

Abstract
Endoplasmic reticulum stress (ERS) is known to play an important role in mediating myocardial ischemic/reperfusion (I/R) injury. Some previous studies have shown that atorvastatin alleviates myocardial I/R injury in animal models, but whether attenuation of ERS-induced apoptosis contributes to this effect remains to be elucidated. Therefore, in this study, we sought to investigate the modulatory effect of atorvastatin on myocardial I/R-induced ERS in rats. Myocardial I/R injury was induced in rats by occlusion of the left anterior descending coronary artery (LAD) for 0.5 h followed by 2 h of reperfusion. Atorvastatin was administered at different dosages (10 mg/kg, 20 mg/kg, and 40 mg/kg) at the onset of reperfusion. The levels of the CK-MB and LDH were detected by ELISA. Myocardial ischemia and infarct size were evaluated by Evans blue and tetrazolium chloride (TTC) staining. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) was used to investigate myocardial cell apoptosis. The expression levels of the genes encoding glucose-regulated protein-78 (GRP78, widely used as a marker of ERS), C/EBP homologous protein (CHOP) and caspase-12 (widely used as markers of ERS-induced apoptosis) were assessed using RT-PCR. The expression levels of the ERS proteins GRP78, CHOP, caspase-12, c-Jun NH2 terminal kinase (JNK) and phosphorylated JNK (p-JNK) were detected by western blot. Our results showed that atorvastatin treatment (20 mg/kg and 40 mg/kg) significantly reduced myocardial infarct size and myocardial cell apoptosis, and decreased the plasma levels of CK-MB and LDH in I/R rats. This treatment also significantly modulated mRNA and protein levels, specifically down-regulating GRP78, CHOP and caspase-12 expression along with JNK activation. These results suggest that the attenuation of ERS-induced apoptosis may be involved in the cardioprotective mechanisms of atorvastatin in myocardial I/R injury.
Keywords: Atorvastatin, ischemia reperfusion injury, endoplasmic reticulum stress, apoptosis
Introduction
In recent years, coronary heart disease (CHD) has led to high rates of morbidity and mortality. Although the development of percutaneous coronary intervention has improved the treatment of acute myocardial infarction patients, myocardial ischemia/reperfusion (I/R) injury during revascularization unavoidably results in clinical complications that lead to poor patient outcomes [1]. Apoptosis of myocardial cells is known to be an important mechanism of I/R injury [2-4]. In addition, accumulating evidence has shown that endoplasmic reticulum stress (ERS) induces myocardial apoptosis in myocardial I/R injury, and some data suggest that attenuation of ERS-induced apoptosis can protect the heart against I/R injury [5-7]. Therefore, these pathways have become the focus of evolving strategies to ameliorate myocardial I/R injury.
ERS is a condition in which unfolded proteins accumulate and aggregate during disruptions of ER homeostasis, such as ischemia, hypoxia, glucose starvation, bulk free radical stimulation, and loss of Ca2+ homeostasis [8]. Glucose-regulated protein-78 (GRP78) is an important ER-resident chaperone. When ERS occurs, cells develop a self-protective strategy, and dissociation of GRP78 from membrane receptors activates and triggers the unfolded protein response (UPR) to restore normal ER function. However, if stress persists or is too severe to repair, the apoptotic pathway is initiated by the UPR to protect the organism [9-12]. Increasing evidence has shown that proteins such as C/EBP homologous protein (CHOP) and caspase-12 and the c-Jun NH2 terminal kinase (JNK)-mediated signaling pathway are involved in ERS-induced apoptosis [13-15]. Thus, therapeutic interventions targeting CHOP, caspase-12 and JNK-mediated apoptosis represent promising strategies for the treatment of ischemic cardiovascular diseases (including CHD).
Atorvastatin, a cholesterol-lowering agent, acts by competitively inhibiting the rate-limiting enzyme of cholesterol biosynthesis, 3-hydroxy-3-methylglutaryl-CoA (HMGCoA) reductase. Statins have drawn significant attention for their benefits in preventing hypercholesterolemia as well as their powerful cardioprotective and other pharmacological effects [16-21]. In addition, animal experiments and clinical studies have indicated that statins can protect the myocardium against myocardial I/R injury [22,23]. Nevertheless, the above studies have not completely elucidated whether attenuation of ERS-induced myocardium apoptosis is involved in the protective mechanism. Thus, we hypothesized that cardio-protection by statins may be associated with alleviation of ERS-mediated apoptosis in myocardial I/R injury. In this study, we sought to examine the effects of atorvastatin on rat myocardial I/R injury in vivo by detecting changes in the gene or protein expression levels of the ERS-mediated apoptosis signal transduction molecules GRP78, CHOP, caspase-12 and JNK. We further analyzed the mechanism involved in the effects of atorvastatin on myocardial I/R injury to provide a theoretical basis and new intervention targets for the prevention and treatment of myocardial I/R injury in the clinic.
