

Abstract
Tuberculosis (TB) is considered a major worldwide health problem with 10 million new cases diagnosed each year. Our understanding of TB immunology has become greater and more refined since the identification of Mycobacterium tuberculosis (MTB) as an etiologic agent and the recognition of new signaling pathways modulating infection. Understanding the mechanisms through which the cells of the immune system recognize MTB can be an important step in designing novel therapeutic approaches, as well as improving the limited success of current vaccination strategies. A great challenge in chronic disease is to understand the complexities, mechanisms, and consequences of host interactions with pathogens. Innate immune responses along with the involvement of distinct inflammatory mediators and cells play an important role in the host defense against the MTB. Several classes of pattern recognition receptors (PRRs) are involved in the recognition of MTB including Toll-Like Receptors (TLRs), C-type lectin receptors (CLRs) and Nod-like receptors (NLRs) linked to inflammasome activation. Among the TLR family, TLR1, TLR2, TLR4, and TLR9 and their down-stream signaling proteins play critical roles in the initiation of the immune response in the pathogenesis of TB. The inflammasome pathway is associated with the coordinated release of cytokines such as IL-1β and IL-18 which also play a role in the pathogenesis of TB. Understanding the cross-talk between these signaling pathways will impact on the design of novel therapeutic strategies and in the development of vaccines and immunotherapy regimes. Abnormalities in PRR signaling pathways regulated by TB will affect disease pathogenesis and need to be elucidated. In this review we provide an update on PRR signaling during M. tuberculosis infection and indicate how greater knowledge of these pathways may lead to new therapeutic opportunities.
Keywords: Tuberculosis, TLRs, inflammasome
Introduction
Tuberculosis (TB) is one of the most common infections worldwide, and in 2012, an estimated 8.6 million people developed TB and 1.3 million died from the disease (including 320,000 deaths among HIV-positive people) [1, 2]. Mycobacterium tuberculosis (MTB) is an intracellular pathogen capable of infecting and surviving within the host’s mononuclear cells particularly macrophages. This involves sequestration of MTB within organized granulomas. Elimination of the microorganism is through a combination of various killing mechanisms including apoptosis of host macrophages [3]. These responses are orchestrated by T helper 1-type (Th1) pro-inflammatory cytokines, which are synthesized by phagocytes upon recognition of pathogen-associated molecular patterns (PAMPs) on MTB by pattern recognition receptors (PRRs). MTB is usually transmitted via aerosols and establishes a stable infectious state in the respiratory system. There, MTB is engulfed by macrophages and dendritic cells (DCs), which serve as host cells for MTB survival and propagation [4]. Binding of MTB ligands to TLR-2, -4 and -9 initiates release of inflammatory mediators, expression of adhesion molecules and further recruitment of macrophages, DCs and PMN to the MTB infected area [5].
Although the host’s innate immune response to MTB infection is critical for the initial defense against bacteria, the adaptive immune response is ultimately required for containment of the infection in the chronic stage of disease. Adaptive immunity to MTB infection is characterized by the appearance of antigen specific CD4+ T-cells that secrete IFN-γ, which, in turn, activates macrophages and other antigen presenting cells (APC) to kill intracellular bacteria [6]. CD8+ T-cells are also important cells for controlling MTB during the chronic phase of infection [7, 8]. In addition, Th17 cells and IL-17 have been reported to be involved in the pathogenesis of MTB [9]. IL-17 is a proinflammatory cytokine produced by Th17 cells and by airway structural cells, which provides IFN-γ-dependent or IFN-γ-independent protection to MTB infection (see Figs. 1 and 2) [10–12].
Fig. 1.
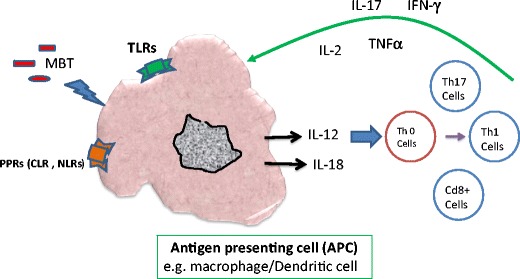
Schematic diagram indicating the role of specific cell types and mediators on the induction of IFNγ by MTB and the subsequent killing of macrophage-resident bacteria. Abbreviations: CLR C-type lectin receptors, MTB Mycobacterium tuberculosis, TLRs Toll like receptors, NLRs NOD-like receptors
Fig. 2.
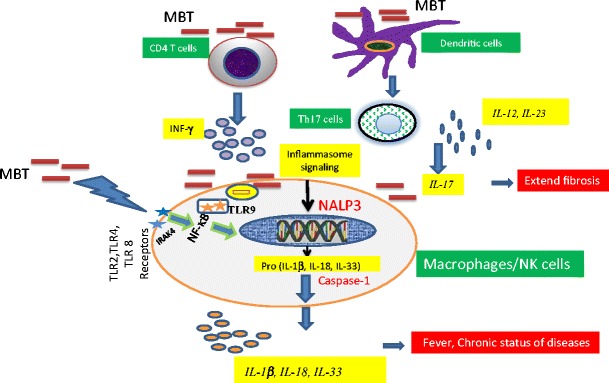
The putative role of TLR/Inflammasome signaling on the regulation of MTB in the cells. Abbreviation: TLR Toll like receptor, MTB Mycobacterium tuberculosis
Indeed, in models of MTB infection, IL-17 and Th17 cells were first implicated in the protective immune response to rapidly growing extracellular bacteria in the lung and gut mucosal surfaces through efficient induction of neutrophil recruitment and tissue repair [13–15]. IL-17 and Th17 cells are important during the initial stages of infection and act upon hematopoietic and non-hematopoietic cells to promote the secretion of antimicrobial peptides such as G-CSF and CXC chemokines. As a consequence of this, DCs migrate to the local lymph nodes and induce the differentiation of both Th1 and Th17 cells. The increased levels of chemokines in the infected lung also promote recruitment of other protective cells such as macrophages and PMN and the formation of mononuclear granulomas. Moreover, an accumulation cytokines such as IL-6 and IL-23 in the lungs can further induce the differentiation and activation of Th17 cells and accelerate the pathogenesis of TB [16].
In this review we focus on the role of signal transduction pathways which have an impact on the pathogenesis of TB. Among these, the generation of ROS and the later activation of PPRs including TLRs and of the inflammasome are highlighted.
Role of Reactive Oxygen Specious (ROS) in Pathogenesis of TB
Reactive oxygen species (ROS) and reactive nitrogen species (RNS) are considered to play important role in the pathogenesis of various inflammatory diseases [17, 18]. Under physiologic conditions; ROS are generated as byproducts of oxygen metabolism [17]. ROS are found in all biological systems and originate from the metabolism of molecular oxygen (O2). Under physiological conditions O2 undergoes reduction by accepting four electrons which results in the formation of water [18]. During this process, reactive intermediates such as the superoxide anion (∙O2), hydrogen peroxide (H2O2) and hydroxyl (∙OH)− radicals are formed [19]. Activated macrophages express two major enzymes, phagocyte oxidase (NOX2/gp91phox) and inducible nitric oxide synthase (NOS2), which are able to generate reactive oxygen intermediates (ROI) and reactive nitrogen intermediates (RNI), respectively. Upon phagocytosis, the preformed NOX2 subunits assemble into an enzymatically active enzyme complex that transfers electrons across the membrane from cytosolic NADPH to molecular oxygen. This produces ∙O2 which dismutate into hydrogen peroxide (H2O2) and thus generate ∙OH radicals which are toxic to MTB [20]. Following inhalation of MTB, alveolar macrophages engulf the bacilli and initiate their killing using a number of mechanisms including the generation of ROI and RNI [21, 22].
