

Abstract
Cholelithiasis is a common problem in the Western world. Recurrent gallstones after cholecystectomy, however, are rare. We describe a case of a young woman with recurrent gallstones after a laparoscopic cholecystectomy leading to cholangitis during pregnancy. Additional testing revealed an ATP-binding cassette B4 (ABCB4) gene mutation. ABCB4 gene mutations leading to a multidrug resistance (MDR)3-P-glycoprotein deficiency are related to, among other diseases, recurrent cholelithiasis. Medical treatment consists of administering oral ursodeoxycholic acid. If untreated, MDR3 deficiency can lead to progressive liver failure requiring liver transplantation.
Background
Only 1.5–1.9%1 2 of patients who have undergone cholecystectomy in the past will suffer from recurrent cholelithiasis, which can lead to severe complications such as pancreatitis and biliary sepsis. Pregnancy and ATP-binding cassette B4 (ABCB4) gene mutations are associated with gallstone disease at young ages.3 4
Case presentation
A 34-year-old woman in her 14th week of pregnancy was admitted to our hospital with biliary colic. Her medical history revealed a diagnosis of biopsy-proven focal nodular hyperplasia of the liver at the age of 25. As a consequence, her oral contraceptives were discontinued. Five years later, at age 30, a laparoscopic cholecystectomy was performed due to symptomatic cholelithiasis. In the years following the cholecystectomy, she had to undergo several endoscopic retrograde cholangiopancreatographies (ERCPs) due to recurrent choledocholithiasis. One of the ERCPs was complicated by a duodenal perforation requiring surgical repair due to the endoscopic removal of a large common bile duct stone.
On presentation, the patient reported upper abdominal pain with nausea. On physical examination, her body temperature was 37.4° Celsius. Palpation of the abdomen revealed slight tenderness of the epigastric region. On the second day of her hospital stay, the patient suffered from recurrent colicky pain and developed jaundice. Biochemical and haematological investigations showed that her serum bilirubin had risen from of 13 to 61 µmol/L (normal <17 µmol/L) and her C reactive protein had risen from 2 mg/L on admission to 158 mg/L (normal <10 mg/L). On admission, ultrasound examination of the abdomen had shown normal calibre bile ducts, although the common bile duct had not been optimally visualised due to intestinal gas. An ERCP was performed, which revealed a purulent cholangitis. Multiple stones (diameter >10 mm) were removed from the common bile duct (see figure 1), and plastic endoprostheses were placed. She recovered quickly and was discharged soon after. Owing to the recurrent episodes of gallstone, the patient's family history was thoroughly reviewed. The patient’s father had suffered from liver disease, and her grandmother had died from cirrhosis. Furthermore, her mother as well as her grandmother had died from colon carcinoma.
Figure1.
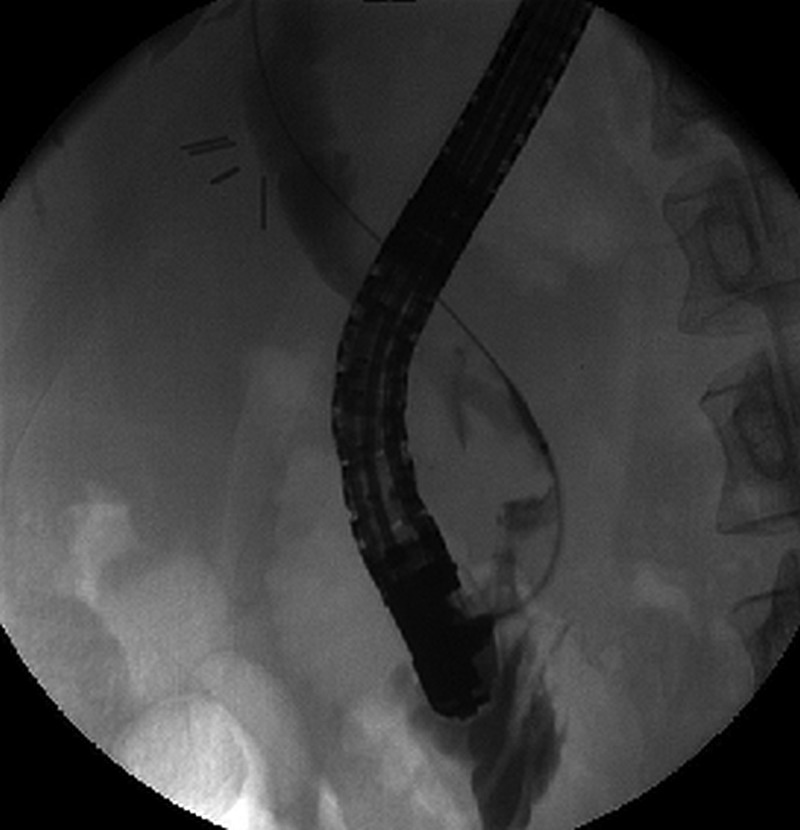
X-ray picture during endoscopic retrograde cholangiopancreatography showing several concretions within the ductus choledochus. In the left upper quadrant the clips can be seen of previous cholecystectomy.