Materials and methods
Drugs and reagents
Atorvastatin was obtained from Pfizer (New York City, NY, USA). The enzyme-linked immunosorbent assay (ELISA) kit for the MB isoenzyme of creatine kinase (CK-MB) and the lactate dehydrogenase (LDH) assay kit were purchased from Sigma Chemical Company (St. Louis, MO, USA). RNA extraction RNeasy Maxi kits were purchased from Qiagen (Valencia, CA, USA). Reverse transcriptase was purchased from Invitrogen (Carlsbad, CA, USA). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody was purchased from Hangzhou Xianzhi Biological Co., LTD (Hang Zhou, China). Polyclonal rabbit antibodies to GRP78, CHOP, caspase-12, JNK, and P-JNK were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Goat anti-rabbit, goat anti-mouse, rabbit anti-goat and rabbit anti-rat horseradish peroxidase (HRP)-labeled secondary antibodies were purchased from Wuhan Boster Biological Engineering Co., LTD (Wuhan, China). Enhanced chemiluminescence (ECL) detection reagents were obtained from Thermo Scientific (Waltham, MA). X-ray film was obtained from Kodak (Tokyo, Japan). Other chemicals and reagents were all of analytical grade and available commercially.
Animals
Male Sprague-Dawley rats weighing 200-220 g were obtained from the Experiment Animal Center of Wuhan University (animal certificate number SCXK (e) 2008-0004). The animals were allowed to move freely, with water and food continually available, and were kept at room temperature. All animal experiments were performed in accordance with the guidelines for laboratory animal care of Medical College of Wuhan University, Hubei, China. Rats were randomly divided into five groups, including the sham group, myocardial ischemia/reperfusion group (I/R), and atorvastatin groups [I/R + Ator: 10 mg/kg (L-Ator), 20 mg/kg (M-Ator) and 40 mg/kg (H-Ator)]. Each group contained 10 animals. Atorvastatin was dissolved in normal saline and administered through tail vein injections to the I/R + Ator groups at the start of reperfusion. In the sham and I/R groups, the same volume of normal saline as in the I/R + Ator groups was administered.
Surgery and experimental design
The myocardial ischemia model was established by occlusion of the left anterior descending coronary artery (LAD) [24]. Briefly, rats were anesthetized with pentobarbital (40 mg/kg), and the trachea was exposed by cutting on the center of the neck line above the chest, 1.5 cm step by step. A scalp acupuncture wire guide was punctured into the trachea via the femoral vein puncture needle core. An endotracheal tube was fixed on the neck skin and retained 1 cm to link the animal to a breathing machine. The right common carotid artery was separated and intubated in connection with a multiple channel physiological signal acquisition system to record artery blood pressure and left heart internal pressure. Thoracotomy was performed on the left side of the sternum. The pericardium was opened, and the heart was exposed. After pericardiotomy, the LAD was occluded with a silk suture between the arterial cone and the left auricle, and the chest was closed immediately. Occlusion was performed for 0.5 h, and recirculation was allowed for 2 h. At 0.5 h after the coronary occlusion, the chest was reopened and the suture was loosened for reperfusion.
Before and during the ischemia and reperfusion periods, heart rate (HR), blood pressure (BP), and standard lead II electrocardiography (ECG) changes were recorded. ST-segment elevation on ECG meant that the I/R model was constructed successfully. Sham-operated animals underwent the same surgical procedures except that the suture around the LAD was not ligatured.
The rats were sacrificed at 2 h after the start of reperfusion, and then heart infarction, apoptosis, and the expression of ERS apoptotic factors were evaluated.
Plasma CK-MB and LDH analysis
Blood samples were obtained from the right common carotid artery using a heparinized syringe and were rapidly centrifuged (3,000 rpm, 10 min) at room temperature. Plasma was prepared and then frozen at -80°C until further analysis. CK-MB and LDH activity were measured by ELISA according to the manufacturer’s protocols, with the change in the rate of absorbance directly proportional to CK-MB or LDH activity.
Myocardial damage and infarct size assessment
The infarct sizes were measured by Evans blue staining and tetrazolium chloride (TTC) staining. Briefly, before the rats were sacrificed, Evans blue stain was injected through the carotid artery. The hearts were then immediately excised and placed in 0.9% saline. Left ventricles were cut into 0.2-mm transverse slices, which were then incubated in 1% 2, 3, 5-triphenyltetrazolium chloride (TTC) (pH 7.4) at 37°C for 15 min and fixed in 10% formalin solution. The area dyed gray was defined as the infarction area, while red indicated ischemic tissue and blue indicated normal myocardium. Each slice was scanned and analyzed using a BI-2000 Medical Image Analysis System. The relative infarct size represented the degree of myocardial infarction.
