

Abstract
Sirtuin-1 (SIRT1) possesses apparently dual roles in regulation of tumor. Previous reports have documented the crosstalk between SIRT1 with signal transducer and activator of transcription 3 (STAT3) and nuclear factor kappa-B (NF-κB) signaling in leukemia, lymphoma and myeloma. In this study, the purpose was to survey the regulatory effects of SIRT1 on gastric cancer (GC) cells (AGS and MKN-45) and the relationships between SIRT1 and activation of STAT3 and NF-κB in GC cells. We found the SIRT1 activator (resveratrol RSV) contributed to the repression of viability and increase of senescence, which were rescued by SIRT1 inhibitor (nicotinamide NA) and SIRT1 depletion by CCK-8 assay and SA-β-gal assay respectively. Further study found SIRT1 activation (RSV supplement) not only inhibited the activation of STAT3 including STAT3 mRNA level, c-myc mRNA level phosphorylated STAT3 (pSTAT3) proteins and acetylizad STAT3 (acSTAT3) proteins, but also repression of pNF-κB p65 and acNF-κB p65. NA reversed the effects of RSV. In addition, either RSV or NA application could not change the cellular viability and senescence in MKN-45 cells with STAT3 knockdown or NF-κB knockdown. Overall, our findings suggested SIRT1 activation could induced the loss of viability and increases of senescence in GC in vitro. Moreover, our observations revealed SIRT1 displayed growth inhibitory activity in GC cells highly associated with causing repression of activation of STAT3 and NF-κB proteins via deacetylation.
Keywords: Sirtuin-1, STAT3, NF-κB, gastric cancer, acetylation, activation
Introduction
Gastric cancer (GC) results in 70 thousand people death every year and has been considered as the second leading mortality related to cancer diseases worldwide [1,2]. Prevalence of GC is particularly serious in Asian countries [3]. Newly diagnosed GC cases from Asian countries accounted for about three-quarters of total cases in 2008 [4]. Up to date, the 5-year survival rate of GC patients is less than 15%, given without effective treatments and early identification for this disease [5]. Owing to the high mortality-to-incidence ratio, exploration of available management of GC is challenging and urgent, including the exploration of new therapeutic targets of GC.
Sirtuin-1 (SIRT1), a class III protein deacetylase, deacetylates proteins to exert diverse physiological functions containing DNA repair, cellular stress resistance, modulating the threshold for cell death and metabolic regulation [6,7]. SIRT1 exerts the potential effects on diabetes, inflammation, and neurodegeneration also [7]. In terms of cancer, SIRT1 possess apparently dual roles [8,9]. On the one hand, loss of SIRT1 function protects from colorectal cancer [10]. Some data also illustrated promotion of cell survival could be directly concerned with tumorigenesis in the presence of stress conditions by SIRT1 [11,12]. On the other hand, SIRT1 prevented DNA from damage and resisted the replicative life span [8,13,14]. It meant SIRT1 was tumor suppressor. Thereby, SIRT1 exploited elaborate network which was depended on cellular and molecular contexts to promote or inhibit tumorigenesis. Regarding of GC, recent reports revealed SIRT1 activator inhibits the growth of GC cells [15,16]. Instead, SIRT1 promoted multidrug resistance in GC cells via binding to activating transcription factor 4 [17]. Moreover upregulation of SIRT1 facilitated murine GC growth promoted by diet-induced obesity [18]. It was controversial whether augmenting SIRT1 levels/activity was in favor of physiological function or not in GC.
At present, numerous regulators have been identified as the substrates of SIRT1, such as ATG APE1, FOXO, Ku70 and so on referring to autophagy, DNA repair activity, oxidative stress and other regulatory mechanisms in cancer [19-22]. Besides, SIRT1 inhibited the signal transducer and activator of transcription 3 (STAT3) and nuclear factor kappa-B (NF-κB) signaling to exhibit anticancer effects in leukemia, lymphoma and myeloma [23-25]. As our known, STAT3 and NF-κB have intrinsic activator effects on cancer inflammation and regulators of the tumor microenvironment [26,27]. All the evidences provided for the hypothesis SIRT1 could play regulatory roles in the important factors (STAT3 and NF-κB) to influence the tumorigenesis of GC. Unfortunately, tumor regulatory functions of SRIT1 were unclear in GC and it is largely lacking whether the regulatory effects of SIRT1 on STAT3 and NF-κB played dominant roles in physiological function of GC cells.
In this research, SIRT1 activator resveratrol and inhibitor nicotinamide were applied to survey the effects of SIRT1 on viability and senescence of GC cells (AGS and MKN-45). Further study focused on the relationships between SIRT1 and activation of STAT3 and NF-κB in GC cells.
