

Abstract
The polymeric fluoropyrimidine F10 displays excellent anti-leukemia activity in pre-clinical models of acute myelogenous leukemia (AML) through dual targeting of thymidylate synthase and DNA topoisomerase 1. Here we report that F10 activates the extrinsic apoptotic pathway in AML cells by enhancing localization of Fas and Fas ligand (FasL) at the plasma membrane and while reducing overall lipid raft levels promotes Fas/FasL co-localization in remaining lipid rafts. The HMG-CoA synthase inhibitor simvastatin was synergistic with F10 and induced cell death via similar apoptotic processes. Our results are consistent with diverse processes activating a common apoptotic pathway characterized by reduced overall levels of lipid rafts and Fas/FasL co-localization in the plasma membrane, including in remaining lipid rafts which may play a role in both cell-survival and cell death signaling.
Keywords: F10, DNA, apoptosis, Fas, lipid rafts
1. Introduction
Thymidylate synthase (TS) is one of the best-validated targets for anti-cancer drugs, including both fluoropyrimidines such as 5-fluorouracil (5-FU) and folate-based TS-inhibitors such as raltitrexed [1]. The general cytotoxic process that cancer cells undergo in response to TS inhibitors is referred to as “thymineless death” [2, 3]. Few studies have investigated thymineless death in leukemia cells because drugs that target TS are not presently used for this indication. Our recent studies demonstrate, however, that acute myelogenous and acute lymphocytic leukemia (AML & ALL) cells are highly sensitive to the novel fluoropyrimidine drug-candidate F10 (Figure 1a) [4–6]. TS is the proximal target for F10, although DNA topoisomerase 1 (Top1) is the ultimate target responsible for generating lethal DNA double strand breaks (DSBs) in AML cells [7, 8].
Figure 1. F10 activates both extrinsic and extrinsic apoptotic pathways.
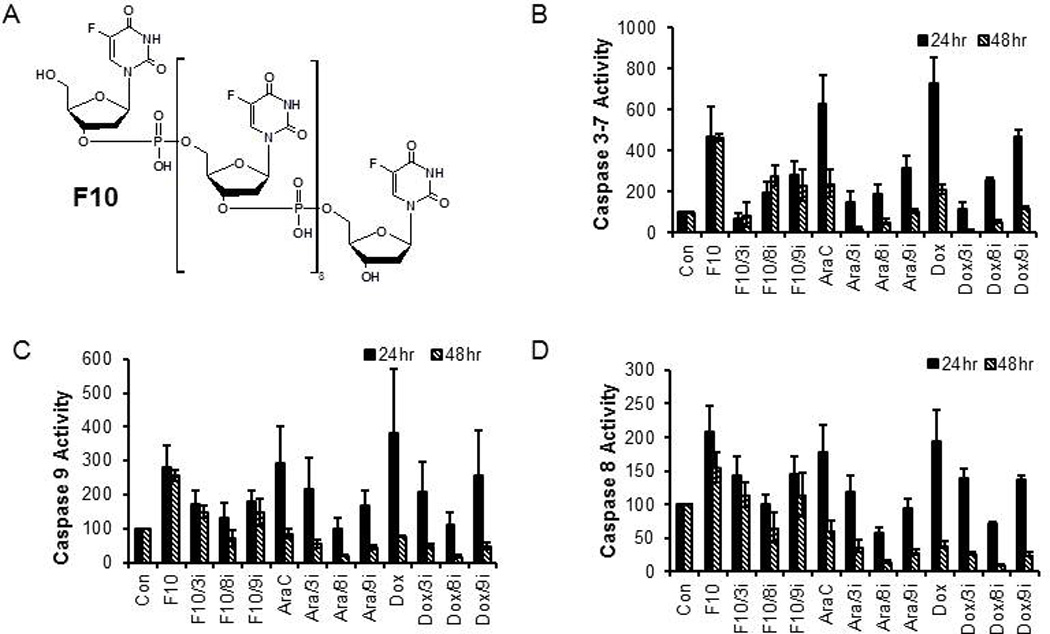
(A) Structure of F10. (B-D) Plots of caspase activity (vehicle-only = 100) for: (B) caspase 3/7; (C) caspase 9; and (D) caspase 8 in response to treatment with F10 (10 nM), AraC (1 µM), or Dox (100 ng/mL) at the times indicated and in the presence of specific inhibitors of caspase 3/7 (DEVD; e.g. F10/3i), caspase 8 (IETD), and caspase 9 (LEHD).
The mechanism linking TS inhibition to Top1 poisoning in F10-treated AML cells involves incorporating 2’-deoxyruridine (dU) and 5-fluoro-2’-deoxyuridine (FdU) into DNA under thymineless conditions. Top1 can cleave, but cannot re-ligate DNA at sites of dU or FdU substitution, and the resulting protein:DNA cleavage complex (e.g. Top1cc) [8] is then processed into a DNA double-strand break [9, 10]. F10-induced Top1cc require cells to undergo DNA replication twice [5]. The first replication occurs under thymineless conditions and promotes FdU and dU misincorporation into DNA while in the second replication Top1 encounters FdU-substituted DNA ahead of the replication fork and becomes trapped. Subsequent collision of the trapped Top1cc with the advancing replication fork generates a DNA double strand break (DSB). This requirement for two rounds of replication contributes to the observed low systemic toxicity for F10.
While the role of Top1cc formation for F10-induced thymineless death has been established, the mechanism by which Top1cc and subsequent DNA damage induce apoptosis in AML cells is not presently known. The extrinsic apoptotic pathway is initiated upon stimulation of death receptors of the tumor necrosis factor (TNF) superfamily, such as CD95 (APO-1/Fas) or TNF-related apoptosis-inducing ligand (TRAIL) which results in activation of the initiator caspase 8 which then activates effector caspases such as caspase 3. Alternatively, the intrinsic or mitochondrial apoptotic pathway can initiate apoptosis through release of cytochrome c and formation of the caspase 9-containing apoptosome complex which then activates caspase 3. In type II cells, initiation of apoptosis via the extrinsic pathway is amplified by cleavage of Bid, a BH3-only member of the Bcl-2 family, which then activates the intrinsic apoptotic pathway [11]. Anticancer drugs may initiate apoptosis via either the intrinsic or extrinsic pathways. Previous studies of thymineless death in colon cancer cells treated with 5-FU and leucovorin demonstrated apoptosis occurred via increased expression of Fas ligand (FasL) via autocrine activation of the Fas death pathway and with the initiation of apoptosis via the extrinsic pathway [2, 12, 13].