TUNEL assay for myocardial tissues
The rate of apoptosis among cells in the myocardium was evaluated by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL). Briefly, after I/R, the anterior wall tissues of the left ventricle were fixed in 4% paraformaldehyde and then embedded in paraffin. The slices were treated according to the manufacturer’s instructions. The labeling reaction was performed with terminal deoxynucleotidyl transferase and fluorescein-dUTP. Labeled myocytes were visualized under a confocal microscope, and photographs were taken under high-power magnification (200 ×). Cells were counted in five random fields on the border of the infarcted area. The apoptotic index (AI) was calculated as the ratio of TUNEL-positive cells to total cells. The results were expressed as the average proportion of positive cells in each group.
RT-PCR analysis
RT-PCR was used to detect the expression of GRP78, caspase-12 and CHOP mRNA. Total RNA was extracted and treated according to the manufacturer’s instructions. The primer sequences for the RT-PCR assay were as follows:
GRP78 forward: 5’-TCTGCTTGATORGTGTGTCCTCTT-3’. reverse: 5’-GTCGTTCACCTTCGTAGACCT-3’. Caspase-12 forward: 5’-TCCTGCTCTTTATORGTCCC-3’. reverse: 5’-CGATORAGCCCAAGGAAGTG-3’. CHOP forward: 5’-GGAGAAGGAGCAGGAGAATORGA-3’. reverse: 5’-AGACAGACAGGAGGTGATORGC-3’. The primers for rat GAPDH were: forward: 5’-CCATORGTTCGTCATORGGGTGTGAACCA-3’. reverse: 5’-GCCAGTAGAGGCAGGGATORGATORGTTC-3’.
Western blot analysis
After the rats were sacrificed, the myocardium was collected and processed according to the manufacturer’s instructions. The primary antibodies were as follows: GRP78 antibody (1:400), caspase-12 antibody (1:300), CHOP antibody (1:300), JNK antibody (1:300), p-JNK antibody (1:300) and GAPDH antibody (1:1,000). Secondary anti-rabbit or anti-goat antibodies (1:50000) were used. The developed signals were visualized using ECL detection kits and analyzed with Quantity One software.
Statistical analysis
Statistical analysis involved the use of SPSS 13.0. The data were expressed as the mean ± standard deviation (SD). Differences were analyzed by one-way analysis of variance (ANOVA) followed by the Newman-Keuls test; values of P < 0.05 were considered statistically significant. The 2-∆∆Ct method was used for the RT-PCR analysis [25].
Results
Effect of atorvastatin on the myocardial infarction size of hearts
To evaluate the direct effect of atorvastatin on myocardial I/R injury, we measured the ischemic area and infarct size in rats using the Evans blue/TTC method (Figure 1A). The ischemic area at risk, corresponding to a percentage of the total left ventricle volume, showed no significant difference among the four experimental groups (Figure 1B). In the I/R group, the myocardial necrotic area constituted 47.8 ± 1.3% of the area at risk. Compared with the I/R group, pre-administration of atorvastatin reduced the infarct size in a dose-dependent manner. The M-Ator and H-Ator groups showed significant reductions in infarct size to 35.5 ± 0.7% (P < 0.05) and 23.2 ± 0.6% (P < 0.01), respectively, whereas the L-Ator groups showed no significant difference (Figure 1C).
Figure 1.
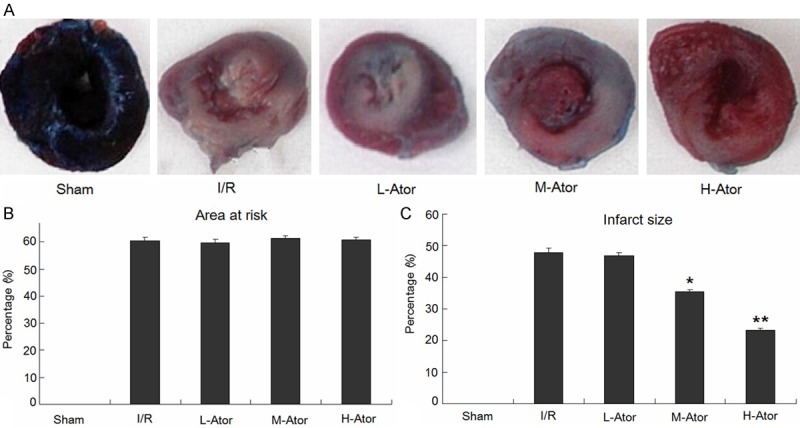
Effects of atorvastatin on myocardial infarct size. A. Representative photographs of Evans blue/TTC-stained sections. B. Ratio of area at risk to total ventricle. C. Ratio of infarct size to area at risk. *Compared with the I/R group P < 0.05, **Compared with the I/R group P < 0.01.