Investigations
The combination of the recurrent cholelithiasis at young age following cholecystectomy and the remarkable family history for liver cirrhosis suggested an inherited factor. Genetic analysis was performed by means of sequencing technique, which revealed a heterozygous ABCB4 gene mutation (c.2177C>T on chromosome 7q21). This mutation leads to a substitution of the amino acid proline with the amino acid leucine at position 726.
Treatment
On discharge, maintenance treatment with ursodeoxycholic acid (UDCA) was initiated, and the dose was increased step wise to 900 mg daily (12 mg/kg body weight per day).5
Outcome and follow-up
The continued course of the patient's pregnancy was uneventful, and she gave birth to a healthy baby boy at 35 weeks and 6 days of gestation. During follow-up we noticed that liver enzyme values had completely normalised, compared to pre-pregnancy values. (table 1).
Table 1.
laboratory values
| Serum liver enzyme functions | Before pregnancy | Following 12 months of UDCA treatment |
|---|---|---|
| Bilirubin | 12 | 6 µmol/L (normal <17 µmol/L) |
| Aspartate amino transferase | 66 | 18 U/L (normal <37 U/L) |
| Alanine amino transferase | 216 | 23 U/L (normal <37 U/L) |
| Alkaline phosphatase | 266 | 107 U/L (normal <120 U/L) |
| y-Glutamyltranspeptidase | 363 | 24 U/L (normal <35 U/L) |
UDCA, ursodeoxycholic acid.
Since the patient had such an excellent clinical response, we did not adjust the treatment dosage of UDCA after the delivery. The plastic endoprostheses were removed 6 months after her delivery. Just recently, 4 years after the admission described here, she gave birth to a second child. The course of the second pregnancy was uneventful.
Discussion
The pregnant patient described here, suffered from cholelithiasis complicated by biliary sepsis. Recurrent gallstone formation had led to repeated ERCPs, which is a potentially hazardous procedure. The natural history of gallstones starts with the formation of gall, which is generated by hepatocytes and excreted into the intestines via the bile ducts. The main functions of gall are to digest fatty substances and to transport nutrients that are not water soluble. The balance between the salts, cholesterol and bilirubin in the primary gall solution has two important consequences. First, sufficient transport of cholesterol is important to prevent the precipitation of sludge and stones. Second, excess bile acids may exert toxic effects, leading to inflammation of the bile ducts and liver.6
A number of transporter proteins regulate the excretion of salts, bilirubin and phospholipids. The last are the main cholesterol carriers. The multidrug resistance (MDR)3P-glycoprotein is responsible for the transport of the phospholipid phosphatidylcholine.7 Reduced or absent transport of phosphatidylcholine (low-phospholipid-associated cholelithiasis syndrome, LPAC), is indeed associated with intrahepatic sludge or stone formation as well as bile salt-induced damage. This relationship explains the association between the gene mutation and the development of fibrosis and even cirrhosis.7–9 Other diseases associated with MDR3 protein deficiency caused by ABCB4 gene mutations are intrahepatic cholestasis of pregnancy (ICP),8 10 progressive familial intrahepatic cholestasis type 3,8 11 benign recurrent intrahepatic cholestasis8 and drug-induced liver injury.8 During pregnancy a marked increase in cholesterol saturation of bile will be noticed. Higher levels of oestrogens stimulate hepatic lipoprotein receptors, increase uptake of dietary cholesterol and increase biliary cholesterol secretion. Furthermore sluggish gallbladder contraction results in impaired gallbladder emptying.12
The ABCB4 gene, which codes for the MDR3 P-glycoprotein, is located on the long arm of chromosome 7 (7q21.1).8 Over 40 mutations of this gene have already been reported. The degree (and types) of symptoms depends on the location of the mutation and whether the mutation is heterozygous or homozygous.11 The mutation detected in our patient has been described in a previous study of 156 patients.3 Most patients in this series had heterogenous missense mutations. The c.2177C>T was reported twice in patients with LPAC, one of them also had ICP. In the case described here, there were no signs of cirrhosis or primary sclerosing cholangitis.
During follow-up for more than 4 years, including an uncomplicated second pregnancy, no periods of recurrent cholangitis were observed. We suggest that, although we have not definitely proven, the administration of UDCA has had a beneficial influence. UDCA is believed to induce an increase in the hydrophilicity of bile acids, to stimulate biliary secretion and to have immune-modulating and anti-inflammatory effects.13–15
Learning points.