Materials and methods
Cell lines, cell culture and treatments
GES-1 (human normal gastric epithelial cell line) was bought from Shanghai Institute of Cell Biology. Human GC cell lines (AGS and MKN-45) were acquired from American Type Culture Collection (ATCC, Rockville, MD, USA). Cells were grown in DMEM (Gibco, Invitrogen, Carlsbad, USA) with 10% FBS in a humidified incubator containing 5% CO2 at 37°C. Cells were subcultured after reached to 80% confluence. Cells were treated with indicated methods after confluence to about 90%. 10 mM resveratrol (RSV) and 5 mM nicotinamide (NA) (Sigma, St. Louis, USA) were performed to incubate cells [28]. SIRT1 knockdown (si) and NF-κB p65 knockdown (NF-κBsi) were performed by using siRNA for SIRT1 and siRNA for NF-κB p65 respectively (Santa Cruz Biotechnology, Santa Cruz, CA, USA). STAT3-specific RNAi was used to conduct the knockdown of human STAT3 (STAT3si) [29]. Non-Target shRNA Control was performed as negative control (si-control) (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Cells were incubated with RSV after SIRT1 knockdown. Total RNA or proteins were extracted after treatments for 24 h. All cellular transfections were conduct by Lipofectamine 2000 (Invitrogen, Carlsbad, USA).
RNA extraction and Quantitative real-time polymerase chain reaction (qRT-PCR) Total RNA was extracted from cells by RNeasy kit (Qiagen, Valencia, CA). qRT-PCR was conducted on ABI 7500 with SYBR green PCR kits (Applied Biosystems). GAPDH gene was named as endogenous control. The data were normalized by 2-ΔΔCt method as relative quantification. The primers were applied as follows. STAT3: F ggg tgg aga agg aca tca gcg gta a, R gcc gac aat act ttc cga atc c. c-Myc: F tag aat tgg att ggg gta aa, R cca acc aaa aat caa cat gaa t. GAPDH: F gga gtc aac gga ttt ggt c, R gga atc att gga aca tgt aaa c.
Viability assay and cell senescence assay
For viability assay, cells with indicated treatments were incubated in CCK-8 (DOJINDO, Kumamoto, Japan). The 450 nm absorbance of each well was assayed after visual color occurrence at indicated time points. All experiments were performed in triplicate. Data were expressed as the percentage of viability from normal cells. Cell senescence were measured by cellular senescence assay kit (Cell Biolabs, San Diego, CA) associated β-gal (SA-β-gal) assay as the manufacture’s protocol. Briefly, cells were treated with RSV, NA or SIRT1 knockdown for 24 h. Then, cells were incubated with freshly prepared fluorometric substrate for 2 h at 37°C in darkness after washed and collected. The fluorescent intensity of each reaction mixture was determined and quantified by Image-J Software (NIH). The average fluorescence intensity was analyzed from five fields for each treatment. Data were expressed as the percentage of fluorescent intensity from normal cells. All experiments were performed in triplicate.
Western blot
Cells were lysed in lysis buffer (20 mM Tris HCL pH 8, 137 mM NaCl, 10% glycerol, 1% NP-40, 2 mM EDTA, protease and phosphatase inhibitors) followed by sonication on ice. Supernatants were fractionated by 10% SDSPAGE gels and electro-transferred onto nitrocellulose membranes (Millipore) after centrifuged at 14,000 g for 30 min at 4°C. The membranes were incubated by primary antibodies. Then, the signals were acquired by using the ECL detection systems (Super Signal West Femto, Pierce) after incubated with horseradish-peroxidase-conjugated secondary antibodies (Pierce). Primary antibodies were used as follows: monoclonal STAT3 antibody (Minneapolis, Minnesota, USA), anti-phosphospecific STAT3 (Tyr705) (Cell Signaling Technology), anti-acetyl-STAT3 (Lys685) (Cell Signaling Technology), anti-acetyl-NF-kB p65 (Lys310), anti-phospho-NF-kB p65 (Ser536) (Cell Signaling Technology), SirT1 primary antibody (Sigma-Aldrich) and β-actin antibody (Sigma-Aldrich).
Data analysis
All data were calculated as Mean ± SEM. Student t-test was used to compare two groups. Comparison among multiple samples was made by ANOVA. The SPSS 18.0 was used for statistical analysis. P < 0.05 was considered as statistically significant difference.