In these studies, we demonstrate that F10 induces thymineless death in AML cells via activation of the extrinsic apoptotic pathway. Our studies revealed Fas activation occurs without an increase in either Fas or FasL expression suggesting Fas activation occurs via altered sub-cellular localization, rather than due to increased expression of FasL. We confirmed by confocal microscopy that Fas and FasL co-localized in the plasma membrane of F10-treated AML cells. Although F10 treatment reduced overall levels of lipid rafts, Fas and FasL co-localization occurred within remaining lipid rafts [14] which may mediate either cell death or survival signaling [15]. The HMGCoA-reductase inhibitor simvastatin [16] also reduced lipid raft levels and induced apoptosis. Further, the combination of F10 + simvastatin was synergistic and highly effective at promoting Fas and FasL co-localization in the plasma membrane and, while reducing overall lipid raft levels, Fas/FasL colocalization occurred in remaining lipid rafts. Our results demonstrate that F10 induces thymineless death through activation of the extrinsic apoptotic pathway through the enhanced co-localization of Fas and FasL in the plasma membrane of AML cells. Altered sub-cellular localization of Fas/FasL is accompanied by reduction in lipid rafts in the plasma membrane which may indicate a role for lipid rafts in cell survival signaling, although colocalization of Fas/FasL in remaining lipid rafts suggest a role in cell death signaling, as well. Our results indicate reduced lipid raft levels and activation of the extrinsic apoptotic pathway is characteristic of diverse apoptosis-inducing stimuli in AML cells including F10-induced thymineless death.
2. Materials and methods
2.1. Materials
All antibodies [17] were purchased from Cell Signaling unless otherwise noted. AlexaFluor 488 goat anti-rabbit (Invitrogen/Life Technologies, Grand Island, NY) was used for flow cytometry and rhodamine-(TRITC-) conjugated donkey anti-rabbit IgG for immunofluorescence (IF) (Jackson ImmunoResearch). All chemicals were from Sigma unless otherwise noted. Caspase 9 inhibitor LEHD and caspase 8 inhibitor IEDT were used at 250 nM and cells were pre-treated 30 min prior to addition of other drugs in all caspase inhibition experiments. Caspase 3 inhibitor (DEVD; 50 nM), NF-κB inhibitor (Bay11-7082; 10 µM), and Fas activating ligand CH-11 (0.25 or 0.5 µL of 0.5 mg/mL) were purchased from Merck/Millipore/Calbiochem, Darmstadt, Germany. Nok1 antibody to Fas ligand was purchased from BD Pharmigen/Bioscience, San Jose, California and was used at either 100 or 200 ng/mL, as indicated.
2.2. Cell culture and apoptosis assays
HL60, THP1, KG1a, OCI-AML3, and Molm13 AML cells (ATCC) were cultured in RPMI 1640 (Gibco) with 10% FBS (Gemini Bio-Products). Cells were plated at a density of 1.5 ×106 cells in 100 mm2 plates and grown overnight. Caspase 3/7, 8, and 9 activity and cell viability assays were performed using Caspase-Glo (Promega) for caspase activity and CellTiter-Glo (Promega) for cell viability. Briefly, 2×105 cells were plated in 24 well plates in triplicate and drug treatments started 20–24 h after plating. Cells were re-suspended before 20 μL aliquots were taken at the indicated times and mixed with an equal volume of assay kit reagent in a 96-well white plate. The plates were then incubated at ambient temperature for 30–60 min per instructions before quantification of lumiescence using a Tecan Genius plate reader. Apoptosis data was normalized for cell number using viability data [18]. Synergy between F10 and simvastatin was determined using the combination index (CI) method of Chou and Talay [19] as implemented in CalcuSyn 2.0 (Biosoft).
Statistical analysis
Assay values were expressed as mean +/− standard error and comparisons between mean values were made using Student’s t-test for paired samples with p ≤ 0.05 (two-tailed) considered significant.
Immunofluorescence and detection of lipid rafts
AML cells were washed to remove all serum and spun onto poly-l-lysine coated 8-well Nunc Lab-Tek II (Thermo-Fisher, Pittsburgh, PA) chambered slides at 150,000 cells per well in serum free media. Cells were spun down at 500 rpm for 5 min on a plate centrifuge, carefully rinsed of media with ice cold 1%BSA/PBS, and then incubated with FITC-labeled Ctx-B (cholera toxin B, Sigma) at 1:2500 in 1%BSA/PBS on ice for 30 min. Subsequent antibody incubations were carried out on ice for 30 min in 1% BSA/PBS with careful rinsing with ice-cold 1% BSA/PBS between. Primary antibodies, Fas (C18C12) or Fas-L (C-178, Santa Cruz Biotechnology) were used at 1:200 dilution. Secondary Rho-TRITC was added at 1:100 followed by counter-staining for DNA with Hoechst dye in some instances. Cells were fixed post-staining with 10% neutral buffered formalin for 5 mins. and visualized using a Zeiss LSM 510 Confocal Microscope (Carl Zeiss, Oberkochen, Germany).
3. Results
3.1. F10 activates both intrinsic and extrinsic apoptotic pathways
Previous studies demonstrated that F10 (Figure 1a) induced apoptosis in AML cells in a p53-independent manner [4]. We investigated the time-dependence of F10-induced apoptosis in AML cells (Figure 1b) to gain insights into the apoptotic mechanism. Relative to doxorubicin (Dox) and cytarabine (AraC), F10-induced apoptosis was more prolonged with enhanced caspase 3/7 activity at both 24 and 48 h, while for Dox and AraC caspase activity peaked near 24 h and markedly decreased by 48 h. The continued induction of caspase activity by F10 is consistent with DNA double strand breaks occurring only in a second cycle of DNA replication that begins 16 h or later after initial treatment [5]. We utilized specific inhibitors of caspase 3 (DEVD), caspase 8 (IETD), and caspase 9 (LEHD) in conjunction with luminescence activity assays for specific caspases to determine the contribution of the intrinsic and extrinsic apoptotic pathways in F10-treated AML cells. In F10-treated HL60 cells, inhibiting either caspase 8 or caspase 9 reduced caspase 3/7 activity consistent with involvement of both the intrinsic and extrinsic apoptotic pathways (Figure 1b). Independent analysis of caspase 9 (Figure 1c) and caspase 8 (Figure 1d) demonstrated that while caspase 8 inhibition substantially reduced caspase 9 activity, the converse was not true and caspase 9 inhibition had a lesser effect on caspase 8. These results are consistent with apoptosis being initiated via the extrinsic pathway with amplification via the intrinsic pathway in a type 2 process. F10 also induced apoptosis in OCI-AML3, Molm13, THP1, and Jurkat (T-cell ALL) cells and in all cases inhibiting caspase 8 reduced caspase 3/7 activities to control levels consistent with extrinsic pathway activation being essential for apoptosis (Supplementary Figure S1).
