

Abstract
Objective: Whether alveolar edema could be cleared by alveolar epithelial is a key to the treatment and prognosis of ALI (acute lung injury). In this study, oleic acid(OA)-induced ALI model was established, the expression of α1 Na+/K+-ATPase (NKA) and β1 Na+/K+-ATPase were performed in vivo to investigate the mechanism of alveolar fluid clearance (AFC) in ALI and the effect of early low doses of dexamethasone on alveolar fluid clearance. Methods: In this study, Male rats were challenged by OA with or without dexamethasone (1 mg/kg, iv) post-treatment. Lung histopathology, blood gas, pulmonary vascular permeability, BALF IL-6, MPO and NKA activity of lung were examined. α1NKA and β1NKA mRNA and protein expression were detected. Results: The results indicated that compared with sham operated group, NKA activity, mRNA and protein expression of α1NKA and β1NKA were decreased in OA treated group, while wet/dry ratio, lung index, IL-6, and MPO activity were increased significantly. Pulmonary edema was obviously seen under light microscope. Those indexes were improved in dexamethasone treated group compared to OA treated group. Conclusion: The expression of NKA to decline for the lung injury is one important mechanism of pulmonary edema. Early low dose of dexamethasone treatment could suppress the expression of inflammatory mediators, improved lung epithelial-endothelial barrier permeability, increased the expressions of α1 NKA and β1 NKA mRNA, α1 NKA and β1 NKA protein level, stimulated NKA activity and decreased pulmonary edema. In conclusion, these observations suggest that early low dose of dexamethasone treatment has a protective effect on OA induced ALI.
Keywords: Dexamethasone, oleic acid, acute lung injury, lung edema, sodium pump
Introduction
Acute lung injury/acute respiratory distress syndrome (ALI/ARDS) is a syndrome with extensive damage to the alveolar-capillary barrier due to variety of clinical disorders, such as trauma, aspiration, shock, serious infection, and is characterized by non-cardiogenic pulmonary edema, severe dyspnea, respiratory distress and refractory hypoxemia [1]. ARDS represents the most severe form and final stage of ALI. Though numerous studies have been made for many years, there is no specific pharmacological measures for prevention and treatment of ALI/ARDS with a high mortality [2,3]. Whether alveolar edema could be cleared by alveolar epithelial is a sign of lung injury degree, and a key to the treatment and prognosis of ALI [4]. Studies have showed that active transport is the main form of alveolar fluid clearance. Sodium channel and sodium pump of the alveolar epithelial play an important role during the removal of water in the lungs [5,6]. However, adjustment mechanism of alveolar epithelial active transport liquid function under pathological conditions remain unclear [7]. Therefore, investigation of the pathogenesis of the early phase of ALI and the effective and specific therapies which could stimulate AFC are still urgently needed.
Because intravenous injection of oleic acid causes pathological changes similar to clinical ALI and ARDS, it has been regarded as a popular experimental model for ALI. Patients with ARDS have obviously increased the blood level of OA [8]. Similarly, higher blood levels of OA were observed in patients developed ARDS [8]. Furthermore, an increase in the proportion of OA incorporated into surfactant phospholipids was found in patients with ARDS and sepsis [9]. These results suggest the blood level of OA as a predictive or prognostic factor for ARDS [10]. Therefore, we study the expression and regulatory mechanism of Na+/K+-ATPase in OA-induced acute lung injury model.
In previous studies, glucocorticoids inhibited the inflammatory response in ALI/ARDS, but its clinical efficacy is still controversial due to different cause of ALI/ARDS, different hormone dosage, application timing of hormone, and the side effects of the drug. Effects of glucocorticoids on acute lung injury induced by oleic acid were seldom reported. Furthermore, few research has been performed to investigate the regulation effects of glucocorticoids on sodium and water transport in lung injury induced by OA. Therefore, in the present study, our purpose was to investigate whether early low dose dexamethasone treatment could alleviate ALI caused by OA through improving lung epithelial-endothelial barrier permeability by stimulating the expression of Na+/K+-ATPase.
Materials and methods
Animals
Adult male Sprague-Dawley rats weighing 235 ± 25 g were purchased from Experimental Animal Center of Chongqing Medical University (Chongqing 400016, China). The rats were housed in a temperature-controlled environment with 12-hour light-dark cycles and were fed standard laboratory diet and water ad libitum. All animal procedures of this study were approved by the Animal Care and Use Committee at the Chongqing Medical University and were in accordance with the Declaration of the National Institutes of Health Guide for Care and Use of Laboratory Animals (Publication No. 85-23, revised 1985).
Reagents
Oleic acid was produced by Sigma-Aldrich (St. Louis, MO). Rat IL-6 ELISA assay kit was supplied by Beijing 4A Biotech Co. (Beijing, China). Protein assay dye reagent was provided by Jiancheng Bioengineering Co. (Nanjing, China). Myeloperoxidase (MPO) detection kit and Na+/K+-ATPase activity assay kit were the products of Jiancheng Biological Co. (Nanjing, China). Total RNA was extracted with TRIpure Reagent (Roche, USA). Reverse transcriptase kit (Promega, USA), PCR kit (TAKARA, Dalian, Chinese). Primary α1-Na+-K+-ATPase monoclonal antibody was provided by NOVUS BIOLOGICALS (USA), primary β1-Na+-K+-ATPase polyclonal antibody was from Santa Cruz (USA), HRP-conjugated anti-mouse IgG and HRP-conjugated anti-rabbit IgG were purchased from GenScrip Co (USA).
Animal model and grouping
Rats were randomly divided into three groups: (1) Sham operated (n = 8); (2) OA treated (n = 8); (3) OA-DEX (dexamethasone) treated (n = 8).
The rats were anaesthetized by intraperitoneal administration of 3.5% Chloral hydrate (1 ml/100 g). The rats in OA treated group were given oleic acid (dose, 0.20 ml/kg) via right jugular vein. The rats in Sham operated group were administered saline (dose, 0.20 ml/kg) via right jugular vein. The rats in OA-DEX group were treated with dexamethasone (dose, 1 mg/kg) via right jugular vein 15 min after oleic acid injection. In the OA treated and Sham operated groups, an equivalent volume of saline was injected instead of dexamethasone. Rats were killed by exsanguination from the carotid artery at 6h after OA injection. Blood sample was collected for blood gas analysis in each animal group.
