

Abstract
22q11.2 Deletion syndrome is one of the most common microdeletional syndromes, with an incidence of 1:4000 live-births, and potentially affects every organ in the body. More than 180 associated clinical features have been reported and not one phenotypic feature is present in 100% of cases. Ocular manifestations reported based on early childhood examinations include eyelid hooding, strabismus, posterior embryotoxon, retinal vessel tortuosity and refractive errors. Keratoconus has been reported once before in association with 22q11.2 deletion syndrome in a young adult. We report the second case of keratoconus in association with 22q11.2 deletion syndrome.
Background
Current recommendations for ophthalmic screenings for 22q11.2 deletion syndrome are at diagnosis and again in early childhood (ages 1–5 years). Our patient had normal ophthalmic paediatric screenings, and presented with no risk factors for the development of keratoconus including no history of eye rubbing and no signs of atopy. Given that keratoconus often does not develop until after puberty, we suggest that patients with 22q11.2 deletion syndrome receive repeat ophthalmic screenings again in late adolescence or early adulthood for detection of keratoconus.
Case presentation
A 23-year-old Caucasian man (figure 1) with no ocular history presented reporting bilateral progressive blurry vision for the past 3 years, which could not be adequately corrected with spectacles. The patient denied any history of eye rubbing. The patient's medical and surgical history were significant for DiGeorge syndrome, scoliosis, surgical repair of ventricular septal defect in 2003, and psychiatric illnesses for which the patient was taking topiramate and aripiprazole. The patient denied allergies. Best corrected visual acuity with spectacles was 20/50 OD and 20/40 OS. External examination revealed Munson's sign. Slit lamp examination demonstrated Fleischer's rings bilaterally. Funduscopic examination was significant for tortuosity of the vasculature (figure 1). Placido disc-based corneal topographic examination revealed bilateral vertical inferior steepening of the corneal curvature, which supported the diagnosis of bilateral keratoconus (figure 2).
Figure 1.

Colour photograph depicting facial features for 22q11.2 deletion syndrome including bulbous nose and micrognathia (A). Colour fundus photograph depicting tortuous retinal vasculature OD (B) and OS (C).
Figure 2.
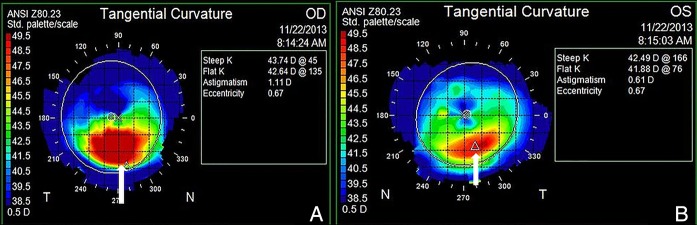
Placido disc-based corneal topography tangential images OD (A) and OS (B) depicting vertical inferior steepening of corneal surface (white arrows) consistent with bilateral keratoconus.
Investigations
The patient was referred for genetic testing to investigate the aetiology of his case of DiGeorge syndrome. High-resolution chromosome analysis and oligo-SNP (oligonucleotide, single nucleotide polymorphism, Affymetrix CytoScan HD) assay were performed in a Clinical Laboratory Improvement Amendments (CLIA)-approved laboratory. High-resolution chromosome analysis resulted in a normal male karyotype, 46XY (Quest Diagnostics, Queens, New York, USA).
The oligo-SNP assay was performed using 2.67 million probes, including 1.9 million copy number probes and 750 000 SNP probes with a median 1150 base pair interprobe distance, and demonstrated a genomic segment with a 2.6 Mb loss at 22q11.2 (Quest Diagnostics, Queens, New York, USA). Thresholds for genome-wide screening were set at >200 kb for gains, >50 kb for losses and 10 Mb for segments of homozygosity. The thresholds may be lower for cytogenetic relevant regions.
For a complete list of genes and a gene map for this region go to: http://genome.ucsc.edu/cgi-bin/hgGateway (accessed 27 April 2014) and search chr22 (18 916 842–21 465 659).
