however, the proliferation of T cells was impaired after ex vivo stimulation with anti-CD3/anti-CD28, which correlated with mortality in postoperative intra-abdominal infection (9). The impaired proliferation was accompanied by reduced production of IL-2, IFN-γ, and tumor necrosis factor-α (TNF-α) by T cells (9, 10). The early response of T cells was shown to directly link the adaptive and innate immune systems (11). In mice, effector memory CD4+ T cells produce significant amounts of IFN-γ during the first 6 h after cecal ligation and puncture (CLP) (12), by which they directly regulate the function of neutrophils (4).
Early during sepsis, CD4+ T cells also upregulate proapoptotic Bim and downregulate antiapoptotic Bcl-2 and Bcl-xL, and a large fraction of T cells goes into apoptosis (13–15). This mainly affects naive CD62Lhi CD44lo T cells (12), depleting potentially protective adaptive immune cells. In addition, regulatory mechanisms of T cells—such as the expression of the negative costimulatory receptor Cytotoxic T-Lymphocyte Antigen 4 (CTLA-4)—are active in the course of sepsis (6, 7). The expression of CTLA-4 correlated with the amount of apoptotic cells (5). Recent studies indicate that, during sepsis, some CD4+ T cells enter a state of exhaustion, characterized by the increased expression of PD-1 (Programmed Cell Death 1), CTLA-4, and GRAIL (Gene Related to Anergy In Lymphocytes), which is accompanied by functional impairment, such as decreased production of effector cytokines, loss of proliferative capacity, as well as decreased cytotoxicity, which in the end results in apoptosis (2).
All these factors may lead to profound suppression of the adaptive immune response during sepsis. In fact, Mohr et al. (16) reported that the generation of antigen-specific antibodies was strongly impaired when mice were primed several days after CLP. Interestingly, the adoptive transfer of naive CD4+ T and B cells did not restore the immune response, implying that not only T-cell intrinsic defects but also active suppression may play a role.
In view of this complex scenario, it is not surprising that discrepant results have been reported concerning the influence of T cells on sepsis survival. Prevention of T-cell apoptosis improved survival and bacterial clearance (17). A protective role of CD4+ T cells in the first 30 h of septic insult was also shown by Martignoni et al. (4). They induced sepsis by CLP in CD4-deficient mice and found increased mortality accompanied by increased bacteremia, as well as functional impairment of neutrophils (4). However, other groups did not find changes in survival rate, bacterial clearance, or inflammation after CD4 T-cell depletion (18, 19); in some cases, even a detrimental role of CD4+ T cells was observed when studying CD4- and TCR-deficient mice after CLP (10, 20). As indicated by a study by Kasten et al. (21), CD4+ T cells are important for modulating the function of neutrophils during early sepsis. Moderately strong antigenic TCR engagement fostered bactericidal functions in neutrophils and improved animal survival, whereas a lack of and, in contrast, excessive activation were both detrimental, the latter being associated with hyperinflammation. The authors conclude that the role of T cells is contextual, depending on both the degree of T-cell activation and the severity of sepsis (12).
Unraveling the complexity of the host response to sepsis—involving the interplay of multiple cell types, a plethora of small molecule mediators, and numerous signaling cascades—requires the use of appropriate animal models. Within the past decade, different surgical approaches mimicking suture failure with subsequent intra-abdominal bacterial invasion have been explored because they best reflect the entire complexity of human sepsis (22–24). The most commonly used model is CLP characterized by intraperitoneal abscess formation (25, 26). In contrast, colon ascendens stent peritonitis (CASP) is a model of diffuse peritonitis. Because the latter may lead to obstruction of the intestine (27 and unpublished observation), we opted for a modification of CASP: the model of acute peritonitis (AP). As originally described by Barrera and colleagues (27), in AP, the stent is implanted into the wall of the cecum rather than the ascending colon as in CASP. A hyperinflammatory state is followed by a hypoinflammatory state similar to the situation in human sepsis (27 and unpublished data).
To elucidate the mechanism of early CD4+ T-cell activation during sepsis, we further used transgenic OTII mice expressing a transgenic TCR, which is specific for an ovalbumin peptide (OVA323-339) in the context of MHC class II molecules (I-A b). In clonotypic CD4+ T cells, the TCR will only be engaged if OVA is applied in vivo. Without OVA, OTII CD4+ T cells can only be activated independent of the TCR. In addition, the influence of antigen-specific activation via the TCR on survival, bacterial load, and inflammatory response was determined.
MATERIALS AND METHODS
Animal experiments and ethics statement
Breeding pairs of OTII mice (B6.Cg-Tg(TcraTcrb)425Cbn/J) were obtained from Jackson Laboratories. Homebred female specific pathogen–free OTII and C57BL/6N wild-type (WT) control mice between the ages of 10 and 12 weeks were used. The animals were cohoused in a conventional temperature-controlled animal facility with a 12-hour light/12-hour dark cycle. All experiments involving animals were performed according to the German animal safety regulations and approved by the animal ethics committee of the local animal protection authority (Landesamt für Landwirtschaft, Lebensmittelsicherheit und Fischerei Mecklenburg-Vorpommern). During the experimental procedures, mice were provided with food and water ad libitum. All possible efforts were made to minimize suffering.
Peritoneal sepsis model—AP
Mice were anaesthetized with ketamine/xylazine (100 mg/10 mg/kg body weight). Acute peritonitis surgery was performed as described by the group of Barrera et al. (27). In brief, an 18-gauge stent was inserted into the cecum opposite the ileocecal valve and fixed. A small amount of bowel contents was extruded through the stent to ensure proper intraluminal position. For fluid replacement, the mice received 0.5 mL 0.9% NaCl intraperitoneally.
Survival was monitored every 3 h for 72 h by an observer who was blinded to the applied treatments. In addition, sepsis severity and body temperature were monitored every 6 h for 72 h. Sepsis severity was determined via general appearance, breathing frequency, and spontaneous and provoked behavior. Scores for healthy (0) to severe alterations (3) for each parameter were summed up (28). Mice reaching a severity score that indicated a point of no return were euthanized by cervical dislocation under deep anesthesia.