Effect of atorvastatin on the plasma concentrations of CK-MB and LDH
In addition, we measured the plasma concentrations of CK-MB and LDH at the end of the I/R period to evaluate the extent of myocardial injury. The plasma levels of CK-MB and LDH were significantly higher in the I/R group than in the sham group (P < 0.01 for both). However, atorvastatin administration attenuated plasma CK-MB and LDH release in a dose-dependent manner during I/R (Figure 2).
Figure 2.
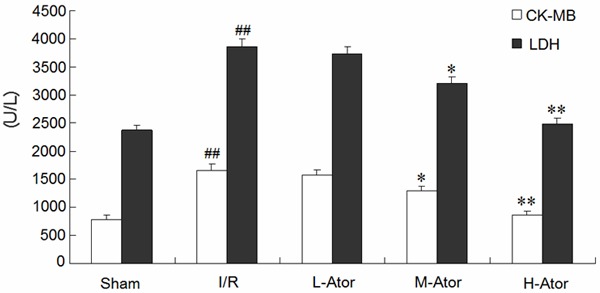
Effects of atorvastatin on the plasma levels of CK-MB and LDH. *Compared with the I/R group P < 0.05, **Compared with the I/R group P < 0.01, ##Compared with the sham group P < 0.01.
Effect of atorvastatin on myocardial cell apoptosis
To explore whether atorvastatin had any effect on myocardial cell apoptosis in rats, we performed the TUNEL assay. The numbers of total cardiomyocytes and TUNEL-positive cells in the specimens were manually counted in five randomly selected fields under light microscopic analysis (Figure 3A-E). The AI was expressed as the ratio of TUNEL-positive cells to total cardiomyocytes, and TUNEL staining showed that the AI was significantly higher in the I/R groups compared with the sham group (68.2 ± 2.0% vs. 6.2 ± 0.6%, P < 0.01). However, atorvastatin administration reduced these values in a dose-dependent manner. As shown in Figure 3, the M-Ator and H-Ator groups showed significant reductions, whereas the L-Ator group showed no significant difference (Figure 3F).
Figure 3.
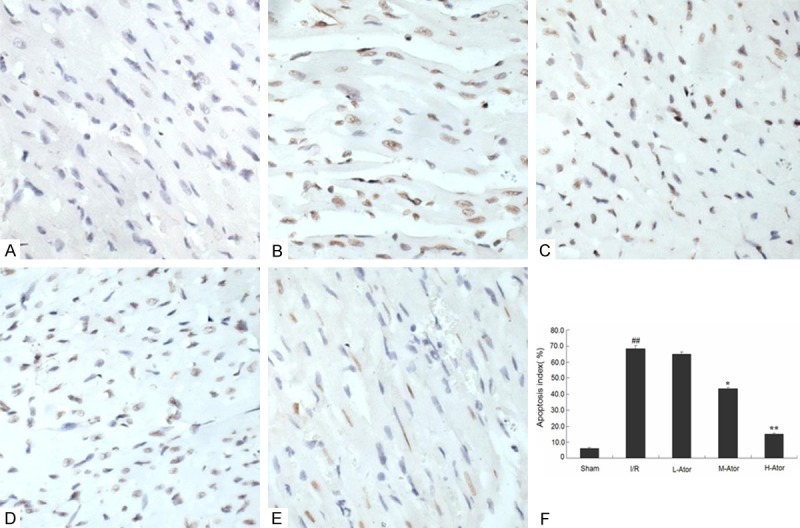
Effect of atorvastatin on I/R-induced myocardial apoptosis in rats. (A-E) Representative autoradiograms of TUNEL staining in the sham (A), I/R(B), L-Ator (C), M-Ator (D) and H-Ator (E) group; apoptotic nuclei appear yellow by TUNEL staining. Bar = 50 μm. (F) Graphs showing the quantification of apoptotic cells in the five groups. *Compared with the I/R group P < 0.05, **Compared with the I/R group P < 0.01, ##Compared with the sham group P < 0.01.
Effect of atorvastatin on myocardial ERS in rats after I/R
To further confirm that ERS was activated after I/R in rats, the central regulator of ER function, GRP78 (widely used as a marker of ERS), was examined by western blot and RT-PCR analysis. The levels of GRP78 mRNA and protein were significantly higher in the I/R group compared with the sham group, as shown in Figure 4. However, compared with the I/R group, the M-Ator and H-Ator groups showed markedly reduced I/R-induced upregulation of GRP78 expression, and the L-Ator group showed a slight but not statistically significant decrease (Figure 4).
