

Abstract
Targeting the prostaglandin (PG) pathway is potentially a critical intervention for the prevention and treatment of cancer. Central to prostaglandin biosynthesis are two isoforms of cyclooxygenase (COX 1 and 2), which produce prostaglandin H2 (PGH2) from plasma membrane stores of fatty acids. COX-1 is constitutively expressed while COX-2 is an inducible isoform upregulated in many cancers. Differences between COX-1 and COX-2 catalytic sites enabled development of selective inhibitors. Downstream of the COX enzymes, prostaglandin E2 synthase converts available PGH2 to prostaglandin E2 (PGE2), which can stimulate cancer progression. Significant research efforts are helping identify more selective targets and fully elucidate the downstream targets of prostaglandin E2 mediated oncogenesis. Nonetheless, as a key rate-limiting control point of PG biosynthesis, COX-2 continues to be an important anticancer target. As we embark upon a new era of individualized medicine, a better understanding of the individual risk/benefit involved in COX-2 selective targeting is rapidly evolving. This review endeavors to summarize developments in our understanding of COX-2 and its downstream targets as vital areas of anti-cancer research and to provide the current status of an exciting aspect of molecular medicine.
Keywords: cyclooxygenase-2, COX-2, prostaglandins E2, PGE2, prevention, cancer, cardiovascular risk
Prostaglandins: a seminal discovery
Proinflammatory lipids play a central role in cancer progression (1) and prostaglandins (PG)s are among the most active of these molecules. As namesake products of the prostate gland, PGs were first isolated from seminal fluid (2) and their discovery established an important area of basic biology(3). PG synthesis is driven by cyclooxygenases (COX)s, also known as prostaglandin H2 synthase (PGHS) or prostaglandin-endoperoxide synthase (PTGS). Cyclooxygenase was purified in 1976 from sheep and bovine seminal vesicles (4, 5). The gene was later cloned (6, 7), but the existence of a single isoform could not account for certain variable characteristics of the enzyme, including: IC50, inhibitor pharmacokinetics, lags in PG synthesis, or rapid increases in PG production (8). Subsequently, these features were explained when COX-2 or PTGS-2 was cloned and found to be inducible by phorbol esters and lipopolysaccharides in human endothelial cells and monocytes (9, 10). These discoveries stimulated significant interest in the development of inhibitors that were selective for each isoform, COX-1 or COX-2 (8).
Immediate-early gene expression
Key aspects of the COX-2 discovery were finding associations with inflammation and immediate-early gene expression (11). This connection led to the discovery that COX-2 was rapidly turned on in rat non-transformed epithelial cells(12). The gene exhibited typical immediate-early response characteristics. Its expression increased within 30 min after exposure to epidermal growth factor or tumor growth factor-α, followed by a return to baseline after 24h. The observation that COX-2 was upregulated by 2–50 fold in human colorectal adenomas and adenocarcinomas helped stimulate intense research activity to understand the association of COX-2 and cancer (13). Subsequently, COX-2 upregulation was observed in the APCmin\+ mouse model, which harbors mutations in the adenomatous polyposis coli gene and serves as a model for familial adenomatous polyposis, FAP (14). Numerous studies later confirmed that COX-2 is consistently upregulated in a significant number of premalignant and malignant tumors (1).
The Eicosanoid Pathway
Although COX molecules are central to the production of prostaglandins, numerous additional control points exist in this pathway (15)(Figure). Upstream of cyclooxygenase lies another rate limiting molecule, a cytosolic isoform of phospholipase A2, which is the predominant enzyme that initiates the calcium dependent release of arachidonic acid (AA) from the sn-2 position of membrane phospholipids (16). Once released, COX enzymes convert the free AA substrate to the precursor molecule prostaglandin H2 (PGH2) that is then acted upon by various synthase molecules to generate the different PGs (8). These synthase molecules include: PGDS, PGES, PGFS, PGIS, and TXAS which are identified by the letter of their respective PG isomer product, D2, E2, F2α, prostacyclin (PGI2), and thromboxane A2 (TXA2). The PGES subclass is heavily involved in inflammation and carcinogenesis, due primarily to the activity of a cytosolic PGE synthase (cPGES) and two membrane bound PGE synthases, mPGES-1 and mPGES-2 (17–19). Although multiple isoforms of PGES exist, it is mPGES-1 that is primarily responsible for increasing the PGE2 levels during inflammation and tumorigenesis (20).
Figure.
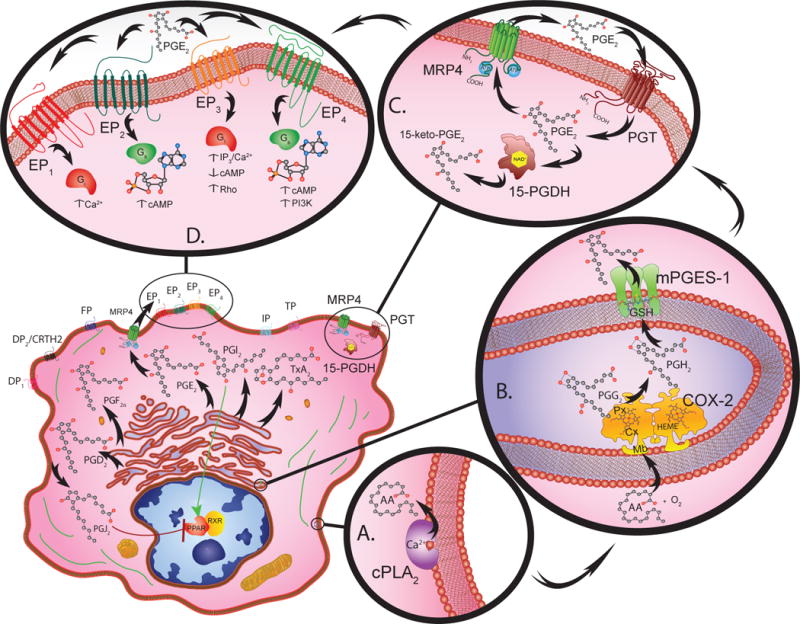
Eicosanoid biosynthesis, metabolism, and signal transduction requires the cooperative interaction between multiple compartments within a given cell (lower left). Cytosolic phospholipase A2 (PLA2) catalyzes the calcium dependent release of arachidonic acid (AA) from membrane phospholipids (A.).