Respiratory rate and arterial blood gas analysis
Under condition of aesthesis, carotid arterial blood sample was collected at 6 h after OA administration. PH, PaO2, and PaCO2 under room air were determined immediately using blood gas analyzer (Nova Biomedical, PHOX PLUS, USA). In each group, respiratory rates were measured at the time of blood sampling.
Lung index, wet/dry ratio determinations
In each group, the whole lung was weighed immediately to get wet weight after its excision. Lung index was obtained by dividing the wet weight of the whole lung by body weight. Subsequently, harvest the right lung lower lobe and weigh as wet weight. The right lung lower lobe was then dried with an oven at 80°C and weighed daily. After 72 h, a constant weight was taken as dry weight. The following formula can be used to determine W/D ratio. W/D ratio = Wet weight/Dry weight × 100%.
Lung permeability index
Six hours after the administration of OA, blood samples were taken from Carotid artery. Open the chest of rat and tracheotomy was performed. After ligation of the right main bronchus with silk thread and endotracheal intubation, the left lung was lavaged five times with 5 mL of normal saline. The bronchoalveolar lavage fluid collection was centrifuged at 3000 rpm for 15 min. The supernatant was removed and kept at -70°C for future analysis. Protein concentration of BALF in the supernatant was determined using Bradford assay. Serum protein concentration of serum was assayed by Automatic biochemical analyzer. The protein concentration in BALF/serum ratio was used as lung permeability index.
IL-6 and MPO determinations
Concentration of IL-6 in bronchoalveolar lavage fluids were measured by using enzyme linked immunosorbent assay with IL-6 ELISA kits for rats according to the manufacturer’s instructions. Lung samples obtained from the right middle lobe were homogenized with 0.9% normal saline and centrifuged. MPO activities were then determined by 1240-UV-Visible Spectrophotometer (Shimadzu, Japan) at 460 nm wavelength with MPO detection kit following the manufacturer's instructions.
Histopathological examination
The upper lobe of the right lung was harvested at 6 h after OA injection and immediately fixed into 4% Paraformaldehyde for 48 h. Lung tissues were embedded in paraffin and sectioned into 5 μm thick pieces for hematoxylin and eosin (H&E) staining. Under light microscope, lung specimens were analyzed by blinded observation to evaluate lung injury.
Measurement of α1-Na+-K+ATPase and β1-Na+-K+ATPase mRNA level in lung tissue
Reverse transcriptase polymerase chain reaction analysis (RT-PCR) was used to measure the level of α1-Na+-K+ATPase and β1-Na+-K+ATPase mRNA in lung tissue. Total RNA was extracted from the right middle lung (about 100 mg) using TRIpure reagent following the instructions provided by the manufacturer. RNA concentration and purity were assessed by spectrophotometry at 260 and 280 nm. One microgram of total RNA synthesized cDNA through reverse transcriptase, followed by amplification with the specific primers. The amplification cycles (30 cycles) were performed as follows: denaturation at 94°C for 30 s, annealing at 58°C (α1-Na+-K+ATPase), 58°C (β1-Na+-K+ATPase) and 55°C (β-actin) for 30 s, extension at 72°C for 1 min, with the last extension at 72°C for 7 minutes. Upon the completion of amplification, RT-PCR products were undergone electrophoresis in 1.5% agarose gels stained with ethidium bromide (EB). Bands were visualized and quantified using Quantity One software (Bio-rad, USA). The relative expression level of target gene was normalized in comparison to β-actin mRNA. The primers used for α1-Na+-K+ATPase, β1-Na+-K+ ATPase and β-actin are demonstrated in Table 1.
Table 1.
Primers used in this study
| Gene | Primer sequence (5’-3’) | Primer length (bp) | Product (bp) | Ata (°C) |
|---|---|---|---|---|
| α1-Na+-K+ATPase | (F) 5’-AGATTTGAGCCGAGGCCTAACACC-3’ | 24 | 418 | 58 |
| (R) 5’-TCCGCCCTTCACCTCCACCAGAT-3’ | 23 | |||
| β1-Na+-K+ATPase | (F) 5’-GCCCCGCCAGGATTGACAC-3’ | 19 | 444 | 58 |
| (R) 5’-CTCATCGCGCTTGCCAGTG-3’ | 19 | |||
| β-actin | (F) 5’-GTACAACCTTCTTGCAGCTCCT-3’ | 22 | 871 | 55 |
| (R) 5’-ACAGGATTCCATACCCAGGAAG-3’ | 22 |
AT = annealing temperature.
Western blot analysis of Na+-K+-ATPase protein expression
Total proteins were extracted from rat lung tissue and immediately homogenized in ice-cold buffer containing T-PER Tissue protein extraction reagent (Pierce, USA). The homogenate was centrifuged for 5 min at 10,000 g and the supernatant was utilized. Protein quantification was performed by bicinchoninic acid assay (Pierce) using bovine serum albumin as standard. Fifty micrograms of proteins were loaded and separated by standard 7.5 or 10% polyacrylamide gels (SDS-PAGE) and transferred onto polyvinylidene fluoride (PVDF) membrane (Bio-Rad). After blocking with 5% nonfat dry milk in TBST buffer [20 mM Tris, pH 7.6; 140 mM NaCl; 0.1% (wt/vol) Tween 20], membranes were incubated overnight at 4°C with diluted specific primary antibodies (α1-Na+-K+-ATPase mouse monoclonal antibody, 1:3000, NOVUS BIOLOGICALS, USA, Cat NO. NB300-146; β1-Na+-K+-ATPase rabbit polyclonal antibody, 1:1000, Santa Cruz, USA, Cat NO. Sc-25709) in 5% BSA in TBST buffer. The membranes were then incubated with secondary antibodies (HRP-conjugated anti-mouse IgG, 1:5000, HRP-conjugated anti-rabbit IgG, 1:5000) for 1.5 h at room temperature in blocking buffer. All washes were in TBST buffer. The membranes were immunoblotted with anti-β-actin antibody as a loading controls. Blots were developed by according to the manufacturer’s instructions of the enhanced chemiluminescent detection kit (Amersham). Blotting bands were scanned and quantified by densitometry using Quantity One image analysis software (Bio-Rad). Expression of sodium transporting proteins was normalized to β-actin expression.
Determination of Na+-K+ ATPase activity in lung tissue
The Na+/K+-ATPase activity was measured by determining the amount of inorganic phosphate (Pi) released from adenosine triphosphate (ATP) using a Na+/K+-ATPase activity assay kit following the instructions of producer. The concentration of Pi was determined by observing absorbance at 660 nm. Na+/K+-ATPase activity was expressed as μmol Pi/mg protein/h.