Figure 4.
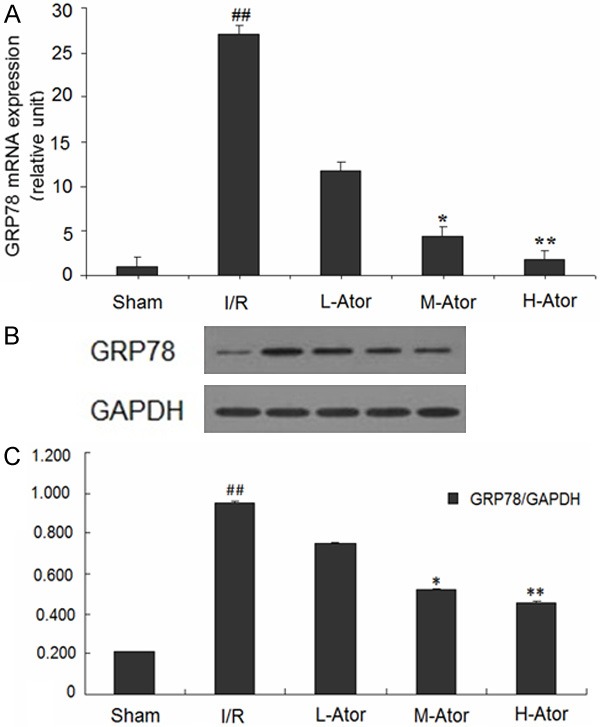
Effect of atorvastatin on myocardial ERS after I/R in rats. A. The levels of GRP78 mRNA in the sham, I/R and atorvastatin-administered groups. B. The levels of GRP78 protein in the five groups. C. Graphs showing the quantification of GRP78 protein in the five groups. *Compared with the I/R group P < 0.05, **Compared with the I/R group P < 0.01, ##Compared with the sham group P < 0.01.
Effect of atorvastatin on the ERS-mediated apoptosis pathway
Signal transduction pathways involving CHOP, caspase-12 and JNK are known to mediate ERS-associated apoptosis. In the present study, the myocardial expression of CHOP, cleaved caspase-12 and p-JNK were determined by western blot and RT-PCR (Figure 5A). Our results showed that I/R markedly increased the levels of CHOP, cleaved caspase-12, and p-JNK in the rat myocardium, whereas treatment with medium- or high-dosage atorvastatin significantly abolished these increased levels of CHOP, caspase-12 and p-JNK. The low-dose atorvastatin group did not show a notable decrease in comparison with the I/R group.
Figure 5.
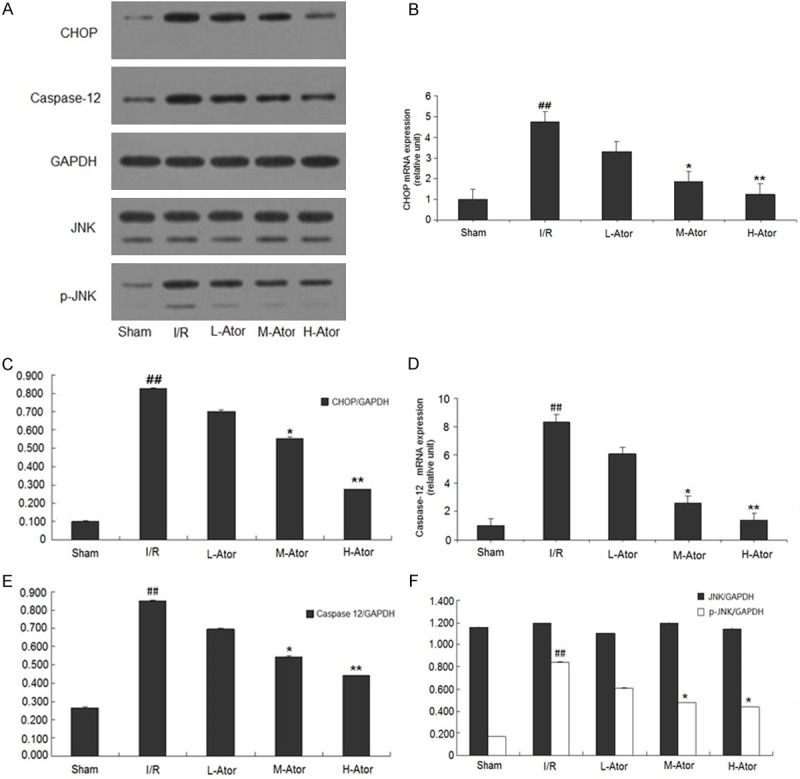
Effect of atorvastatin on the ERS-mediated apoptosis pathway in rats after I/R injury. A. Graphs showing CHOP, caspase-12, JNK and p-JNK protein expression in the sham, I/R and atorvastatin-administered groups. B. Levels of CHOP mRNA in the five groups. C. Graphs showing the quantification of CHOP protein in the five groups. D. Levels of caspase-12 mRNA in the five groups. E. Graphs showing the quantification of caspase-12 protein in the five groups. F. Graphs showing the quantification of JNK and p-JNK protein in the five groups. *Compared with the I/R group P < 0.05, **Compared with the I/R group P < 0.01, ##Compared with the sham group P < 0.01.