Free AA serves as a substrate for COX-2 (72kDa) monomer subunits that form functional homodimer complexes (B.). Each monomer has a membrane-binding domain (Mb) that anchors the protein into the membrane of the endoplasmic reticulum or nuclear envelope. The catalytic domain contains cyclooxygenase (Cx) and peroxidase (Px) active sites that are organized on either side of a heme (HEME) prosthetic group. The cyclooxygenase site converts arachidonic acid to hydroperoxy-endoperoxide prostaglandin G2 (PGG2) through the addition of two O2 molecules. The peroxidase site then reduces PGG2 to PGH2.
Once PGH2 is generated, various synthase molecules convert it to bioactive PGs. Of these synthases, mPGES-1 is primarily responsible for increasing the PGE2 levels that promote inflammation, and tumorigenesis (B.). mPEGS-1 exists as a 16kDa monomer that forms active homotrimer complexes by interacting with glutathione (GSH) in perinuclear or endoplasmic reticulum membranes.
Prostanoids are transported into the extracellular microenvironment by specific multidrug resistance associated proteins (MRP). These MRP molecules contain a 12-membrane spanning domain structure that contains two cytosolic ATP-binding/hydrolysis sites. Among these transmembrane molecules, MRP4 is a 160kDa protein that acts as the primary transporter for PGs (C.).
The PG receptors, DP1, DP2, EP1-4, FP, IP and TP are G-protein coupled receptors classified according to their ligand specificity (D.). There are four EP receptors that rely on G-stimulatory (Gs) or G-inhibitory (Gi) proteins to activate second messengers such as cAMP, Ca2+, and inositol phosphates to initiate downstream signaling. More specifically, EP1 regulates Ca2+ flux; EP2 and EP4 both increase cAMP levels; whereas EP3 decreases cAMP, increases IP3/Ca2+, and activates Rho. Peroxisome proliferator-activated receptors (PPAR) also bind PGs and complex with retinoic X receptors (RXR) to initiate gene transcription.
The catabolism of PG involves two-steps, uptake and inactivation (C.). PGs are taken up by a 12 transmembrane domain glycoprotein known as a PG transporter (PGT). After PGE2 is transported across the plasma membrane it is enzymatically catabolized by NAD+ dependent 15-hydroxyprostaglandin dehydrogenase (15-PGDH) causing inactivation. The NAD+-15-PGDH monomers (29 kDa) dimerize into enzymatically active complexes, which form 15-keto inacitve metabolites.
Once the various PGs are synthesized they are exported into the extracellular microenvironment by specific multidrug resistance associated proteins (MRP)(21, 22). MRPs are expressed in virtually all tissues and cell types and facilitate ATP dependent unidirectional transport of lipids and other organic anionic molecules. The transport of eicosanoids by the MRP/ABCC subfamily member proteins for the PGs is primarily by MRP4/ABCC4 (21, 22)(Figure).
After export into the external microenvironment, the various prostanoid molecules bind to the appropriate G-protein coupled receptor (GPCR; Figure)(23). Similar to the designation for the PG synthases, the eicosanoid binding GPCRs are identified by the letter of their respective PG ligand, which includes two DP1, DP2, four EP1-4, FP, IP and TP plasma membrane bound cell surface receptors(23). These GPCRs can be activated following autocrine or paracrine stimulus in the tumor microenvironment. Among these GPCRs, EP2 and EP4 are primarily responsible for mediating PGE2 driven proinflammatory and pro-malignant signals downstream. In addition to the PG binding GPCRs on the cell surface, certain nuclear receptors belonging to the peroxisome proliferator activated receptor (PPAR) family exist in the cell nucleus, which bind certain PGs (24).
The catabolism of PGs involves a two-step process. PGs are taken up by a different subset of membrane transport molecules and then acted upon by catabolic enzymes (Figure). Active uptake by organic anion transporter peptides (OATP) predominantly occurs by prostaglandin transporter (PGT), a subclass of OATP molecule (25–27). Once taken up by cells, a number of PGs are enzymatically catabolized and inactivated by NAD+ dependent 15-hydroxyprostaglandin dehydrogenase (Figure) (28–30).
COX-2: a key molecular target
Although much work is underway to identify targets downstream of COX-2, as the primary rate-limiting factor in this pathway it remains the key target(15). An essential aspect of COX-2 biology is its sufficiency as a single molecule to cause cancer formation in numerous transgenic mouse models (31–33). In many of these mouse experiments, this occurs in the absence of the typical genetic modifications that accompany tumor progression. This lends an epigenetic aspect to the downstream impact of upregulating the COX-2 pathway.