Statistical analysis
Statistical analysis was performed using SPSS 11.5 statistical analysis software. Data were presented as mean ± standard deviation (SD). One-way analysis of variance (ANOVA) was used for multiple comparisons and least significant difference test (LSD-t) was used for intragroup comparison. A value of P < 0.05 was considered statistically significant.
Results
Respiratory rate and blood gas analysis
Hypoxemia was much more marked in the OA treated group, compared with the sham operated group and the dexamethasone treated group, as Figure 1 demonstrated. Significantly lower PaO2 was observed in the OA treated group compared to the sham operated group (56.09 ± 2.48 mmHg versus 100.37 ± 5.17 mmHg, P < 0.01). The PaO2/FIO2 was also significantly lower in the OA treated group than the sham operated group (267.10 ± 11.79 versus 477.95 ± 24.62, P < 0.01). Respiratory rate was evidently increased in the OA group compared with the sham operated group (145.50 ± 6.19 beat/min versus 81.13 ± 2.23 beat/min, P < 0.01). Compared to OA treated group, dexame-thasone increased PaO2 (83.23 ± 4.65 mmHg versus 56.09 ± 2.48 mmHg, P < 0.01), PaO2/FIO2 (396.33 ± 22.15 versus 267.10 ± 11.79) and reduced respiratory rate (108.38 ± 3.20 beat/min versus 145.50 ± 6.19 beat/min, P < 0.01), indicating that dexamethasone ameliorated OA-induced pulmonary injury (Figure 1).
Figure 1.
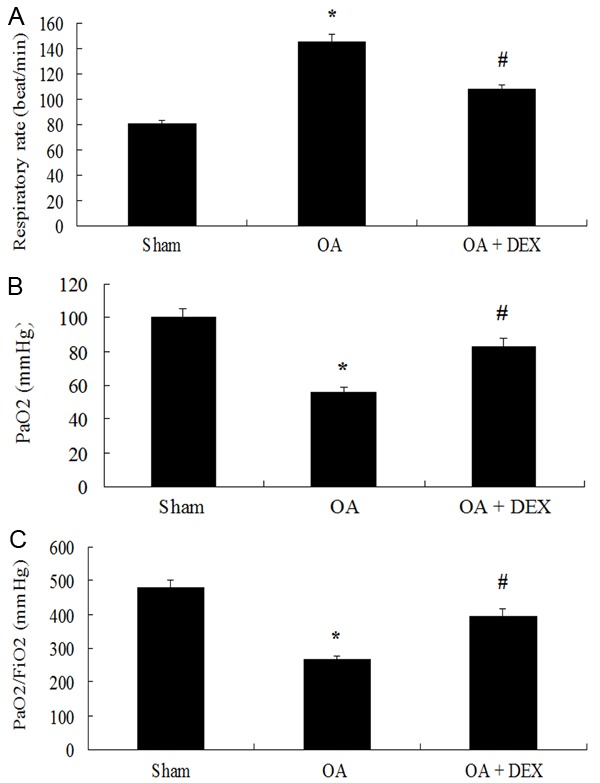
Comparison of Respiratory rate (A), PaO2 (B) and PaO2/FiO2 (C) between groups. *P < 0.01 compared with sham operated; #P < 0.01 compared with OA treated.
Determination of lung water (lung index and wet/dry weight ratio)
The pulmonary edema is viewed as a hallmark of ALI, so we determined lung index and wet-to-dry lung weight ratio (W/D) in various groups of experimental animals (Figure 2). Lung index (1.13% ± 0.17% versus 0.54% ± 0.26%, P < 0.01) and W/D ratio (6.37 ± 0.62 versus 4.46 ± 0.15, P < 0.01) obviously increased following injection of oleic acid compared to the sham operated group. However, the dexamethasone treated group showed a significantly decreased lung index (0.81% ± 0.06% versus 1.13% ± 0.17%, P < 0.01) and W/D ratio (5.07 ± 0.22 versus 6.37 ± 0.62, P < 0.01) compared to OA treated group.
Figure 2.
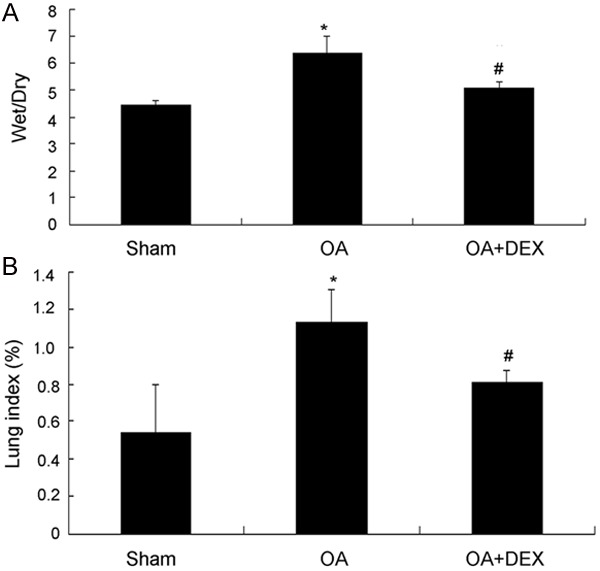
Comparison of Wet/Dry weight ratio (A) and Lung index (B) between groups. *P < 0.01 compared with sham operated; #P < 0.01 compared with OA treated.
Measurement of lung permeability index
As shown in Figure 3, LPI in the OA treated group was increased obviously compared with the sham operated group (4.75 × 10-3 ± 0.14 × 10-3 versus 1.36 × 10-3 ± 0.06 × 10-3, P < 0.01). No obvious changes were observed in lung permeability index in the sham operated group. Dexamethasone significantly reduced LPI comared with the OA treated group (2.73 × 10-3 ± 0.08 × 10-3 versus 4.75 × 10-3 ± 0.14 × 10-3, P < 0.01).
Figure 3.
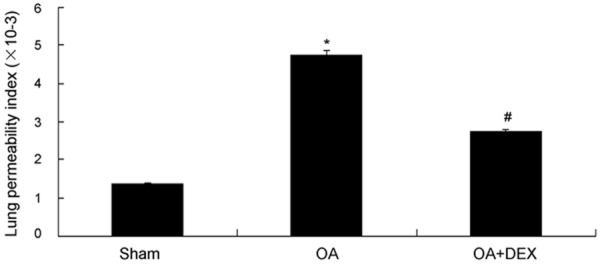
Comparison of lung permeability index (LPI) between three groups, *P < 0.01 compared with sham operated; #P < 0.01 compared with OA treated.