The rapid generation of ROS is critical in host defense against many bacteria and fungi, and ROS has broad signaling functions [23]. For example, the NADPH oxidase protein complexes generate the superoxide anion and downstream ROS. NADPH oxidase is the principal source of ROS generation in activated neutrophils and macrophages. Thus, NADPH oxidase has an important role in host defense against MTB and any patients with a loss of function mutation in genes encoding components of this enzyme complex could be deficient in killing bacilli. Indeed, mutations in the CYBB gene encoding the gp91 (phox) subunit of the phagocyte NADPH oxidase is associated with MTB [21]. In addition, a hemizygous splice mutation in intron 5 of CYBB was linked to the concomitant occurrence of chronic granulomatous disease (CGD) with MTB [24].
IFN-γ induces NOS2 and its product nitric oxide (NO) which in turn can be broken down to nitrite and nitrate. Under acidic conditions, such as within the phagosomes of IFN-γ activated macrophages, nitrite forms nitrous acid, which dismutates to NO and the toxic radical, nitrogen dioxide [25]. NO can synergize with superoxide, produced by the macrophage or generated as byproduct of respiratory metabolism by the pathogen, to form the highly poisonous peroxynitrite (ONOO−) radical [26]. These ROI and RNI react with a wide range of molecules, including nucleic acids, proteins, lipids and carbohydrates, resulting in the killing of MTB. To counteract these actions, MTB uses a variety of molecules to either detoxify ROI and RNI before they can inflict damage or to repair the damage they cause [27]. In particular, the presence of MTB results in glucose-6-phosphate (G6P) being oxidised by NADP-dependent and F420-dependent (FGD1) dehydrogenases to generate NADPH, an important source of electrons, and thereby overcome oxidative stress [28]. In addition, MTB uses a combination of its cell surface alpha-ketoacid dehydrogenase complexes to form a NADH-dependent peroxidase and peroxynitrite reductase [29].
Role of Toll Like Receptors (TLRs) in Pathogenesis of MTB
Pattern recognition receptors (PRRs) are a group of receptors which sense the presence of bacteria, fungi and viruses. PRRs are also responsible for recognizing endogenous molecules released from damaged cells, which are named damage associated molecular patterns (DAMPs) [30, 31]. To date, four different types of PRR families have been characterized [32]. These families include transmembrane proteins such as the TLRs and c-type lectin receptors (CTLRs), as well as cytoplasmic proteins such as the Retinoic acid-inducible gene (RIG)-I-like receptors (RLRs) and NOD-like receptors (NLRs) [33].
TLRs are a family of single membrane-spanning receptors of which 10 have been characterized in man and 13 in mouse [34–36]. TLRs play a critical role in both innate resistance and the initiation of adaptive immunity to infectious agents [37–40]. They act by recognizing pathogen-associated molecular patterns (PAMPs) or endogenous inflammation-associated molecules [36, 41, 42]. These are distinct molecular structures on microbes and different sets of TLRs have been associated with the response to different classes of microorganisms e.g. recognition of viruses by TLR3, TLR7, TLR8 and TLR9 [36, 41, 43–46].
Bacterial DNA which contains unmethylated CpG oligonucleotides (ODN) motifs also acts as important regulators of human neutrophil functions via TLR9. For example, stimulation of the TLR9 pathway by CpGODN induces CXCL8 production by human neutrophils via the generation of ONOO− [47, 48]. TLR-ligand binding can induce two signaling pathways, the myeloid differentiation primary response gene 88 (MyD88)-dependent and MyD88-independent pathways, which induce the production of both pro-inflammatory cytokines and type I IFNs [36, 49, 50]. MyD88 is used by all TLRs except TLR3. These two distinct responses are mediated via the selective use of adaptor molecules recruited to the Toll/IL-1 receptor (TIR) domains of TLRs after ligand binding. Four adaptor molecules have been identified to date: MyD88, TIR-associated protein (TIRAP), TIR domain-containing adaptor protein-inducing IFN-β (TRIF) and TRIF related adaptor molecules (TRAM) [51].
MyD88 and TIRAP are responsible for the induction of pro-inflammatory genes, and TRIF and TRAM induce IFNs. In MyD88-dependent signaling, MyD88 is recruited to, and associates with, the cytoplasmic domain of the TLRs upon ligand binding. Then IL-1R-associated kinase 4 (IRAK-4) and IRAK- 1 are subsequently recruited and activated by phosphorylation. Activated IRAK-4 phosphorylates IRAK-1, which then, in turn, associates with tumor necrosis factor receptor (TNFR)-associated factor 6 (TRAF6). TRAF6 activates transforming growth factor (TGF) activating kinase 1 (TAK1) [36], which, in turn, phosphorylates IKK-β and mitogen-activated protein kinase (MAPK) kinase6 (MKK6), leading to degradation of I-κB, nuclear translocation of NF-κB and induction of inflammatory genes [52].
As a result TLR activation upregulates the transcription of proinflammatory cytokines including IL-1β, TNF-α and IL-6 which are essential for the recruitment of immune cells to the site of infection and controlling MTB infection [53–55]. Activation of the MyD88-dependent pathway also results in the activation of mitogen-activated protein (MAP) kinases (MAPK) such as p38 and JNK, which leads to the activation of AP-1 [56]. During MyD88-independent signaling TLR4 activation triggers the induction of a type 1 IFN response, leading to the induction of IFN-α and IFN-inducible genes [55]. The TLRs known to be involved in recognition of MTB are TLR2, TLR4, TLR9, and possibly TLR8 [36, 41, 43–46]. Four primary immunodeficiencies (PIDs) involving mutations in MyD88, IRAK4, NEMO and IKBA are associated with altered susceptibility to M. tuberculosis [57–59].
TLR2 can form heterodimers with both TLR1 and TLR6. These heterodimers have been implicated in the recognition of mycobacterial cell wall glycolipids including lipoarabinomannan (LAM), lipomannan (LM), 38-kDa and 19-kD mycobacterial glycoproteins, phosphatidylinositol mannoside (PIM), triacylated (TLR2/TLR1) or diacylated (TLR2/TLR6) lipoproteins [49, 60, 61]. TLR2 and TLR1 act together to mediate responses to M. tuberculosis [62, 63] and the role of TLR1/2 gene variants in the predisposition to tuberculosis has been investigated. Most studies have focused on TLR2 variants and only weak and non-replicated associations have been reported to date [62, 63]. TLR2 is believed to be important in the initiation of the innate host defense against MTB [61, 64]. In addition, IL-1β production is dependent upon TLR2 and TLR6, but not TLR4 or TLR9, stimulation [65]. TLR2 is also important for IL-12 release in macrophages, but not in DCs [66]. TLR2−/− mice show defective granuloma formation following MTB infection and have a greatly enhanced susceptibility to infection compared to the WT mice [53, 67]. In addition, TLR2−/− mice are unable to control chronic infection with MTB [67, 68]. Mice lacking TLR9 also succumb earlier to MTB infection than wild-type animals [61, 66, 69–72].
The role of other TLRs, such as TLR4 and TLR9 in the pathogenesis of MTB has not studied in such detail [65, 73, 74]. Mice deficient in the TLR/IL-1R family receptor adaptor molecule MyD88 have been shown to be highly susceptible to infection with MTB, which suggests a major role for this pathway in the innate defense against the MTB [68, 75–86]. In addition, TLR2-induced ROS production plays a crucial role in the expression of CXCL8 and CCL2 in human monocytes requiring the activation of both p38 and ERK1/2 MAPK pathways [78]. Overexpression of both TLR2 and TLR4 are important for viable MTB infection in human cell lines [79].