Antibodies for flow cytometry
Peritoneal lavage and splenocytes were obtained as described previously (6). The following fluorochrome-labeled antibodies were purchased from BD Biosciences (Heidelberg, Germany): αCD4 (L3T4 and RM4-5), αB220 (RA3-6B2), αCD11b (M1/70), αCD11c (HL3), αCD69 (H1.2F3), αCTLA-4 (UC10-4F10-11), and αLy6G (1A8). A biotinylated cocktail of antibodies against Armenian and Syrian hamster IgG was purchased from Biolegend (San Diego, Calif). Antibodies directed against CD19 (1D3), CD25 (PC61.5), and MHCII (M5/114.15.2) were acquired from eBioscience (Hatfield, UK). For intranuclear Foxp3 staining, we used αFoxp3 antibodies (3G3) and the Foxp3-Staining Buffer Set from Miltenyi Biotech (Bergisch Gladbach, Germany). Streptavidin-PE and streptavidin-PE-Cy7 were obtained from eBioscience.
Adoptive cell transfer
Splenocytes from TCR-transgenic OTII mice were obtained as described previously, and erythrocytes were lysed with ammonium chloride buffer containing of 0.01 M KHCO3, 0.155 M NH4Cl, and 0.1 mM EDTA. Cells (5 × 107) were labeled with carboxyfluorescein succinimidyl ester (CFSE) using the CellTrace CFSE Cell Proliferation Kit by Invitrogen (Darmstadt, Germany) according to the manufacturer’s instructions. Afterward, CD4+ T cells were negatively isolated using the EasySep Mouse CD4+ T Cell Enrichment Kit by StemCell Technologies (Grenoble, France).
CD4+ CFSE-labeled OTII cells (1 × 107) were adoptively transferred intravenously into the retro-orbital venous plexus of C57BL/6 WT mice. Twenty-four hours later, AP was induced. Either during AP induction or 72 h later, one group of mice received 200 μg OVA in 100 μL PBS, whereas controls were treated with NaCl only.
Proliferation analysis
Proliferation of the CFSE-labeled TCR-transgenic OTII CD4+ T cells was determined in the spleen by flow cytometry 3 days after OVA application. There was no interference from autofluorescence (not shown). Splenocytes were harvested as described previously and stained with antibodies against CD4, CD19, and CD11b (see above). Proliferation was analyzed using the FlowJo Proliferation tool (TreeStar Inc., Ashland, Oreg).
Cytokine expression
To determine serum concentrations of IL-2, IL-4, IL-6, IL-10, IL-12p70, IL-17a, IFN-γ, MCP-1, and TNF-α, the Mouse Th1/Th2/Th17 Cytokine Kit or the Mouse Inflammation Kit (BD, San Diego, Calif) were used according to the manufacturer’s instructions.
Determination of bacterial load
Twenty-four hours after sepsis induction by AP, mice were anesthetized and killed by cervical dislocation. Blood was collected by retro-orbital puncture and anticoagulated with heparin. For peritoneal lavage, 10 mL of sterile PBS (ice-cold) was injected and the cell suspension was recovered.
Lung and spleen were explanted. The homogenized organ suspensions, blood, and lavage were incubated on Columbia blood agar (Becton Dickinson, Heidelberg, Germany) for 22 h at 37°C. Bacterial colonies were enumerated and related to organ weight and volume.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 5 for Windows (GraphPad Software, San Diego, Calif). Survival was analyzed using Kaplan-Meier survival curves and the log-rank test. Comparison of two groups (e.g., OTII versus WT C57BL/6) was performed using the Mann-Whitney U test. For multiple comparisons of more than two groups (e.g., untreated and septic OTII versus WT C57BL/6), one-way analysis of variance with Bonferroni multiple comparison test was performed. Values of P ≤ 0.05 were considered to be statistically significant.
RESULTS
Differential expression of activation markers on CD4+ T cells early during sepsis because of antigen recognition
To answer the question of how CD4+ T cells become activated during early sepsis in vivo, we used OTII mice. These mice express a transgenic TCR specific for an OVA peptide (OVA323–339) in the context of MHCII (I-A b). As a consequence, in the clonotypic CD4+ T cells, the TCR will only be engaged during sepsis if OVA is applied in vivo. Without OVA, clonotypic T cells, which are the majority, can only be activated in a TCR-independent manner.
To determine the activation status of CD4+ T cells during sepsis, we measured the expression of activation markers CD69, CD25, and CTLA-4 on CD4+ Foxp3− effector T cells (Teffs) and CD4+ Foxp3+ regulatory T cells (Tregs) in both OTII and WT mice 24 h after sepsis induction; untreated animals served as controls.
As shown in Figure 1, Foxp3+ Tregs had higher basal levels of all activation markers than Teffs. The expression of the early activation marker CD69 was significantly increased on Teffs and Tregs in septic WT and OTII animals (Fig. 1, A and B), confirming previous results of our group (6, 7) and Kasten et al. (21). This was true for the level of expression per cell indicated by the geometric mean of fluorescence intensity (gMFI) as well as for the percentage of CD69-positive cells (Figure, Supplemental Digital Content 1, at http://links.lww.com/SHK/A240). Regulatory T cells additionally increased the expression of CTLA-4 (Fig. 1, C and D). Full activation of T cells further requires an increase in CD25 expression on Tregs and Teffs. This, however, took place only in WT mice, whereas, in OTII mice, CD25 expression remained unchanged on Tregs during sepsis and it even decreased on Teffs (Fig. 1, E and F), indicating incomplete activation of T cells in the absence of the antigen. On the other hand, the fact that WT CD4+ Teffs and Tregs, which comprise a broad TCR repertoire, induced CD25 and were hence fully activated, implies that TCR-mediated canonical antigen recognition occurs during sepsis in WT animals.
Fig. 1.