Western blots also revealed Bid cleavage occurred at these earlier timepoints consistent with apoptosis occurring via a type 2 process [11] (Supplementary Figure S2a). A similar process was observed for TNFα/CHX, a positive-control for extrinsic pathway activation (Supplementary Figure S2a). Interestingly, etoposide, a Top2-poison and putative positive-control for initiating apoptosis via the intrinsic pathway also induced potent Bid cleavage in HL60 cells, and caspase 3/7 activity was reduced to near control levels by caspase 8 inhibition with caspase 9 inhibition having a lesser effect consistent with HL-60 cells undergoing a common apoptotic process in response to diverse pro-apoptotic treatments (Supplementary Figure S2b).
3.2. F10 sensitizes AML cells to Fas activation
Fas activation occurs via trimerization that is promoted through binding of activating ligands such as FasL or the anti-Fas agonistic antibody CH-11 [20]. We investigated whether stimulation of the Fas death pathway with CH-11 induced apoptosis in AML cells and, if so, whether Fas stimulation synergized with F10 treatment. CH-11 had minimal pro-apoptotic effects as a single agent, however it strongly stimulated F10-induced apoptosis and reduced viability in both OCI-AML3 (Figure 2a and b) and HL60 cells (Figure 2c and d).
Figure 2. F10 induced apoptosis is enhanced by Fas death pathway stimulation.
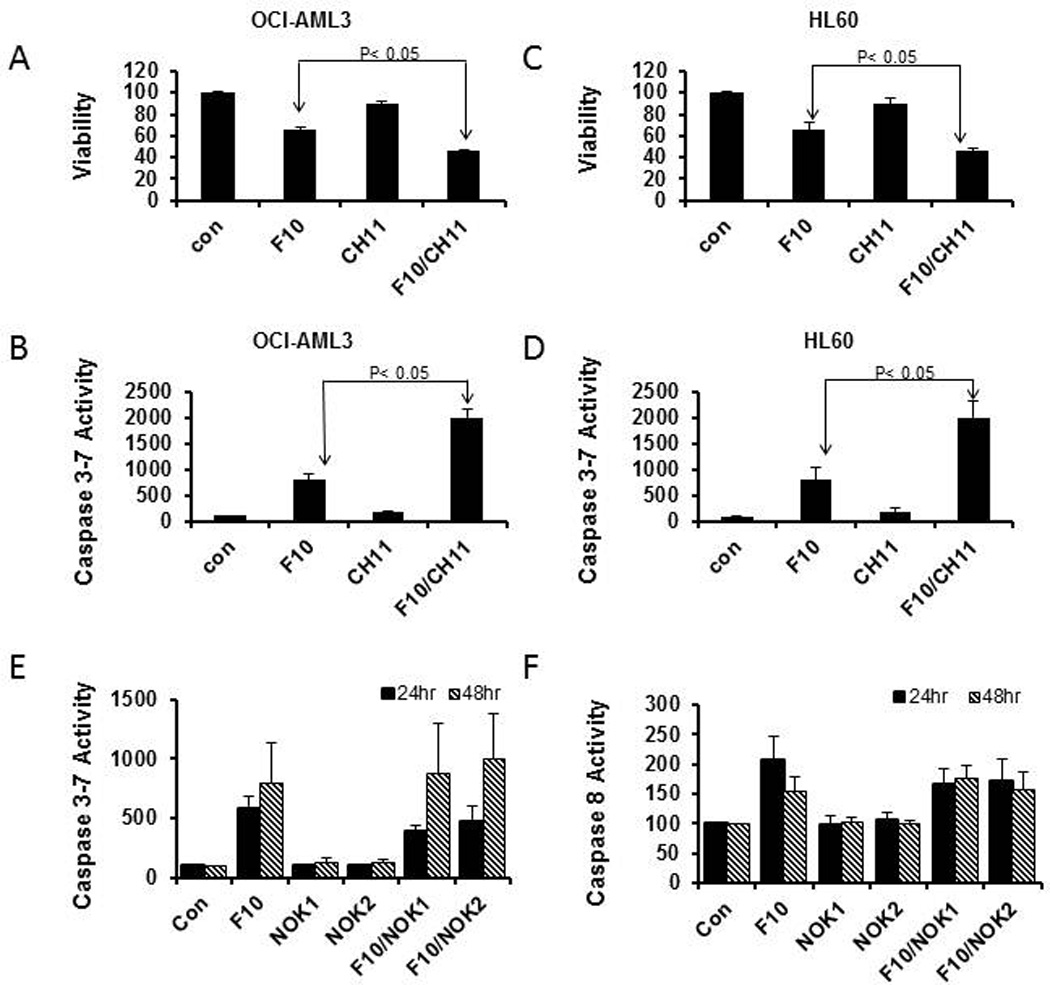
(A) F10 decreases viability (%-control) and induces apoptosis in OCI-AML3 cells (A,B) and HL-60 cells (C,D) and these effects are enhanced by co-treatment with the anti-Fas agonistic mAb CH-11. (E,F) Plots of caspase 3/7 (E) and caspase 8 (F) activity induced by F10 in HL60 cells in the presence and absence of NOK, an antibody to FasL, at 100 ng/mL (NOK1) or 200 ng/mL (NOK2). FasL sequestration partly reduced caspase 8 activity at 24 h but not at 48 h; effects on caspase 3/7 were not significant.