Determination of MPO activity in lung tissues and IL-6 levels in BALF
MPO activity (6.81 ± 0.25 u/g versus 1.74 ± 0.14 u/g, P < 0.01) in lung tissue and IL-6 levels of BALF (280.07 ± 20.84 pg/ml versus 67.08 ± 7.51 pg/ml, P < 0.01) were significantly increased in the OA treated group compared with the sham operated group. However, dexamethasone administration resulted in an evident decrease of MPO activity (3.62 ± 0.19 u/g versus 6.81 ± 0.25 u/g, P < 0.01) in lung tissue and IL-6 levels of BALF (147.98 ± 14.24 pg/ml versus 280.07 ± 20.84 pg/ml, P < 0.01) compared with the OA treated group (Figure 4).
Figure 4.
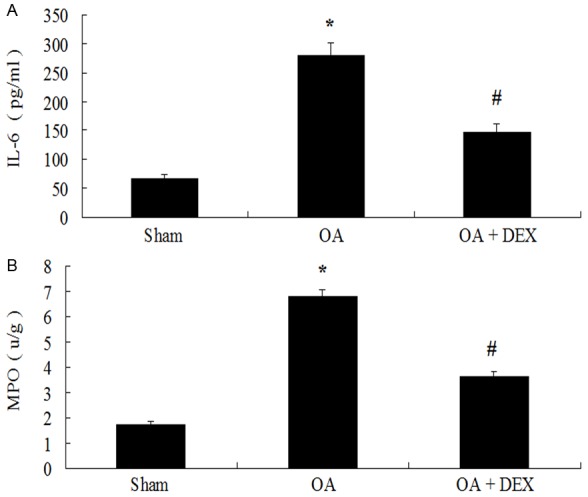
Comparison of levels of IL-6 and MPO between three groups *P < 0.01 compared with sham operated; #P < 0.01 compared with OA treated.
Histological examination
As shown in Figure 5A, in the sham operated group, observing the lung tissue slice of HE staining, significant secretion and inflammatory cell infiltration were not observed in the pulmonary alveolus cavity, the structure of pulmonary alveolus wall is normal.
Figure 5.
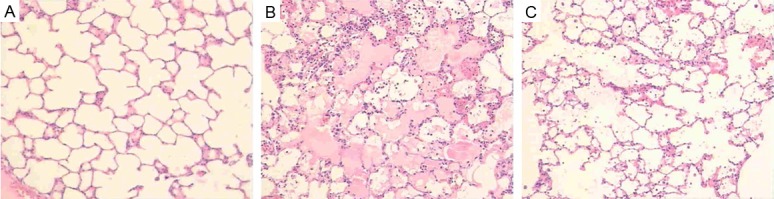
Histopathological changes of lung tissue in three groups (A-C). (A) Sham operated group (HE × 100). (B) OA treated group (HE × 100). (C) OA-DEX (dexamethasone) treated group (HE × 100).
Under the light microscope, evident lung injury resembling those seen in lung of patients with ALI/ARDS were seen in the OA treated group, including obvious infiltration of inflammatory cells, pulmonary interstitial and alveolar edema, large quantities of protein-rich percolate in alveolar space and destruction of alveolar structure (Figure 5B). However, dexamethasone treatment obviously attenuated these histological changes induced by OA injection (Figure 5C).
Levels of Na+/K+-ATPase mRNA in lung tissue
The level of Na+/K+-ATPase mRNA was evaluated by RT-PCR. As shown in Figure 6, the level of α1-Na+/K+-ATPase (0.13 ± 0.01 versus 1.12 ± 0.02, P < 0.01) and β1-Na+/K+-ATPase mRNA (0.28 ± 0.02 versus 1.11 ± 0.04, P < 0.01) in oleic acid model group was decreased obviously compared with those of the sham operated group. Compared to OA treated group, the level of α1-Na+/K+-ATPase mRNA (0.50 ± 0.02 versus 0.13 ± 0.01, P < 0.01) and β1-Na+/K+-ATPase (0.52 ± 0.02 versus 0.28 ± 0.02, P < 0.01) was increased obviously in dexamethasone treated group.
Figure 6.
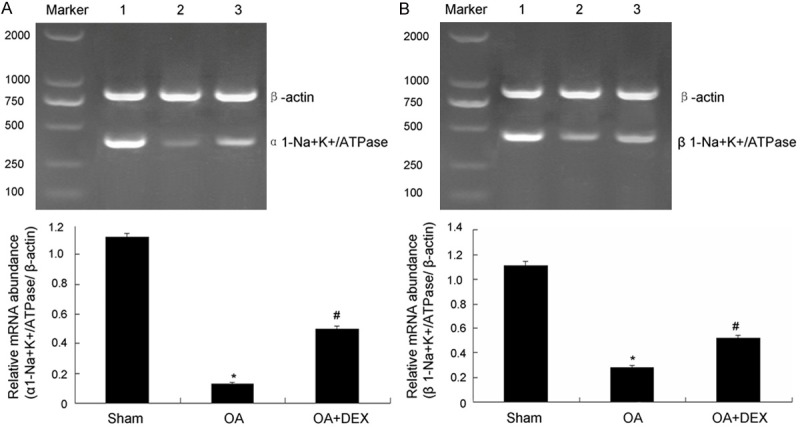
Measurement of ATPase mRNA level. A. Measurement of α1-Na+-K+-ATPase mRNA level in lung tissue and the statistical analysis. B. Measurement of β1-Na+-K+-ATPase mRNA level in lung tissue and the statistical analysis. 1: Sham operated group; 2: OA treated group; 3: OA-DEX (dexamethasone) treated group. *P < 0.01 compared with sham operated; #P < 0.01 compared with OA treated.
Expression of Na+-K+-ATPase protein in lung tissue
The expression of Na+/K+-ATPase protein in lung tissue was determined by Western blot. As shown in Figure 7, the protein expression of α1-Na+/K+-ATPase (0.06 ± 0.02 versus 0.31 ± 0.02, P < 0.01) and β1-Na+/K+-ATPase in lung tissue (0.32 ± 0.01 versus 0.77 ± 0.03, P < 0.01) in oleic acid model group declined obviously compared with those of the sham operated group. Compared to OA treated group, dexamethasone stimulated obviously the protein expression of α1-Na+/K+-ATPase (0.18 ± 0.03 versus 0.06 ± 0.02, P < 0.01) and β1-Na+/K+-ATPase (0.47 ± 0.02 versus 0.32 ± 0.01 P < 0.01).