Other studies in mice with inactivated TLR genes indicated that TLR2 is important in controlling and surviving MTB infection [68, 73]. However, other studies suggested that TLR4 is critical for surviving MTB infection [80, 87]. The importance of TLR4 may depend on the dose of MTB used for challenge [88] or the mouse strain used [81]. Human studies show that polymorphisms in both TLR2 and TLR4 are associated with increased susceptibility to microbial infections possibly by changing the Th1/Th2 response [82–85]. Interestingly, Fenhalls et al. reported that the expression of TLRs in TB lung granulomas related to the presence or absence of immunohistologically detectable IL-4 [86].
Changes in TLR expression and/or their down-stream activation state might represent useful markers of the immunological status of TB patients and their contacts. The TLR distribution in TB granulomas lesions indicates that TLR1, TLR2, and TLR4 are expressed in both immune cells and non-immune cells; however TLR9 is only detectable in immune cells [89]. Furthermore, in an animal model of TB, TLR8-deficient mice succumb more rapidly to infection with M. tuberculosis, despite efficiently controlling the number of viable bacilli in different organs. Although no changes in CD4+ and CD8+ T-cells were observed there were increases in lung neutrophils and macrophages. Exaggerated mortality was due to massive liver necrosis and was reversed by a combination of blocking antibodies to IL-1 and TNF-α. Thus, in this model of MTB infection, TLR8 plays a key role in dampening inflammation and tissue damage [90]. Overall, Recognition of MTB by TLRs triggers various intracellular signaling cascades ultimately resulting in the production of cytokines, chemokines and antimicrobial molecules [91, 92].
In humans, the association of TLR polymorphisms with susceptibility to TB remains to be confirmed [92]. Different polymorphisms in the human TLR2 gene were reported to associate with increased susceptibility to TB in some studies [93–97] but not others [98–101]. Furthermore, a MAL/TIRAP functional variant, affecting signaling through TLR2, was shown to be protective in TB [102]. Genetic polymorphisms in TLR4 were linked to an increased susceptibility and severity of pulmonary TB in an Asian population in India [103] but not in Indian or Chinese TB patients in Gambia [101, 102, 104]. This discrepancy might be due to a dynamic host-pathogen interplay between genetic and pathogen phenotypes [102].
Role of Pentraxin 3 (PTX3) in Pathogenesis of MTB
Pentraxin 3 (PTX3), or TNF-stimulated gene 14 (TSG-14), is a 42-kDa soluble pattern-recognition receptor produced by phagocytes and non-immune cells at sites of inflammation or injury and plays an important role in female fertility and vascular biology [103]. PTX3 shows up to 28 % sequence identity to human C-reactive protein (CRP) and serum amyloid P-component (SAP) [104, 105]. It is a member of the pentraxin family which are involved in the acute phase response to injury, trauma and infection [106]. PTX3 is rapidly secreted into the serum of mice and humans from extra-hepatic sources after LPS, IL-1 or TNF-α stimulation [107].
PTX3 binds to the complement component C1q [108] and to microorganisms, including Pseudomonas aeruginosa, Salmonella typhimurium and Aspergillus fumigates to induce innate immune responses [109, 110] and to drive a protective adaptive immunity [111]. Since whole mycobacteria and mycobacterial lipoarabinomannan strongly induce PTX3 production by human mononuclear phagocytes [112] a role for PTX3 in the immunobiology of mycobacterial infection has been inferred. Interestingly, PTX3 receptor gene variants are associated with an increased risk of pulmonary tuberculosis in West Africans [113]. Furthermore, PTX3, levels are significantly correlated with the severity of clinical presentation at diagnosis and of lung involvement in disease and may represent a good biomarker for inflammation and disease activity during MTB infection [112].
Role of Inflammasome Signaling in MTB
There are two classes of innate immune receptors described: a) TLRs, located on cell membranes or intracellularly, and b) NLRs located in the cytoplasm [114–118]. Both classes of receptors are programmed to recognize microbial PAMPs and danger-associated molecular patterns (DAMPs) and switch cells for activation to releasing of proinflammatory and chemokines. The importance of two receptors in pathogenesis of chronic lung disease has elicited much attention [119, 120]. In the next section, we describe the regulation of inflammasome signaling and discuss whether abnormalities in NLRP3 inflammasome function may be associated with MTB.
The inflammasome consists of a multimeric cytosolic complex comprising the adaptor protein apoptosis associated speck-like protein containing a caspase recruiting domain (ASC), a sensor protein such as NRLP3 together with the effector proteins caspase-1 and caspase 5 [119]. Several NLRs function in immunity through the formation of a multi-protein complex known as an inflammasome which play critical roles in the 0 pathogenesis of chronic disorders [119–121].
NLRs exist in three families; the NODs, the NLRPs and the (IPAFs). Stimulation of cells with PAMPs, or by DAMPs, leads to increased expression of IL-1β and other IL-1 cytokine family members, such as IL-18 and IL-33 [120]. Proinflammatory cytokines of the IL-1 family may play an important role in anti-mycobacterial host defense mechanisms [121]. Moreover, MTB stimulates inflammatory cells to release IL-1β through pathways involving TLR2/TLR6 and NOD2 receptors [122]. Recognition of MTB by TLR and NOD2 leads to increased transcription of pro-IL-1β through mechanisms involving ERK, p38 and Rip2, but not JNK. Interestingly, although caspase-1 is necessary for the processing of pro-IL-1β, activation of caspase-1 is not dependent on the stimulation of cells by MTB [123]. In human THP-1 macrophages, MTB activation results in secretion of IL-1β in an ASC/NLRP3-dependent manner [124]. In addition, Mycobacterium marinum activates IL-1β production in an NLRP3- and caspase-1-dependent manner in vitro highlighting the potential importance of inflammasome signaling in the pathogenesis of MBT [125, 126].
Inflammasome-mediated IL-1β secretion is triggered by a combination of signal transduction pathways activated via TLRs and purinergic (P2X7) receptors. In turn, IL-1β induces the release of GM-CSF which leads to the activation and increased survival of monocytes/macrophages and enhanced oxidative burst in the lungs, thus maintaining and prolonging inflammatory reactions [127]. The purinergic P2X7 receptor is the key driver of ATP-mediated inflammasome maturation and release of IL-1β [122, 128, 129]. Pro-inflammatory cytokine regulation by the inflammasome may be critical to long-term survival of MTB infection since experiments in IL-1α/β, IL-1R and IL-18 knockout (KO) mice have shown that these cytokines play a role in limiting bacterial burden in the lung, in regulating the subsequent expression of other cytokines, in controlling NO production and in the formation of granulomas [122, 128, 129].
Caspase-1 independent IL-1β production may also be critical for host resistance to MTB and this occurs independently of TLR signaling in vivo. Furthermore, although IL-1β induction by MTB in vitro depends on TLR triggering and the inflammasome, both triggers are dispensable for IL-1β production in mice infected with the pathogen in vivo [130]. Thus, although recent data established that IL-1β plays a critical component in innate resistance to MTB, the pathways involved in the expression and regulation of IL-1β induction following MTB infection in vivo are complex and may involve mechanisms that do not fit the classical paradigms of TLR recognition and inflammasome-mediated caspase-1 processing seen with other infections or in the response to MTB observed in vitro [130].
MTB-induced IL-1β secretion in human and mouse macrophages in vitro and this process was dependent on ASC, caspase-1, and NLRP3, but not NLCR4 [130]. In vivo, murine ASC helps protect the host from death during chronic MTB infection whilst the effects of Casp-1 and NLP3 were negligible. The inability of ASC KO mice to form organized granulomas and the reduced presence of lung dendritic cells indicates a breakdown in host defense against MTB. Thus, ASC was identified as a critical protein involved in the host response to MTB infection in an inflammasome-independent manner [75]. Other cytokines activated by the inflammasome have also been reported to play a role in the pathogenesis of MTB. Thus, IL-12 and IL-18 produced by dendritic cells and macrophages induce NK-cell activity and skew the immune response towards an IFN-γ-dependent Th1 response, which is considered critical for protection against MTB [8].