The potential role of the Fas death pathway for apoptosis in F10-treated AML cells stimulated our investigating the extent that transcription factors previously implicated in FasL upregulation in drug-treated cells were activated upon F10-treatment in AML cells. NF-κB and AP-1 have each been implicated in regulating FasL expression in other systems including 5-FU treatment in colon cancer cells [13]. We performed nuclear fractionation of p65/Rel to determine to what extent F10-treatment resulted in NF-κB activation and nuclear translocation in AML cells. These studies demonstrated F10-induced NF-κB activation (Figure 3a). F10 treatment also induced phosphorylation of c-Jun (Figure 3b), a component of AP-1 together with c-Fos, consistent with AP-1 activation. Inhibiting NF-κB with Bay11-7082 did not however decrease F10-induced apoptosis, and actually stimulated apoptosis, particularly at later timepoints (Figure 3c). Although both NF-κB and c-Jun are activated in response to F10 treatment in AML cells there is no apparent change in protein levels of either Fas or FasL (Figure 3d) indicating Fas death pathway activation does not occur via FasL upregulation.
Figure 3. F10 treatment induces activation of NF-κB promotes survival in HL-60 cells.
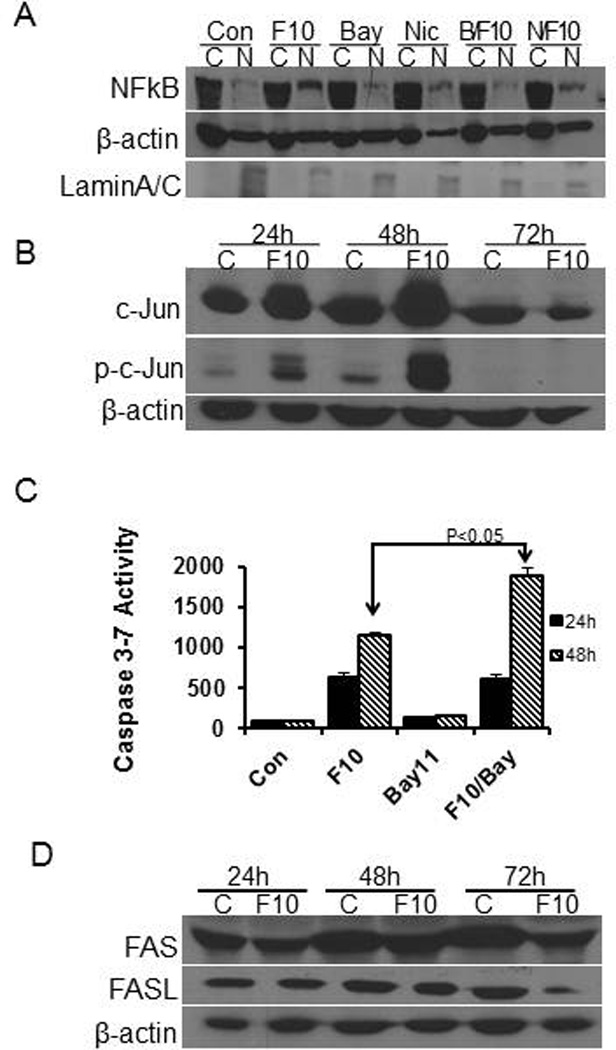
(A) Western blot for NF-kB p65 sub-unit evaluating cytoplasmic/nuclear ((C)/(N)) expression under the treatment conditions as indicated. (B) Western blot for c-Jun and p-c-Jun in control and F10-treated HL-60 cells at the indicated times. c-Jun is activated at 24 and 48 h consistent with AP-1 activation in response to F10 treatment. (C) F10-induced activation of caspase 3/7 activity is significantly enhanced at 48 h by NF-κB inhibition. (D) Western blot showing no change in expression for either Fas or FasL in HL-60 cells treated with F10 for 24, 48, or 72 h.
To further explore the role of autocrine Fas/FasL signaling for F10-induced activation of the extrinsic pathway, we evaluated whether NOK1, an antibody that sequesters FasL, might decrease F10-induced Caspase 8 activation. The effects of NOK1 were not significant for inhibiting Caspase 3/7 at either 24 or 48 h. NOK1 treatment did significantly reduce caspase 8 activity at 24 h; however the reduction was only about 10% of total caspase 8 activity (Figure 2e and f). These studies indicate the Fas pathway may be involved in F10-induced apoptosis in AML cells (based on F10+CH-11 synergy), but are not consistent with upregulation of FasL and autocrine activation of the Fas pathway as being the predominant death-inducing process in F10-treated AML cells as observed in colon cancer cells treated with 5-FU/LV [2, 12, 13].
3.3. Fas/FasL Clustering in Lipid Rafts
Confocal microscopy studies were undertaken in HL60 cells to determine whether F10-treatment altered the sub-cellular localization of the Fas death receptor and FasL. F10 induced clustering of Fas (Figure 4) and FasL (Figure 5), although Western blot did not indicate a substantial change in global levels for either protein (Figure 3d). We also investigated whether F10-treatment resulted in co-localization of Fas and FasL at the plasma membrane and within lipid rafts which were detected by localization of FITC-labeled cholera toxin B subunit (CtxB), a marker of glycolipoprotein microdomain-1 (GM1), a marker for lipid rafts. Lipid rafts are regions of the plasma membrane that are rich in cholesterol and glycosphingolipids and that may facilitate tightpacking of death receptor signaling molecules, thus promoting trimerization [21]. Lipid rafts may also play a role in cell-survival signaling [15]. F10 treatment caused clustering of Fas in the plasma membrane and decreased levels of lipid rafts, although some cells displayed colocalization of Fas with remaining lipid rafts (Figure 4). Analogous experiments detecting FasL and lipid rafts using confocal microscopy revealed an identical pattern for FasL as for Fas consistent with Fas/FasL co-localization (Figure 5). These studies demonstrate that F10-induces co-localization of Fas and FasL at the plasma membrane, including within remaining lipid rafts which are largely depleted by F10 treatment. Thus F10 treatment activates the extrinsic apoptotic pathway and this is accompanied by changes in the composition of the plasma membrane that promote Fas/FasL co-localization. Inhibition of NF-κB had no detectable effect on Fas/FasL localization in the plasma membrane of F10-treated AML cells (Supplementary Figure S3) indicating the altered sub-cellular distribution of Fas/FasL is independent of NF-κB activation, consistent with a predominantly anti-apoptotic role for NF-κB in response to F10-treatment in AML cells in contrast to a proapoptotic role in 5-FU-treated colon cancer cells.
Figure 4. F10 treatment results in clustering of Fas and co-localization with lipid rafts in treated cells.