Figure 7.
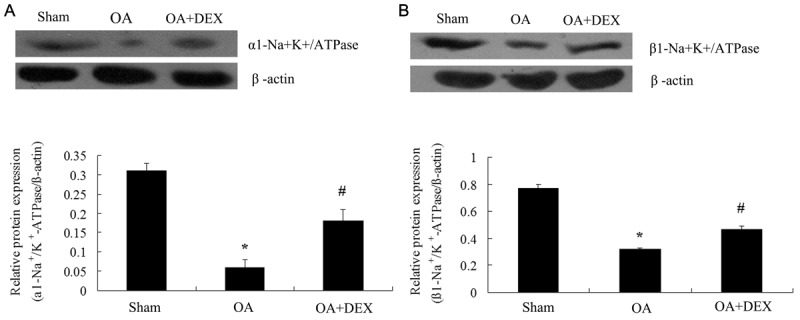
The protein expression of ATPase protein in lung tissues by western blot. A. The protein expression of α1-Na+-K+-ATPase in lung tissues were determined by Western blot and statistical analysis. B. The protein expression of β1-Na+-K+-ATPase in lung tissues was determined by Western blot and statistical analysis. *P < 0.01 compared with sham operated; #P < 0.01 compared with OA treated.
Changes of Na+-K+-ATPase activity in lung tissues
As shown in Figure 8, compared to sham operated group, Na+-K+-ATPase activity in lung tissue was decreased significantly in OA treated group (3.05 ± 0.18 μmolPi/mg protein/h versus 4.74 ± 0.10 μmolPi/mg protein/h, P < 0.01). Compared to OA treated group, Na+-K+-ATPase activity in lung tissue was increased significantly in dexamethasone treated group (4.06 ± 0.12 μmolPi/mg protein/h versus 3.05 ± 0.18 μmolPi/mg protein/h, P < 0.01).
Figure 8.
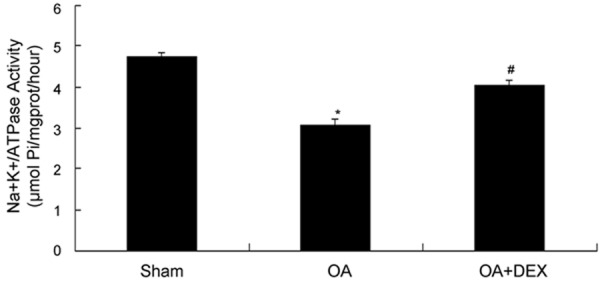
Comparison of Na+-K+-ATPase activity in lung tissues between groups. *P < 0.01 compared with sham operated; #P < 0.01 compared with OA treated.
Discussion
Increased lung water is one character of acute lung injury. Whether alveolar edema could be cleared by alveolar epithelial is a key to the treatment and prognosis of ALI. However, the adjustment mechanism of AFC under pathological condition sill remains unclear. ALI/ARDS remains a major challenge in critically ill patients. The application of glucocorticoids in ALI/ARDS has always been controversial. Studies have shown that high-dose and short-term glucocorticoid treatment can neither reduce mortality of in patients with early ARDS nor prevent the occurrence of ARDS with high-risk groups [11]. Similarly, patients with early-phase ARDS receiving high-dose glucocorticoid were ineffective [12]. Meduri et al. [13] reported that low-dose and prolonged glucocorticoid treatment was benefit for patients with early severe ARDS, which have been demonstrated to improve pulmonary and extrapulmonary organ function, to shorten the duration of mechanical ventilation, and to alleviated systemic inflammation. Lefort et al. [14] reported that dose effect of dexamethasone (1, 5 and 25 mg/kg) pretreatment on lung injury induced by endotoxin was obviously different. Low doses of dexamethasone markedly suppressed bronchopulmonary hyperreactivity, pulmonary neutrophil sequestration, and alveolocapillary dysfunction, whereas higher doses were inactive or aggravating. It may be explained for the high-dose steroid side effects, the release of inflammatory mediators and other factors. These different results may be accounted for different cause of ALI/ARDS, different hormone dosage, application timing of hormone, the side effects of the drug. Therefore, in the present study, ALI model which induced by intravenous injection of oleic acid was established to investigate the mechanism of AFC in ALI and the effect of early application of low dose of dexamethasone on acute lung inflammatory factors.
This study showed that OA induced lung epithelial-endothelial barrier damage, resulting in increased pulmonary edema. Early low dose of dexamethasone suppressed the expression of inflammation factor, improved lung epithelial-endothelial barrier permeability, increased the mRNA levels of α1 Na+-K+-ATPase and β1 Na+-K+-ATPase, stimulated protein expression of α1 Na+-K+-ATPase and β1 Na+-K+-ATPase and Na+-K+-ATPase activity, and decreased pulmonary edema, which suggest that early low dose of dexamethasone has a protective effect on oleic acid induced acute lung injury.
As a classical experimental model of ALI, the histopathological changes of lung injury induced by oleic acid are similar to those in clinical ALI and ARDS. This animal model does not depend on the activity of inflammatory cells and their products, which contributes to research the role of activity of inflammatory cells and their products in ALI/ARDS, contribute to research the occurrence and development process of pulmonary edema [15]. In the present study, we found that markedly pathological changes were observed in the OA-induced lung injury, which simulated the pathological character of human ALI/ARDS. At the same time, our experimental data showed that injection of OA significantly increased the lung index and wet/dry weight ratio, decreased PaO2/FiO2 compared to the sham operated group. Thus, these results indicated that the model of OA-induced ALI animal was established successfully, as reported previously [16,17]. The results showed that dexamethasone significantly improved oxygenation index, lung wet dry weight ratio and lung histopathological changes of lung injury induced by oleic acid, indicating that early low-dose dexamethasone treatment early has a protective effect on ALI.
The pathological basis of ALI is high pulmonary capillary permeability mediated by inflammatory cells (such as neutrophils, monocytes, giant cells), plasma protein and water into the extravascular space and the alveoli. Thus, the degree of lung damage can be accurately reflected by the changes of pulmonary capillary permeability. LPI is an important indicator which assesses pulmonary microvascular endothelial barrier function and pulmonary edema degree [18]. In this study, pulmonary vascular permeability was evaluated by LPI, W/D, lung water content and LI [19]. Compared to OA treated group, the expression of lung wet/dry ratio, LI and LPI were significantly decreased in dexamethasone treatment group. These indicators are consistent with the pathological changes of lung tissue. Thus, it confirmed that dexamethasone can improve pulmonary vascular permeability and inhibit the formation of pulmonary edema. Inflammatory response is very important in the development of ALI/ARDS.