Data in MyD88-deficient mice which are highly susceptible to MTB and succumb very rapidly to infection supports a role for MyD88 in regulating MTB infection [75, 131]. MyD88, however, plays a role in both inflammasome and TLR signaling and this raises the possibility that a lack of IL-1β or IL-18 is responsible for the heightened susceptibility of MyD88 KO mice to MTB infection [122]. Fremond et al. reported that IL-1R-signalling is important for protection against MTB whilst IL-18R-signalling is not [128]. In contrast, Schneider et al has reported a similar degree of susceptibility to MTB infection to that observed in MyD88 KO mice in IL-18 KO mice [129]. IL-18 KO mice succumbed much more readily to experimental MTB infection than WT or TLR-2/-4 double KO (TLR-2/-4 DKO) mice. In the absence of IL-18, immunity to MTB was hampered by decreased Th1 responses and PMN-dominated lung immunopathology concomitant with unrestrained growth of the tubercle bacilli. Thus, some controversy still remains as to the precise role of IL-18 in the protective immunity against MTB infection [129].
Conclusion and Future Outlook
TB remains one of the leading causes of death from a single infectious agent worldwide. In order to generate better protective strategies we need to further define the pathological mechanisms underlying the immune response to MTB. Whilst inflammasome and TLR cross talk does not seem to be essential for the primary control of MTB infection, recent data suggests a critical role of these pathways in the persistence of MTB. Activation of these pathways results in the release of inflammatory mediators that recruit protective cells to the infected area. However, there is a down side to this effect. Excessive production of IL-23 and IL-17 causes pathology due to excessive recruitment and phenotypic changes in inflammatory cells. Hence, there is a fine balance between Th1 and inflammasome/TLRs responses that is central in defining the outcome of MTB infection. The role and mechanisms underpinning PTX3 and other PPRs in the immune response to MTB still requires further elucidation however.
In addition, it is critical to further define the mechanisms associated with the cross talk between TLRs and the inflammasome and to use this knowledge to generate rational protective strategies that promote a balanced acquired immune response with minimal collateral damage. Determination of key nodes within the pathways involved in the pathogenesis of MTB may provide new therapeutic targets to prevent the persistence of disease.
Abbreviations
- APC
Antigen-presenting cell
- BAL
Bronchoalveolar lavage
- cDC
Conventional dendritic cell
- COPD
Chronic obstructive pulmonary disease
- CXCL
CXC-chemokine ligand
- CTL
Cytotoxic T lymphocytes
- CMR
Cell-mediated reactions
- CLR
C-type lectin receptors
- DTH
Delayed-type hypersensitivity
- DCs
Dendritic cells
- IFN-γ
Interferon gamma
- IL
Interleukin
- IPAF
IL-1β converting enzyme (ICE) protease activating factor
- KO
Knockout
- LAM
Lipoarabinomannan
- LM
Lipomannan
- LPS
Lipopolysaccharide
- MTB
Mycobacterium tuberculosis
- MMPs
Matrix metalloproteinase
- NLRP
NACHT, LRR and PYD domain-containing protein
- PTX3
Pentraxin 3
- PAMPs
Pathogen-associated molecular patterns
- pDC
Plasmacytoid dendritic cell
- PMN
Polymorphonuclear cells
- PRRs
Pattern recognition receptors
- ROS
Reactive oxygen species
- TLR
Toll-like receptor
- TB
Tuberculosis
References
- 1.WHO Report—Global tuberculosis control. World Health Organization; Geneva: http://www.who.int/tb/publications/global_report/en/ (2013).
- 2.Incidence of tuberculosis (per 100,000 people) http://data.worldbank.org/indicator/SH.TBS.INCD.
- 3.Van Crevel R, Ottenhoff TH, van der Meer JW. Innate immunity to Mycobacterium tuberculosis. Clin Microbiol Rev. 2002;15:294–309. doi: 10.1128/CMR.15.2.294-309.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Giacomini E, Iona E, Ferroni L, Miettinen M, Fattorini L, Orefici G, et al. Infection of human macrophages and dendritic cells with Mycobacterium tuberculosis induces a differential cytokine gene expression that modulates T cell response. J Immunol. 2001;166:7033–41. doi: 10.4049/jimmunol.166.12.7033. [DOI] [PubMed] [Google Scholar]
- 5.Ahmad S. Pathogenesis, immunology, and diagnosis of latent Mycobacterium tuberculosis infection. Clin Dev Immunol. 2011;2011:814943. doi: 10.1155/2011/814943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan J, Xing Y, Magliozzo RS, Bloom BR. Killing of virulent Mycobacterium tuberculosis by reactive nitrogen intermediates produced by activated murine macrophages. J Exp Med. 1992;175(4):1111–22. doi: 10.1084/jem.175.4.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Pinxteren LA, Cassidy JP, Smedegaard BH, Agger EM, Andersen P. Control of latent Mycobacterium tuberculosis infection is dependent on CD8 T cells. Eur J Immunol. 2000;30(12):3689–98. doi: 10.1002/1521-4141(200012)30:12<3689::AID-IMMU3689>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 8.Ulrichs T, Kaufmann SH. New insights into the function of granulomas in human tuberculosis. J Pathol. 2006;208(2):261–9. doi: 10.1002/path.1906. [DOI] [PubMed] [Google Scholar]
- 9.Zhang M, Wang Z, Graner MW, Yang L, Liao M, Yang Q, et al. B cell infiltration is associated with the increased IL-17 and IL-22 expression in the lungs of patients with tuberculosis. Cell Immunol. 2011;270(2):217–23. doi: 10.1016/j.cellimm.2011.05.009. [DOI] [PubMed] [Google Scholar]
- 10.Khader SA, Bell GK, Pearl JE, et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol. 2007;8(4):369–77. doi: 10.1038/ni1449. [DOI] [PubMed] [Google Scholar]
- 11.Wozniak TM, Saunders BM, Ryan AA, Britton WJ. Mycobacterium bovis BCG-specific Th17 cells confer partial protection against Mycobacterium tuberculosis infection in the absence of gamma interferon. Infect Immun. 2010;78:4187–94. doi: 10.1128/IAI.01392-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Torrado E, Cooper AM. IL-17 and Th17 cells in tuberculosis. Cytokine Growth Factor Rev. 2010;21(6):455–62. doi: 10.1016/j.cytogfr.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ye P, Garvey PB, Zhang P, Nelson S, Bagby G, Summer WR, et al. Interleukin-17 and lung host defense against Klebsiella pneumoniae infection. Am J Respir Crit Care Med. 2001;25:335–40. doi: 10.1165/ajrcmb.25.3.4424. [DOI] [PubMed] [Google Scholar]
- 14.Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, et al. Requirement of Interleukin-17 receptor signalling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194:519–27. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Happel K, Dubin P, Zheng M, Ghilardi N, Lockhart C, Quinton L, et al. Divergent roles of IL-23 and IL-12 in host defense against Klebsiella pneumoniae. J Exp Med. 2005;202:761–9. doi: 10.1084/jem.20050193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khader SA, Gopal R. IL-17 in protective immunity to intracellular pathogens. Virulence. 2010;1:423–7. doi: 10.4161/viru.1.5.12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Langen RC, Korn SH, Wouters EF. ROS in the local and systemic pathogenesis of COPD. Free Radic Biol Med. 2003;35(3):226–35. doi: 10.1016/s0891-5849(03)00316-2. [DOI] [PubMed] [Google Scholar]