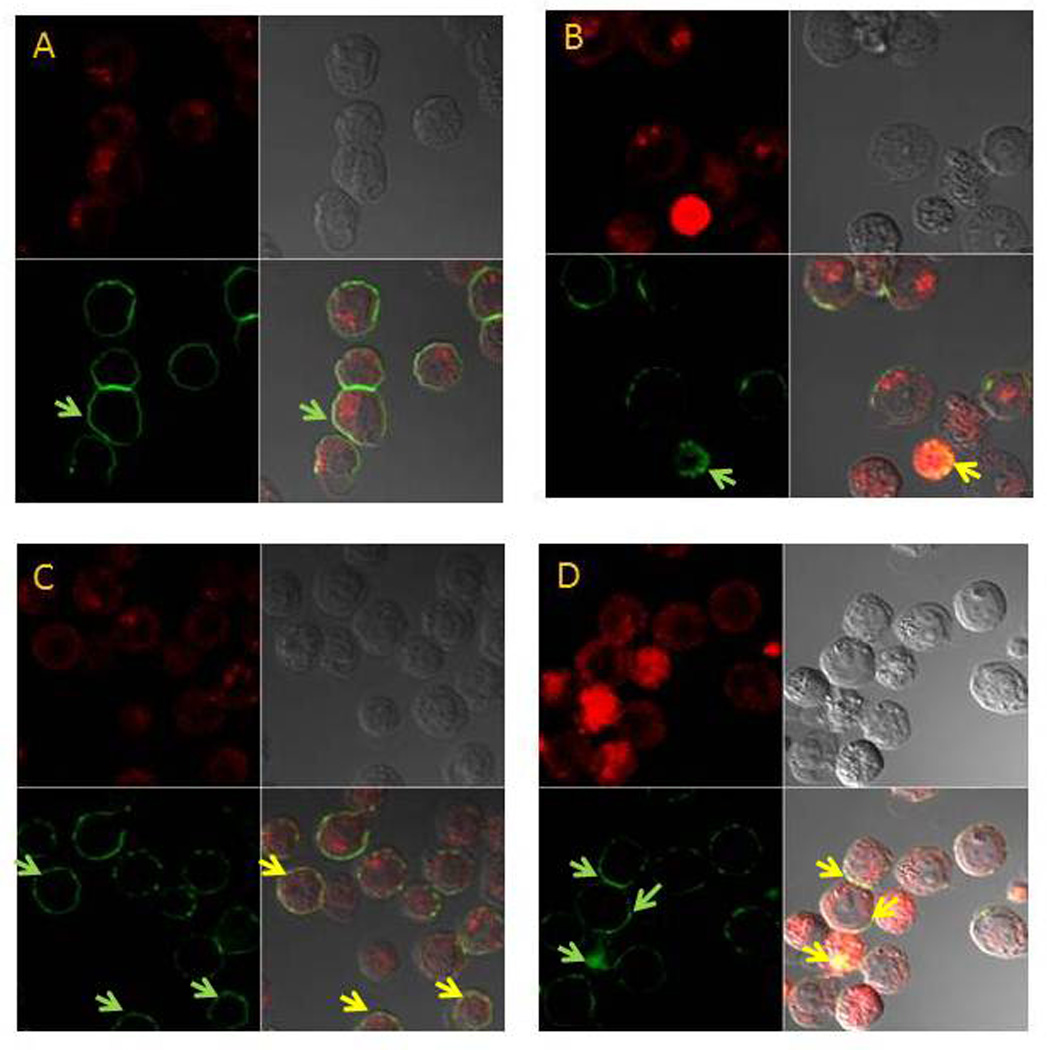
Confocal microscopy images for Fas in HL-60 cells (A) no treatment; (B) F10 treated; (C) Simvastatin treatment; (D) F10+simvastatin treatment. Confocal images: Fas – upper left (red); DIC image (upper right); cholera toxin (lipid rafts) – lower left (green; green arrow shows lipid rafts); Fas + DIC + cholera toxin merged (co-localized = yellow; yellow arrow shows Fas co-localized with lipid rafts).
Figure 5. F10 treatment results in clustering of FasL and co-localization with lipid rafts in treated cells.
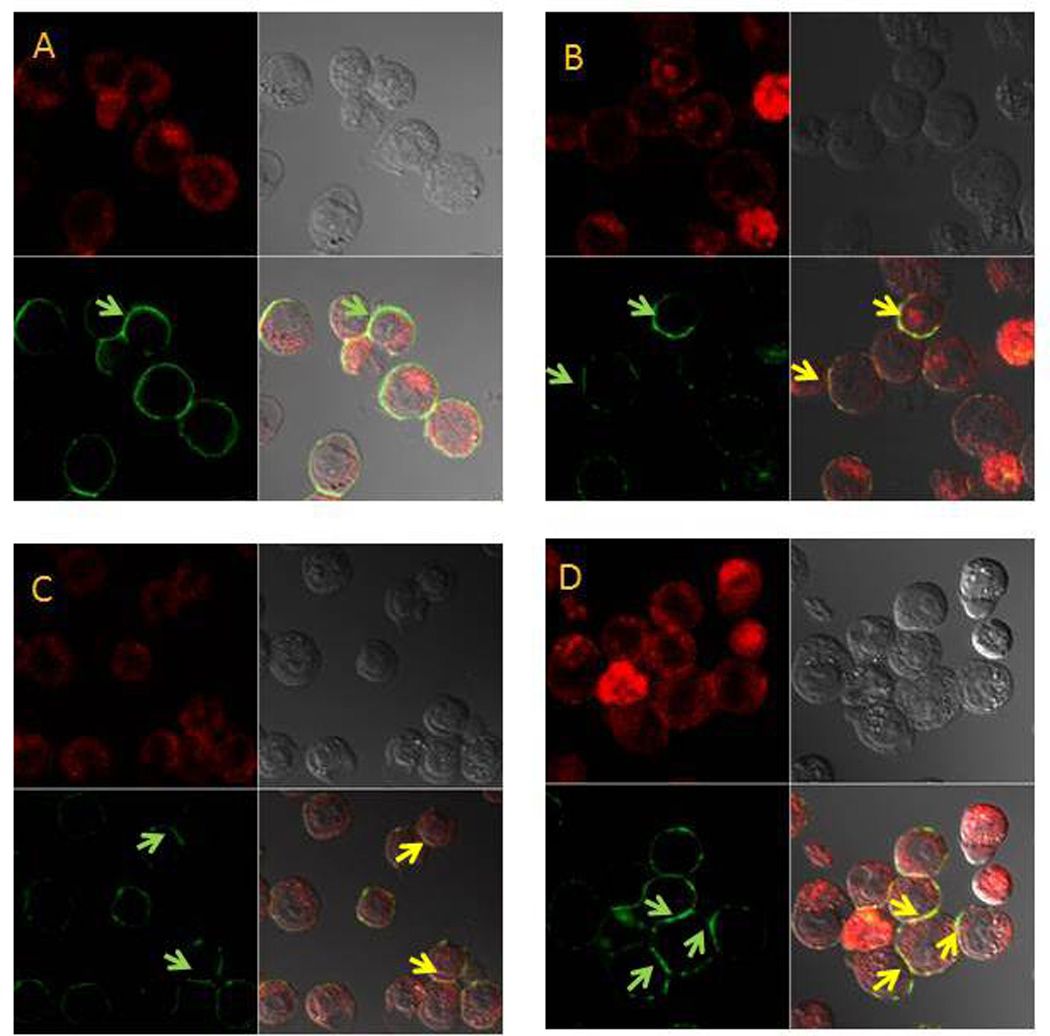
Confocal microscopy images for FasL in HL-60 cells (A) no treatment; (B) F10 treated; (C) Simvastatin treatment; (D) F10+simvastatin treatment. Confocal images: FasL – upper left (red); DIC image (upper right); cholera toxin (lipid rafts) – lower left (green; green arrow shows lipid rafts); FasL + DIC + cholera toxin merged (colocalized = yellow; yellow arrow shows FasL co-localized with lipid rafts).