Over-expression of inflammatory factors and their interaction is the main reason for occurrence of acute lung injury, regulation of inflammatory response is the principal means of ALI/ARDS treatment. Inflammatory stimuli activated effector cells in vivo which release IL-6, TNF-α and other proinflammatory cytokines. IL-6 is a major inflammatory factor in acute-phase of inflammatory reaction which induce accumulation and activation of pulmonary PMN and lung injury. Myeloperoxidase (MPO) is a peroxide enzyme released from the azurophilic granules of neutrophils, which can catalyze hydrogen peroxide reacts with chlorine to produce hypochlorous acid (HOCl). Hypochlorous acid, a potent bactericidal agent as well as a neutrophil oxidant, can cause tissue injury and inflammation. MPO activity reflects the degree of PMN accumulation in the lung, which shows a good linear relationship between the two factors [20,21]. In this study, the underlying mechanism of OA-induced ALI is involved in cytokines such as IL-6 and other inflammatory factors.
Massive release of these inflammatory factors made a large number of PMN infiltration in the damaged lung tissue, and increased MPO activity of lung. Compared to OA-group, the activity of MPO in lung tissue and the levels of inflammatory IL-6 in BALF were decreased obviously in dexamethasone treatment group. These results indicate that early low dose of dexamethasone can suppress the production of inflammatory cytokines, reduce accumulation and activation of pulmonary neutrophils, and prevent from lung injury.
Dexamethasone decreased the production of inflammatory cytokines, alleviated alveolar epithelial and endothelial cell damage, and reduced pulmonary edema. Thus, these results meant that dexamethasone improve the alveolar epithelium-endothelial barrier function. However, attenuation of pulmonary edema may be related to up-regulation of alveolar liquid clearance. Several studies demonstrated that active sodium transport is regarded as the main mechanism that contributes to alveolar fluid clearance. Na+ diffuse into the cell via apically located epithelial Na+ channel, then is extruded into the serosal space through Na+, K+-ATPase located at the basolateral membrane. This transport hereby produces an osmotic gradient that favors water reabsorption from the alveolar to the interstitium and finally to the blood circulation. Alveolar epithelial Na+, K+-ATPase plays a key role in the alveolar fluid clearance [22]. The Na+, K+-ATPase is a transmembrane heterodimer protein consisting of a large α-subunit and smaller β-subunit. The α-subunit, the catalytic component of the enzyme, contains the binding sites for Na, K, and ATP. The β-subunit, a glycosylated transmembrane protein, controls heterodimer assembly and insertion of the enzyme into the plasma membrane [23]. The α1- and β1-subunits is the predominant isoforms expressed in alveolar type II cells. Inhibition of the Na+, K+-ATPase by ouabain has been reported to reduce ion transport in cultured alveolar type II cells and dropsy clearance in isolated lungs [24]. Looney [25] reported that decreased expression of both the α1- and α2-subunits of the Na+, K+-ATPase impairs maximal lung liquid clearance. Overexpression of β1-Na, K-ATPase in the lung improves pulmonary fluid clearance and protects against lung edema [22]. The experimental results showed that compared with the sham operated group, expression of pro-inflammatory cytokines in lung tissue and pulmonary vascular permeability in oleic acid model group increased significantly, expression of α1-Na+-K+-ATPase mRNA, β1-Na+-K+-ATPase mRNA, α1-Na+-K+-ATPase protein, β1-Na+-K+-ATPase protein and NKA activity in lung tissue were decreased obviously. There is evident pulmonary edema, suggesting that the decreased expression of α1-Na+-K+-ATPase mRNA and protein, β1-Na+-K+-ATPase mRNA and protein, and Na+-K+-ATPase activity is one important mechanisms of pulmonary edema.
In previous studies, dexamethasone has been shown to regulate the Na+-K+-ATPase in lung. Compared to sham operated group, dexamethasone up-regulated alveolar fluid clearance by 80%, stimulated Na+-K+-ATP enzyme expression of rat alveolar epithelial cells [26]. Dagenais et al. reported that dexamethasone treatment does not up-regulate the mRNA levels of α1-Na+-K+-ATPase in alveolar epithelial cells but can increase the expression of β1-Na+-K+-ATPase mRNA [27]. However, Chalaka et al. [28] found that dexamethasone elevates α1-Na+-K+-ATPase mRNA expression in a fetal rat lung epithelial cell line. These different results may be explained for the experimental models, drug delivery, drug dosage, observation time and other relevant factor.
The effects of glucocorticoids on acute lung injury induced by oleic acid were seldom reported. Furthermore, few research has been performed to investigate the regulation effects of glucocorticoids on sodium and water transport in lung injury induced by OA. The experimental results showed that dexamethasone can inhibit the decreased expression of α1-Na+-K+-ATPase mRNA, β1-Na+-K+-ATPase mRNA, α1-Na+-K+-ATPase protein, β1-Na+-K+-ATPase protein and Na+-K+-ATPase activity in lung tissue to alleviate lung edema. Thus, these data suggest that early low dose of dexamethasone treatment has protective effect on oleic acid induced acute lung injury, thereby promoting the absorption of alveolar fluid.
Dexamethasone can regulated the Na+-K+-ATPase expression of alveolar epithelial in acute lung injury, and its mechanism might relate to the following aspects. 1) It has been demonstrated that hypoxia decrease alveolar fluid clearance by inhibiting active transepithelial Na+ transport [29]. In alveolar epithelial cells, hypoxia causes a significant decrease of Na-K-ATPase activity, as well as the expression of α1- and β1 Na-K-ATPase mRNA [30]. However, dexamethasone can effectively improve hypoxemia, alleviate lung injury. 2) Under pathological condition, in response to proinflammatory cytokines, activated neutrophils and macrophages can localize in the lung, produce and release inflammatory mediators, such as oxygen free radicals and NO. Inflammatory cells and inflammatory mediators interact to promote the inflammatory response signal gradually enlarge, damage the alveolar cells, causing the destruction of alveolar epithelial barrier, reduce the Na+-K+-ATPase activity, and result in pulmonary edema [31]. Dexamethasone can inhibit the expression of inflammatory factors, reduce NO production, scavenging oxygen free radicals to alleviate acute lung injury [32].