- 18.Andrade DR, Jr, Souza RB, Santos SA, Andrade DR. Oxygen free radicals and pulmonary disease. J Bras Pneumol. 2005;31(1):60–8. [Google Scholar]
- 19.Mak JC. Pathogenesis of COPD. Part II. Oxidative-anti-oxidative imbalance. Int J Tuberc Lung Dis. 2008;12(4):368–74. [PubMed] [Google Scholar]
- 20.Hartmann P, Becker R, Franzen C, Schell-Frederick E, Romer J, Jacobs M, et al. Phagocytosis and killing of Mycobacterium avium complex by human neutrophils. J Leukocyte Biol. 2001;69:397. [PubMed] [Google Scholar]
- 21.Azfar SF, Islam N. Suppression of Mycobacterium tuberculosis induced reactive oxygen species and tumor necrosis factor-alpha activity in human monocytes of systemic lupus erythematosus patients by reduced glutathione. Oman Med J. 2012;27(1):11–9. doi: 10.5001/omj.2012.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zuñiga J, Torres-García D, Santos-Mendoza T, Rodriguez-Reyna TS, Granados J, Yunis EJ. Cellular and humoral mechanisms involved in the control of tuberculosis. Clin Dev Immunol. 2012;2012:193923. doi: 10.1155/2012/193923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar A, Farhana A, Guidry L, Saini V, Hondalus M, Steyn AJ. Redox homeostasis in mycobacteria: the key to tuberculosis control? Expert Rev Mol Med. 2011;13:e39. doi: 10.1017/S1462399411002079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bustamante J, Aksu G, Vogt G, de Beaucoudrey L, Genel F, Chapgier A, et al. BCG-osisandtuberculosis in a child with chronic granulomatous disease. J Allergy Clin Immunol. 2007;120(1):32–8. doi: 10.1016/j.jaci.2007.04.034. [DOI] [PubMed] [Google Scholar]
- 25.Nathan C, Shiloh MU. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc Natl Acad Sci U S A. 2000;97(16):8841–8. doi: 10.1073/pnas.97.16.8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990;87(4):1620–4. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Montgomery LG, Lemon WS. The cellular reaction of the pleura to infection with Mycobacterium tuberculosis. J Thorac Cardiovasc Surg. 1993;2:429. [Google Scholar]
- 28.Gurumurthy M, Rao M, Mukherjee T, Rao SP, Boshoff HI, Dick T, et al. A novel F(420)—dependent anti-oxidant mechanism protects Mycobacterium tuberculosis against oxidative stress and bactericidal agents. Mol Microbiol. 2013;87(4):744–55. doi: 10.1111/mmi.12127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bryk R, Lima CD, Erdjument-Bromage H, Tempst P, Nathan C. Metabolic enzymes of mycobacteria linked to antioxidant defense by a thioredoxin-like protein. Science. 2002;295(5557):1073–7. doi: 10.1126/science.1067798. [DOI] [PubMed] [Google Scholar]
- 30.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–84. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 31.Chen GY, Nuñez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10:826–37. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meylan E, Tschopp J, Karin M. Intracellular pattern recognition receptors in the host response. Nature. 2006;442:39–44. doi: 10.1038/nature04946. [DOI] [PubMed] [Google Scholar]
- 33.Akira S, Uematsu S, Takeuchi O. Pathogen recognition andinnate immunity. Cell. 2006;12:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 34.Cui S, Eisenacher K, Kirchhofer A, Brzozka K, Lammens A, Lammens K, et al. The C-terminal regulatory domain is the RNA 50 -triphosphate sensor of RIG-I. Mol Cell. 2008;29:169–79. doi: 10.1016/j.molcel.2007.10.032. [DOI] [PubMed] [Google Scholar]
- 35.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–50. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 36.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–20. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 37.Casanova JL, Abel L, Quintana-Murci L. Human TLRs and IL-1Rs in host defense: natural insights from evolutionary, epidemiological, and clinical genetics. Annu Rev Immunol. 2011;29:447–91. doi: 10.1146/annurev-immunol-030409-101335. [DOI] [PubMed] [Google Scholar]
- 38.Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–7. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- 39.Medzhitov R, Preston-Hurlburt P, Janeway CA. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–7. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 40.Doyle SL, O’Neill LA. Toll-like receptors: from the discovery of NFkappaB to new insights into transcriptional regulations in innate immunity. Biochem Pharmacol. 2006;72:1102–13. doi: 10.1016/j.bcp.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 41.Gan L, Li L. Regulations and roles of the interleukin-1 receptor associated kinases (IRAKs) in innate and adaptive immunity. Immunol Res. 2006;35:295–302. doi: 10.1385/IR:35:3:295. [DOI] [PubMed] [Google Scholar]
- 42.Akira S, Hemmi H. Recognition of pathogen-associated molecular patterns by TLR family. Immunol Lett. 2003;85:85–95. doi: 10.1016/s0165-2478(02)00228-6. [DOI] [PubMed] [Google Scholar]
- 43.Kurt-Jones EA, Popova L, Kwinn L, Haynes LM, et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol. 2000;1:398–401. doi: 10.1038/80833. [DOI] [PubMed] [Google Scholar]
- 44.Kay E, Scotland RS, Whiteford JR. Toll-like receptors: role in inflammation and therapeutic potential. Biofactors. 2013 doi: 10.1002/biof.1156. [DOI] [PubMed] [Google Scholar]
- 45.Liu Y, Yin H, Zhao M, Lu Q. TLR2 and TLR4 in autoimmune diseases: a comprehensive review. Clin Rev Allergy Immunol. 2013;47(2):136–47. doi: 10.1007/s12016-013-8402-y. [DOI] [PubMed] [Google Scholar]
- 46.Hossain MM, Norazmi MN. Pattern recognition receptors and cytokines in Mycobacterium tuberculosis infection-the double-edged sword. Biomed Res Int. 2013;2013:179174. doi: 10.1155/2013/179174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–5. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 48.Mortaz E, Adcock IM, Ito K, Kraneveld AD, Nijkamp FP, Folkerts G. Cigarette smoke induces CXCL8 production by human neutrophils via activation of TLR9 receptor. Eur Respir J. 2010;36(5):1143–54. doi: 10.1183/09031936.00062209. [DOI] [PubMed] [Google Scholar]
- 49.El Kebir D, József L, Pan W, Wang L, Filep JG. Bacterial DNA activates endothelial cells and promotes neutrophil adherence through TLR9 signaling. J Immunol. 2009;182(7):4386–94. doi: 10.4049/jimmunol.0803044. [DOI] [PubMed] [Google Scholar]
- 50.Means TK, Jones BW, Schromm AB, et al. Differential effects of a Toll-like receptor antagonist on Mycobacterium tuberculosis-induced macrophage responses. J Immunol. 2001;166(6):4074–82. doi: 10.4049/jimmunol.166.6.4074. [DOI] [PubMed] [Google Scholar]