3.4. Simvastatin decreases lipid rafts and promotes apoptosis without inducing Fas clustering
The quantities of lipid rafts and their activity in promoting death receptor activation can be modulated by agents that affect cholesterol levels such as β-cyclodextrin [16], or inhibitors of HMG-CoA reductase (e.g. statins) [22]. Simvastatin treatment reduced lipid rafts in AML cells (Figure 3) and also reduced viability and induced apoptosis in HL-60 (Figure 6a and b) and in OCI-AML3 cells (Supplementary Figure S4) in a dosedependent manner. Simvastatin also induced clustering of Fas (Figure 4) and FasL (Figure 5) in the plasma membrane including localization within remaining lipid rafts (Figure 4). Simvastatin treatment also enhanced F10-induced apoptosis (Figure 6a and b) and cotreatment of HL-60 cells with simvastatin and F10 was synergistic with combination index values 0.77 (ED50), 0.73 (ED75), and 0.73 (ED90). Interestingly, while both F10 and simvastatin independently decreased lipid raft levels, the combination of F10 and simvastatin did not display a further decrease in lipid raft levels, however the clustering of Fas and FasL into remaining lipid rafts was markedly enhanced for the combination (Figures 4,5). These results indicate that combining statins or other metabolic inhibitors with F10 enhances death receptor clustering in AML cells and that lipid rafts likely contribute to cell death signaling while likely playing a role in cell-survival signaling, as well.
Figure 6. Simvastatin is synergistic with F10 for AML treatment and both reduces lipid raft levels and increases Fas clustering in lipid rafts.
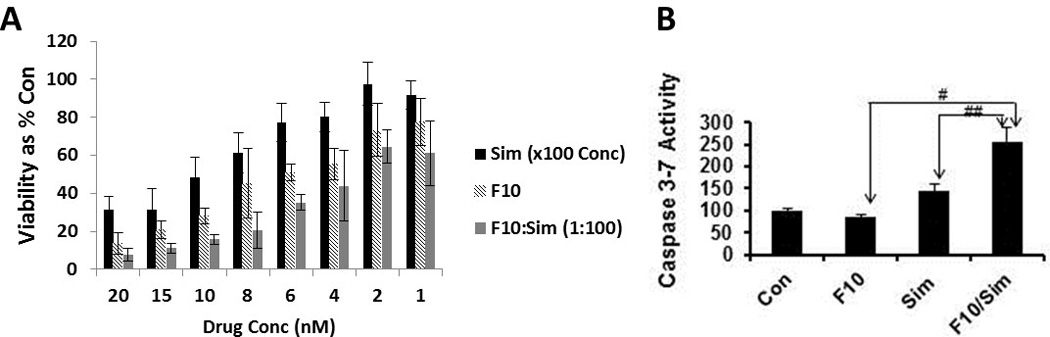
(A,B) Effects of simvastatin and F10+simvastatin on the viability (A) and caspase 3/7 activity (B) of HL-60 cells.
4. Discussion
Our previous studies demonstrated TS and Top1 were dual targets for F10 and that F10-induced thymineless AML cell death was predominantly apoptotic [4, 5]. The present studies demonstrate that F10-activates the extrinsic apoptotic pathway in AML cells including HL60 (Figure 1) as well as OCI-AML3, Molm13, THP1, and Jurkat (Supplementary Figure S1). F10-treatment enhanced Fas localization in the plasma membrane, including in lipid rafts, (Figure 3) and enhanced activation of Fas without upregulating either Fas or FasL protein expression (Figure 3D). These findings have therapeutic consequences as agents that stimulate the Fas death pathway, i.e. the agonistic Fas antibody CH-11 in the present studies, synergize with F10 and enhance treatment efficacy.
The mechanism by which F10 induces thymineless death following Top1cc formation in AML cells is currently not known. Previous studies with alternative fluoropyrimidine drugs (e.g. FdU and 5-FU) [23] as well as folate-based TS inhibitors [24] demonstrated thymineless cell death was consequent to DNA damage. While both the intrinsic [25] and extrinsic [13] apoptotic pathways have been implicated previously in 5-FU-induced apoptosis, our studies indicate the extrinsic pathway initiates apoptosis in F10-treated AML cells. Our studies show the Fas death pathway is activated in AML cells in response to F10-treatment (Figures 2–5) which provides new insights into how this agent may be used clinically. We also demonstrate that simvastatin treatment promotes Fas and FasL colocalization consistent with Fas-mediated death signaling in response to a common cellstress pathway rather than a specific consequence of Top1cc formation and DNA DSBs. Further, the combination of F10 and simvastatin is synergistic (Figure 6) indicating that the convergence of multiple inputs into this cell death signaling pathway has positive therapeutic consequences. Activation of the Fas pathway via F10 and/or simvastatin treatment may also sensitize leukemic cells to activated cytotoxic T-lymphocytes that express FasL, enhancing the immune anti-leukemia response and positively affecting treatment outcomes.