An ATP-binding cassette B4 (ABCB4) gene mutation should be considered in patients who suffer from symptomatic cholelithiasis at a young age (under 40 years old), recurrent biliary symptoms despite cholecystectomy and concretions or sludge in the intrahepatic and extrahepatic biliary system.
Several diseases, including low-phospholipid-associated cholelithiasis syndrome, intrahepatic cholestasis of pregnancy, liver cirrhosis, progressive familial intrahepatic cholestasis type 3, benign recurrent intrahepatic cholestasis and drug-induced liver injury, have been linked to ABCB4 gene mutations.
Ursodeoxycholic acid is helpful in order to preventing stone formation and bile duct damage.
Footnotes
Contributors: JHE and AD wrote the final version of the manuscript. All authors participated in the preparation of the manuscript.
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Von Schönfels W, Buch S, Wölk M et al. Recurrence of gallstones after cholecystectomy is associated with ABCG5/8 genotype. J Gastroenterol 2013;48:391–6. 10.1007/s00535-012-0639-3 [DOI] [PubMed] [Google Scholar]
- 2.Konsten J, Gouma DJ, von Meyenfeldt MF et al. Long-term follow-up after open cholecystectomy. Br J Surg 1993;80:100–2. 10.1002/bjs.1800800132 [DOI] [PubMed] [Google Scholar]
- 3.Poupon R, Rosmorduc O, Boelle PY et al. Genotype-phenotype relationships in the low-phospholipid-associated cholelithiasis syndrome: a study of 156 consecutive patients. Hepatol 2013;58:1105–10. 10.1002/hep.26424 [DOI] [PubMed] [Google Scholar]
- 4.Sundaram SS, Sokol RJ. The multiple facets of ABCB4 (MDR3) deficiency. Curr Treat Options Gastroenterol 2007;10:495–503. 10.1007/s11938-007-0049-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.European Association for the Study of the Liver. EASL clinical practice guidelines: management of cholestatic liver diseases. J Hepatol 2009;51:237–67. 10.1016/j.jhep.2009.04.009 [DOI] [PubMed] [Google Scholar]
- 6.Boyer JL. Bile formation and secretion. Compr Physiol 2013;3:1035–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosmorduc O, Poupon R. Low phospholipid associated cholelithiasis: association with mutation in the MDR3/ABCB4 gene. Orphanet J Rare Dis 2007;2:29 10.1186/1750-1172-2-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davit-Spraul A, Gonzales E, Baussan C et al. The spectrum of liver diseases related to ABCB4 gene mutations: pathophysiology and clinical aspects. Semin Liver Dis 2010;30:134–46. 10.1055/s-0030-1253223 [DOI] [PubMed] [Google Scholar]
- 9.Rosmorduc O, Hermelin B, Poupon R. MDR3 gene defect in adults with symptomatic intrahepatic and gallbladder cholesterol cholelithiasis. Gastroenterology 2001;120:1459–67. 10.1053/gast.2001.23947 [DOI] [PubMed] [Google Scholar]
- 10.Gotthart D, Runz H, Keitel V et al. A mutation in the canalicular phospholipid transporter gene, ABCB4, is associated with cholelithiasis, ductopenia, and cirrhosis in adults. Hepatoloy 2008;48:1157–66. 10.1002/hep.22485 [DOI] [PubMed] [Google Scholar]
- 11.van der Woerd WL, van Mil SW, Stapelbroek JM et al. Familial cholelithiasis: progressive familial intrahepatic cholestasis, benign recurrent intrahepatic cholestasis and intrahepatic cholestasis of pregnancy. Best Pract Res Clin Gastroenterol 2010;24:541–53. 10.1016/j.bpg.2010.07.010 [DOI] [PubMed] [Google Scholar]
- 12.Longo DL, Fauci AS, Kasper DL et al. Harrison's principles of internal medicine. 18th edn New York: McGraw-Hill Medical Publishing Division, 2011:2618. [Google Scholar]
- 13.Poupon R. Ursodeoxycholic acid and bile-acid mimetics as therapeutic agents for cholestatic liver diseases: an update of their mechanisms of action. Clin Res Hepatol Gastroenterol 2012;36(Suppl1):S3 10.1016/S2210-7401(12)70015-3 [DOI] [PubMed] [Google Scholar]
- 14.Beuers U. Drug insight: mechanisms and sites of action of ursodeoxycholic acid in cholestasis. Nat Clin Pract Gastroenterol Hepatol 2006;3:318–28. 10.1038/ncpgasthep0521 [DOI] [PubMed] [Google Scholar]
- 15.Roma MG, Toledo FD, Boaglio AC et al. Ursodeoxycholic acid in cholestasis: linking action mechanisms to therapeutic applications. Clin Sci (Lond) 2011;121:523–44. 10.1042/CS20110184 [DOI] [PubMed] [Google Scholar]