Results
SIRT1 reduced the viability of GC cells
Two GC cell lines (AGS and MKN-45) and human normal gastric epithelial cell line (GES-1) were applied in our study. The SIRT1 activator resveratrol (RSV 100 µM) and SIRT1 inhibitor nicotinamide (NA 5 mM) were performed to analyze the influences of SIRT1 on viability of GC cells. Treatment with RSV or NA could not significantly change the viability of GES-1 (Figure 1A). Interestingly, RSV remarkably inhibited viability of AGS cells as well as MKN-45 cells (80.96% for AGS, P = 0.007 and 85.52% for MKN-45, P = 0.006, respectively), when compared to the C group, as shown in Figure 1B and 1C. In two GC cell lines, the presence of SIRT1 inhibitor NA augmented the amount of viability, by compared to normal cells (109.02% ± 9.90% for AGS and MKN-45, P = 0.049). We also silenced the expression of SIRT1 to further examine the effects of SIRT1. The efficiency of SIRT1 expression was confirmed by western plot (Figure 1D). The results displayed that the effects of SIRT1 knockdown were similar to the NA (P > 0.05) and RSV did not repressed viability in cells with SIRT1 knockdown, when compared to Si groups (112.37% ± 5.42% VS 109.93% ± 1.95% for AGS and 114.14% ± 7.57% vs. 111.71% ± 6.60% for MKN-45) as shown in Figure 1B and 1C. These data demonstrated that SIRT1 activator contributed to the repression of viability, which were rescued by SIRT1 inhibitor and SIRT1 depletion.
Figure 1.
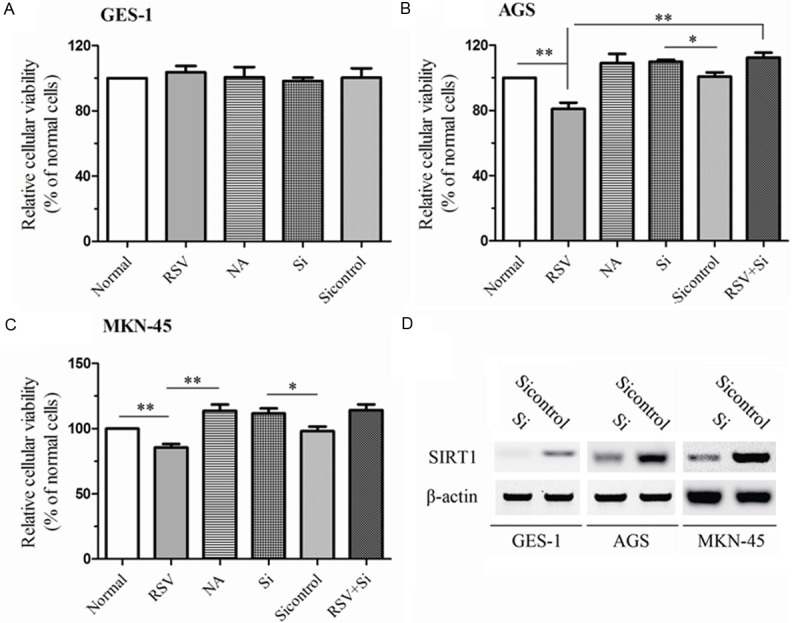
SIRT1 exerted the performances in influencing the cellular viability in normal gastric cells and in GC cells. Cells (GES-1, AGS and MKN-45) were silenced with siRNA for SIRT1 knockdown. The efficiency of knockdown was examined by western plot (D). Then, Cells or cells with silenced SIRT1 were preincubated with NA or RSV for 24 h. Normal, Si and Si control groups stood for the cells without treatment, cells with silenced SIRT1 and cells with Non-Target shRNA respectively. The viability of GES-1 (A), AGS (B) and MKN-45 (C) were measured by CCK-8 assay. All experiments were performed in triplicate. Data were expressed as the percentage of viability from normal cells. The data represented the mean ± SEM. The statistical results are indicated by asterisks and **represents P < 0.01.
SIRT1 promoted the senescence of GC cells
Previous study had identified SIRT1 leaded to G1-phase arrest rather than apoptosis in GC cells [16]. As SIRT1 had the important antioxidative effects on combating oxidative stress and senescence, further we were interested in whether SIRT1 could reduce the senescence in GC cells. In GES-1 cells, SIRT1 could not change the cellular senescence obviously (Figure 2A). Of note, supplement of RSV induced senescence rather than repressed senescence in AGS cells as well as in MKN-45 cells, when compared with C groups (185.70% ± 23.65% for AGS and 219.68% ± 37.53% for MKN-45, P < 0.001) as shown in Figure 2B and 2C. Moreover, addition of 5 mM NA could abolish the increase of β-gal activity induced by RSV in AGS cells, when compared to RSV groups (P < 0.001). The effects of SIRT1 knockdown were similar to NA and SIRT1 knockdown blocked the cellular senescence induced by RSV in AGS cells, by contrast to RSV group (P < 0.001) (Figure 2B). Similar results were acquired from MKN-45 cells (Figure 2C). These data illustrated SIRT1 promoted the senescence of GC cells.
Figure 2.