Our studies unexpectedly showed F10 treatment reduced lipid raft levels in AML cells which could indicate a role for lipid rafts in cell-survival signaling as lipid rafts have been shown to contribute to both cell death and survival signaling [15]. HMG-CoA inhibition with simvastatin induced similar effects on lipid raft levels as for F10. Interestingly, both treatments, as well as the synergistic co-treatment, caused clustering of Fas/FasL in remaining lipid rafts consistent with a role in cell death signaling as well. Other studies have demonstrated involvement of lipid rafts in Fas death pathway activation. For example, treatment of CD44+ AML cells with the antibody A3D8 induced clustering of Fas into lipid rafts independent of changes in Fas expression and initiated apoptosis via the extrinsic pathway [26]. The anti-tumor lipid edelfosine also promoted lipid raft formation and Fas localization into lipid rafts in T-cell ALL Jurkat cells and stimulated activation of the extrinsic apoptotic pathway [27]. Hence, the use of F10 or other strategies directed towards DNA damage may be complemented by use of edelfosine or other pro-apoptotic lipids that stimulate Fas localization into lipid rafts by direct interaction with the plasma membrane.
The pathways that culminate with Fas localization into lipid rafts in response to F10 treatment require further clarification. As previous studies demonstrated NF-κB and AP-1 modulated FasL expression in other systems [13, 28, 29], we determined to what extent components of these transcription factor complexes are activated in response to F10 treatment. F10 induced nuclear translocation of p65/Rel in AML cells as well as phosphorylation of c-Jun (Figure 3), a component together with c-Fos of AP-1. F10 did not, however, alter overall cellular levels of Fas or FasL in our cells indicating direct transcriptional upregulation of FasL is not important for F10-induced apoptosis in AML cells. Further, inhibiting NF-κB enhanced rather than reduced F10-induced apoptosis consistent with NF-κB being predominantly involved in pro-survival signaling in response to F10-treatment. These results are consistent with F10-treatment activating pro-apoptotic signaling resulting in lipid raft depletion and Fas/FasL co-localization in the plasma membrane, including in remaining lipid rafts. The signaling process(es) responsible for Fas/FasL co-localization in the plasma membrane and the pro-survival and pro-death signaling processes mediated via lipid rafts in F10-treated AML cells are under further investigation in our laboratory.
Supplementary Material
Highlights.
F10, a promising drug-candidate for AML, activates the extrinsic apoptotic pathway.
F10 depletes AML cells of lipid rafts and promotes clustering of Fas and Fas ligand
Lipid rafts may contribute to both cell survival and cell death signaling in AML cells
F10 is synergistic with statin drugs which also deplete AML cells of lipid rafts
Diverse treatments activate a common apoptotic pathway in AML cells
Acknowledgement
Author’s contributions: W.G. designed experiments, analyzed data, and wrote the paper. J.G. helped design and performed experiment. T.S.P. helped with review and revision of the manuscript.
Funding: This work was supported in parts by NIH (U01CA102532; W.H. Gmeiner) and (P30; CA012197); Doug Coley Foundation for Leukemia Research (T. Pardee), the Frances P Tutwiler Fund (T. Pardee) and the Translational Science Institute of WFUCCC (T. Pardee).
The authors thank Ken Grant for assistance with confocal microscopy studies, and Brandi Brickford for collecting microscopy images of NAD+ autofluorescence.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
W.H. Gmeiner and T.S. Pardee have ownership interest and managerial roles in Salzburg Therapeutics which holds licenses to F10 technology. J. Jennings-Gee reports no competing interests.
References
- 1.Gmeiner WH. Novel chemical strategies for thymidylate synthase inhibition. Curr Med Chem. 2005;12(2):191–202. doi: 10.2174/0929867053363432. [DOI] [PubMed] [Google Scholar]
- 2.Houghton JA, Harwood FG, Tillman DM. Thymineless death in colon carcinoma cells is mediated via fas signaling. Proc Natl Acad Sci U S A. 1997;94(15):8144–8149. doi: 10.1073/pnas.94.15.8144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gmeiner WH. Genetic Determinants of Fluoropyrimidine Chemotherapy. Drug Development Res. 2006;67:119–129. [Google Scholar]
- 4.Pardee TS, Gomes E, Jennings-Gee J, Caudell D, Gmeiner WH. Unique dual targeting of thymidylate synthase and topoisomerase 1 by FdUMP[10] results in high efficacy against AML and low toxicity. Blood. 2012;119:3561–3570. doi: 10.1182/blood-2011-06-362442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jennings-Gee J, Pardee TS, Gmeiner WH. Replication-dependent irreversible topoisomerase 1 poisoning is responsible for FdUMP[10] anti-leukemic activity. Exp Hematol. 2013;41(2):180–188. e4. doi: 10.1016/j.exphem.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pardee TS, Stadelman K, Jennings-Gee J, Caudell DL, Gmeiner WH. The Poison Oligonucleotide F10 is Highly Proliferative Against Acure Lympoblastic Leukemia While Sparing Normal Hemtopoietic Cells. Oncotarget. 2014 doi: 10.18632/oncotarget.1937. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gmeiner WH, Trump E, Wei C. Enhanced DNA-directed effects of FdUMP[10] compared to 5FU. Nucleosides Nucleotides Nucleic Acids. 2004;23(1–2):401–410. doi: 10.1081/ncn-120028336. [DOI] [PubMed] [Google Scholar]
- 8.Liao ZY, Sordet O, Zhang HL, Kohlhagen G, Antony S, Gmeiner WH, Pommier Y. A novel polypyrimidine antitumor agent FdUMP[10] induces thymineless death with topoisomerase I-DNA complexes. Cancer Res. 2005;65(11):4844–4851. doi: 10.1158/0008-5472.CAN-04-1302. [DOI] [PubMed] [Google Scholar]
