

Abstract
We describe a rare case of light chain immunoglobulin amyloid (AL) accumulation in the central and lower pole renal calyces. Our patient, a woman aged 60, presented with several episodes of gross haematuria. Radiological imaging detected a filling defect in the left renal pelvis. Rigid ureteroscopy showed a corresponding mucosal abnormality resembling transitional cell carcinoma. A definitive preoperative tissue diagnosis could not be reached. Laparoscopic-assisted left nephroureterectomy was indicated. Histopathological examination excluded malignancy, revealing congophilic deposits of submucosal amyloid. A constellation of findings confirmed localised or primary amyloidosis with an AL immunophenotype but no evidence of clonal B-cell disease in the amyloid-associated lymphoplasmacytic cell infiltrate. Investigation for systemic plasma cell dyscrasia and echocardiography and scintigraphy for visceral amyloid deposits were negative for systemic disease. At a follow-up period of 30 months, there is no recurrence. However, our patient was diagnosed with breast cancer 21 months ago.
Background
Amyloid disease (amyloidosis) exhibits a variety of clinical manifestations depending on whether it is limited to a specific site or a wide range of body organ systems. The defining characteristic of amyloid is the presence of misfolded variants of normal proteins. These are arranged in β-pleated sheets when viewed by X-ray diffraction or as fibrils under electron microscopy. It is believed that certain long chain immunoglobulins can become a central nidus for growth for creating amyloid fibrils which act as templates for others in a chain reaction.1 These fibrils form an insoluble extracellular substance that results in progressive disruption of normal tissue form or function.
The only way to achieve a definitive diagnosis is by tissue biopsy where deposits appear as a distinctive ‘glossy’, ‘waxy’ or ‘amorphous’ hyaline-like eosinophilic substance. The diagnosis is confirmed using Congo red, a diazo dye that generates a classical yellow–green birefringence when viewed between crossed polarisers.2
The precursor protein that constitutes the amyloid can be further subclassified by immunohistochemical labelling. For instance, overproduction of protein A by the liver, secondary to chronic inflammatory states, results in the systemic deposition of protein A amyloid (AA). Primary amyloidosis is composed of AL and usually coincides with a neoplastic proliferation of plasma cells. Amyloid composed of mutated transthyretinin is inherited and shows familial clustering with a distinct pattern of organ involvement.
In practice the most useful clinical guide for amyloidosis is to split cases into localised or systemic forms. This creates two prognostic groups that differ according to natural history and aggressiveness. Systemic amyloidosis progresses rapidly and fatally, whereas localised disease is static and benign.
The paucity of case reports in the medical literature reflects the fact that idiopathic primary amyloidosis of the urinary tract is rare and upper tract lesions are exceptional. We re-iterate the potential of LA of the genitourinary tract to masquerade as malignancy and highlight the challenge of reaching a definitive preoperative diagnosis. We report no progression, recurrence or systemic disease after a 30-month follow-up period. Our patient also developed an apparently unrelated carcinoma of the left breast (pT2, pN0, pM0), 9 months postnephrectomy.
Case presentation
A 60-year-old woman presented with two episodes of macroscopic haematuria over a period of 18 months. Urine cultures were sterile and each episode resolved with antibiotic therapy. Significant medical history included investigation for supraventricular tachycardia in the 1990s and cervical intraepithelial neoplasia. Ophthalmological treatment included extirpation of a vitreous body, retinal detachment, cataract due to posterior capsule opacification, full thickness macular hole right eye and peripheral lattice degeneration of retina related to high myopia. She had a lifelong smoking habit. No familial disease was reported.
Multimodality imaging of the urinary tract was performed and flexible cystoscopy showed incidental endoscopic features of squamous metaplasia. Ultrasonography of the urinary tract was normal. A computer tomogram excretion urogram (figure 1) showed irregular thickening of the mucosa in the left collecting system consistent with transitional cell carcinoma. Renal fluoroscopic imaging concurred (figure 2). Direct visualisation was achieved via rigid ureteroscopy (figure 3). However, cytological washings and biopsy under direct vision were unsuccessful. There was a strong clinicoradiological suspicion of an upper tract malignancy and urgent laparoscopic-assisted nephroureterectomy was undertaken. Inflammatory-type adhesions were encountered around the renal pelvis/and at the pelviureteric junction. This necessitated conversion to an open midline approach as it became impossible to proceed safely. The kidney was mobilised and the ureter excised down to the level of the bladder.