Cyclooxygenase Molecules
The structural differences between COX-1 and COX-2 facilitated differential targeting(8). A COX-3 variant was also described as a splice variant of COX-1, but its biological function remains in question (34). The active enzyme complex consists of functional homodimers (Figure). Each subunit contains a cyclooxygenase site that converts free arachidonic acid to prostaglandin G2 (PGG2) and a peroxidase containing heme group that reduces PGG2 to PGH2. Although the catalytic sites are highly homologous, the COX-2 catalytic site contains a more open and spacious substrate cavity. The cavity forms along the membrane-binding domain and projects into the center of the protein. This cavity is regulated by a protein gate complex, which mediates both substrate and inhibitor entry. The functional cellular enzyme complexes are localized in both the endoplasmic reticulum and nuclear envelope.
COX-1 and COX-2 have different normal tissue distribution profiles. COX-1 is highly expressed in most tissues including platelets, lung, prostate, brain, GI tract, kidney, liver, and spleen. In recent autopsy/biopsy studies, COX-1 was found in blood vessels, interstitial cells, smooth muscle cells, platelets and mesothelial cells (35). In contrast, COX-2 exhibited basal expression levels in macrophages, endothelial cells, coronary artery, heart, prostate, lung, placenta, pancreas, brain, and kidney (35).
In functional studies, certain underappreciated properties were associated with COX activity. For example, endogenously produced endocannabinoids and free fatty acids were discovered to be metabolic substrates of COX enzymes (36). These new found enzymatic functions occurred more efficiently through COX-2 activity than COX-1 (36). In the case of one endocannabinoid, anandamide metabolism by COX-2 was involved in producing D-type prostaglandins that caused the death of certain tumorigenic keratinocytes (37). In a different set of functional enzyme studies, recent evidence has shown that fatty acid-mediated cross-talk occurs between monomers of cyclooxygenase homodimers that is allosterically regulated (38). These new findings suggest that the distribution, function, and regulation of both COX-1 and COX-2 is much more complex than first believed.
NSAIDs and COXIBs
It is becoming increasingly clear that COX-1 and COX-2 regulation is very complex and exerts a diverse impact on biology and physiology. A variety of side effects have now been associated with long-term use of non steroidal anti-inflammatory drugs (NSAIDs) and COX-2 selective inhibitors (COXIBs) that likely reflect on-target effects (39). Prolonged use of nonselective NSAIDs, such as aspirin, ibuprofen, and naproxen, is associated with COX-1 specific side effects such as the induction of GI complications and the promotion of bleeding through the inhibition of platelet activation. The development of COXIBs helped to alleviate these complications by selectively eliminating COX-2 activity while sparing COX-1 mediated biological effects. The COXIB drugs include: celecoxib, rofecoxib, valdecoxib, parecoxib and etoricoxib. The degree of gastrointestinal benefit versus cardiovascular toxicity that results from chronic COXIB use seems to associate with the degree of specificity for COX-2 (39, 40).
NSAIDs/COXIBs and cancer
Extensive information from population studies and clinical trials indicates that regular intake of various NSAIDs reduces the risk of cancer (1) in multiple organ sites (41–46) and several randomized trials showed significant benefit from the use of aspirin. In contrast to COX-2 selective inhibitors which act competitively, aspirin is non-selective and irreversibly inactivates both COX1 and COX2 (36). Regular aspirin use significantly reduces the incidence of various cancers (47). For example, prospective cohort studies involving 82,911 women enrolled in the Nurses’ Health Study (48), or 47,363 male health professionals (49) showed that regular long-term use of aspirin was associated with a significantly reduced incidence of CRC. In prospective studies that examined the recurrence of adenomatous polyps in patients with a history of resected colon cancer, daily use of aspirin was associated with a significant reduction in the incidence of colorectal adenomas (50). Another prospective study compared low dose (81mg qd) and high dose (325mg qd) aspirin with placebo to reduce adenoma formation (51). In this study, the most benefit was observed in the low-dose aspirin group (38 % reduction in adenoma formation), compared to the placebo group (47 %) and the high dose aspirin group (45 %). In multiple breast cancer trials, regular aspirin use was also associated with a reduction in the incidence of cancer (52, 53). Multiple studies have also shown a potential reduction in lung cancer risk in association with regular aspirin use (54, 55). The benefits of aspirin use in cancer prevention have led to a recent international consensus statement to highlight the advantages (56) and the initiation of a trial focused on preventing esophageal cancer that combines aspirin with esomeprazole (a proton pump inhibitor) AspECT, to limit any gastric side effects (57).
Although the cancer preventive properties of aspirin remain encouraging, a number of case/control studies support the use of COXIBs in a prevention setting as being more selective with fewer GI side effects. For example, the Adenoma Prevention with Celecoxib (APC) trial, for example, clearly demonstrated the efficacy of celecoxib in preventing formation of adenomatous polyps, a precursor of colon cancer (58). The effects on advanced adenoma formation were particularly significant, 21.3% incidence in patients taking placebo, 12.5% incidence (P < 0.0001) in patients taking low dose celecoxib (200mg bid) and 15.8% (P < 0.0001) in patients taking high-dose celecoxib (400 mg bid) (59, 60). In follow up studies on the APC trial, patients with variants in the cytochrome P450 2C9 (CYP2C9) gene exhibited impaired metabolism of celecoxib. Impaired metabolism, associated with two variants in particular, CYP2C9*2 (R144C) and CYP2C9*3 (I359L), influenced the dose-related response or toxicity of celecoxib (61). In a separate trial that employed celecoxib once daily, known as the Prevention of Colorectal Sporadic Adenomatous Polyps (PreSAP) trial, the treatment group showed significantly reduced occurrence of colorectal adenomas within three years after polypectomy (62). In yet another randomized trial using rofecoxib called Adenomatous Polyp Prevention on Vioxx (APPROVe), the discovery of increased cardiovascular risk overshadowed the findings of a significantly reduced risk of developing advanced colorectal adenomas (63–65). The associations between the use of COXIBs and a reduction in cancer risk are not limited to colon cancer. Lung cancer incidence is also reduced through the use of COXIBs (66). Further, in a prostate cancer study, COX-2 inhibitors delayed or prevented disease progression (67). Collectively, these studies illustrate the potential benefits associated with the use of COXIBs, particularly a reduction in cancer incidence in high-risk populations.