Discussion
Apoptosis is a vital pathophysiological mechanism associated with myocardial I/R injury, and our results showed that atorvastatin provided cardioprotective effects in rats after myocardial I/R by inhibiting cell apoptosis. In our study, myocardial tissues showed high levels of apoptosis in the I/R group, whereas the AI was markedly attenuated in the groups pretreated with atorvastatin, suggesting that inhibition of apoptosis may be the mechanism that enables atorvastatin to protect against myocardial I/R injury. This suppression of apoptosis was also correlated with lower plasma CK-MB and LDH activities and a reduced infarct size.
Furthermore, this study suggests that ERS-mediated apoptosis may be involved in the protective mechanism of atorvastatin on myocardial I/R injury. The mRNA and protein levels of ERS markers such as GRP78, CHOP, and caspase-12 were enhanced by I/R, whereas atorvastatin pretreatment reversed these effects. Moreover, the results of our study are similar to those of previous reports. GRP78, a main ER molecular chaperone, is associated with protein folding, protein translocation and protein secretion. The expression of GRP78 is increased during ischemia and early reperfusion and is associated with the inhibition of excessive ER stress [26,27]. CHOP is an important mediator with a dominant role in the response to ERS, and CHOP deficiency has been shown to attenuate myocardial apoptosis after reperfusion [28,29]. Caspase-12 normally exists in an inactive pro-caspase form, but once activated initiates downstream apoptotic pathways. Indeed, caspase-12 deficiency was shown to inhibit ERS-induced apoptosis [30-32]. Therefore, down-regulation of CHOP and caspase-12 expression and subsequent inhibition of CHOP and caspase-12 signaling-mediated apoptosis may explain the protective effect of atorvastatin in myocardial I/R injury.
In addition to CHOP and caspase-12, we assessed the protein levels of JNK and P-JNK to further explore the possible protective mechanism of atorvastatin. The JNK signal transduction pathway also plays a vital role in mediating ERS-induced apoptosis, and a previous study reported that JNK mediates apoptotic signals in response to ERS [33]. We found that p-JNK expression was downregulated in the atorvastatin-treated groups, whereas JNK expression was not altered. Thus, we believe that decreased expression of p-JNK contributes to the cardioprotective effect of atorvastatin against myocardial I/R.
In conclusion, our results indicate that atorvastatin pretreatment can ameliorate myocardial injury by inhibiting the expression of CHOP, caspase-12 and p-JNK and reducing ERS-induced apoptosis. Thus, these findings may provide novel therapeutic strategies for myocardial I/R injury.
Acknowledgements
This study was supported by National Natural Science Foundation of China (No. 81170133) and Research Project of Hubei Provincial Department of Education (B20111201).
Disclosure of conflict of interest
None.