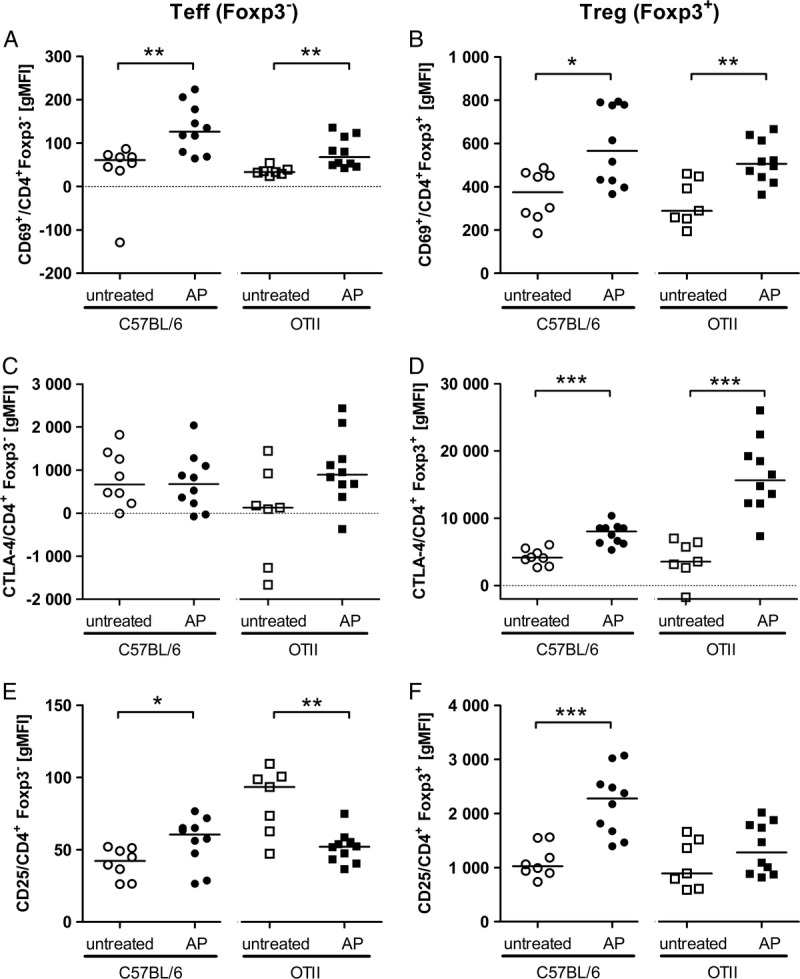
Activation markers on CD4+ T cells early during sepsis. OTII mice and C57BL/6 WT control mice were either left untreated (open symbols) or sepsis was induced by AP surgery (filled symbols). Twenty-four hours after AP, splenocytes were isolated and CD4+ Foxp3− Teffs (A, C, E) as well as CD4+ Foxp3+ Tregs (B, D, F) were analyzed for expression of activation markers CD69 (A, B), CTLA-4 (C, D), and CD25 (E, F) by flow cytometry. Values are given as gMFI (geometric mean of fluorescence intensity) minus gMFI for an isotype control. Medians are indicated. Combined results of two independent experiments with similar results are shown. Sample sizes were seven to 12 animals per group. Septic mice were compared with untreated mice of the same strain using the Mann-Whitney U test. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
During sepsis, CD4+ T cells require a specific antigen for induction of proliferation
To analyze T-cell proliferation in vivo, we adoptively transferred CFSE-labeled OTII CD4+ T cells into C57BL/6 WT mice. Proliferation was measured by flow cytometric determination of the CFSE content per cell. There was no interference from autofluorescence; without transfer of labeled cells, fluorescence was negative (not shown). In nonseptic mice that received transferred cells, the cognate antigen OVA was required to induce proliferation in CD4+ T cells (Figure, Supplemental Digital Content 2, at http://links.lww.com/SHK/A241). To test whether the flooding with immune stimulatory bacterial compounds during sepsis would override antigen dependence of T-cell proliferation, sepsis was induced 24 h after T-cell transfer. At the time point of sepsis induction by AP, mice received OVA or the same volume of saline solution, and proliferation of the transferred T cells was determined by flow cytometry 3 days later.
Although the transferred CD4+ T cells could be partially activated during sepsis in a TCR-independent manner (Fig. 1), their proliferation remained TCR dependent. This was obvious by the strong proliferative response when OVA was applied during sepsis induction (AP + OVA; Fig. 2). Early during sepsis, CD4+ T cells responded strongly to antigen recognition, as reflected by their expression of activation markers and subsequent proliferation.
Fig. 2.

In sepsis, CD4+ T cells fail to proliferate in the absence of the specific antigen. Transgenic OTII CD4+ cells were CFSE labeled and adoptively transferred into C57BL/6 WT mice. Twenty-four hours later, mice were subjected to AP and received 200 μg of OVA or the same volume of vehicle (NaCl) intraperitoneally. Proliferation of transferred CFSE+ CD4+ T cells was determined by flow cytometry 3 days after AP induction. One representative result of each group is shown. n = 7 to 8.
In the absence of the OVA antigen, merely 1% of transferred cells proliferated (Fig. 2). These were presumably derived from T cells expressing TCRs of other specificities, which comprise up to 20% of CD4+ T cells in OTII mice, which are Rag sufficient.
Early during sepsis, antigen-specific proliferation of CD4+ T cells was amplified, although it was later attenuated
In this study, the response of CD4+ T cells to their specific antigen was intact during sepsis, whereas other groups reported impairment of CD4+ T-cell activation (29, 30). To clarify this, we repeated the experiment but delayed application of the antigen OVA by 72 h after sepsis induction. Proliferation was determined 3 days after OVA application. When septic mice received OVA at the time point of AP induction, the antigen-induced proliferation of transferred transgenic OTII T cells was even increased compared with nonseptic animals (Fig. 3, A, C, and D). However, CD4+ T cells that were stimulated with OVA 3 days after the septic insult showed impaired proliferation (Fig. 3, B – D). Hence, in agreement with other reports, sepsis-induced alteration of the T-cell response takes place also in the AP model, albeit with delayed onset.
Fig. 3.