Figure 1.
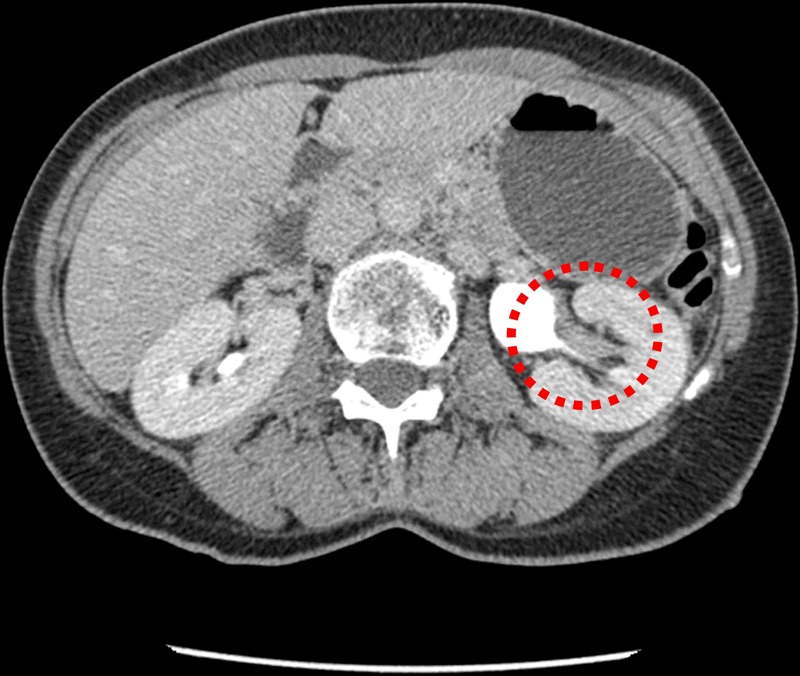
Enhanced axial computer tomography urogram shows thickening of the mucosa in the left lower pole calyx (dashed red circle).
Figure 2.
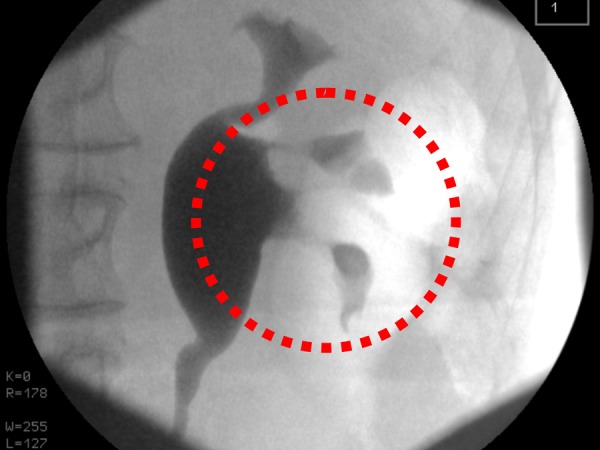
Video fluoroscopy showing abnormal mucosa in the lower pole calyces (dashed red circle).
Figure 3.
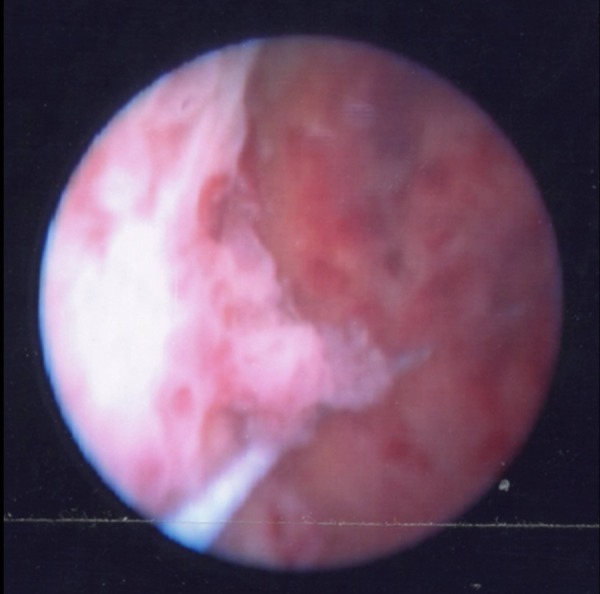
Rigid ureteroscopy shows papillary excrescences resembling urothelial malignancy.