References
- 1.Prasad A, Stone GW, Holmes DR, Gersh B. Reperfusion injury, microvascular dysfunction, and cardioprotection: the “dark side” of reperfusion. Circulation. 2009;120:2105–2112. doi: 10.1161/CIRCULATIONAHA.108.814640. [DOI] [PubMed] [Google Scholar]
- 2.Gottlieb RA, Burleson KO, Kloner RA, Babior BM, Engler RL. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J Clin Invest. 1994;94:1621–1628. doi: 10.1172/JCI117504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saraste A, Pulkki K, Kallajoki M, Henriksen K, Parvinen M, Voipio-Pulkki LM. Apoptosis in human acute myocardial infarction. Circulation. 1997;95:320–323. doi: 10.1161/01.cir.95.2.320. [DOI] [PubMed] [Google Scholar]
- 4.Freude B, Masters TN, Robicsek F, Fokin A, Kostin S, Zimmermann R, Ullmann C, Lorenz-Meyer S, Schaper J. Apoptosis is initiated by myocardial ischemia and executed during reperfusion. J Mol Cell Cardiol. 2000;32:197–208. doi: 10.1006/jmcc.1999.1066. [DOI] [PubMed] [Google Scholar]
- 5.Martindale JJ, Fernandez R, Thuerauf D, Whittaker R, Gude N, Sussman MA, Glembotski CC. Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circ Res. 2006;98:1186–1193. doi: 10.1161/01.RES.0000220643.65941.8d. [DOI] [PubMed] [Google Scholar]
- 6.Natarajan R, Salloum FN, Fisher BJ, Smithson L, Almenara J, Fowler AA 3rd. Prolyl hydroxylase inhibition attenuates post-ischemic cardiac injury via induction of endoplasmic reticulum stress genes. Vascul Pharmacol. 2009;51:110–118. doi: 10.1016/j.vph.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 7.Khan M, Meduru S, Mostafa M, Khan S, Hideg K, Kuppusamy P. Trimetazidine, administered at the onset of reperfusion, ameliorates myocardial dysfunction and injury by activation of p38 mitogen-activated protein kinase and Akt signaling. J Pharmacol Exp Ther. 2010;333:421–429. doi: 10.1124/jpet.109.165175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pahl HL. Signal transduction from the endoplasmic reticulum to the cell nucleus. Physiol Rev. 1999;79:683–701. doi: 10.1152/physrev.1999.79.3.683. [DOI] [PubMed] [Google Scholar]
- 9.Kozutsumi Y, Segal M, Normington K, Gething MJ, Sambrook J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature. 1988;332:462–464. doi: 10.1038/332462a0. [DOI] [PubMed] [Google Scholar]
- 10.Okada K, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S, Hirata A, Fujita M, Nagamachi Y, Nakatani T, Yutani C, Ozawa K, Ogawa S, Tomoike H, Hori M, Kitakaze M. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation. 2004;110:705–712. doi: 10.1161/01.CIR.0000137836.95625.D4. [DOI] [PubMed] [Google Scholar]
- 11.Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880–885. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bravo R, Parra V, Gatica D, Rodriguez AE, Torrealba N, Paredes F, Wang ZV, Zorzano A, Hill JA, Jaimovich E, Quest AF, Lavandero S. Endoplasmic reticulum and the unfolded protein response: dynamics and metabolic integration. Int Rev Cell Mol Biol. 2013;301:215–290. doi: 10.1016/B978-0-12-407704-1.00005-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miyazaki Y, Kaikita K, Endo M, Horio E, Miura M, Tsujita K, Hokimoto S, Yamamuro M, Iwawaki T, Gotoh T, Ogawa H, Oike Y. C/EBP homologous protein deficiency attenuates myocardial reperfusion injury by inhibiting myocardial apoptosis and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:1124–1132. doi: 10.1161/ATVBAHA.111.224519. [DOI] [PubMed] [Google Scholar]
- 14.Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 15.Romero L, Andrews K, Ng L, O’Rourke K, Maslen A, Kirby G. Human GSTA1-1 reduces c-Jun N-terminal kinase signalling and apoptosis in Caco-2 cells. Biochem J. 2006;400:135–141. doi: 10.1042/BJ20060110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Di Napoli P, Maggi A, Spina R, Barsotti L, Taccardi AA, Stuppia L, Vianale G, Palka G, Barsotti A. Simvastatin and ischemia-reperfusion damage: its effects on apoptotic myocyte death and on the endothelial expression of nitric-oxide synthetase in an experimental model of the isolated rat heart. Cardiologia. 1999;44:69–74. [PubMed] [Google Scholar]