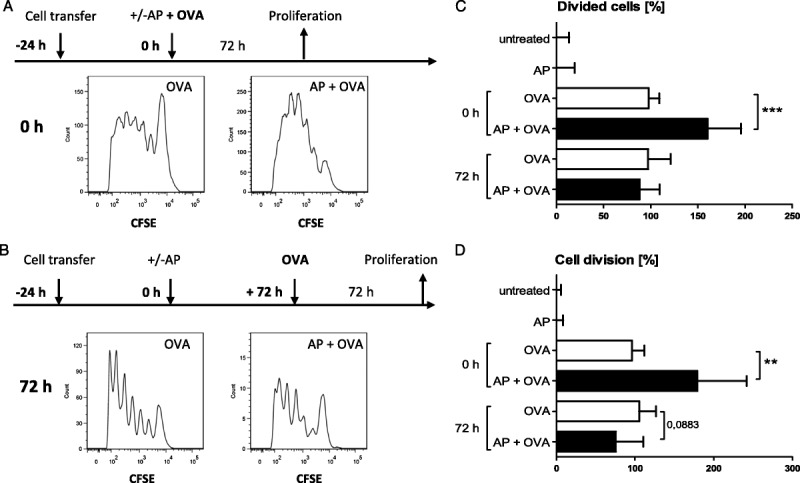
Early during sepsis, antigen-specific proliferation of CD4+ T cells was amplified, although it was later attenuated. Transgenic OTII CD4+ cells were CFSE labeled and adoptively transferred into C57BL/6 WT mice 24 h before mice were subjected to AP (−24 h). Mice either received 200 μg of OVA intraperitoneally at the time point of AP (0 h, A) or 72 h later (B). Proliferation of transferred CFSE+ CD4+ T cells was determined by flow cytometry 3 days after OVA application. Two control groups received adoptive cell transfer and were subjected to AP or left untreated but did not receive OVA. T-cell division was negligible in these controls. One representative result of each animal group receiving OVA is shown. C, Percentages of transferred cells that underwent cell division. Values were normalized (nonseptic animals treated with OVA, 100%). D, Average numbers of cell divisions in CFSE-labeled cells. Values were normalized (nonseptic mice treated with OVA, 100%). Medians and interquartile ranges are indicated. Sample sizes were seven to 15 animals per group. AP + OVA were compared with OVA alone using Mann-Whitney U test. ***P ≤ 0.001; **P ≤ 0.01.
Sepsis survival was reduced in OTII mice
As OTII CD4+ T cells appeared to be dysfunctional during sepsis in the absence of OVA, we monitored sepsis survival for 3 days. OTII mice showed significantly increased mortality (83%) compared with WT mice (50%; Fig. 4A; P = 0.0154; WT, n = 22; OTII, n = 30).
Fig. 4.
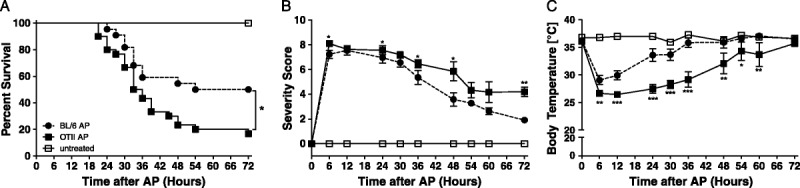
Higher mortality and prolonged recovery phase in septic OTII mice compared with WT. OTII mice and C57BL/6 control mice were subjected to AP or left untreated. Survival was monitored for 3 days (A). Sepsis severity was determined as described in Materials and Methods (B). In addition, body temperature was measured at close intervals (C). Sample sizes were 22 WT, 30 OTII per group. B, C, Data are expressed as the mean ± SEM. Septic OTII mice were compared with septic WT animals using the log-rank test. *P ≤ 0.05.
Initially, sepsis severity, as indicated by the sum severity score, was moderately but significantly increased in OTII mice compared with WT animals (Fig. 4B). Moreover, surviving OTII mice went through a prolonged recovery phase compared with WT animals (Fig. 4B; Figure, Supplemental Digital Content 3A, at http://links.lww.com/SHK/A242). Nonsurviving animals succumbed during the corresponding time period from 24 h after sepsis induction (Figure, Supplemental Digital Content 3B, at http://links.lww.com/SHK/A242).
The measurement of body temperature during the course of disease gave similar results. Although WT and OTII mice experienced a comparable drop in body temperature early after sepsis onset, the phase of recovery was prolonged in OTII animals (Fig. 4C). Surviving OTII mice had prolonged hypothermia compared with WT controls, whereas nonsurviving animals in both groups showed similar severe degrees of hypothermia (Figure, Supplemental Digital Content 3C, D, at http://links.lww.com/SHK/A242).
In summary, the very high degree of physiological stress immediately after sepsis induction was moderately but significantly increased in OTII mice—even more impressive was their inability to recover from the insult.
Cytokine profiles during sepsis in the presence and absence of TCR signaling
We next determined whether TCR signaling influences cytokine responses in the periphery. Proinflammatory and anti-inflammatory cytokines were measured in sera of septic OTII and WT mice 24 h after AP. Serum concentrations of TNF-α, IFN-γ, and IL17-A were in tendency increased in OTII mice compared with WT animals. The two groups had comparable levels of IL-6 and IL-10, as shown in Figure 5. There were no differences in IL-2, IL-4, IL-12p70, and MCP-1 concentrations (Figure, Supplemental Digital Content 4, at http://links.lww.com/SHK/A243). Interestingly, only those OTII mice whose cytokine levels remained low survived sepsis, whereas some WT mice tolerated higher concentrations of inflammatory cytokines (Fig. 5; Figure, Supplemental Digital Content 4, at http://links.lww.com/SHK/A243).
Fig. 5.
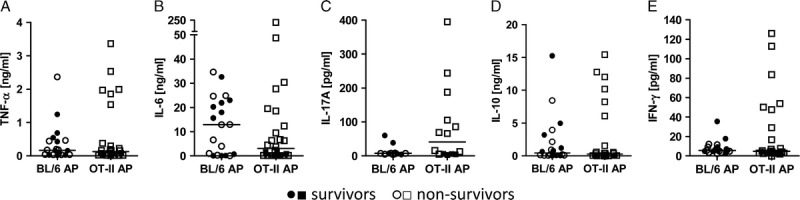
Cytokine profiles during sepsis in the presence and absence of TCR signaling. OTII and C57BL/6 control mice were subjected to AP. After 24 h, serum levels of TNF-α (A), IL-6 (B), IL-17A (C), IL-10 (D), and IFN-γ (E) were determined by cytometric bead array. The medians are shown. Nonsurviving animals are depicted as open symbols. The sample sizes were 20 WT, 26 OTII per group. Septic OTII and WT mice were compared using the Mann-Whitney U test, showing no significant differences.