Glucocorticoid treatment may induce or exacerbate infection. Previous studies have shown that high dose of corticosteroids increased incidence of infectious complications in patients suffered from severe sepsis or septic shock. In an eight-dose study, Weigelt et al. [33] found that high-dose corticosteroids have failed to show benefit in patients with ARDS, and even significantly increased incidence of second infection compared to the control group. It may be explained for high-dose corticosteroids reduce the response to infection. Therefore rational use of corticosteroids is very important. Low-dose and prolonged glucocorticoid treatment-induced down-regulation of systemic inflammation in early severe ARDS is associated with a significant improvement in pulmonary and extrapulmonary organ dysfunction and without increasing infection rate. In ALI/ARDS trial, Low-dose of corticosteroids has been shown to improve the morbidity and mortality, without increasing side effects. Silva et al. [34] reported that early short-term, low-dose methylprednisolone is as effective as prolonged methylprednisolone in the treatment of ALI.
In summary, it is suggested that Na+-K+-ATPase expression is one important mechanism of AFC. Early low dose of dexamethasone treatment suppressed the expression of inflammation factor, improved lung epithelial-endothelial barrier permeability, increased the mRNA levels of α1-Na+-K+-ATPase and β1-Na+-K+-ATPase, and stimulated protein expression of α1-Na+-K+-ATPase and β1-Na+-K+-ATPase and Na+-K+-ATPase activity, decreased pulmonary edema. These observations suggest that early low dose of dexamethasone treatment has protective effect on oleic acid induced acute lung injury .It might be helpful to use low dose of dexamethasone in those patients with early stage of ALI/ARDS.
Acknowledgements
This study was supported by grants from National Natural Science Foundation of China (Grant No. 81270141).
Disclosure of conflict of interest
None.
References
- 1.Carnesecchi S, Dunand-Sauthier I, Zanetti F, Singovski G, Deffert C, Donati Y, Cagarelli T, Pache T, Pache JC, Krause KH, Reith W, Barazzone-Argiroffo C. NOX1 is responsible for cell death through STAT3 activation in hyperoxia and is associated with the pathogenesis of acute respiratory distress syndrome. Int J Clin Exp Pathol. 2014;7:537–551. [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson ER, Matthay MA. Acute lung injury: epidemiology, pathogenesis, and treatment. J Aerosol Med Pulm Drug Deliv. 2010;23:243–252. doi: 10.1089/jamp.2009.0775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lomas-Neira J, Chung CS, Ayala A. RNA interference as a potential therapeutic treatment for inflammation associated lung injury. Int J Clin Exp Med. 2008;1:154–160. [PMC free article] [PubMed] [Google Scholar]
- 4.Jaitovich A, Sznajder JI. Improving survival by increasing lung edema clearance: is airspace delivery of dopamine a solution? Crit Care. 2008;12:135. doi: 10.1186/cc6825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sartori C, Mattay MA, Scherrer U. Transepithelial sodium and water transport in lung. Major player and novel therapeutic target in pulmonary edema. Adv Exp Med Biol. 2001;502:315–338. doi: 10.1007/978-1-4757-3401-0_21. [DOI] [PubMed] [Google Scholar]
- 6.Nie HG, Chen L, Han DY, Li J, Song WF, Wei SP, Fang XH, Gu X, Matalon S, Ji HL. Regulation of epithelial sodium channels by cGMP/PKGII. J Physiol. 2009;587:2663–2676. doi: 10.1113/jphysiol.2009.170324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson MD, Bao HF, Helms MN, Chen XJ, Tigue Z, Jian L, Dobbs LG, Eaton DC. Functional ion channels in pulmonary alveolar type I cells support a role for type I cells in lung transport. Proc Natl Acad Sci U S A. 2006;103:4964–4969. doi: 10.1073/pnas.0600855103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quinlan GJ, Lamb NJ, Evans TW, Gutteridge JM. Plasma fatty acid changes and increased lipid peroxidation in patients with adult respiratory distress syndrome. Crit Care Med. 1996;24:241–246. doi: 10.1097/00003246-199602000-00010. [DOI] [PubMed] [Google Scholar]
- 9.Gunther A, Schmidt R, Harodt J, Schmehl T, Walmrath D, Ruppert C, Grimminger F, Seeger W. Bronchoscopic administration of bovine natural surfactant in ARDS and septic shock: impact on biophysical and biochemical surfactant properties. Eur Respir J. 2002;19:797–804. doi: 10.1183/09031936.02.00243302. [DOI] [PubMed] [Google Scholar]
- 10.Vadász I, Morty RE, Kohstall MG, Olschewski A, Grimminger F, Seeger W, Ghofrani HA. Oleic acid inhibits alveolar fluid reabsorption: A role in acute respiratory distress syndrome? Am J Respir Crit Care Med. 2005;171:469–479. doi: 10.1164/rccm.200407-954OC. [DOI] [PubMed] [Google Scholar]
- 11.Thompson BT. Glucocorticoids and acute lung injury. Crit Care Med. 2003;31:253–257. doi: 10.1097/01.CCM.0000057900.19201.55. [DOI] [PubMed] [Google Scholar]
- 12.Bernard GR, Luce JM, Sprung CL, Rinaldo JE, Tate RM, Sibbald WJ, Kariman K, Higgins S, Bradley R, Metz CA. High-dose corticosteroids in patients with the adult respiratory distress syndrome. N Engl J Med. 1987;317:1565–1570. doi: 10.1056/NEJM198712173172504. [DOI] [PubMed] [Google Scholar]
- 13.Meduri GU, Golden E, Freire AX, Taylor E, Zaman M, Carson SJ, Gibson M, Umberger R. Methylprednisolone infusion in early severe ARDS: results of a randomized controlled trial. Chest. 2007;131:954–963. doi: 10.1378/chest.06-2100. [DOI] [PubMed] [Google Scholar]