Pathological examination showed a segment of granular haemorrhagic mucosa lining the central and lower calyx (figure 4A, B). There was no evidence of urothelial malignancy. Instead, light microscopic examination of H&E stained tissue sections showed extensive amorphous, eosinophilic material in the submucosa (figures 5–7). Congo red positive amyloid exhibited apple green birefringence under cross polarised light (figure 8). There were amyloid associated plasma cells that expressed polytypic light chains using an in situ hybridisation immunohistochemical technique (figures 9–11A, B). Supplementary molecular studies confirmed polyclonal immunoglobulin and T-cell receptor rearrangements in these intralesional plasma cells. The location of the submucosal amyloid deposits matched the filling defects seen on imaging.
Figure 4.
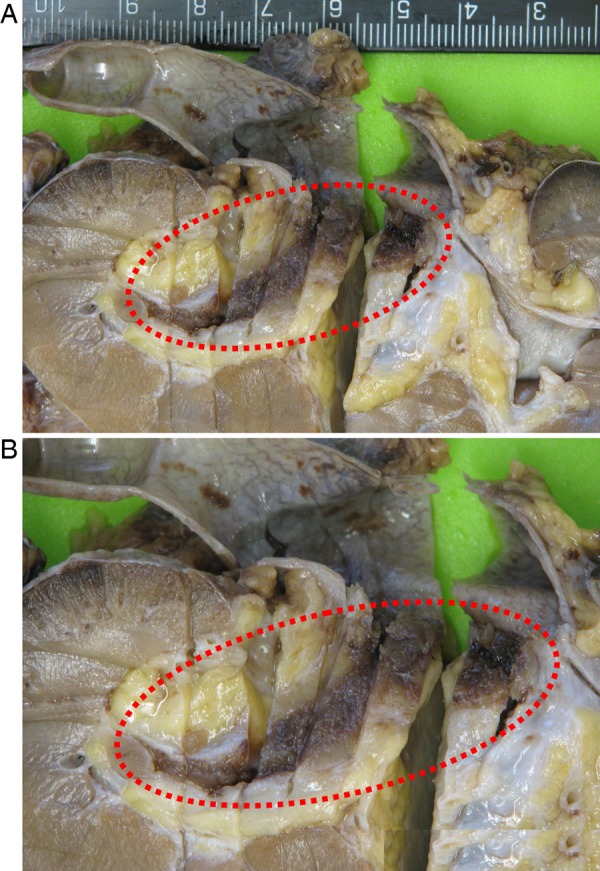
(A) Formalin-fixed nephroureterectomy specimen with granular haemorrhagic mucosa highlighted (dashed red ellipse). (B) Close up of highlighted region.
Figure 5.
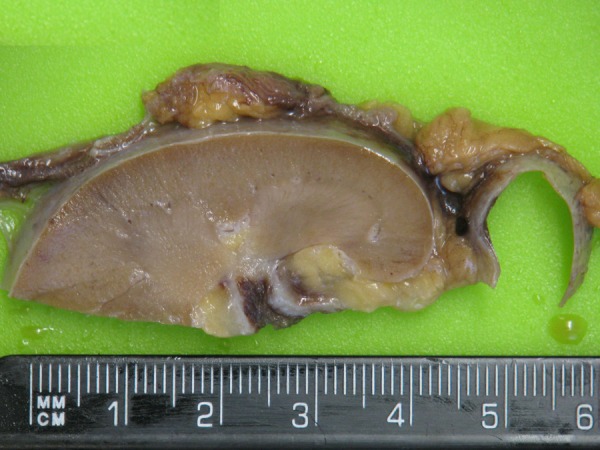
Tissue slice of abnormal pelvicalyceal mucosa.
Figure 6.
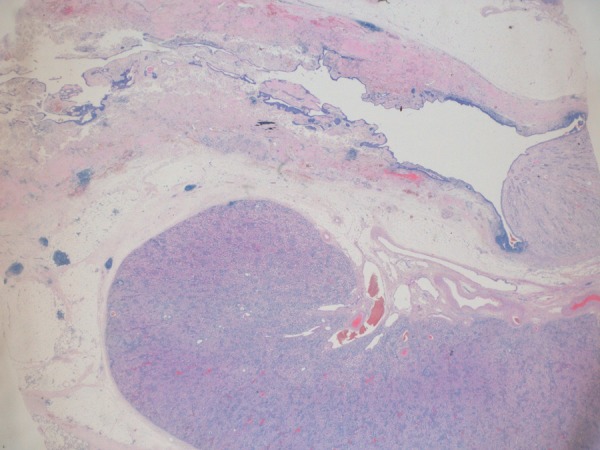
Corresponding stained section of tissue slice from figure 5 (H&E ×2.5).