Genetic polymorphisms in COX-2
Genetic variation in the COX molecules may play a role in the development of cancer. In the breast, a decrease in cancer risk was observed among women who reported using aspirin (53). In contrast, in those individuals not using NSAIDs in the same study, a COX-2 polymorphism (rs2143416) was found to be significantly associated with the development of breast carcinoma (53). In another breast cancer study, the incidence of T8473C genetic polymorphisms present in the COX-2 gene influenced risk (52). In an aspirin trial of colorectal carcinoma, two single-nucleotide polymorphisms in rs5277 and rs4648310 COX-2 alleles were associated with increased adenoma recurrence (68). In basal cell carcinoma (BCC) by contrast, polymorphisms did not influence response to NSAIDs, however, individuals harboring a variant allele of COX-2, T8473C, were at 2.27-fold higher risk of developing BCC compared to wild-type (69). In the case of lung cancer, a T8473C polymorphism in COX-2 in non-smokers was associated with a 5.75-fold higher risk of developing lung cancer compared to wild type allele carriers (70). Collectively these studies suggest that polymorphisms in the COX-2 gene can enhance cancer risk in a variety of cancers. Since enhanced risk was typically seen in the non-treatment groups, these data suggest that aspirin potentially may help overcome the risk associated with these COX-2 polymorphisms.
Understanding the cardiovascular risks
Cardiovascular toxicity, typically defined as stroke, myocardial infarction and other thromboembolic events, has emerged as an important risk factor to consider during the regular use of coxibs (71). The first study to raise concern regarding the use of COXIBs was the Vioxx Gastrointestinal Outcomes Research (VIGOR) trial (72). This study compared naproxen (500 mg bid) with rofecoxib (50 mg/day) to examine GI safety in patients with rheumatoid arthritis. The VIGOR study showed a fourfold lower risk for myocardial infarction (MI) in patients treated with naproxen compared to those treated with rofecoxib (72). In another study known as the Celecoxib Long-Term Arthritis Safety Study (CLASS) trial, patients with either osteo- or rheumatoid arthritis were evaluated for the GI safety of celecoxib (73). The CLASS study showed no difference in the incidence of cardiovascular events (CV) between drug regimens in patients treated with celecoxib (400 mg bid) versus ibuprofen (800 mg tid) or diclofenac (75 mg bid) (73). The APC colon cancer prevention trial used doses of celecoxib at 200 or 400 mg bid vs placebo (58, 73). This trial showed a dose dependent increase in the risk ratio for CV events in the celecoxib arms compared with placebo (58). In follow up studies, the influence of celecoxib on cardiovascular events was found to be associated with preexisting atherosclerotic heart disease (60). Cardiovascular risk assessment continues to be a concern with COXIB use and will require developing accurate predictive measures that are mechanism based and take into account the overall health status of a patient.
One potential mechanism that influences the CV risk associated with COXIB use is thought to involve shifting the haemostatic balance between anti-thrombotic prostacyclin (PGI2) and pro-thrombotic thromboxane A2 (TxA2) in the circulation (71). Platelets survive for 8–12 day in circulation and lack the capacity for protein synthesis. Thus once they are shed into circulation, platelets must rely on existing levels of COX-1 to initiate pro-thombotic-TxA2 metabolism via thromboxane synthase. In contrast, nucleated endothelial cells lining the blood vessels continually synthesize COX-2 to produce anti-thrombotic-PGI2, via prostacyclin synthase. Chronic use of COXIBs at high concentrations is necessary to shift the equilibrium in favor of pro-thrombotic-TxA2 because endothelial cells have the ability to synthesize new COX-2 and replace depleted downstream anti-thrombotic-PGI2. This process is greatly amplified in patients that harbor atherosclerotic disease.
Other approaches for the clinical use of COXIBs
COXIBs may also be effective in slowing the progression of established cancer when used in combination with established cancer therapies. It was recently shown that progression free survival was longer in patients with NSCLC that expressed high levels of COX-2 in their tumors when celecoxib treatment was combined with erlotinib (an epidermal growth factor inhibitor; EGFR) (74). The rationale for combining celecoxib with erlotinib is supported by preclinical data that showed a reduction in adenomatous polyp formation in APCmin\+ mice with both treatments (75). In another study, NSCLC patients were treated with either celecoxib or zileuton (5-lipoxygenase inhibitor) alone or in combination in addition to carboplatin and gemcitabine (76). Patients whose tumors had moderate to high COX-2 expression had longer survival when celecoxib was added to chemotherapy compared to those whose tumors did not over-express COX-2 (76).
Returning to the Future
Since the earliest attempts at cancer prevention there have been success stories regarding NSAID and COXIB use (77). Accurately assessing the risk versus the benefit that accompanies any drug is key to its safe and effective use. Unfortunately, the successful determination of risk-response profiles may take years of follow up to properly evaluate (49). This might be particularly true with regard to targeting the eicosanoid pathway due to the many potential branch points that require analysis. However, the advent of modern molecular profiling methods may provide an opportunity to evaluate risk versus response pathways relatively soon. These molecular profiling approaches are likely to include evaluating polymorphisms that drive response profiles (78) as well as evaluation of cancer risk polymorphisms (79) and cardiovascular and GI risk polymorphisms (80–83). Remaining mindful of the need to proactively integrate risk response evaluations into clinical trials is critical. To this end, successfully developing modern molecular profiling technologies that enable rapid assessment of risk are essential. Such technologies, when combined with standard clinical risk profiles, will help us fully understand how to best use NSAIDs and COXIBs for cancer prevention and treatment by identifying patients most likely to benefit and/or excluding those individuals at highest risk of significant toxicity.