Influence of TCR signaling on bacterial clearance and peritoneal immune cell composition
Next, we determined the bacterial load in different organs 24 h after AP. OTII mice had an increased bacterial burden (1–2 log ranges) in all examined organs, which, however, did not reach significance because of large variances (Fig. 6A). Correspondingly, the peritoneal neutrophil influx was in tendency increased in OTII animals, whereas there were no differences between OTII and WT mice in terms of other peritoneal immune cell populations, such as B cells, CD4+ T cells, dendritic cells, and macrophages (Fig. 6B). Taken together, this suggests that the inability to mount an antigen-specific T-cell response to the infectious agents impaired the ability to control bacterial dissemination, resulting in an increased (and/or prolonged) inflammatory response.
Fig. 6.
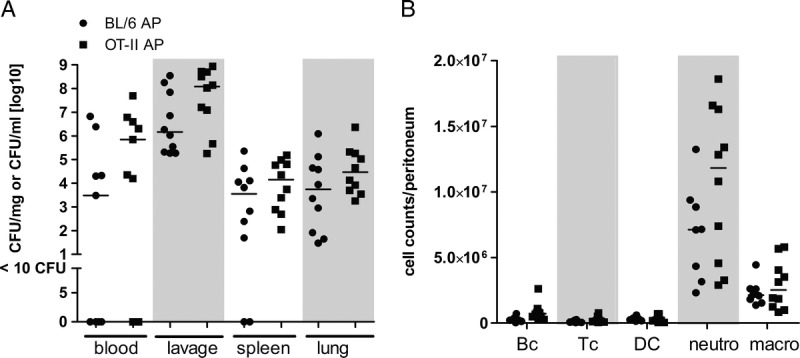
Influence of TCR signaling on bacterial clearance and peritoneal immune cell composition. OTII and C57BL/6 control mice were subjected to AP. A, After 24 h, peritoneal lavage was performed, blood was collected, and spleen and lung were recovered. Lavage, anticoagulated blood, spleen, and lung tissue homogenates were incubated on Columbia blood agar for 22 h at 37°C to determine bacterial load. Colony-forming units (CFU) were related to fluid volume or organ weight. B, Cell counts of B cells (Bc), CD4+ T cells (Tc), dendritic cells (DC), neutrophils (neutro), and macrophages (macro) were determined by flow cytometry as described in Materials and Methods. Medians are indicated. Sample sizes were nine to 10 per group. Septic OTII and WT mice were compared using the Mann-Whitney U test, showing no significant differences.
The fraction of Foxp3+ Tregs was low in OTII mice
Compared with WT animals, septic OTII mice exhibited decreased sepsis survival with a prolonged recovery phase. Although changes in inflammatory parameters were moderate, OTII mice were clearly less resilient to inflammation. Because we previously showed a role of Tregs in the recovery from sepsis (7), we determined the Treg:Teff ratio in the spleen 24 h after AP surgery. In nonseptic animals, total CD4+ T-cell counts were comparable in OTII and WT mice and they decreased in a similar way after sepsis onset (Fig. 7, A and B). In contrast, both septic and nonseptic OTII mice showed significantly reduced percentages of CD4+ Foxp3+ Tregs compared with WT (nonseptic, 11% vs. 5%, P = 0.0003; septic, 13% vs. 7.5%, P < 0.0001). Absolute Treg numbers tended to decrease as well, as shown in Figure 7, but this was not statistically significant.
Fig. 7.

Reduced numbers of Tregs in OTII mice. OTII mice and C57BL/6 control mice were either left untreated or sepsis was induced by AP surgery. After 24 h, splenocytes were isolated and cell counts and percentages of CD4+ T cells (A, B) and CD4+ Foxp3+ Tregs (C, D) were determined by flow cytometry. Means are indicated. Sample sizes were seven to 10 per group. Septic and untreated OTII mice were compared with WT animals using one-way ANOVA with Bonferroni post hoc test. *P ≤ 0.05; ***P ≤ 0.001.
DISCUSSION
Our data show that, early during sepsis, antigen-induced activation and proliferation of CD4+ T cells were not impaired. In septic mice, transferred transgenic OTII T cells proliferated even more vigorously than in nonseptic animals, if the antigen OVA was applied during AP surgery. Later during the disease course, however, antigen-triggered T-cell proliferation was impaired. T-cell receptor engagement by antigen was clearly necessary for full activation and proliferation of the T cells in sepsis. In the absence of antigen, such as in septic OTII mice, the early activation marker CD69 was rapidly upregulated on both Teffs and Tregs; in addition, Tregs increased CTLA-4 expression. However, there was no induction of CD25 expression on CD4+ T cells nor were the cells able to proliferate.
The findings of the present study support the model of T-cell readiness on infection, which was proposed by Jiang et al. (31) based on their observations in Listeria monocytogenes–infected mice: most T cells were induced to express CD69 regardless of their antigen specificity, probably via type I interferons or toll-like receptor engagement on T cells, whereas only antigen-specific T cells were fully activated to express CD25 and proliferate. Sepsis is characterized by the systemic release of large amounts of microbe-associated molecular patterns or MAMPs. This can explain the partial activation in most T cells because CD4+ T cells are known to upregulate CD69 in response to lipopolysaccharide, CpG, or poly I:C (20, 32–34). Nonetheless, in septic animals, T cells failed to upregulate CD25 or proliferate if they were not given an antigen-specific stimulus. Because only antigen-specific activation will save T cells from apoptosis (35), presumably T cells that are partially activated in the absence of antigen will be prone to cell death in sepsis, as it has been shown in L. monocytogenes infection (31). This would reconcile the regular observations of both T-cell activation and cell death in sepsis.
In contrast to our results using the AP model of sepsis, Unsinger et al. (30) did not observe proliferation of transferred CD4+ T cells in WT animals after CLP and concluded that T cells were not fully activated in response to sepsis. Compared with the present study, the experimenters transferred a much smaller number of CD4+ T cells, which, in addition, had not been selected for antigen specificity, such that the antigen-triggered proliferation may have been missed because of limited sensitivity. In AP, antigen-specific activation of transferred OVA-specific T cells proceeded unimpaired and was clearly measurable after 3 days. Moreover, approximately 1% of transferred CD4+ OTII T cells proliferated during sepsis even in the absence of OVA. Because, in Rag-sufficient OTII mice, about 20% of the CD4+ T cells bear nonclonotypic specificities (36), we assume that the proliferating cells were recognizing microbial antigens. In a pilot study, we observed approximately 15% of splenic CD4+ T cells to express Ki-67 as early as 24 h after AP, whereas, in OTII mice, only 7% were Ki-67+ (unpublished data), indicating proliferation of WT CD4+ T cells early during sepsis.