Figure 7.
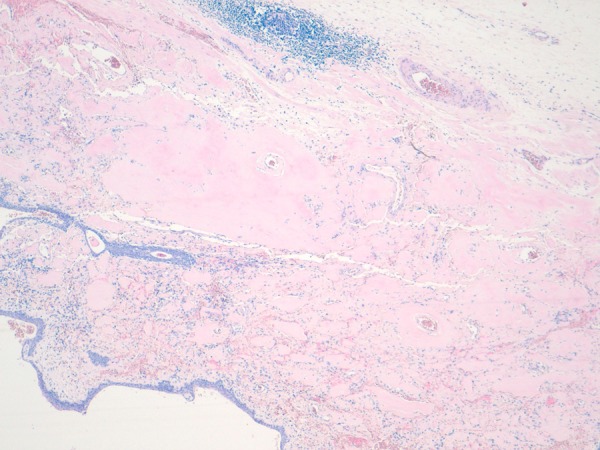
Photomicrograph showing massive submucosal amyloid deposits (H&E ×100).
Figure 8.
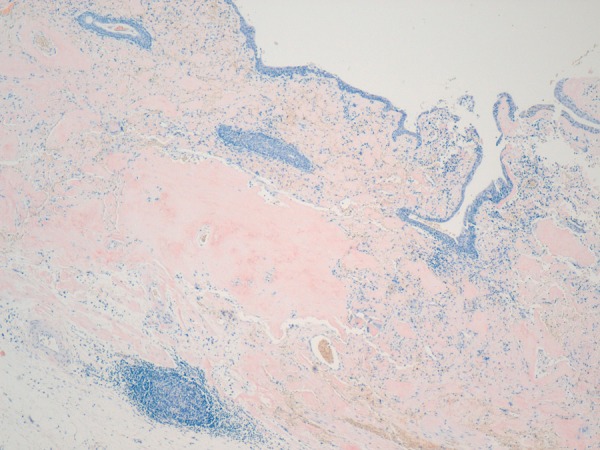
Suburothelial amyloid (Congo red ×100).
Figure 9.
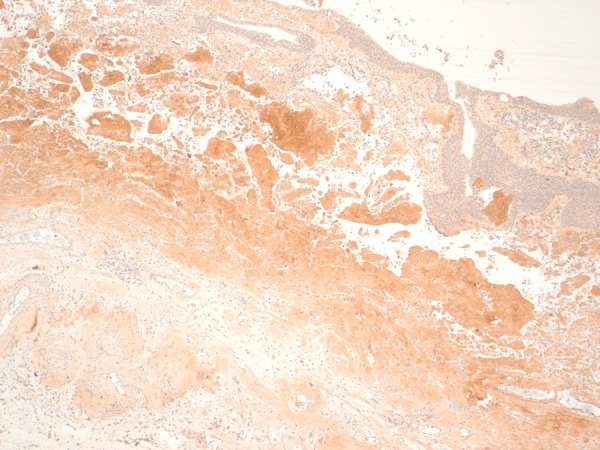
Immunohistochemistry for light chain immunoglobulins (immunoperoxidase ×100).
Figure 10.
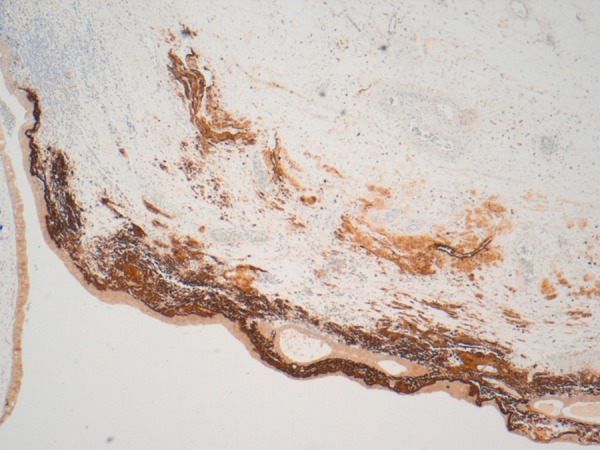
Amyloid with numerous plasma cells and intermingled inflammatory infiltrate (CD138 ×100).
Figure 11.