References
- 1.Wang D, DuBois RN. The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene. 2009 doi: 10.1038/onc.2009.421. in prress. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Von Euler U. Über die spezifische blutdrucksenkende Substanz des menschlichen Prostata- und Samenblasensekrets. Wien Klin Wochenschr. 1935;14:1182–3. [Google Scholar]
- 3.Oates JA. The 1982 Nobel Prize in Physiology or Medicine. Science. 1982;218:765–8. doi: 10.1126/science.6753151. [DOI] [PubMed] [Google Scholar]
- 4.Miyamoto T, Ogino N, Yamamoto S, Hayaishi O. Purification of prostaglandin endoperoxide synthetase from bovine vesicular gland microsomes. J Biol Chem. 1976;251:2629–36. [PubMed] [Google Scholar]
- 5.Hemler M, Lands W, Smith W. Purification of the cyclooxygenase that forms prostaglandins: demonstration of two forms of iron in the holoenzyme. J Biol Chem. 1976;251:5575–9. [PubMed] [Google Scholar]
- 6.Hla T, Farrell M, Kumar A, Bailey JM. Isolation of the cDNA for human prostaglandin H synthase. Prostaglandins. 1986;32:829–45. doi: 10.1016/0090-6980(86)90093-6. [DOI] [PubMed] [Google Scholar]
- 7.Merlie JP, Fagan D, Mudd J, Needleman P. Isolation and characterization of the complementary DNA for sheep seminal vesicle prostaglandin endoperoxide synthase (cyclooxygenase) J Biol Chem. 1988;263:3550–3. [PubMed] [Google Scholar]
- 8.Marnett LJ. The COXIB experience: a look in the rearview mirror. Annu Rev Pharmacol Toxicol. 2009;49:265–90. doi: 10.1146/annurev.pharmtox.011008.145638. [DOI] [PubMed] [Google Scholar]
- 9.Fu JY, Masferrer JL, Seibert K, Raz A, Needleman P. The induction and suppression of prostaglandin H2 synthase (cyclooxygenase) in human monocytes. J Biol Chem. 1990;265:16737–40. [PubMed] [Google Scholar]
- 10.Hla T, Neilson K. Human cyclooxygenase-2 cDNA. Proc Natl Acad Sci U S A. 1992;89:7384–8. doi: 10.1073/pnas.89.16.7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eberhart CE, Dubois RN. Eicosanoids and the gastrointestinal tract. Gastroenterology. 1995;109:285–301. doi: 10.1016/0016-5085(95)90296-1. [DOI] [PubMed] [Google Scholar]
- 12.DuBois RN, Awad J, Morrow J, Roberts LJ, 2nd, Bishop PR. Regulation of eicosanoid production and mitogenesis in rat intestinal epithelial cells by transforming growth factor-alpha and phorbol ester. J Clin Invest. 1994;93:493–8. doi: 10.1172/JCI116998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eberhart CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S, DuBois RN. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology. 1994;107:1183–8. doi: 10.1016/0016-5085(94)90246-1. [DOI] [PubMed] [Google Scholar]
- 14.Williams CS, Luongo C, Radhika A, et al. Elevated cyclooxygenase-2 levels in Min mouse adenomas. Gastroenterology. 1996;111:1134–40. doi: 10.1016/s0016-5085(96)70083-5. [DOI] [PubMed] [Google Scholar]
- 15.Cha YI, DuBois RN. NSAIDs and cancer prevention: targets downstream of COX-2. Annu Rev Med. 2007;58:239–52. doi: 10.1146/annurev.med.57.121304.131253. [DOI] [PubMed] [Google Scholar]
- 16.Burke JE, Dennis EA. Phospholipase A2 structure/function, mechanism, and signaling. J Lipid Res. 2009;50(Suppl):S237–42. doi: 10.1194/jlr.R800033-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jakobsson PJ, Thoren S, Morgenstern R, Samuelsson B. Identification of human prostaglandin E synthase: a microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc Natl Acad Sci U S A. 1999;96:7220–5. doi: 10.1073/pnas.96.13.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koeberle A, Werz O. Inhibitors of the Microsomal Prostaglandin E(2) Synthase-1 as Alternative to Non Steroidal Anti-inflammatory Drugs (NSAIDs) – A Critical Review. Curr Med Chem. 2009;16:4274–96. doi: 10.2174/092986709789578178. [DOI] [PubMed] [Google Scholar]
- 19.Samuelsson B, Morgenstern R, Jakobsson PJ. Membrane prostaglandin E synthase-1: a novel therapeutic target. Pharmacol Rev. 2007;59:207–24. doi: 10.1124/pr.59.3.1. [DOI] [PubMed] [Google Scholar]
- 20.Murakami M, Kudo I. Prostaglandin E synthase: a novel drug target for inflammation and cancer. Curr Pharm Des. 2006;12:943–54. doi: 10.2174/138161206776055912. [DOI] [PubMed] [Google Scholar]
- 21.Zhou SF, Wang LL, Di YM, et al. Substrates and inhibitors of human multidrug resistance associated proteins and the implications in drug development. Curr Med Chem. 2008;15:1981–2039. doi: 10.2174/092986708785132870. [DOI] [PubMed] [Google Scholar]
- 22.Russel FG, Koenderink JB, Masereeuw R. Multidrug resistance protein 4 (MRP4/ABCC4): a versatile efflux transporter for drugs and signalling molecules. Trends Pharmacol Sci. 2008;29:200–7. doi: 10.1016/j.tips.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 23.Sugimoto Y, Narumiya S. Prostaglandin E receptors. J Biol Chem. 2007;282:11613–7. doi: 10.1074/jbc.R600038200. [DOI] [PubMed] [Google Scholar]