- 51.Kawai T, Akira S. TLR signaling. Cell Death Differ. 2006;13:816–25. doi: 10.1038/sj.cdd.4401850. [DOI] [PubMed] [Google Scholar]
- 52.Albiger B, Dahlberg S, Henriques-Normark B, Normark S. Role of the innate immune system in host defence against bacterial infections: focus on the toll-like receptors. J Intern Med. 2007;261:511–28. doi: 10.1111/j.1365-2796.2007.01821.x. [DOI] [PubMed] [Google Scholar]
- 53.Takeda K, Akira S. TLR signaling pathways. Semin Immunol. 2004;16(1):3–9. doi: 10.1016/j.smim.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 54.Bean AG, Roach DR, Briscoe H, France MP, Korner H, Sedgwick JD, et al. Structural deficiencies in granuloma formation in TNF gene-targeted mice underlie the heightened susceptibility to aerosol Mycobacterium tuberculosis infection, which is not compensated for by lymphotoxin. Immunology. 1999;162(6):3504–11. [PubMed] [Google Scholar]
- 55.Yamada H, Mizumo S, Horai R, Iwakura Y, Sugawara I. Protective role of interleukin-1 in mycobacterial infection in IL-1 alpha/beta double-knockout mice. Lab Investig. 2000;80(5):759–67. doi: 10.1038/labinvest.3780079. [DOI] [PubMed] [Google Scholar]
- 56.Akira S. Toll-like receptor signaling. J Biol Chem. 2003;278(40):38105–8. doi: 10.1074/jbc.R300028200. [DOI] [PubMed] [Google Scholar]
- 57.Jo EK. Mycobacterial interaction with innate receptors: TLRs, C-type lectins, and NLRs. Curr Opin Infect Dis. 2008;21(3):279–86. doi: 10.1097/QCO.0b013e3282f88b5d. [DOI] [PubMed] [Google Scholar]
- 58.Courtois G, Smahi A, Reichenbach J, et al. A hypermorphic IkappaBalpha mutation is associated with autosomal dominant anhidrotic ectodermal dysplasia and T cell immunodeficiency. J Clin Invest. 2003;112(7):1108–15. doi: 10.1172/JCI18714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Döffinger R, Smahi A, Bessia C, et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat Genet. 2001;27(3):277–85. doi: 10.1038/85837. [DOI] [PubMed] [Google Scholar]
- 60.Picard C, Puel A, Bonnet M, et al. Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science. 2003;299(5615):2076–9. doi: 10.1126/science.1081902. [DOI] [PubMed] [Google Scholar]
- 61.Bulut Y, Michelsen KS, Hayrapetian L, et al. Mycobacterium tuberculosis heat shock proteins use diverse toll-like receptor pathways to activate pro-inflammatory signals. J Biol Chem. 2005;280(22):20961–7. doi: 10.1074/jbc.M411379200. [DOI] [PubMed] [Google Scholar]
- 62.Bafica A, Scanga CA, Feng CG, Leifer C, Cheever A, Sher A. TLR9 regulates Th1 responses and cooperates with TLR2 in mediating optimal resistance to Mycobacterium tuberculosis. J Exp Med. 2005;202(12):1715–24. doi: 10.1084/jem.20051782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol. 2011;30(1):16–34. doi: 10.3109/08830185.2010.529976. [DOI] [PubMed] [Google Scholar]
- 64.Drage MG, Pecora ND, Hise AG, Febbraio M, Silverstein RL, et al. TLR2 and its co-receptors determine responses of macrophages and dendritic cells to lipoproteins of Mycobacterium tuberculosis. Cell Immunol. 2009;258:29–37. doi: 10.1016/j.cellimm.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Underhill DM, Ozinsky A, Smith KD, Aderem A. Toll-like receptor-2 mediates mycobacteria-induced proinflammatory signaling in macrophages. Proc Natl Acad Sci U S A. 1999;96(25):14459–63. doi: 10.1073/pnas.96.25.14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kleinnijenhuis J, Joosten LA, van de Veerdonk FL, Savage N, van Crevel R, Kullberg BJ, et al. Transcriptional and inflammasome-mediated pathways for the induction of IL-1beta production by Mycobacterium tuberculosis. Eur J Immunol. 2009;39(7):1914–22. doi: 10.1002/eji.200839115. [DOI] [PubMed] [Google Scholar]
- 67.Pompei L, Jang S, Zamlynny B, et al. Disparity in IL-12 release in dendritic cells and macrophages in response to Mycobacterium tuberculosis is due to use of distinct TLRs. J Immunol. 2007;178(8):5192–9. doi: 10.4049/jimmunol.178.8.5192. [DOI] [PubMed] [Google Scholar]
- 68.Drennan MB, Nicolle D, Quesniaux VJF, et al. Toll-like receptor 2-deficient mice succumb to Mycobacterium tuberculosis infection. Am J Pathol. 2004;164:49–57. doi: 10.1016/S0002-9440(10)63095-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Reiling N, Hölscher C, Fehrenbach A, et al. Cutting edge: toll-like receptor (TLR)2- and TLR4-mediated pathogen recognition in resistance to airborne infection with Mycobacterium tuberculosis. J Immunol. 2002;169(7):3480–4. doi: 10.4049/jimmunol.169.7.3480. [DOI] [PubMed] [Google Scholar]
- 70.Quesniaux V, Fremond C, Jacobs M, Parida S, Nicolle D, Yeremeev V, et al. Toll-like receptor pathways in the immune responses to mycobacteria. Microbes Infect. 2004;6(10):946–59. doi: 10.1016/j.micinf.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 71.Ladel CH, Blum C, Dreher A, Reifenberg K, Kopf M, Kaufmann SH. Lethal tuberculosis in interleukin-6-deficient mutantmice. Infect Immun. 1997;65:4843–9. doi: 10.1128/iai.65.11.4843-4849.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kleinnijenhuis J, Oosting M, Joosten LA, Netea MG, Van Crevel R. Innate immune recognition of Mycobacterium tuberculosis. Clin Dev Immunol. 2011;2011:405310. doi: 10.1155/2011/405310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sugawara I, Yamada H, Li C, Mizuno S, Takeuchi O, et al. Mycobacterial infection in TLR2 and TLR6 knockout mice. Microbiol Immunol. 2003;47:327–36. doi: 10.1111/j.1348-0421.2003.tb03404.x. [DOI] [PubMed] [Google Scholar]
- 74.Abel B, Thieblemont N, Quesniaux VJ, Brown N, Mpagi J, et al. Toll-like receptor 4 expression is required to control chronic Mycobacterium tuberculosis infection in mice. J Immunol. 2002;169:3155–62. doi: 10.4049/jimmunol.169.6.3155. [DOI] [PubMed] [Google Scholar]
- 75.Fremond CM, Togbe D, Doz E, Rose S, Vasseur V, Maillet I, et al. IL-1 receptor-mediated signal is an essential component of MyD88-dependent innate response to Mycobacterium tuberculosis infection. J Immunol. 2007;179(2):1178–89. doi: 10.4049/jimmunol.179.2.1178. [DOI] [PubMed] [Google Scholar]
- 76.Fremond CM, Yeremeev V, Nicolle DM, Jacobs M, Quesniaux VF, Ryffel B. Fatal Mycobacterium tuberculosis infection despite adaptive immune response in the absence of MyD88. J Clin Invest. 2004;114(12):1790–9. doi: 10.1172/JCI21027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Scanga CA, Bafica A, Feng CG, Cheever AW, Hieny S, Sher A. MyD88-deficient mice display a profound loss in resistance to Mycobacterium tuberculosis associated with partially impaired Th1 cytokine and nitric oxide synthase 2 expression. Infect Immun. 2004;4:2400–4. doi: 10.1128/IAI.72.4.2400-2404.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lee HM, Shin DM, Kim KK, Lee JS, Paik TH, Jo EK. Roles of reactive oxygen species in CXCL8 and CCL2 expression in response to the 30-kDa antigen of Mycobacterium tuberculosis. J Clin Immunol. 2009;29(1):46–56. doi: 10.1007/s10875-008-9222-3. [DOI] [PubMed] [Google Scholar]