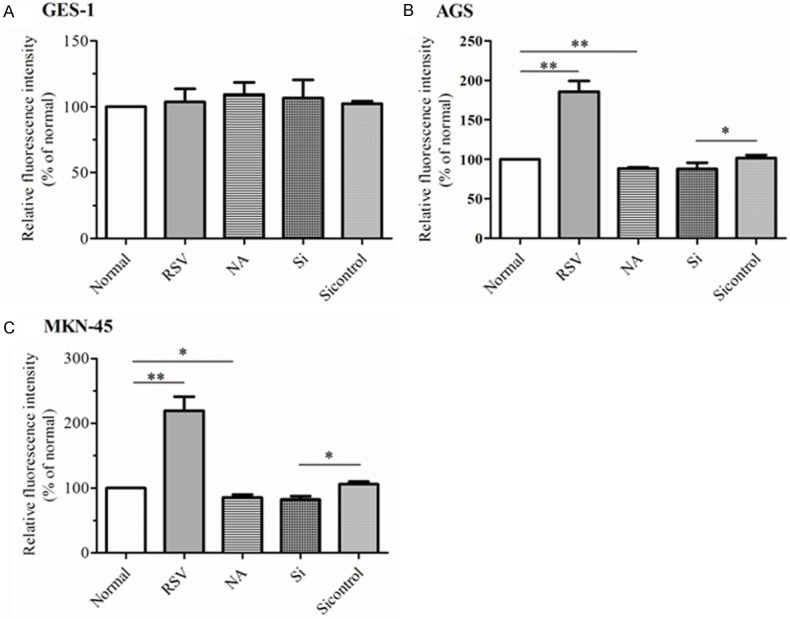
SIRT1 had the properties of modulating cellular senescence in GC cells. GES-1 (A), AGS (B) and MKN-45 (C) were treated with indicated methods. Then, cellular senescence was assayed by SA-β-gal assay. The relative fluorescence intensity was normalized as the percentage of β-gal activity from normal cells. All experiments were performed in triplicate. The data represented the mean ± SEM. The statistical results were indicated by asterisks and **represents P < 0.01.
SIRT1 counteracted the activation of STAT3 and NF-κB in GC in vitro
STAT3 and NF-κB have been considered as the substrates of SIRT1, which is a class III protein deacetylase [23-25]. Acetylation of NF-κB was necessary to exploit its transcriptional activity [30]. Moreover, SIRT1 activation had been reported to have anti-myeloma, anti-lymphoma, and anti-leukemia effects which was involved in STAT3 and NF-κB activity [31-33]. Thereby, we preferred to know whether SIRT1 was concerned with activation of STAT3 and NF-κB in GC. The AGS cells were applied in these experiments. First, the relative expression of STAT3 and was examined by qRT-PCR. RSV significantly downregulated the STAT3 mRNA, compared to the C group (P = 0.024). NA increased the expression of total STAT3 mRNA dramatically, by contrast to C group (P = 0.041) as shown in Figure 3A. Moreover, c-myc mRNA level was compared to analyze the activation of STAT3 and NF-κB as the c-myc gene has been identified as co-dependent target of STAT3 and NF-κB [34]. RSV inhibited the expression of c-Myc mRNA (0.65 ± 0.10, P = 0.004) and there was significant difference in expression of c-Myc mRNA expression between in NA group and C group (3.13 ± 1.10, P = 0.028) as shown in Figure 3B. Following, we examined the proteins of total STAT3 (tSTAT3), phosphorylated STAT3 (pSTAT3), total NF-κB p65, phosphorylated NF-κB (pNF-κB), acetylated NF-κB (acNF-κB) and acetylated STAT3 (acSTAT3) in various groups by western plot as shown in Figure 3C. RSV repressed the expression of total STAT3 proteins and pSTAT3 proteins as well as acSTAT3 proteins. Whereas, NA preincubation promoted expression of three kinds of proteins, including tSTAT3, pSTAT3 and acSTAT3. Excitingly, SIRT1 activation inhibited the expression of pNF-κB and acNF-κB, also.
Figure 3.