- 24.Wang D, DuBois RN. Inflammatory mediators and nuclear receptor signaling in colorectal cancer. Cell Cycle. 2007;6:682–5. doi: 10.4161/cc.6.6.4030. [DOI] [PubMed] [Google Scholar]
- 25.Hagenbuch B, Gui C. Xenobiotic transporters of the human organic anion transporting polypeptides (OATP) family. Xenobiotica. 2008;38:778–801. doi: 10.1080/00498250801986951. [DOI] [PubMed] [Google Scholar]
- 26.Chi Y, Khersonsky SM, Chang YT, Schuster VL. Identification of a new class of prostaglandin transporter inhibitors and characterization of their biological effects on prostaglandin E2 transport. J Pharmacol Exp Ther. 2006;316:1346–50. doi: 10.1124/jpet.105.091975. [DOI] [PubMed] [Google Scholar]
- 27.Holla VR, Backlund MG, Yang P, Newman RA, DuBois RN. Regulation of prostaglandin transporters in colorectal neoplasia. Cancer Prev Res (Phila Pa) 2008;1:93–9. doi: 10.1158/1940-6207.CAPR-07-0009. [DOI] [PubMed] [Google Scholar]
- 28.Änggård E, Samuelsson B. Purification and properties of a 15-hydroxyprostaglandin dehydrogenase from swine lung. Ark Kemi. 1966;25:293–300. [Google Scholar]
- 29.Backlund MG, Mann JR, Holla VR, et al. 15-Hydroxyprostaglandin dehydrogenase is down-regulated in colorectal cancer. J Biol Chem. 2005;280:3217–23. doi: 10.1074/jbc.M411221200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tai HH, Cho H, Tong M, Ding Y. NAD+-linked 15-hydroxyprostaglandin dehydrogenase: structure and biological functions. Curr Pharm Des. 2006;12:955–62. doi: 10.2174/138161206776055958. [DOI] [PubMed] [Google Scholar]
- 31.Martin Sanz P, Hortelano S, Bosca L, Casado M. Cyclooxygenase 2: understanding the pathophysiological role through genetically altered mouse models. Front Biosci. 2006;11:2876–88. doi: 10.2741/2016. [DOI] [PubMed] [Google Scholar]
- 32.Colby JK, Klein RD, McArthur MJ, et al. Progressive metaplastic and dysplastic changes in mouse pancreas induced by cyclooxygenase-2 overexpression. Neoplasia. 2008;10:782–96. doi: 10.1593/neo.08330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu CH, Chang SH, Narko K, et al. Overexpression of cyclooxygenase-2 is sufficient to induce tumorigenesis in transgenic mice. J Biol Chem. 2001;276:18563–9. doi: 10.1074/jbc.M010787200. [DOI] [PubMed] [Google Scholar]
- 34.Li S, Dou W, Tang Y, Goorha S, Ballou LR, Blatteis CM. Acetaminophen: antipyretic or hypothermic in mice? In either case, PGHS-1b (COX-3) is irrelevant. Prostaglandins Other Lipid Mediat. 2008;85:89–99. doi: 10.1016/j.prostaglandins.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zidar N, Odar K, Glavac D, Jerse M, Zupanc T, Stajer D. Cyclooxygenase in normal human tissues – is COX-1 really a constitutive isoform, and COX-2 an inducible isoform? J Cell Mol Med. 2008 doi: 10.1111/j.1582-4934.2008.00430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rouzer CA, Marnett LJ. Cyclooxygenases: structural and functional insights. J Lipid Res. 2009;50(Suppl):S29–34. doi: 10.1194/jlr.R800042-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van Dross RT. Metabolism of anandamide by COX-2 is necessary for endocannabinoid-induced cell death in tumorigenic keratinocytes. Mol Carcinog. 2009 doi: 10.1002/mc.20515. [DOI] [PubMed] [Google Scholar]
- 38.Yuan C, Sidhu RS, Kuklev DV, et al. Cyclooxygenase Allosterism, Fatty Acid-mediated Cross-talk between Monomers of Cyclooxygenase Homodimers. J Biol Chem. 2009;284:10046–55. doi: 10.1074/jbc.M808634200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Farkouh ME, Greenberg BP. An evidence-based review of the cardiovascular risks of nonsteroidal anti-inflammatory drugs. Am J Cardiol. 2009;103:1227–37. doi: 10.1016/j.amjcard.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 40.Hunt RH, Lanas A, Stichtenoth DO, Scarpignato C. Myths and facts in the use of anti-inflammatory drugs. Ann Med. 2009:1–16. doi: 10.1080/07853890902887295. [DOI] [PubMed] [Google Scholar]
- 41.Harris RE. Cyclooxygenase-2 (cox-2) blockade in the chemoprevention of cancers of the colon, breast, prostate, and lung. Inflammopharmacology. 2009;17:55–67. doi: 10.1007/s10787-009-8049-8. [DOI] [PubMed] [Google Scholar]