Other groups including Unsinger’s have reported impairment of CD4+ T-cell function in sepsis (10, 30, 37). Corroborating these findings, in our hands, CD4+ T-cell function was impaired in the later course of sepsis. At sepsis onset, however, these cells could be fully activated in an antigen-dependent manner and proliferated even more vigorously than in nonseptic animals. This adjuvant effect of sepsis appears to contribute to an effective immune response and bacterial clearance. Gurung et al. (38) showed that naive apoptotic cells induce a state of tolerance, whereas activated apoptotic cells support robust proliferation of T cells. Hence, besides the microbe-associated molecular patterns derived from the invasive bacteria, cellular activation and cell damage may act as danger-associated molecular signals contributing to the observed adjuvant effect early in sepsis.
In summary, the presented data show that, early during sepsis, antigen-specific CD4+ T cells are fully activated and proliferate. In addition, nonspecific stimuli lead to preactivation of a broad mass of CD4+ T cells, as indicated by the induction of CD69.
To study the impact of the antigen-specific activation of CD4+ T cells on sepsis outcome, we compared the survival of OTII and WT mice in AP. Corroborating the findings obtained by Kasten et al. (21) in CLP, the AP-induced mortality was increased in OTII mice. Initially, OTII and WT mice showed moderately but significantly higher disease severity; even more impressive, their recovery phase was significantly prolonged, and, from day 2 onward, more animals died. In CLP, Kasten et al. (21) observed more bacteria and more neutrophils in the peritoneum as well as increased systemic IL-6 concentrations in OTII mice compared with WT controls. They were able to rescue animals by antigenic T-cell stimulation, implying that antigen recognition by T cells was required for neutrophil function and efficient clearance of bacteria (21). In the present study, using the AP model, the bacterial loads in different organs and the systemic cytokine concentrations 24 h after sepsis induction were 10- to 100-fold increased in OTII as compared with WT mice. This suggests that the inability to mount an antigen-specific T-cell response to the infectious agents impaired the ability to control bacterial dissemination, resulting in an increased and/or prolonged inflammatory response. It was shown before that mice exhibiting the highest cytokine levels are prone to die from sepsis (39), and OTII mice were less resilient to inflammation. In contrast to WT animals, not a single OTII mouse with very high cytokine serum concentrations survived.
Because Tregs are required for control of inflammation in infection (40), we investigated the Treg compartment. It is well documented that, during CLP, relative Treg numbers increase, whereas their absolute number remains unchanged (e.g., 7). This is most likely caused by a relative resistance to apoptosis compared with Teffs (41). Moreover, Tregs and Teffs were activated within the first 24 h after sepsis. In this study, we corroborated these findings in the AP sepsis model. Notably, in both nonseptic and septic OTII mice, the percentages of splenic Foxp3+ Tregs were reduced to about 50% of that of WT animals. For the development of natural Tregs, antigen recognition in the thymus is required (42, 43). In the absence of OVA, some Tregs still develop in Rag-sufficient OTII mice. They originate from nonclonotypic T-cell precursors harboring endogenous Tcrb and/or Tcra rearrangements (44). Therefore, they are not OVA specific but recognize other antigens. However, their TCR repertoire is narrowed to about 50% of that of WT animals, reducing their ability to control experimental graft-versus-host disease (44). In a similar manner, the reduced numbers and narrowed repertoire of natural Tregs in OTII mice may have impaired their function in AP. In sepsis, Foxp3+ Tregs develop enhanced suppressive properties (7, 19). They are required for recovery and restitution of immune homeostasis after septic inflammation, as our group has previously demonstrated by Treg depletion (7). However, Tregs in sepsis may be a double-edged sword (40). Besides their role in limiting inflammation and promoting recovery from sepsis, their strong activation may account for the immunosuppression observed in septic patients, a major problem in clinical settings (45). We are currently analyzing their specific role during the immune suppressive state observed later in the course of sepsis.
It is known that OTII mice have a 4-fold increase in the CD4:CD8 T-cell ratio with a strongly reduced CD8+ T-cell population in the periphery (46). This, however, is unlikely to explain the increased mortality in these mice because depletion of CD8+ T cells was beneficial in murine sepsis (47). It appears that CD8+ T cells contribute to progression of inflammation by contribution to neutrophil recruitment as well as intestinal and liver inflammation (48, 49).
We conclude that there are several mutually nonexclusive explanations for the reduced survival of OTII mice in acute sepsis: first, impairment of antimicrobial functions of antigen-specific Teffs; second, the lack of sufficient Treg numbers to ensure restitution of immune homeostasis; and third, less likely, alterations in the CD8+ T-cell population.
In summary, the present study demonstrates that CD4+ T cells are activated differentially during early sepsis. The TCR-independent stimuli preactivate these cells that need TCR-specific stimuli to become fully activated and proliferate. Adjuvant effects at the beginning of sepsis enhanced the antigen-specific T-cell activation, whereas it was attenuated in the later course of the disease. The antigen-specific T cells appear to influence the outcome of sepsis in a highly contextual manner: rapid exertion of effector functions may contribute to bacterial clearance but can also drive hyperinflammation, whereas Treg activity is required for reconstitution of immune homeostasis but may carry the risk of prolonged immune suppression.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Susanne Neumeister for expert technical help and Sebastian Stentzel for inspiring discussions and for critical reading of the manuscript. Kate Splieth eliminated some grammatical errors and typos; the responsibility for those remaining is ours.
Footnotes
This work was financially supported by the Deutsche Forschungsgemeinschaft (GRK-840 and CRC-TRR34).
The authors declare no financial or commercial conflict of interest.
Supplemental digital content is available for this article. Direct URL citation appears in the printed text and is provided in the HTML and PDF versions of this article on the journal’s Web site (www.shockjournal.com).