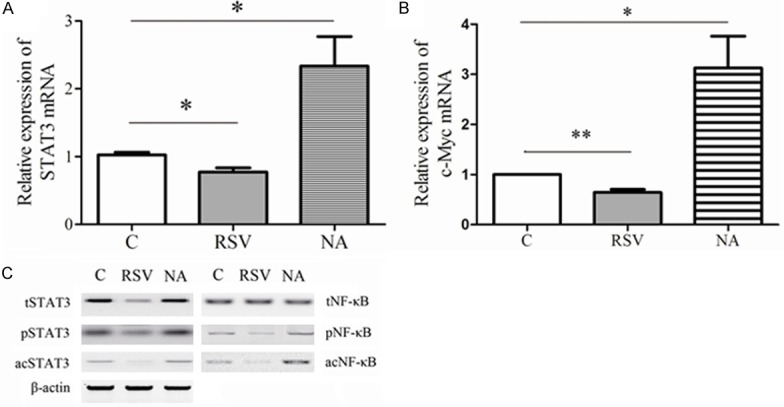
SIRT1 modulated activation of STAT3 and NF-κB by deacetylation. AGS cells were incubated with RSV (RSV group) or NA (NA group) for 24 h. C group represented the AGS cells without treatment. Then, STAT3 mRNA (A) and c-Myc mRNA (B) were isolated from cells and measured by qRT-PCR. The data were normalized by 2-ΔΔCt method as relative quantification and expressed as mean ± SEM. The statistical results were indicated by asterisks and **represents P < 0.01. Total STAT3 proteins (tSTAT3), phosphorylated STAT3 proteins (pSTAT3), acetylated STAT3 proteins (acSTAT3), tNF-κB, pNF-κB and acNF-κB were assessed by western plot (C) in three groups.
To reconfirm the crosstalk between SIRT1 with STAT3 and NF-κB, the cells were treated with STAT3 knockdown or NF-κB p65 knockdown. Figure 4C showed the efficiency of knockdown. Following, the cellular viability and senescence were assessed (Figure 4A, 4B). According to application of RSV and NA, we found either RSV or NA could not change the cellular viability and senescence in MKN-45 cells with STAT3 knockdown or NF-κB knockdown. All the data revealed two mechanisms. The one was deacetylation inhibited the activation of STAT3 and NF-κB in AGS cells. The other was SIRT1 counteracted the activation of STAT3 and NF-κB in GC in vitro.
Figure 4.
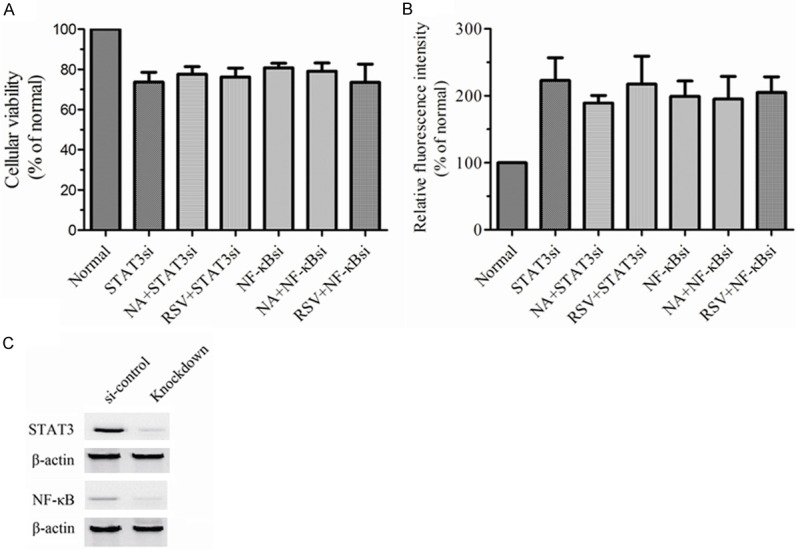
The effects of SIRT1 on viability and senescence were involved in STAT3 and NF-κB. AGS cells were knocked down by using siRNA for STAT3 (STAT3si) and siRNA for NF-κB p65 (NF-κBsi) respectively. The effectiveness of genes knockdown was assessed by western plot (C). The si-control group represented the cells with Non-Target shRNA. Then AGS cells or AGS cells with indicated treatments were incubated with RSV or NA for 24 h. Cellular viability (A) and senescence (B) were measured by CCK-8 assay and SA-β-gal assay respectively in differ groups. All experiments were performed in triplicate. The data represented the mean ± SEM.
Discussion
It was controversial whether activation or inhibition of SIRT1 possessed more anticancer activity. SIRT1 might develop elaborate network to promote or inhibit tumorigenesis depending on cellular and molecular contexts [8,9]. It was largely unknown whether SRIT1 had tumor regulatory functions in GC. In order to investigate the functions of SRIT1, we supplied resveratrol or nicotinamide for SRIT1 activation or its inhibition [28]. Cellular viability possesses key effects on mechanism for neoplastic progression [35]. In this study, we illustrated SIRT1 activator contributed to the repression of viability, which were rescued by SIRT1 inhibitor and Sirt1 depletion in GC cells (AGS and MKN-45) (Figure 1B-D). We found there was no remarkable change in viability of normal gastric epithelial cells (GES-1) in the presence of SRIT1 activation or its inhibition (Figure 1A and 1D) as well. The process of cellular senescence stands for an important barrier which combats cancer initiation and development [36,37]. In our report, NA could abolish the increase of β-gal activity induced by RSV in GC cells (Figure 2B and 2C). It demonstrated SIRT1 accelerated the senescence of GC cells. Our data not only supported the reports of Yang et al about the anti-tumor activity of RSV [15], but also further improve the understanding that SIRT1 inhibitor and SIRT1 depletion reversed the effects of RSV on anti-GC.