- 42.Jafari S, Etminan M, Afshar K. Nonsteroidal anti-inflammatory drugs and prostate cancer: a systematic review of the literature and meta-analysis. Can Urol Assoc J. 2009;3:323–30. doi: 10.5489/cuaj.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cole BF, Logan RF, Halabi S, et al. Aspirin for the chemoprevention of colorectal adenomas: meta-analysis of the randomized trials. J Natl Cancer Inst. 2009;101:256–66. doi: 10.1093/jnci/djn485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abnet CC, Freedman ND, Kamangar F, Leitzmann MF, Hollenbeck AR, Schatzkin A. Non-steroidal anti-inflammatory drugs and risk of gastric and oesophageal adenocarcinomas: results from a cohort study and a meta-analysis. Br J Cancer. 2009;100:551–7. doi: 10.1038/sj.bjc.6604880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takkouche B, Regueira-Mendez C, Etminan M. Breast cancer and use of nonsteroidal anti-inflammatory drugs: a meta-analysis. J Natl Cancer Inst. 2008;100:1439–47. doi: 10.1093/jnci/djn324. [DOI] [PubMed] [Google Scholar]
- 46.Khuder SA, Herial NA, Mutgi AB, Federman DJ. Nonsteroidal antiinflammatory drug use and lung cancer: a metaanalysis. Chest. 2005;127:748–54. doi: 10.1378/chest.127.3.748. [DOI] [PubMed] [Google Scholar]
- 47.Bosetti C, Gallus S, La Vecchia C. Aspirin and cancer risk: an updated quantitative review to 2005. Cancer Causes Control. 2006;17:871–88. doi: 10.1007/s10552-006-0033-7. [DOI] [PubMed] [Google Scholar]
- 48.Chan AT, Giovannucci EL, Meyerhardt JA, Schernhammer ES, Curhan GC, Fuchs CS. Long-term use of aspirin and nonsteroidal anti-inflammatory drugs and risk of colorectal cancer. JAMA. 2005;294:914–23. doi: 10.1001/jama.294.8.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chan AT, Giovannucci EL, Meyerhardt JA, Schernhammer ES, Wu K, Fuchs CS. Aspirin dose and duration of use and risk of colorectal cancer in men. Gastroenterology. 2008;134:21–8. doi: 10.1053/j.gastro.2007.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sandler RS, Halabi S, Baron JA, et al. A randomized trial of aspirin to prevent colorectal adenomas in patients with previous colorectal cancer. N Engl J Med. 2003;348:883–90. doi: 10.1056/NEJMoa021633. [DOI] [PubMed] [Google Scholar]
- 51.Baron JA, Cole BF, Sandler RS, et al. A randomized trial of aspirin to prevent colorectal adenomas. N Engl J Med. 2003;348:891–9. doi: 10.1056/NEJMoa021735. [DOI] [PubMed] [Google Scholar]
- 52.Shen J, Gammon MD, Terry MB, Teitelbaum SL, Neugut AI, Santella RM. Genetic polymorphisms in the cyclooxygenase-2 gene, use of nonsteroidal anti-inflammatory drugs, and breast cancer risk. Breast Cancer Res. 2006;8:R71. doi: 10.1186/bcr1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gallicchio L, McSorley MA, Newschaffer CJ, et al. Nonsteroidal antiinflammatory drugs, cyclooxygenase polymorphisms, and the risk of developing breast carcinoma among women with benign breast disease. Cancer. 2006;106:1443–52. doi: 10.1002/cncr.21763. [DOI] [PubMed] [Google Scholar]
- 54.Slatore CG, Au DH, Littman AJ, Satia JA, White E. Association of nonsteroidal anti-inflammatory drugs with lung cancer: results from a large cohort study. Cancer Epidemiol Biomarkers Prev. 2009;18:1203–7. doi: 10.1158/1055-9965.EPI-08-1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Van Dyke AL, Cote ML, Prysak G, Claeys GB, Wenzlaff AS, Schwartz AG. Regular adult aspirin use decreases the risk of non-small cell lung cancer among women. Cancer Epidemiol Biomarkers Prev. 2008;17:148–57. doi: 10.1158/1055-9965.EPI-07-0517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cuzick J, Otto F, Baron JA, et al. Aspirin and non-steroidal anti-inflammatory drugs for cancer prevention: an international consensus statement. Lancet Oncol. 2009;10:501–7. doi: 10.1016/S1470-2045(09)70035-X. [DOI] [PubMed] [Google Scholar]
- 57.Das D, Chilton AP, Jankowski JA. Chemoprevention of oesophageal cancer and the AspECT trial. Recent Results Cancer Res. 2009;181:161–9. doi: 10.1007/978-3-540-69297-3_15. [DOI] [PubMed] [Google Scholar]
- 58.Bertagnolli MM, Eagle CJ, Zauber AG, et al. Celecoxib for the prevention of sporadic colorectal adenomas. N Engl J Med. 2006;355:873–84. doi: 10.1056/NEJMoa061355. [DOI] [PubMed] [Google Scholar]
- 59.Dubois RN. New, long-term insights from the Adenoma Prevention with Celecoxib Trial on a promising but troubled class of drugs. Cancer Prev Res (Phila Pa) 2009;2:285–7. doi: 10.1158/1940-6207.CAPR-09-0038. [DOI] [PubMed] [Google Scholar]
- 60.Bertagnolli MM, Eagle CJ, Zauber AG, et al. Five-year efficacy and safety analysis of the Adenoma Prevention with Celecoxib Trial. Cancer Prev Res (Phila Pa) 2009;2:310–21. doi: 10.1158/1940-6207.CAPR-08-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chan AT, Zauber AG, Hsu M, et al. Cytochrome P450 2C9 Variants Influence Response to Celecoxib for Prevention of Colorectal Adenoma. Gastroenterology. 2009 doi: 10.1053/j.gastro.2009.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Arber N, Eagle CJ, Spicak J, et al. Celecoxib for the prevention of colorectal adenomatous polyps. N Engl J Med. 2006;355:885–95. doi: 10.1056/NEJMoa061652. [DOI] [PubMed] [Google Scholar]