- 9.Lin CP, Ban Y, Lyu YL, Desai SD, Liu LF. A ubiquitin-proteasome pathway for the repair of topoisomerase I-DNA covalent complexes. J Biol Chem. 2008;283(30):21074–21083. doi: 10.1074/jbc.M803493200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang YW, Regairaz M, Seiler JA, Agama KK, Doroshow JH, Pommier Y. Poly(ADP-ribose) polymerase and XPF-ERCC1 participate in distinct pathways for the repair of topoisomerase I-induced DNA damage in mammalian cells. Nucleic Acids Res. 2011;39(9):3607–3620. doi: 10.1093/nar/gkq1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scaffidi C, Schmitz I, Zha J, Korsmeyer SJ, Krammer PH, Peter ME. Differential modulation of apoptosis sensitivity in CD95 type I and type II cells. J Biol Chem. 1999;274(32):22532–22538. doi: 10.1074/jbc.274.32.22532. [DOI] [PubMed] [Google Scholar]
- 12.Tillman DM, Petak I, Houghton JA. A Fas-dependent component in 5-fluorouracil/leucovorin-induced cytotoxicity in colon carcinoma cells. Clin Cancer Res. 1999;5(2):425–430. [PubMed] [Google Scholar]
- 13.Harwood FG, Kasibhatla S, Petak I, Vernes R, Green DR, Houghton JA. Regulation of FasL by NF-kappaB and AP-1 in Fas-dependent thymineless death of human colon carcinoma cells. J Biol Chem. 2000;275(14):10023–10029. doi: 10.1074/jbc.275.14.10023. [DOI] [PubMed] [Google Scholar]
- 14.Mollinedo F, Gajate C. Fas/CD95 death receptor and lipid rafts: new targets for apoptosis-directed cancer therapy. Drug Resist Updat. 2006;9(1–2):51–73. doi: 10.1016/j.drup.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 15.Reis-Sobreiro M, Roue G, Moros A, Gajate C, de la Iglesia-Vicente J, Colomer D, Mollinedo F. Lipid raft-mediated Akt signaling as a therapeutic target in mantle cell lymphoma. Blood Cancer J. 2013;3:e118. doi: 10.1038/bcj.2013.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li YC, Park MJ, Ye SK, Kim CW, Kim YN. Elevated levels of cholesterol-rich lipid rafts in cancer cells are correlated with apoptosis sensitivity induced by cholesteroldepleting agents. Am J Pathol. 2006;168(4):1107–1118. doi: 10.2353/ajpath.2006.050959. quiz 1404-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Daniel RA, Rozanska AL, Thomas HD, Mulligan EA, Drew Y, Castelbuono DJ, Hostomsky Z, Plummer ER, Boddy AV, Tweddle DA, Curtin NJ, Clifford SC. Inhibition of poly(ADP-ribose) polymerase-1 enhances temozolomide and topotecan activity against childhood neuroblastoma. Clin Cancer Res. 2009;15(4):1241–1249. doi: 10.1158/1078-0432.CCR-08-1095. [DOI] [PubMed] [Google Scholar]
- 18.Sharpe R, Pearson A, Herrera-Abreu MT, Johnson D, Mackay A, Welti JC, Natrajan R, Reynolds AR, Reis-Filho JS, Ashworth A, Turner NC. FGFR signaling promotes the growth of triple-negative and basal-like breast cancer cell lines both in vitro and in vivo. Clin Cancer Res. 17(16):5275–5286. doi: 10.1158/1078-0432.CCR-10-2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 20.Huang DC, Hahne M, Schroeter M, Frei K, Fontana A, Villunger A, Newton K, Tschopp J, Strasser A. Activation of Fas by FasL induces apoptosis by a mechanism that cannot be blocked by Bcl-2 or Bcl-x(L) Proc Natl Acad Sci U S A. 1999;96(26):14871–14876. doi: 10.1073/pnas.96.26.14871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eramo A, Sargiacomo M, Ricci-Vitiani L, Todaro M, Stassi G, Messina CG, Parolini I, Lotti F, Sette G, Peschle C, De Maria R. CD95 death-inducing signaling complex formation and internalization occur in lipid rafts of type I and type II cells. Eur J Immunol. 2004;34(7):1930–1940. doi: 10.1002/eji.200324786. [DOI] [PubMed] [Google Scholar]
- 22.Wu H, Jiang H, Lu D, Xiong Y, Qu C, Zhou D, Mahmood A, Chopp M. Effect of simvastatin on glioma cell proliferation, migration, and apoptosis. Neurosurgery. 2009;65(6):1087–1096. doi: 10.1227/01.NEU.0000360130.52812.1D. discussion 1096-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lawrence TS, Davis MA, Tang HY, Maybaum J. Fluorodeoxyuridine-mediated cytotoxicity and radiosensitization require S phase progression. Int J Radiat Biol. 1996;70(3):273–280. doi: 10.1080/095530096145003. [DOI] [PubMed] [Google Scholar]
- 24.Yin MB, Guimaraes MA, Zhang ZG, Arredondo MA, Rustum YM. Time dependence of DNA lesions and growth inhibition by ICI D1694, a new quinazoline antifolate thymidylate synthase inhibitor. Cancer Res. 1992;52(21):5900–5905. [PubMed] [Google Scholar]
- 25.Ge X, Wang Y, Li Q, Yu H, Ji G, Miao L. NK4 regulates 5-fluorouracil sensitivity in cholangiocarcinoma cells by modulating the intrinsic apoptosis pathway. Oncol Rep. 2013;30(1):448–454. doi: 10.3892/or.2013.2427. [DOI] [PubMed] [Google Scholar]
- 26.Qian H, Xia L, Ling P, Waxman S, Jing Y. CD44 ligation with A3D8 antibody induces apoptosis in acute myeloid leukemia cells through binding to CD44s and clustering lipid rafts. Cancer Biol Ther. 2012;13(13):1276–1283. doi: 10.4161/cbt.21784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gajate C, Gonzalez-Camacho F, Mollinedo F. Involvement of raft aggregates enriched in Fas/CD95 death-inducing signaling complex in the antileukemic action of edelfosine in Jurkat cells. PLoS One. 2009;4(4):e5044. doi: 10.1371/journal.pone.0005044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peng Y, Gallagher SF, Haines K, Baksh K, Murr MM. Nuclear factor-kappaB mediates Kupffer cell apoptosis through transcriptional activation of Fas/FasL. J Surg Res. 2006;130(1):58–65. doi: 10.1016/j.jss.2005.07.030. [DOI] [PubMed] [Google Scholar]
- 29.Kasibhatla S, Brunner T, Genestier L, Echeverri F, Mahboubi A, Green DR. DNA damaging agents induce expression of Fas ligand and subsequent apoptosis in T lymphocytes via the activation of NF-kappa B and AP-1. Mol Cell. 1998;1(4):543–551. doi: 10.1016/s1097-2765(00)80054-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.