STAT3 activation is characterized by STAT3 phosphorylation, which causes upregualtion of its gene and regulation of its downstream genes to take intrinsic and extrinsic functions [26]. Constitutive STAT3 activation promotes gastric tumourigenesis in both human adenocarcinomas and mouse models [38]. It has been confirmed acetylation is critical and active for STAT3 phosphorylation. In addition, acetylation of NF-κB p65 at Lys-310 exhibited enhanced phosphorylation and transcriptional activity [39]. Activation of deacetylase SIRT1 inhibited the STAT3 and NF-κB activity to exert the anti-myeloma, anti-lymphoma, and anti-leukemia effects [23-25]. So we were interested in whether SIRT1 activator and inhibitor could influence activity of STAT3 and NF-κB by deacetylation and acetylation in AGS cell line. Our data illustrated RSV induced the repression of the STAT3 and NF-κB transcriptional activity in protein levels, as well as reduction of acSTAT3 and acNF-κB. The NA displayed adverse effects of RSV (Figure 3C). The c-myc gene was the target and downstream gene of STAT3 activation [26,40]. STAT3 and NF-κB were interdependent. pro-oncogenic transcription factor c-myc gene was the target of STAT3 activation as well as the downstream factor of NF-κB [34]. Thus, we assayed the expression of c-myc to assess the transcriptional activity of STAT3 and NF-κB. It found RSV significantly reduced the c-myc mRNA level and NA abolished the reduction of c-myc mRNA level induced by RSV in AGS (Figure 3B). These results demonstrated SIRT1 counteracted the activation of STAT3 and NF-κB by deacetylation. Deacetylation of SIRT1 was ubiquitination [8,9,41]. It meant SIRT1 had deacetylase activity affecting lots of transcription factors, besides STAT3 and NF-κB. Previous researches verified SIRT1 had developed elaborate network to take diverse activities. So, whether the regulation of STAT3 and NF-κB mediated by SIRT1 were dominant in physiological function of GC cells needed to been further investigated. According to silenced STAT3 and NF-κB p65 genes(Figure 4C), we found either STAT3 knockdown or NF-κB p65 knockdown could significant induce the loss of viability, but supplements of RSV and NA didn’t change the effects of STAT3 or NF-κB P65 knockdown significantly in AGS (Figure 4A and 4B). These data further supported SIRT1 counteracted the activation of STAT3 and NF-κB to repress the GC growth.
Above all, our findings suggested SIRT1 activation could induced the loss of viability and increases of senescence in GC in vitro. Moreover, our observations revealed SIRT1 displayed growth inhibitory activity in GC cells highly associated with causing repression of activation of STAT3 and NF-κB proteins via deacetylation. Our work provided detailed evidences for the anti-tumor mechanism of SIRT1 in GC. We proposed application of SIRT1 activators might be new therapeutic targets of GC.
Acknowledgements
This study was granted by Gansu Provincial Hospital.
Disclosure of conflict of interest
None.
References
- 1.Zabaleta J. Multifactorial etiology of gastric cancer. Methods Mol Biol. 2012;863:411–435. doi: 10.1007/978-1-61779-612-8_26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brenner H, Rothenbacher D, Arndt V. Epidemiology of stomach cancer. Methods Mol Biol. 2009;472:467–477. doi: 10.1007/978-1-60327-492-0_23. [DOI] [PubMed] [Google Scholar]
- 3.Leung WK, Wu MS, Kakugawa Y, Kim JJ, Yeoh KG, Goh KL, Wu KC, Wu DC, Sollano J, Kachintorn U, Gotoda T, Lin JT, You WC, Ng EK, Sung JJ Asia Pacific Working Group on Gastric Cancer. Screening for gastric cancer in Asia: current evidence and practice. Lancet Oncol. 2008;9:279–287. doi: 10.1016/S1470-2045(08)70072-X. [DOI] [PubMed] [Google Scholar]
- 4.Shen L, Shan YS, Hu HM, Price TJ, Sirohi B, Yeh KH, Yang YH, Sano T, Yang HK, Zhang X, Park SR, Fujii M, Kang YK, Chen LT. Management of gastric cancer in Asia: resource-stratified guidelines. Lancet Oncol. 2013;14:e535–547. doi: 10.1016/S1470-2045(13)70436-4. [DOI] [PubMed] [Google Scholar]