- 79.Means TK, Wang S, Lien E, Yoshimura A, Golenbock DT, Fenton MJ. Human Toll-like receptors mediate cellular activation by Mycobacterium tuberculosis. J Immunol. 1999;163:3920–7. [PubMed] [Google Scholar]
- 80.Branger J, Knapp S, Weijer S, Leemans JC, Pater JM, Speelman P, et al. Role of Toll-like receptor 4 in Gram-positive and Gram-negative pneumonia in mice. Infect Immun. 2004;72:788–94. doi: 10.1128/IAI.72.2.788-794.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Krutzik SR, Ochoa MT, Sieling PA, Uematsu S, Ng YW, Legaspi A, et al. Activation and regulation of Toll-like receptors 2 and 1 in human leprosy. Nat Med. 2003;9:525–32. doi: 10.1038/nm864. [DOI] [PubMed] [Google Scholar]
- 82.Alexopoulou L, Thomas V, Schnare M, Lobet Y, Anguita J, Schoen RT, et al. Hyporesponsiveness to vaccination with BorreliaburgdorferiOspA in humans and in TLR1- and TLR2- deficient mice. Nat Med. 2002;8:878–84. doi: 10.1038/nm732. [DOI] [PubMed] [Google Scholar]
- 83.Arbour NC, Lorenz E, Schutte BC, Zabner J, Kline JN, Jones M, et al. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet. 2000;25:187–91. doi: 10.1038/76048. [DOI] [PubMed] [Google Scholar]
- 84.Kang TJ, Lee SB, Chae GT. A polymorphism in the Toll-like receptor 2 is associated with IL-12 production from monocyte in lepromatous leprosy. Cytokine. 2002;20:56–62. doi: 10.1006/cyto.2002.1982. [DOI] [PubMed] [Google Scholar]
- 85.Lorenz E, Mira JP, Cornish KL, Arbour NC, Schwartz DA. A novel polymorphism in the Toll-like receptor 2 gene and its potential association with staphylococcal infection. Infect Immun. 2000;68:6398–401. doi: 10.1128/iai.68.11.6398-6401.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fenhalls G, Squires GR, Stevens-Muller L, Bezuidenhout J, Amphlett G, Duncan K, et al. Associations between Toll-like receptors and interleukin-4 in the lungs of patients with tuberculosis. Am J Respir Cell Mol Biol. 2003;29:28–38. doi: 10.1165/rcmb.2002-0163OC. [DOI] [PubMed] [Google Scholar]
- 87.Reiling N, Holscher C, Fehrenbach A, Kroger S, Kirschning CJ, et al. Toll-like receptor (TLR)2- and TLR4-mediated pathogen recognition in resistance to airborne infection with Mycobacterium tuberculosis. J Immunol. 2002;169:3480–4. doi: 10.4049/jimmunol.169.7.3480. [DOI] [PubMed] [Google Scholar]
- 88.Kamath AB, Alt J, Debbabi H, Behar SM. Toll-like receptor 4-defective C3H/HeJ mice are not more susceptible than other C3H substrains to infection with Mycobacterium tuberculosis. Infect Immun. 2003;71:4112–8. doi: 10.1128/IAI.71.7.4112-4118.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rook GA, Dheda K, Zumla A. Opinion: immune responses to tuberculosis in developing countries: implications for new vaccines. Nat Rev Immunol. 2005;5:661–7. doi: 10.1038/nri1666. [DOI] [PubMed] [Google Scholar]
- 90.Davila S, Hibberd ML, HariDass R, Wong HE, Sahiratmadja E, Bonnard C, et al. Genetic association and expression studies indicate a role of Toll-like receptor 8 in pulmonary tuberculosis. PLoS Genet. 2008;4:e1000218. doi: 10.1371/journal.pgen.1000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kumar H, Kawai T, Akira S. Toll-like receptors and innate immunity. Biochem Biophys Res Commun. 2009;388(4):621–5. doi: 10.1016/j.bbrc.2009.08.062. [DOI] [PubMed] [Google Scholar]
- 92.Songane M, Kleinnijenhuis J, Netea MG, van Crevel R. The role of autophagy in host defence against Mycobacterium tuberculosis infection. Tuberculosis (Edinb) 2012;92(5):388–96. doi: 10.1016/j.tube.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 93.Ogus AC, Yoldas B, Ozdemir T, Uguz A, Olcen S, et al. The Arg753GLn polymorphism of the human toll-like receptor 2 gene in tuberculosis disease. Eur Respir J. 2004;23:219–23. doi: 10.1183/09031936.03.00061703. [DOI] [PubMed] [Google Scholar]
- 94.Thuong NT, Hawn TR, Thwaites GE, Chau TT, Lan NT, et al. A polymorphism in human TLR2 is associated with increased susceptibility to tuberculous meningitis. Genes Immunol. 2007;8:422–8. doi: 10.1038/sj.gene.6364405. [DOI] [PubMed] [Google Scholar]
- 95.Velez DR, Wejse C, Stryjewski ME, Abbate E, Hulme WF, et al. Variants in toll-like receptors 2 and 9 influence susceptibility to pulmonary tuberculosis in Caucasians, African-Americans, and West Africans. Hum Genet. 2010;127:65–73. doi: 10.1007/s00439-009-0741-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yim JJ, Lee HW, Lee HS, Kim YW, Han SK, et al. The association between microsatellite polymorphisms in intron II of the human Toll-like receptor 2 gene and tuberculosis among Koreans. Genes Immunol. 2006;7:150–5. doi: 10.1038/sj.gene.6364274. [DOI] [PubMed] [Google Scholar]
- 97.Zhang Y, Jiang T, Yang X, Xue Y, Wang C, Liu J, et al. Toll-like receptor-1, -2, and -6 polymorphisms and pulmonary tuberculosis susceptibility: a systematic review and meta-analysis. PLoS One. 2013;8(5):e63357. doi: 10.1371/journal.pone.0063357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Biswas D, Gupta SK, Sindhwani G, Patras A. TLR2 polymorphisms, Arg753Gln and Arg677Trp, are not associated with increased burden of tuberculosis in Indian patients. BMC Res Notes. 2009;2:162. doi: 10.1186/1756-0500-2-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ma MJ, Xie LP, Wu SC, Tang F, Li H, et al. Toll-like receptors, tumor necrosis factor-alpha, and interleukin-10 gene polymorphisms in risk of pulmonary tuberculosis and disease severity. Hum Immunol. 2010;71:1005–10. doi: 10.1016/j.humimm.2010.07.009. [DOI] [PubMed] [Google Scholar]
- 100.Selvaraj P, Harishankar M, Singh B, Jawahar MS, Banurekha VV. Toll like receptor and TIRAP gene polymorphisms in pulmonary tuberculosis patients of South India. Tuberculosis. 2010;90:306–10. doi: 10.1016/j.tube.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 101.Xue Y, Zhao ZQ, Wang HJ, Jin L, Liu CP, et al. Toll-like receptors 2 and 4 gene polymorphisms in a southeastern Chinese population with tuberculosis. Int J Immunogenet. 2010;37:135–8. doi: 10.1111/j.1744-313X.2009.00892.x. [DOI] [PubMed] [Google Scholar]
- 102.Khor CC, Chapman SJ, Vannberg FO, Dunne A, Murphy C, et al. A Mal functional variant is associated with protection against invasive pneumococcal disease, bacteremia, malaria and tuberculosis. Nat Genet. 2007;39:523–8. doi: 10.1038/ng1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Najmi N, Kaur G, Sharma SK, Mehra NK. Human Toll-like receptor 4 polymorphisms TLR4 Asp299Gly and Thr399Ile influence susceptibility and severity of pulmonary tuberculosis in the Asian Indian population. Tissue Antigens. 2010;76:102–9. doi: 10.1111/j.1399-0039.2010.01481.x. [DOI] [PubMed] [Google Scholar]