REFERENCES
- 1. Slade E, Tamber PS, Vincent JL: The Surviving Sepsis Campaign: raising awareness to reduce mortality. Crit Care 7( 1): 1– 2, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boomer JS, Shuherk-Shaffer J, Hotchkiss RS, Green JM: A prospective analysis of lymphocyte phenotype and function over the course of acute sepsis. Crit Care 16( 3): R112, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR: Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med 29( 7): 1303– 1310, 2001. [DOI] [PubMed] [Google Scholar]
- 4. Martignoni A, Tschop J, Goetzman HS, Choi LG, Reid MD, Johannigman JA, Lentsch AB, Caldwell CC: CD4-expressing cells are early mediators of the innate immune system during sepsis. Shock 29( 5): 591– 597, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Roger PM, Hyvernat H, Ticchioni M, Kumar G, Dellamonica J, Bernardin G: The early phase of human sepsis is characterized by a combination of apoptosis and proliferation of T cells. J Crit Care 27( 4): 384– 393, 2012. [DOI] [PubMed] [Google Scholar]
- 6. Busse M, Traeger T, Potschke C, Billing A, Dummer A, Friebe E, Kiank C, Grunwald U, Jack RS, Schutt C, et al. : Detrimental role for CD4+ T lymphocytes in murine diffuse peritonitis due to inhibition of local bacterial elimination. Gut 57( 2): 188– 195, 2008. [DOI] [PubMed] [Google Scholar]
- 7. Kuhlhorn F, Rath M, Schmoeckel K, Cziupka K, Nguyen HH, Hildebrandt P, Hunig T, Sparwasser T, Huehn J, Potschke C, et al. : Foxp3+ regulatory T cells are required for recovery from severe sepsis. PLoS One 8( 5): e65109, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nakanishi K: Innate and acquired activation pathways in T cells. Nat Immunol 2( 2): 140– 142, 2001. [DOI] [PubMed] [Google Scholar]
- 9. Heidecke CD, Hensler T, Weighardt H, Zantl N, Wagner H, Siewert JR, Holzmann B: Selective defects of T lymphocyte function in patients with lethal intraabdominal infection. Am J Surg 178( 4): 288– 292, 1999. [DOI] [PubMed] [Google Scholar]
- 10. Carson WFt, Cavassani KA, Ito T, Schaller M, Ishii M, Dou Y, Kunkel SL: Impaired CD4+ T-cell proliferation and effector function correlates with repressive histone methylation events in a mouse model of severe sepsis. Eur J Immunol 40( 4): 998– 1010, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thiel M, Caldwell CC, Kreth S, Kuboki S, Chen P, Smith P, Ohta A, Lentsch AB, Lukashev D, Sitkovsky MV: Targeted deletion of HIF-1alpha gene in T cells prevents their inhibition in hypoxic inflamed tissues and improves septic mice survival. PLoS One 2( 9): e853, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kasten KR, Tschop J, Adediran SG, Hildeman DA, Caldwell CC: T cells are potent early mediators of the host response to sepsis. Shock 34( 4): 327– 336, 2010. [DOI] [PubMed] [Google Scholar]
- 13. Hildeman DA, Zhu Y, Mitchell TC, Bouillet P, Strasser A, Kappler J, Marrack P: Activated T cell death in vivo mediated by proapoptotic bcl-2 family member bim. Immunity 16( 6): 759– 767, 2002. [DOI] [PubMed] [Google Scholar]
- 14. Schwulst SJ, Muenzer JT, Peck-Palmer OM, Chang KC, Davis CG, McDonough JS, Osborne DF, Walton AH, Unsinger J, McDunn JE, et al. : Bim siRNA decreases lymphocyte apoptosis and improves survival in sepsis. Shock 30( 2): 127– 134, 2008. [DOI] [PubMed] [Google Scholar]
- 15. Weber SU, Schewe JC, Lehmann LE, Muller S, Book M, Klaschik S, Hoeft A, Stuber F: Induction of Bim and Bid gene expression during accelerated apoptosis in severe sepsis. Crit Care 12( 5): R128, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mohr A, Polz J, Martin EM, Griessl S, Kammler A, Potschke C, Lechner A, Broker BM, Mostbock S, Mannel DN: Sepsis leads to a reduced antigen-specific primary antibody response. Eur J Immunol 42( 2): 341– 352, 2012. [DOI] [PubMed] [Google Scholar]
- 17. Hotchkiss RS, Tinsley KW, Swanson PE, Chang KC, Cobb JP, Buchman TG, Korsmeyer SJ, Karl IE: Prevention of lymphocyte cell death in sepsis improves survival in mice. Proc Natl Acad Sci U S A 96( 25): 14541– 14546, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Enoh VT, Lin SH, Etogo A, Lin CY, Sherwood ER: CD4+ T-cell depletion is not associated with alterations in survival, bacterial clearance, and inflammation after cecal ligation and puncture. Shock 29( 1): 56– 64, 2008. [DOI] [PubMed] [Google Scholar]
- 19. Scumpia PO, Delano MJ, Kelly KM, O’Malley KA, Efron PA, McAuliffe PF, Brusko T, Ungaro R, Barker T, Wynn JL, et al. : Increased natural CD4+CD25+ regulatory T cells and their suppressor activity do not contribute to mortality in murine polymicrobial sepsis. J Immunol 177( 11): 7943– 7949, 2006. [DOI] [PubMed] [Google Scholar]
- 20. Enoh VT, Lin SH, Lin CY, Toliver-Kinsky T, Murphey ED, Varma TK, Sherwood ER: Mice depleted of alphabeta but not gammadelta T cells are resistant to mortality caused by cecal ligation and puncture. Shock 27( 5): 507– 519, 2007. [DOI] [PubMed] [Google Scholar]
- 21. Kasten KR, Tschop J, Goetzman HS, England LG, Dattilo JR, Cave CM, Seitz AP, Hildeman DA, Caldwell CC: T-cell activation differentially mediates the host response to sepsis. Shock 34( 4): 377– 383, 2010. [DOI] [PubMed] [Google Scholar]
- 22. Buras JA, Holzmann B, Sitkovsky M: Animal models of sepsis: setting the stage. Nat Rev Drug Discov 4( 10): 854– 865, 2005. [DOI] [PubMed] [Google Scholar]
- 23. Dejager L, Pinheiro I, Dejonckheere E, Libert C: Cecal ligation and puncture: the gold standard model for polymicrobial sepsis? Trends Microbiol 19( 4): 198– 208, 2011. [DOI] [PubMed] [Google Scholar]