- 5.Correa P. Is gastric cancer preventable? Gut. 2004;53:1217–1219. doi: 10.1136/gut.2004.039834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yuan Z, Zhang X, Sengupta N, Lane WS, Seto E. SIRT1 regulates the function of the Nijmegen breakage syndrome protein. Mol Cell. 2007;27:149–162. doi: 10.1016/j.molcel.2007.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587–591. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bosch-Presegue L, Vaquero A. The dual role of sirtuins in cancer. Genes Cancer. 2011;2:648–662. doi: 10.1177/1947601911417862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuan H, Su L, Chen WY. The emerging and diverse roles of sirtuins in cancer: a clinical perspective. Onco Targets Ther. 2013;6:1399–1416. doi: 10.2147/OTT.S37750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lo Sasso G, Ryu D, Mouchiroud L, Fernando SC, Anderson CL, Katsyuba E, Piersigilli A, Hottiger MO, Schoonjans K, Auwerx J. Loss of sirt1 function improves intestinal anti-bacterial defense and protects from colitis-induced colorectal cancer. PLoS One. 2014;9:e102495. doi: 10.1371/journal.pone.0102495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen X, Hokka D, Maniwa Y, Ohbayashi C, Itoh T, Hayashi Y. Sirt1 is a tumor promoter in lung adenocarcinoma. Oncol Lett. 2014;8:387–393. doi: 10.3892/ol.2014.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liang Z, Yang Y, Wang H, Yi W, Yan X, Yan J, Li Y, Feng Y, Yu S, Yang J, Jin Z, Duan W, Chen W. Inhibition of SIRT1 Signaling Sensitizes the Antitumor Activity of Silybin against Human Lung Adenocarcinoma Cells In Vitro and In Vivo. Mol Cancer Ther. 2014;13:1860–1872. doi: 10.1158/1535-7163.MCT-13-0942. [DOI] [PubMed] [Google Scholar]
- 13.Hwang BJ, Madabushi A, Jin J, Lin SY, Lu AL. Histone/protein deacetylase SIRT1 is an anticancer therapeutic target. Am J Cancer Res. 2014;4:211–221. [PMC free article] [PubMed] [Google Scholar]
- 14.Ming M, Soltani K, Shea CR, Li X, He YY. Dual role of SIRT1 in UVB-induced skin tumorigenesis. Oncogene. 2015;34:357–63. doi: 10.1038/onc.2013.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang Q, Wang B, Zang W, Wang X, Liu Z, Li W, Jia J. Resveratrol inhibits the growth of gastric cancer by inducing G1 phase arrest and senescence in a Sirt1-dependent manner. PLoS One. 2013;8:e70627. doi: 10.1371/journal.pone.0070627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang Q, Wang B, Gao W, Huang S, Liu Z, Li W, Jia J. SIRT1 is downregulated in gastric cancer and leads to G1-phase arrest via NF-kappaB/Cyclin D1 signaling. Mol Cancer Res. 2013;11:1497–1507. doi: 10.1158/1541-7786.MCR-13-0214. [DOI] [PubMed] [Google Scholar]
- 17.Zhu H, Xia L, Zhang Y, Wang H, Xu W, Hu H, Wang J, Xin J, Gang Y, Sha S, Xu B, Fan D, Nie Y, Wu K. Activating transcription factor 4 confers a multidrug resistance phenotype to gastric cancer cells through transactivation of SIRT1 expression. PLoS One. 2012;7:e31431. doi: 10.1371/journal.pone.0031431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li HJ, Che XM, Zhao W, He SC, Zhang ZL, Chen R, Fan L, Jia ZL. Diet-induced obesity promotes murine gastric cancer growth through a nampt/sirt1/c-myc positive feedback loop. Oncol Rep. 2013;30:2153–2160. doi: 10.3892/or.2013.2678. [DOI] [PubMed] [Google Scholar]
- 19.Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, Tsokos M, Alt FW, Finkel T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A. 2008;105:3374–3379. doi: 10.1073/pnas.0712145105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 21.Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M, Guarente L. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551–563. doi: 10.1016/s0092-8674(04)00126-6. [DOI] [PubMed] [Google Scholar]
- 22.Jeong J, Juhn K, Lee H, Kim SH, Min BH, Lee KM, Cho MH, Park GH, Lee KH. SIRT1 promotes DNA repair activity and deacetylation of Ku70. Exp Mol Med. 2007;39:8–13. doi: 10.1038/emm.2007.2. [DOI] [PubMed] [Google Scholar]
- 23.Nie Y, Erion DM, Yuan Z, Dietrich M, Shulman GI, Horvath TL, Gao Q. STAT3 inhibition of gluconeogenesis is downregulated by SirT1. Nat Cell Biol. 2009;11:492–500. doi: 10.1038/ncb1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bernier M, Paul RK, Martin-Montalvo A, Scheibye-Knudsen M, Song S, He HJ, Armour SM, Hubbard BP, Bohr VA, Wang L, Zong Y, Sinclair DA, de Cabo R. Negative regulation of STAT3 protein-mediated cellular respiration by SIRT1 protein. J Biol Chem. 2011;286:19270–19279. doi: 10.1074/jbc.M110.200311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yuan ZL, Guan YJ, Chatterjee D, Chin YE. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science. 2005;307:269–273. doi: 10.1126/science.1105166. [DOI] [PubMed] [Google Scholar]