Discussion
Keratoconus is a bilateral, asymmetric, non-inflammatory corneal ectasia, characterised by progressive corneal protrusion and thinning, resulting in myopic refractive error and irregular astigmatism.1 Incidence is estimated at 1.3–25/100 000. Onset is often after puberty with progression until the corneal curvature stabilises in the third to fourth decade of life.2 Visual impairment and the resultant decrease in quality of life for the patient are significant. The treatment of keratoconus causes a considerable economic burden to society.3
Multiple genes and gene loci have been shown to be associated with keratoconus.1 Prior to genetic testing, the physician must identify an indication for the test, which is often based on the patient's history and phenotypic presentation. Genetic counsellors should also be included in counselling the patient on the risks, benefits and cost of the requested test.4 Insurance companies will often cover the cost of the test if medical necessity and potential to treat can be demonstrated by the physician. However, if necessary, patients can obtain genetic testing at reduced or no charge through programmes such as eyeGENE, funded by the National Eye Institute and National Institute of Health.5 The risks of genetic tests include potential psychological strain that may develop following receiving unanticipated test results. The benefits include identification and management of associated clinical features, which may also be able to limit the financial burden to the patient and society.4
Systemic diseases associated with keratoconus include Down syndrome, Leber's congenital amaurosis and connective tissue diseases such as osteogenesis imperfecta and Ehlers-Danlos syndrome.1 Keratoconus has been reported once before in association with 22q11.2 deletion syndrome in a young adult.6 22q11.2 Deletion syndrome, which commonly includes DiGeorge syndrome, velocardiofacial syndrome (VCFS) and conotruncal anomaly face syndrome, is one of the most common microdeletional syndromes, with an incidence of 1:4000 live births, and potentially affects every organ in the body.7 Diagnosis is confirmed by detecting the DNA microdeletion from chromosome 22q11.2. Inheritance has been proven to be autosomal dominant; however, most mutations are de novo with neither parent being affected. While penetrance is 100%, phenotypic expression is extremely heterogeneous with a broad spectrum of clinical morbidity. 22q11.2 Deletion syndrome has more than 180 associated clinical features that have been reported and not one phenotypic feature is present in 100% of cases. Common systemic findings include conotruncal cardiac anomalies, hypernasal speech, characteristic facial features, psychiatric illness and immunodeficiencies.8 Ocular manifestations include eyelid hooding, strabismus, posterior embryotoxon, retinal vessel tortuosity and refractive errors.7
Management of patients with 22q11.2 deletion syndrome is complex, requiring multidisciplinary intervention. No cure for 22q11.2 deletion syndrome exists and long-term studies are still needed to gauge the estimated lifespan of affected patients. Shprintzen,8 however, reported studying several affected patients in their seventh decade of life. Bassett et al9 proposed a multidisciplinary management protocol that recommends specialty-specific screenings for select age groups to best anticipate the multitude of common and rarely associated findings that present at particular stages of life. Current recommendations for ophthalmic screenings are at diagnosis and again in early childhood (1–5 years).9 While we are reporting only the second case of keratoconus in association with 22q11.2 deletion syndrome, we suspect that other undetected cases exist. Our patient had a normal ocular examination during his paediatric screening process, and per current protocols was not screened again. Given that keratoconus often does not develop until after puberty, we suggest that patients with 22q11.2 deletion syndrome receive repeat ophthalmic screenings again in late adolescence or early adulthood for detection of keratoconus.
Learning points.
22q11.2 Deletion syndrome is a common microdeletional syndrome that potentially affects every organ in the body, with more than 180 documented phenotypic features.
Current ophthalmic screening protocols for 22q11.2 deletion syndrome recommend screenings at diagnosis and again between the ages of 1 and 5 years of life.
Keratoconus does not present until after puberty and may be an ocular manifestation of 22q11.2 deletion syndrome.
We recommend an additional ophthalmic screening examination for patients with 22q11.2 deletion syndrome in late adolescence or early adulthood for the detection of keratoconus.
Footnotes
Contributors: NS oversaw all work conducted for the completion of this manuscript and guarantees its integrity.
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Chang HY, Chodosh J. The genetics of keratoconus. Semin Ophthalmol 2013;28:275–80. 10.3109/08820538.2013.825295 [DOI] [PubMed] [Google Scholar]
- 2.Vazirani J, Basu S. Keratoconus: current prospectives. Clin Ophthalmol 2013;7:2019–30. 10.2147/OPTH.S50119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rebinitsch RL, Kymes SM, Walline J et al. The lifetime economic burden of keratoconus: a decision analysis using a Markov model. Am J Ophthalmolol 2011;151:768–73.e2. 10.1016/j.ajo.2010.10.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zanollia MT, Khetana V, Dotan G et al. Should patients with ocular genetic disorders have genetic testing? Curr Opin Ophthalmol 2014;25:359–65. 10.1097/ICU.0000000000000083 [DOI] [PubMed] [Google Scholar]
- 5.Capasso JE. The cost of genetic testing for ocular disease: who pays? Curr Opin Ophthalmol 2014;25:394–9. 10.1097/ICU.0000000000000085 [DOI] [PubMed] [Google Scholar]
- 6.Bassett AS, Hodgkinson K, Chow EWC et al. 22q11 Deletion syndrome in adults with schizophrenia. Am J Med Genet 1998:328–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Casteels I, Casaer P, Gewillig M et al. Ocular findings in children with a microdeletion in chromosome 22q11.2. Eur J Pediatr 2008;167:751–5. 10.1007/s00431-007-0582-0 [DOI] [PubMed] [Google Scholar]
- 8.Shprintzen RJ. Velo-cardio-facial syndrome: 30 years of study. Dev Disabil Res Rev 2008;14:3–10. 10.1002/ddrr.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bassett AS, McDonald-McGinn DM, Devriendt K et al. Practical guidelines for managing patients with 22q11.2 deletion syndrome. J Pediatr 2011;159:332–9.el. 10.1016/j.jpeds.2011.02.039 [DOI] [PMC free article] [PubMed] [Google Scholar]