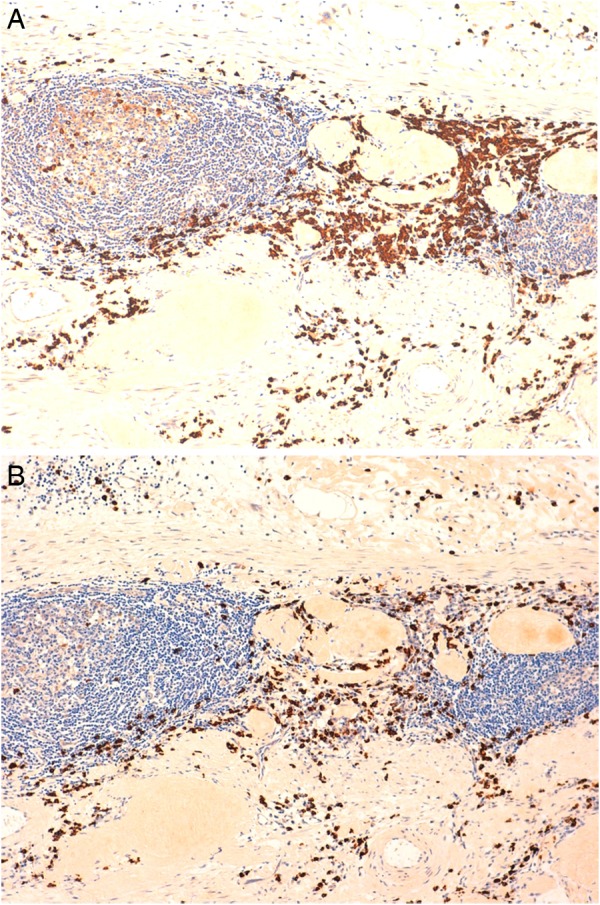
Polyclonal expression in bystander plasma cell population (A) κ, (B) λ (immunoperoxidase ×200).
Full haematological investigation including bone marrow trephine, serum-free light chains and urine analysis for Bence Jones proteins were negative. Systemic screening with serum amyloid protein scintigraphy for visceral involvement and echocardiography for cardiac amyloidosis were negative.
Thirty months have elapsed since the original diagnosis of LA and there is no disease recurrence. However, after 9 months of follow-up our patient was diagnosed with a large (50 mm maximum diameter) grade 3 invasive ductal carcinoma of the left breast (Alred score for oestrogen and progesterone receptors =4/8, Her-2 negative). Stains for amyloid associated with the mammary primary were negative. Mastectomy and axillary nodal clearance were performed with adjuvant radiotherapy to the chest wall and oestrogenic blockade (arimidex).
Differential diagnosis
Transitional cell carcinoma of the upper urinary tract.
Treatment
Laparoscopic nephrectomy.
Outcome and follow-up
There is no recurrence of amyloidosis after a follow-up period of 30 months. Our patient was diagnosed with breast cancer 21 months ago.
Discussion
A MEDLINE search using the terms ‘renal pelvis amyloidosis’ found 23 case reports in the medical literature to date,3–24 excluding two cases of AA amyloidosis secondary to Addison's disease and hereditary amyloidosis. Vital statistics collected from over half of these reports showed a mean age at presentation of 64 and a male preponderance (M:F ratio=9:5).
Idiopathic AL amyloidosis has been described in the lungs, trachea, gut and nervous system but the organs most commonly reported are the bladder, lungs, skin and larynx.25 Primary amyloidosis affecting the renal pelvis is rare and was originally described at necropsy in 1927 where ‘warty nodules’ were found.26
Macroscopic findings similar to our case have been described at cystoscopy in LA.26–29 The renal pelvis in our patient's kidney contained papilliform excrescences when viewed under direct vision using a rigid ureteroscope, but a granular haemorrhagic mucosal surface in the formalin-fixed resection specimen.
Our case highlights the lack of immunospecificity for AL amyloid. Amyloid subtyping was performed by the National Amyloid Centre. Using antibodies against protein A, AA amyloid was excluded, as virtually all cases of protein AA will label immunohistochemically. In contrast, there was no reaction elicited by a battery of monospecific antibodies directed against the majority of known AL epitopes (κ and λ immunoglobulin light chains). It has been proposed that this is due to occlusion of epitope binding sites during fibril formation and tissue fixation which is estimated to occur in approximately 20% of AL amyloid deposits.30 A definitive result of non-AA amyloid with a confident presumptive diagnosis of AL amyloid was made.