- 17.Chen MS, Xu FP, Wang YZ, Zhang GP, Yi Q, Zhang HQ, Luo JD. Statins initiated after hypertrophy inhibit oxidative stress and prevent heart failure in rats with aortic stenosis. J Mol Cell Cardiol. 2004;37:889–896. doi: 10.1016/j.yjmcc.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 18.Ostadal P, Alan D, Hajek P, Vejvoda J, Mates M, Blasko P, Veselka J, Kvapil M, Kettner J, Wiendl M, Aschermann O, Slaby J, Nemecek E, Holm F, Rac M, Macek M, Cepova J. Fluvastatin in the therapy of acute coronary syndrome: Rationale and design of a multicenter, randomized, double-blind, placebo-controlled trial (The FACS Trial) [ISRCTN81331696] . Curr Control Trials Cardiovasc Med. 2005;6:4. doi: 10.1186/1468-6708-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kapur NK, Musunuru K. Clinical efficacy and safety of statins in managing cardiovascular risk. Vasc Health Risk Manag. 2008;4:341–353. doi: 10.2147/vhrm.s1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu ZH, Chen YQ, Zhao SP. Simvastatin inhibits ox-LDL-induced inflammatory adipokines secretion via amelioration of ER stress in 3T3-L1 adipocyte. Biochem Biophys Res Commun. 2013;432:365–369. doi: 10.1016/j.bbrc.2013.01.094. [DOI] [PubMed] [Google Scholar]
- 21.Amin KA, Abd El-Twab TM. Oxidative markers, nitric oxide and homocysteine alteration in hypercholesterolimic rats: role of atorvastatine and cinnamon. Int J Clin Exp Med. 2009;2:254–65. [PMC free article] [PubMed] [Google Scholar]
- 22.Di Napoli P, Taccardi AA, Grilli A, De Lutiis MA, Barsotti A, Felaco M, De Caterina R. Chronic treatment with rosuvastatin modulates nitric oxide synthase expression and reduces ischemia-reperfusion injury in rat hearts. Cardiovasc Res. 2005;66:462–471. doi: 10.1016/j.cardiores.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 23.Balakumar P, Mahadevan N. Interplay between statins and PPARs in improving cardiovascular outcomes: a double-edged sword? Br J Pharmacol. 2012;165:373–9. doi: 10.1111/j.1476-5381.2011.01597.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maulik N, Engelman RM, Rousou JA, Flack JE 3rd, Deaton D, Das DK. Ischemic preconditioning reduces apoptosis by upregulating anti-death gene Bcl-2. Circulation. 1999;100:II369–375. doi: 10.1161/01.cir.100.suppl_2.ii-369. [DOI] [PubMed] [Google Scholar]
- 25.Pinteaux E, Rothwell NJ, Boutin H. Neuroprotective actions of endogenous interleukin-1 receptor antagonist (IL-1ra) are mediated by glia. Glia. 2006;53:551–556. doi: 10.1002/glia.20308. [DOI] [PubMed] [Google Scholar]
- 26.Chen JC, Wu ML, Huang KC, Lin WW. HMG-CoA reductase inhibitors activate the unfolded protein response and induce cytoprotective GRP78 expression. Cardiovasc Res. 2008;80:138–150. doi: 10.1093/cvr/cvn160. [DOI] [PubMed] [Google Scholar]
- 27.Urban P, Pavlíková M, Sivonová M, Kaplán P, Tatarková Z, Kaminska B, Lehotský J. Molecular analysis of endoplasmic reticulum stress response after global forebrain ischemia/ reperfusion in rats: effect of neuroprotectant simvastatin. Cell Mol Neurobiol. 2009;29:181–192. doi: 10.1007/s10571-008-9309-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Masciarelli S, Fra AM, Pengo N, Bertolotti M, Cenci S, Fagioli C, Ron D, Hendershot LM, Sitia R. CHOP-independent apoptosis and pathway-selective induction of the UPR in developing plasma cells. Mol Immunol. 2010;47:1356–1365. doi: 10.1016/j.molimm.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miyazaki Y, Kaikita K, Endo M, Horio E, Miura M, Tsujita K, Hokimoto S, Yamamuro M, Iwawaki T, Gotoh T, Ogawa H, Oike Y. C/EBP homologous protein deficiency attenuates myocardial reperfusion injury by inhibiting myocardial apoptosis and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:1124–1132. doi: 10.1161/ATVBAHA.111.224519. [DOI] [PubMed] [Google Scholar]
- 30.McGuckin MA, Eri RD, Das I, Lourie R, Florin TH. ER stress and the unfolded protein response in intestinal inflammation. Am J Physiol Gastrointest Liver Physiol. 2010;298:G820–832. doi: 10.1152/ajpgi.00063.2010. [DOI] [PubMed] [Google Scholar]
- 31.Kim DS, Kwon DY, Kim MS, Kim HK, Lee YC, Park SJ, Yoo WH, Chae SW, Chung MJ, Kim HR, Chae HJ. The involvement of endoplasmic reticulum stress in flavonoid-induced protection on cardiac cell death caused by ischaemia/reperfusion. J Pharm Pharmacol. 2010;62:197–204. doi: 10.1211/jpp.62.02.0007. [DOI] [PubMed] [Google Scholar]
- 32.Song XJ, Yang CY, Liu B, Wei Q, Korkor MT, Liu JY, Yang P. Atorvastatin inhibits myocardial cell apoptosis in a rat model with post-myocardial infarction heart failure by downregulating ER stress response. Int J Med Sci. 2011;8:564–572. doi: 10.7150/ijms.8.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murakami Y, Aizu-Yokota E, Sonoda Y, Ohta S, Kasahara T. Suppression of endoplasmic reticulum stress-induced caspase activation and cell death by the overexpression of Bcl-xL or Bcl-2. J Biochem. 2007;141:401–410. doi: 10.1093/jb/mvm044. [DOI] [PubMed] [Google Scholar]