- 14.Lefort J, Motref L, Vargaftig BB. Airway administration of Eschefichia coli endotoxin to mice induce glucocorticoid-resistaut bronchoconstriction and vasopermeation. Am J Respir Cel Mol Biol. 2001;24:345–351. doi: 10.1165/ajrcmb.24.3.4289. [DOI] [PubMed] [Google Scholar]
- 15.Matue-Bello G, Frevert CW, Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Physiol. 2008;295:379–399. doi: 10.1152/ajplung.00010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li TS, Zhao B, Wang C, Wang H, Liu Z, Li W, Jin H, Tang C, Du J. Regulatory effects of hydrogen sulfide on il-6, il-8 and il-10 levels in the plasma and pulmonary tissue of rats with acute lung injury. Exp Biol Med. 2008;233:1081–1087. doi: 10.3181/0712-RM-354. [DOI] [PubMed] [Google Scholar]
- 17.He XL, Han B, Mura M, Xia S, Wang S, Ma T, Liu M, Liu Z. Angiotensin-converting enzyme inhibitor captopril prevents oleic acid induced severe acute lung injury in rats. Shock. 2007;28:106–111. doi: 10.1097/SHK.0b013e3180310f3a. [DOI] [PubMed] [Google Scholar]
- 18.Liu KX, Wu WK, He W, Liu CL. Ginkgo biloba extract (EGb 761) attenuates lung injury induced by intestinal ischemia/reperfusion in rats: roles of oxidative stress and nitric oxide. World J Gastroenterol. 2007;13:299–305. doi: 10.3748/wjg.v13.i2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maddison B, Giudici R, Calzia E, Wollf C, Hinds C, Radermacher P, Pearse RM. Extravascular lung water volume measurement by a novel lithium-thermal indicator dilution method: comparison of three techniques to post-mortem gravimetry. Intensive Care Med. 2008;34:2106–2111. doi: 10.1007/s00134-008-1207-4. [DOI] [PubMed] [Google Scholar]
- 20.Muhling J, Fuchs M, Campos M, Gonter J, Sablotzki A, Engel J, Welters ID, Wolff M, Matejec R, Dehne MG, Menges T, Krull M, Hempelmann G. Effects of ornithine on neutrophil (PMN) free amino acid and alpha-keto acid profiles and immune functions in vitro. Amino Acids. 2004;27:313–319. doi: 10.1007/s00726-004-0126-0. [DOI] [PubMed] [Google Scholar]
- 21.Basher F, Fan H, Zingarelli B, Borg KT, Luttrell LM, Tempel GE, Halushka PV, Cook JA. beta-Arrestin 2: a Negative Regulator of Inflammatory Responses in Polymorphonuclear Leukocytes. Int J Clin Exp Med. 2008;1:32–41. [PMC free article] [PubMed] [Google Scholar]
- 22.Azzam ZS, Dumasius V, Saldias FJ, Adir Y, Sznajder JI, Factor P. Na, K-ATPase overexpression improves alveolar fluid clearance in a rat model of elevated left atrial pressure. Circulation. 2002;105:497–501. doi: 10.1161/hc0402.102848. [DOI] [PubMed] [Google Scholar]
- 23.Blanco G, Mercer RW. Isozymes of the Na-K-ATPase: heterogeneity in structure, diversity in function. Am J Physiol. 1998;275:633–650. doi: 10.1152/ajprenal.1998.275.5.F633. [DOI] [PubMed] [Google Scholar]
- 24.Sznajder JI, Olivera WG, Ridge KM, Rutschman DH. Mechanisms of lung liquid clearance during hyperoxia in isolated rat lungs. Am J Respir Crit Care Med. 1995;151:1519–1525. doi: 10.1164/ajrccm.151.5.7735609. [DOI] [PubMed] [Google Scholar]
- 25.Looney MR, Sartori C, Chakraborty S, Jame PF, Lingrel JB, Matthay MA. Decreased expression of both the alpha1- and alph2-subunits of the Na-K-ATPase reduces maximal alveolar epithelial fluid clearance. Am J Physiol Lung Cell Mol Physiol. 2005;289:104–110. doi: 10.1152/ajplung.00464.2004. [DOI] [PubMed] [Google Scholar]
- 26.Folkesson HG, Norlin A, Wang Y, Abedinpour P, Matthay MA. Dexamethasone and thyroid hormone pretreatment upregulate alveolar epithelial fluid clearance in adult rats. J Appl Physiol. 2000;88:416–424. doi: 10.1152/jappl.2000.88.2.416. [DOI] [PubMed] [Google Scholar]
- 27.Dagenais A, Denis C, Vires MF, Girouard S, Masse C, Nguyen T, Yamagata T, Grygorczyk C, Kothary R, Berthiaume Y. Modulation of alpha-ENaC and alpha1-Na+-K+-ATPase by cAMP and dexamethasone in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2001;281:217–230. doi: 10.1152/ajplung.2001.281.1.L217. [DOI] [PubMed] [Google Scholar]
- 28.Chalaka S, Ingbar DH, Sharma R, Zhau Z, Wendt CH. Na(+)-K(+)-ATPase gene regulation by glucocorticoids in a fetal lung epithelial cell line. Am J Physiol. 1999;277:197–203. doi: 10.1152/ajplung.1999.277.1.L197. [DOI] [PubMed] [Google Scholar]
- 29.Zhao X, Jin Y, Li H, Wang Z, Zhang W, Feng C. Hypoxia-inducible factor 1 alpha contributes to pulmonary vascular dysfunction in lung ischemia-reperfusion injury. Int J Clin Exp Pathol. 2014;7:3081–3088. [PMC free article] [PubMed] [Google Scholar]
- 30.Clerici C, Matthay MA. Hypoxia regulates gene expression of alveolar epithelial transport proteins. J Appl Physiol. 2000;88:1890–1896. doi: 10.1152/jappl.2000.88.5.1890. [DOI] [PubMed] [Google Scholar]
- 31.Althaus M, Pichl A, Clauss WG, Seeger W, Fronjus M, Morty RE. Nitric oxide inhibits highly selective sodium channels and the Na+/K+-ATPase in H441 cells. Am J Respir Cell Mol Biol. 2011;44:53–65. doi: 10.1165/2009-0335OC. [DOI] [PubMed] [Google Scholar]
- 32.Yu Z, Ouyang JP, Li YP. Dexamethasone attenuated endotoxin-induced acute lung injury through inhibiting expression of inducible nitric oxide synthase. Clin Hemorheol Microcirc. 2009;41:117–125. doi: 10.3233/CH-2009-1162. [DOI] [PubMed] [Google Scholar]
- 33.Weigelt JA, Norcoross JF, Borman KR, Snyder WH. Early steroid therapy for respiratory failure. Arch Surg. 1985;120:536–540. doi: 10.1001/archsurg.1985.01390290018003. [DOI] [PubMed] [Google Scholar]
- 34.Silva PL, Garcia CS, Maronas PA, Caqido VR, Negri EM, Damaceno-Rodrigues NR, Ventura GM, Bozza PT, Zin WA, Capelozzi VL, Pelosi P, Rocco PR. Early short-term versus prolonged low-dose methylprednisolone therapy in acute lung injury. Eur Respir J. 2009;33:634–645. doi: 10.1183/09031936.00052408. [DOI] [PubMed] [Google Scholar]