Approximately two-thirds of all cases of amyloid disease are of AL subtype. Of these, the majority are attributed to a subtle and non-proliferating plasma cell disorder analogous to monoclonal gammopathy of unknown significance (MGUS). In the remainder, a full blown plasma cell dyscrasia is detected. One-fifth of amyloid disease is both AL subtype and localised with no recognised cause and no detectable systemic disease. The pathogenesis of primary idiopathic LA is unexplained, although speculative theories have been proposed. One postulates that an occult plasma cell neoplasia only becomes apparent, up to a decade after the initial diagnosis.31 Alternatively inflammation can trigger proliferation of a lymphoplasmacytic clone which secrete excess light chain immunoglobulin and create AL amyloid deposits.32 Our case does not offer support for either mechanism as no local or systemic clonal or neoplastic proliferation could be identified, despite extensive investigation.
Our case illustrates the surgical dilemma in managing primary amyloidosis arising from the renal pelvis. Imitation of urothelial malignancy and the fact that endoscopic biopsy of upper urinary tract urothelial carcinoma has a significant false-negative rate can be misleading when determining appropriate treatment.33 In our case, a combination of forceps biopsy and ureteroscopic washings failed to provide a definitive preoperative diagnosis. Access to the upper tract can be challenging due to the small calibre of the ureter which limits the size of the biopsy forceps that can be passed through the ueteroscope. Our case also emphasises the importance of submucosal tissue sampling for accurate diagnosis. Furthermore, it reinforces the reality that LA of the renal pelvis is rarely managed conservatively due to inherent difficulties in sampling of upper renal tract lesions and providing a definitive preoperative tissue diagnosis.
In our case, LA of the renal pelvis mucosa preceded a high-grade mammary carcinoma by 9 months. The differential diagnosis of amyloid deposition can include occult neoplasia of either epithelial or non-epithelial origin. The biochemical composition of tumour-related amyloid is usually light chain immunoglobulin and acute-phase reactants.
Localised amyloid deposition has been reported at the site of a wide variety of epithelial and non-epithelial neoplasms, including female genital tract malignancies.34–36 Malignancy may also precipitate body wide deposition37 38 or secondary amyloidosis and was seen in as many as one-quarter of all cases from one autopsy series.39 The mechanism is mediated by the acute-phase reactant, serum amyloid A (SAA), secreted by the liver in response to tumour inflammation. Elevated SAA occurs across tumour types, including breast malignancy.40
In our case, the pelvicalyceal amyloid was subtyped as AL and histological, immunohistochemical and molecular genetic studies excluded a monoclonal immunoproliferation of deranged plasma cells in the associated lymphoplasmacytic inflammatory infiltrate. In addition examination of the bone marrow excluded the possibility of an underlying plasma cell disorder or primary amyloidosis.
Furthermore, an occult breast primary would have been expected to result in secondary amyloidosis but there was no progression to systemic disease over time. LA is 10 times less common than systemic amyloidosis and it is possible that systemic progression may have been curtailed by early detection and treatment of the breast primary.
Learning points.
Our case clinically and radiographically masqueraded as an upper tract urothelial malignancy and the patient was nephrectomised. Conservative management was precluded as it was not possible to obtain a definitive preoperative diagnosis of amyloidosis.
Our case highlights the ever present risk of a false diagnosis of LA of the renal pelvis due to clinical, radiological and endoscopic overlap with urothelial malignancy. In contrast to urothelial neoplasia, it is crucial for the endoscopist to obtain adequate subepithelial mesenchymal tissue to accurately diagnose amyloidosis and avoid overtreatment.
Intraoperative adhesions were encountered during surgical dissection around the renal pelvis and this necessitated conversion to an open procedure. A polytypic inflammatory infiltrate was demonstrated in association with the renal pelvis amyloid and there was no evidence of a systemic plasma cell dyscrasia.
Idiopathic AL type primary renal pelvis amyloidosis was confirmed by monospecific immunohistochemical staining. Investigations to evaluate the extent of organ involvement were negative. Haematological monitoring during a follow-up period of 30 months has shown no evidence of multiple myeloma or other lymphoid malignancy.
Interestingly, our patient developed a coincidental, apparently unrelated, locally advanced breast carcinoma 9 months after the initial diagnosis of AL LA. There is no precedent in the medical literature and we have addressed the question of whether this might have been an early manifestation of an occult primary epithelial malignancy. Amyloid was missing at the site of the breast primary and no systemic amyloidosis was shown even after extensive investigation and follow-up.