- 24. Rittirsch D, Hoesel LM, Ward PA: The disconnect between animal models of sepsis and human sepsis. J Leukoc Biol 81( 1): 137– 143, 2007. [DOI] [PubMed] [Google Scholar]
- 25. Schabbauer G: Polymicrobial sepsis models: CLP versus CASP. Drug Discovery Today: Disease Models 9( 1): e17– e21, 2012. [Google Scholar]
- 26. Maier S, Traeger T, Entleutner M, Westerholt A, Kleist B, Huser N, Holzmann B, Stier A, Pfeffer K, Heidecke CD: Cecal ligation and puncture versus colon ascendens stent peritonitis: two distinct animal models for polymicrobial sepsis. Shock 21( 6): 505– 511, 2004. [DOI] [PubMed] [Google Scholar]
- 27. Barrera G, Landoni V, Martire-Greco D, Chiarella P, Meiss R, Gomez SA, Alves-Rosa F, Rearte B, Isturiz M, Palermo MS, et al. : Model of polymicrobial peritonitis that induces the proinflammatory and immunosuppressive phases of sepsis. Infect Immun 79( 3): 1280– 1288, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zantl N, Uebe A, Neumann B, Wagner H, Siewert JR, Holzmann B, Heidecke CD, Pfeffer K: Essential role of gamma interferon in survival of colon ascendens stent peritonitis, a novel murine model of abdominal sepsis. Infect Immun 66( 5): 2300– 2309, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Aziz M, Yang WL, Matsuo S, Sharma A, Zhou M, Wang P: Upregulation of GRAIL is associated with impaired CD4 T cell proliferation in sepsis. J Immunol 192( 5): 2305– 2314, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Unsinger J, Herndon JM, Davis CG, Muenzer JT, Hotchkiss RS, Ferguson TA: The role of TCR engagement and activation-induced cell death in sepsis-induced T cell apoptosis. J Immunol 177( 11): 7968– 7973, 2006. [DOI] [PubMed] [Google Scholar]
- 31. Jiang J, Lau LL, Shen H: Selective depletion of nonspecific T cells during the early stage of immune responses to infection. J Immunol 171( 8): 4352– 4358, 2003. [DOI] [PubMed] [Google Scholar]
- 32. Sprent J, Zhang X, Sun S, Tough D: T-cell proliferation in vivo and the role of cytokines. Philos Trans R Soc Lond B Biol Sci 355( 1395): 317– 322, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tough DF, Sun S, Sprent J: T cell stimulation in vivo by lipopolysaccharide (LPS). J Exp Med 185( 12): 2089– 2094, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vilanova M, Tavares D, Ferreira P, Oliveira L, Nobrega A, Appelberg R, Arala-Chaves M: Role of monocytes in the up-regulation of the early activation marker CD69 on B and T murine lymphocytes induced by microbial mitogens. Scand J Immunol 43( 2): 155– 163, 1996. [DOI] [PubMed] [Google Scholar]
- 35. Boise LH, Thompson CB: Hierarchical control of lymphocyte survival. Science 274( 5284): 67– 68, 1996. [DOI] [PubMed] [Google Scholar]
- 36. Barnden MJ, Allison J, Heath WR, Carbone FR: Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol 76( 1): 34– 40, 1998. [DOI] [PubMed] [Google Scholar]
- 37. Hotchkiss RS, Monneret G, Payen D: Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol 13( 12): 862– 874, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gurung P, Kucaba TA, Ferguson TA, Griffith TS: Activation-induced CD154 expression abrogates tolerance induced by apoptotic cells. J Immunol 183( 10): 6114– 6123, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Osuchowski MF, Connett J, Welch K, Granger J, Remick DG: Stratification is the key: inflammatory biomarkers accurately direct immunomodulatory therapy in experimental sepsis. Crit Care Med 37( 5): 1567– 1573, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Belkaid Y, Rouse BT: Natural regulatory T cells in infectious disease. Nat Immunol 6: 353– 360, 2005. [DOI] [PubMed] [Google Scholar]
- 41. Boomer JS, Green JM, Hotchkiss RS: The changing immune system in sepsis: is individualized immuno-modulatory therapy the answer? Virulence 5( 1): 45– 56, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fontenot JD, Rudensky AY: Molecular aspects of regulatory T cell development. Semin Immunol 16( 2): 73– 80, 2004. [DOI] [PubMed] [Google Scholar]
- 43. DiPaolo RJ, Shevach EM: CD4+ T-cell development in a mouse expressing a transgenic TCR derived from a Treg. Eur J Immunol 39( 1): 234– 240, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fohse L, Suffner J, Suhre K, Wahl B, Lindner C, Lee CW, Schmitz S, Haas JD, Lamprecht S, Koenecke C, et al. : High TCR diversity ensures optimal function and homeostasis of Foxp3+ regulatory T cells. Eur J Immunol 41( 11): 3101– 3113, 2011. [DOI] [PubMed] [Google Scholar]
- 45. Hotchkiss RS, Monneret G, Payen D: Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis 13( 3): 260– 268, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Leung S, Smith D, Myc A, Morry J, Baker JR, Jr.: OT-II TCR transgenic mice fail to produce anti-ovalbumin antibodies upon vaccination. Cell Immunol 282( 2): 79– 84, 2013. [DOI] [PubMed] [Google Scholar]
- 47. van Schaik SM, Abbas AK: Role of T cells in a murine model of Escherichia coli sepsis. Eur J Immunol 37( 11): 3101– 3110, 2007. [DOI] [PubMed] [Google Scholar]
- 48. Sherwood ER, Enoh VT, Murphey ED, Lin CY: Mice depleted of CD8+ T and NK cells are resistant to injury caused by cecal ligation and puncture. Lab Invest 84( 12): 1655– 1665, 2004. [DOI] [PubMed] [Google Scholar]
- 49. Wesche-Soldato DE, Chung CS, Gregory SH, Salazar-Mather TP, Ayala CA, Ayala A: CD8+ T cells promote inflammation and apoptosis in the liver after sepsis: role of Fas-FasL. Am J Pathol 171( 1): 87– 96, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.