- 26.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7:41–51. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- 27.Yu H, Jove R. The STATs of cancer--new molecular targets come of age. Nat Rev Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 28.Hori YS, Kuno A, Hosoda R, Horio Y. Regulation of FOXOs and p53 by SIRT1 modulators under oxidative stress. PLoS One. 2013;8:e73875. doi: 10.1371/journal.pone.0073875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holtick U, Vockerodt M, Pinkert D, Schoof N, Sturzenhofecker B, Kussebi N, Lauber K, Wesselborg S, Loffler D, Horn F, Trumper L, Kube D. STAT3 is essential for Hodgkin lymphoma cell proliferation and is a target of tyrphostin AG17 which confers sensitization for apoptosis. Leukemia. 2005;19:936–944. doi: 10.1038/sj.leu.2403750. [DOI] [PubMed] [Google Scholar]
- 30.Chen LF, Mu Y, Greene WC. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO J. 2002;21:6539–6548. doi: 10.1093/emboj/cdf660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhardwaj A, Sethi G, Vadhan-Raj S, Bueso-Ramos C, Takada Y, Gaur U, Nair AS, Shishodia S, Aggarwal BB. Resveratrol inhibits proliferation, induces apoptosis, and overcomes chemoresistance through down-regulation of STAT3 and nuclear factor-kappaB-regulated antiapoptotic and cell survival gene products in human multiple myeloma cells. Blood. 2007;109:2293–2302. doi: 10.1182/blood-2006-02-003988. [DOI] [PubMed] [Google Scholar]
- 32.Li T, Wang W, Chen H, Li T, Ye L. Evaluation of anti-leukemia effect of resveratrol by modulating STAT3 signaling. Int Immunopharmacol. 2010;10:18–25. doi: 10.1016/j.intimp.2009.09.009. [DOI] [PubMed] [Google Scholar]
- 33.Estrov Z, Shishodia S, Faderl S, Harris D, Van Q, Kantarjian HM, Talpaz M, Aggarwal BB. Resveratrol blocks interleukin-1beta-induced activation of the nuclear transcription factor NF-kappaB, inhibits proliferation, causes S-phase arrest, and induces apoptosis of acute myeloid leukemia cells. Blood. 2003;102:987–995. doi: 10.1182/blood-2002-11-3550. [DOI] [PubMed] [Google Scholar]
- 34.Han SS, Yun H, Son DJ, Tompkins VS, Peng L, Chung ST, Kim JS, Park ES, Janz S. NF-kappaB/STAT3/PI3K signaling crosstalk in iMyc E mu B lymphoma. Mol Cancer. 2010;9:97. doi: 10.1186/1476-4598-9-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411:342–348. doi: 10.1038/35077213. [DOI] [PubMed] [Google Scholar]
- 36.Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, Baer R, Gu W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell. 2012;149:1269–1283. doi: 10.1016/j.cell.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A, Iken M, Vucur M, Weiss S, Heikenwalder M, Khan S, Gil J, Bruder D, Manns M, Schirmacher P, Tacke F, Ott M, Luedde T, Longerich T, Kubicka S, Zender L. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479:547–551. doi: 10.1038/nature10599. [DOI] [PubMed] [Google Scholar]
- 38.Giraud AS, Menheniott TR, Judd LM. Targeting STAT3 in gastric cancer. Expert Opin Ther Targets. 2012;16:889–901. doi: 10.1517/14728222.2012.709238. [DOI] [PubMed] [Google Scholar]
- 39.Chen LF, Williams SA, Mu Y, Nakano H, Duerr JM, Buckbinder L, Greene WC. NF-kappaB RelA phosphorylation regulates RelA acetylation. Mol Cell Biol. 2005;25:7966–7975. doi: 10.1128/MCB.25.18.7966-7975.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kiuchi N, Nakajima K, Ichiba M, Fukada T, Narimatsu M, Mizuno K, Hibi M, Hirano T. STAT3 is required for the gp130-mediated full activation of the c-myc gene. J Exp Med. 1999;189:63–73. doi: 10.1084/jem.189.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kume S, Haneda M, Kanasaki K, Sugimoto T, Araki S, Isshiki K, Isono M, Uzu T, Guarente L, Kashiwagi A, Koya D. SIRT1 inhibits transforming growth factor beta-induced apoptosis in glomerular mesangial cells via Smad7 deacetylation. J Biol Chem. 2007;282:151–158. doi: 10.1074/jbc.M605904200. [DOI] [PubMed] [Google Scholar]