Footnotes
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Wetzel R. Kinetics and thermodynamics of amyloid fibril assembly. Acc Chem Res 2006;39:671–9. [DOI] [PubMed] [Google Scholar]
- 2.Gertz MA, Comenzo R, Falk RH et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, France, 18–22 April 2004. Am J Hematol 2005;79:319–28. [DOI] [PubMed] [Google Scholar]
- 3.Borza T, Shah RB, Faerber GJ et al. Localized amyloidosis of the upper urinary tract: a case series of three patients managed with reconstructive surgery or surveillance. J Endourol 2010;24:641–4. [DOI] [PubMed] [Google Scholar]
- 4.Chisholm GD, Cooter NB, Dawson JM. Primary amyloidosis of the renal pelvis. BMJ 1967;1:736–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dias R, Fernandes M, Patel RC et al. Amyloidosis of renal pelvis and urinary bladder. Urology 1979;14:401–4. [DOI] [PubMed] [Google Scholar]
- 6.Fox M, Hammond JC, Knox R et al. Localised primary amyloidosis of the renal pelvis. Br J Urol 1984;56:223–4. [DOI] [PubMed] [Google Scholar]
- 7.Gelbard M, Johnson S. Primary amyloidosis of renal pelvis and renal cortical adenoma. Urology 1980;15:614–17. [DOI] [PubMed] [Google Scholar]
- 8.German KA, Morgan RJ. Primary amyloidosis of the renal pelvis and upper ureter. Br J Urol 1994;73:99–100. [DOI] [PubMed] [Google Scholar]
- 9.Iida S, Chujyo T, Nakata Y et al. A case of amyloidosis of the renal pelvis. Hinyokika Kiyo 2003;49:423–6. [PubMed] [Google Scholar]
- 10.Kirkpantur A, Baydar DE, Altun B et al. Concomitant amyloidosis, renal papillary carcinoma and ipsilateral pelvicalyceal urothelial carcinoma in a patient with familial Mediterranean fever. J Amyloid 2009;16:54–9. [DOI] [PubMed] [Google Scholar]
- 11.Gardner KD Jr, Castellino RA, Kempson R et al. Primary amyloidosis of the renal pelvis. N Engl J Med 1971;284:1196–8. [DOI] [PubMed] [Google Scholar]
- 12.Gilbert LW, McDonald Jr. Primary amyloidosis of the renal pelvis and ureter: report of case. J Urol 1952;68:137–9. [DOI] [PubMed] [Google Scholar]
- 13.Merrimen JL, Alkhudair WK, Gupta R. Localized amyloidosis of the urinary tract: case series of nine patients. Urology 2006;67:904–9. [DOI] [PubMed] [Google Scholar]
- 14.Monge M, Chauveau D, Cordonnier C et al. Localized amyloidosis of the genitourinary tract: report of 5 new cases and review of the literature. Medicine (Baltimore) 2011;90:212–12. [DOI] [PubMed] [Google Scholar]
- 15.Murphy MN, Alguacil-Garcia A, MacDonald RG. Primary amyloidosis of renal pelvis with duplicate collecting system. Urology 1986;27:470–3. [DOI] [PubMed] [Google Scholar]
- 16.Paidy S, Unold D, Catanzano TM. AIRP best cases in radiologic-pathologic correlation: localized amyloidosis of the renal pelvis. Radiographics 2012;32:2025–30. [DOI] [PubMed] [Google Scholar]
- 17.Pan DL, Na YQ. Amyloidosis of the unilateral renal pelvis, ureter and urinary bladder: a case report. Chin Med Sci J 2011;26:197–200. [DOI] [PubMed] [Google Scholar]
- 18.Rauschmeier H, Hofstädter F, Jakse G. Ossified amyloidosis of the kidney pelvis. Urologe A 1983;22:255–9. [PubMed] [Google Scholar]
- 19.Savareux L, Guy L, Essamet W et al. Pyelo-ureteric amyloidosis. Prog Urol 2004;14:406–10. [PubMed] [Google Scholar]
- 20.Shi LX, Li G, Zhang L et al. Primary localized AA type amyloidosis of the renal pelvis, ureter and bladder. Pak J Med Sci 2012;28:530–2. [Google Scholar]
- 21.Shiramizu M, Nakamura K, Baba S et al. Primary localized amyloidosis of the renal pelvis coexisting with transitional cell carcinoma: a case report. Hinyokika Kiyo 1992;38:699–702. [PubMed] [Google Scholar]