- 104.Newport MJ, Allen A, Awomoyi AA, Dunstan SJ, McKinney E, et al. The toll-like receptor 4 Asp299Gly variant: no influence on LPS responsiveness or susceptibility to pulmonary tuberculosis in The Gambia. Tuberculosis (Edinb) 2004;84:347–52. doi: 10.1016/j.tube.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 105.Garlanda C, Bottazzi B, Bastone A, Mantovani A. Pentraxins at the crossroads between innate immunity, inflammation, matrix deposition, and female fertility. Annu Rev Immunol. 2005;23:337–66107. doi: 10.1146/annurev.immunol.23.021704.115756. [DOI] [PubMed] [Google Scholar]
- 106.Lee GW, Lee TH, Vilcek J. TSG-14, a tumor necrosis factor- and IL-1-inducible protein, is a novel member of the pentaxin family of acute phase proteins. J Immunol. 1993;150(5):1804–12. [PubMed] [Google Scholar]
- 107.Breviario F, d’Aniello EM, Golay J, Peri G, Bottazzi B, Bairoch A, et al. Interleukin-1-inducible genes in endothelial cells. Cloning of a new gene related to C-reactive protein and serum amyloid P component. J Biol Chem. 1992;267(31):22190–7. [PubMed] [Google Scholar]
- 108.Mantovani A, Garlanda C, Bottazzi B. Pentraxin 3, a non-redundant soluble pattern recognition receptor involved in innate immunity. Vaccine. 2003;21(Suppl 2):S43–7. doi: 10.1016/s0264-410x(03)00199-3. [DOI] [PubMed] [Google Scholar]
- 109.Introna M, Alles VV, Castellano M, Picardi G, De Gioia L, Bottazzai B, et al. Cloning of mouse ptx3, a new member of the pentraxingene family expressed at extrahepatic sites. Blood. 1996;87(5):1862–72. [PubMed] [Google Scholar]
- 110.Lee GW, Goodman AR, Lee TH, Vilcek J. Relationship of TSG-14 protein to the pentraxin family of major acute phase proteins. J Immunol. 1994;153(8):3700–7. [PubMed] [Google Scholar]
- 111.Bottazzi B, Vouret-Craviari V, Bastone A, De Gioia L, Matteucci C, Peri G, et al. Multimer formation and ligand recognition by the long pentraxin PTX3. Similarities and differences with the short pentraxins C-reactive protein and serum amyloid P component. J Biol Chem. 1997;272(52):32817–23. doi: 10.1074/jbc.272.52.32817. [DOI] [PubMed] [Google Scholar]
- 112.Garlanda C, Hirsch E, Bozza S, Salustri A, et al. Non-redundant role of the long pentraxin PTX3 in anti-fungal innate immune response. Nature. 2002;420(6912):182–6. doi: 10.1038/nature01195. [DOI] [PubMed] [Google Scholar]
- 113.Souza DG, Soares AC, Pinho V, et al. Increased mortality and inflammation in tumor necrosis factor-stimulated gene-14 transgenic mice after ischemia and reperfusion injury. Am J Pathol. 2002;160:1755–65. doi: 10.1016/s0002-9440(10)61122-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Vouret-Craviari V, Matteucci C, Peri G, Poli G, Introna M, Mantovani A. Expression of a long pentraxin, PTX3, by monocytes exposed to the mycobacterial cell wall component lipoarabinomannan. Infect Immun. 1997;65(4):1345–50. doi: 10.1128/iai.65.4.1345-1350.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Olesen R, Wejse C, Velez DR, Bisseye C, et al. DC-SIGN (CD209), pentraxin 3 and vitamin D receptor gene variants associate with pulmonary tuberculosis risk in West Africans. Genes Immunol. 2007;8(6):456–67. doi: 10.1038/sj.gene.6364410. [DOI] [PubMed] [Google Scholar]
- 116.Azzurri A, Sow OY, Amedei A, Bah B, Diallo S, Peri G, et al. IFN-gamma-inducible protein 10 and pentraxin 3 plasma levels are tools for monitoring inflammation and disease activity in Mycobacterium tuberculosis infection. Microbes Infect. 2005;7(1):1–8. doi: 10.1016/j.micinf.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 117.Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2(8):675–80. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 118.de Kluijver J, Grünberg K, Pons D, de Klerk EP, Dick CR, Sterk PJ, et al. Interleukin-1b and interleukin-1ra levels in nasal lavages during experimental rhinovirus infection in asthmatic and non-asthmatic subjects. Clin Exp Allergy. 2003;33:1415–8. doi: 10.1046/j.1365-2222.2003.01770.x. [DOI] [PubMed] [Google Scholar]
- 119.Nakae S, Komiyama Y, Yokoyama H, Nambu A, Umeda M, Iwase M, et al. IL-1 is required for allergen-specific Th2 cell activation and the development of airway hypersensitivity response. Int Immunol. 2003;15(4):483–90. doi: 10.1093/intimm/dxg054. [DOI] [PubMed] [Google Scholar]
- 120.Ting JP, Lovering RC, Alnemri ES, Bertin J, Boss JM, et al. The NLR gene family: a standard nomenclature. Immunity. 2008;28(3):285–7. doi: 10.1016/j.immuni.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fietta P, Delsante G. The inflammasomes: the key regulators of inflammation. Riv Biol. 2009;102(3):365–84. [PubMed] [Google Scholar]
- 122.Koo IC, Wang C, Raghavan S, Morisaki JH, Cox JS, et al. ESX-1-dependent cytolysis in lysosome secretion and inflammasome activation during mycobacterial infection. Cell Microbiol. 2008;10:1866–78. doi: 10.1111/j.1462-5822.2008.01177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mariathasan S, Monack DM. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol. 2007;7:31–40. doi: 10.1038/nri1997. [DOI] [PubMed] [Google Scholar]
- 124.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–26. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 125.Castaño D, Barrera LF, Rojas M. Mycobacterium tuberculosis alters the differentiation of monocytes into macrophages in vitro. Cell Immunol. 2011;268(2):60–7. doi: 10.1016/j.cellimm.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 126.Mayer-Barber KD, Barber DL, Shenderov K, et al. Caspase-1 independent IL-1beta production is critical for host resistance to mycobacterium tuberculosis and does not require TLR signaling in vivo. J Immunol. 2010;184(7):3326–30. doi: 10.4049/jimmunol.0904189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mishra BB, Moura-Alves P, Sonawane A, Hacohen N, Griffiths G, et al. Mycobacterium tuberculosis protein ESAT-6 is a potent activator of the NLRP3/ASC inflammasome. Cell Microbiol. 2010;12:1046–63. doi: 10.1111/j.1462-5822.2010.01450.x. [DOI] [PubMed] [Google Scholar]
- 128.Master SS, Rampini SK, Davis AS, Keller C, Ehlers S, et al. Mycobacterium tuberculosis prevents inflammasome activation. Cell Host Microbe. 2008;3:224–32. doi: 10.1016/j.chom.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–50. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 130.Shi S, Blumenthal A, Hickey CM, Gandotra S, Levy D, Ehrt S. Expression of many immunologically important genes in Mycobacterium tuberculosis-infected macrophages is independent of both TLR2 and TLR4 but dependent on IFN-alphabeta receptor and STAT1. J Immunol. 2005;175:3318–28. doi: 10.4049/jimmunol.175.5.3318. [DOI] [PubMed] [Google Scholar]
- 131.Schneider BE, Korbel D, Hagens K, Koch M, Raupach B, Enders J, et al. A role for IL-18 in protective immunity against Mycobacterium tuberculosis. Eur J Immunol. 2010;40(2):396–405. doi: 10.1002/eji.200939583. [DOI] [PMC free article] [PubMed] [Google Scholar]