- 22.Sparwasser C, Gilbert P, Mohr W et al. Unilateral extended amyloidosis of the renal pelvis and ureter: a case report. Urol Int 1991;46:208–10. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki T, Kikuchi K, Saito S et al. A case of a primary localized amyloidosis (amyloid tumor) of the renal pelvis and ureter. Gan No Rinsho 1987;33:1494–500. [PubMed] [Google Scholar]
- 24.Ullmann AS. Primary amyloidosis of the renal pelvis: a case report and review of literature. Mich Med 1973;72:29–33. [PubMed] [Google Scholar]
- 25.Kyle RA, Greipp PR. Amyloidosis (AL). Clinical and laboratory features in 229 cases. Mayo Clin Proc 1983;58:665–83. [PubMed] [Google Scholar]
- 26.Akimoto K. Ober amyloidartige Eiweissniederschlage im Nierenbecken. Beitr Path Anat 1927;78:239 (Japanese/Eng. translation). [Google Scholar]
- 27.Ferch R, Haskell R, Farebrother T. Primary amyloidosis of the urinary bladder and ureters. Br J Urol 1997;80:953–4. [DOI] [PubMed] [Google Scholar]
- 28.Livingstone RR, Sarembock LA, Barnes RD et al. Colchicine therapy in primary amyloidosis of the bladder: a case report. J Urol 1989;142:1570–1. [DOI] [PubMed] [Google Scholar]
- 29.Malek RS, Greene LF, Farrow GM. Amyloidosis of the urinary bladder. Br J Urol 1971;43:189–200. [DOI] [PubMed] [Google Scholar]
- 30.Kyle RA, Gertz MA. Primary systemic amyloidosis. Clinical and laboratory features in 474 cases. Semin Hematol 1995;32:45–59. [PubMed] [Google Scholar]
- 31.Kyle RA, Bayrd ED. ‘Primary’ systemic amyloidosis and myeloma. Discussion of relationship and review of 81 cases. Arch Intern Med 1961;107:344–53. [DOI] [PubMed] [Google Scholar]
- 32.Agarwal SK, Walmsley BH, Marley NJ. Primary amyloidosis of urinary bladder. J R Soc Med 1995;88:171–2. [PMC free article] [PubMed] [Google Scholar]
- 33.Keeley FX, Kulp DA, Bibbo M et al. Diagnostic accuracy of ureteroscopic biopsy in upper tract transitional cell carcinoma. J Urol 1997;157:33–7. [PubMed] [Google Scholar]
- 34.Goteri G, Ranaldi R, Pileri SA et al. Localized amyloidosis and gastrointestial lymphoma: a rare association. Histopathology 1998;32:348–55. [DOI] [PubMed] [Google Scholar]
- 35.Kimball KG. Amyloidosis in association with neoplastic disease: Report of an unusual case and clinicopathological experience at Memorial Center for Cancer and Allied Disease during eleven years (1948–1958). Ann Intern Med 1961;55:958–74. [DOI] [PubMed] [Google Scholar]
- 36.Prathap K, Looi LM, Prasad O. Localized amyloidosis in nasopharyngeal carcinoma. Histopathology 1984;8:27–34. [DOI] [PubMed] [Google Scholar]
- 37.Agha I, Mahoney R, Beardslee M et al. Systemic amyloidosis associated with pleomorphic sarcoma of the spleen and remission of nephrotic syndrome after removal of the tumor. Am J Kidney Dis 2002;40:411–41. [DOI] [PubMed] [Google Scholar]
- 38.Jaakkola H, Tornroth T, Groop P et al. Renal failure and nephrotic syndrome associated with gastrointestinal stromal tumour (GIST)—a rare cause of AA amyloidosis. Nephrol Dial Transplant 2001;16:1517–18. [DOI] [PubMed] [Google Scholar]
- 39.Dahlin DC. Secondary amyloidosis. Ann Intern Med 1949;31:105–19. [DOI] [PubMed] [Google Scholar]
- 40.Pierce BL, Ballard-Barbash R, Bernstein L et al. Elevated biomarkers of inflammation are associated with reduced survival among breast cancer patients. J Clin Oncol 2009;27:3437–44. [DOI] [PMC free article] [PubMed] [Google Scholar]