- 63.Baron JA, Sandler RS, Bresalier RS, et al. Cardiovascular events associated with rofecoxib: final analysis of the APPROVe trial. Lancet. 2008;372:1756–64. doi: 10.1016/S0140-6736(08)61490-7. [DOI] [PubMed] [Google Scholar]
- 64.Bresalier RS, Sandler RS, Quan H, et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med. 2005;352:1092–102. doi: 10.1056/NEJMoa050493. [DOI] [PubMed] [Google Scholar]
- 65.Baron JA, Sandler RS, Bresalier RS, et al. A randomized trial of rofecoxib for the chemoprevention of colorectal adenomas. Gastroenterology. 2006;131:1674–82. doi: 10.1053/j.gastro.2006.08.079. [DOI] [PubMed] [Google Scholar]
- 66.Harris RE, Beebe-Donk J, Alshafie GA. Reduced risk of human lung cancer by selective cyclooxygenase 2 (COX-2) blockade: results of a case control study. Int J Biol Sci. 2007;3:328–34. doi: 10.7150/ijbs.3.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pruthi RS, Derksen JE, Moore D, et al. Phase II trial of celecoxib in prostate-specific antigen recurrent prostate cancer after definitive radiation therapy or radical prostatectomy. Clin Cancer Res. 2006;12:2172–7. doi: 10.1158/1078-0432.CCR-05-2067. [DOI] [PubMed] [Google Scholar]
- 68.Barry EL, Sansbury LB, Grau MV, et al. Cyclooxygenase-2 polymorphisms, aspirin treatment, and risk for colorectal adenoma recurrence–data from a randomized clinical trial. Cancer Epidemiol Biomarkers Prev. 2009;18:2726–33. doi: 10.1158/1055-9965.EPI-09-0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vogel U, Christensen J, Wallin H, Friis S, Nexo BA, Tjonneland A. Polymorphisms in COX-2, NSAID use and risk of basal cell carcinoma in a prospective study of Danes. Mutat Res. 2007;617:138–46. doi: 10.1016/j.mrfmmm.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 70.Vogel U, Christensen J, Wallin H, et al. Polymorphisms in genes involved in the inflammatory response and interaction with NSAID use or smoking in relation to lung cancer risk in a prospective study. Mutat Res. 2008;639:89–100. doi: 10.1016/j.mrfmmm.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 71.Antman EM, Bennett JS, Daugherty A, Furberg C, Roberts H, Taubert KA. Use of nonsteroidal antiinflammatory drugs: an update for clinicians: a scientific statement from the American Heart Association. Circulation. 2007;115:1634–42. doi: 10.1161/CIRCULATIONAHA.106.181424. [DOI] [PubMed] [Google Scholar]
- 72.Bombardier C, Laine L, Reicin A, et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. N Engl J Med. 2000;343:1520–8. doi: 10.1056/NEJM200011233432103. 2 p following 8. [DOI] [PubMed] [Google Scholar]
- 73.White WB, Faich G, Whelton A, et al. Comparison of thromboembolic events in patients treated with celecoxib, a cyclooxygenase-2 specific inhibitor, versus ibuprofen or diclofenac. Am J Cardiol. 2002;89:425–30. doi: 10.1016/s0002-9149(01)02265-2. [DOI] [PubMed] [Google Scholar]
- 74.Fidler MJ, Argiris A, Patel JD, et al. The potential predictive value of cyclooxygenase-2 expression and increased risk of gastrointestinal hemorrhage in advanced non-small cell lung cancer patients treated with erlotinib and celecoxib. Clin Cancer Res. 2008;14:2088–94. doi: 10.1158/1078-0432.CCR-07-4013. [DOI] [PubMed] [Google Scholar]
- 75.Buchanan FG, Holla V, Katkuri S, Matta P, DuBois RN. Targeting cyclooxygenase-2 and the epidermal growth factor receptor for the prevention and treatment of intestinal cancer. Cancer Res. 2007;67:9380–8. doi: 10.1158/0008-5472.CAN-07-0710. [DOI] [PubMed] [Google Scholar]
- 76.Edelman MJ, Watson D, Wang X, et al. Eicosanoid modulation in advanced lung cancer: cyclooxygenase-2 expression is a positive predictive factor for celecoxib + chemotherapy–Cancer and Leukemia Group B Trial 30203. J Clin Oncol. 2008;26:848–55. doi: 10.1200/JCO.2007.13.8081. [DOI] [PubMed] [Google Scholar]
- 77.Lippman SM, Hawk ET. Cancer Prevention: From 1727 to Milestones of the Past 100 Years. Cancer Res. 2009 doi: 10.1158/0008-5472.CAN-09-1750. [DOI] [PubMed] [Google Scholar]
- 78.Lurje G, Nagashima F, Zhang W, et al. Polymorphisms in cyclooxygenase-2 and epidermal growth factor receptor are associated with progression-free survival independent of K-ras in metastatic colorectal cancer patients treated with single-agent cetuximab. Clin Cancer Res. 2008;14:7884–95. doi: 10.1158/1078-0432.CCR-07-5165. [DOI] [PubMed] [Google Scholar]
- 79.Poole EM, Bigler J, Whitton J, Sibert JG, Potter JD, Ulrich CM. C-reactive protein genotypes and haplotypes, polymorphisms in NSAID-metabolizing enzymes, and risk of colorectal polyps. Pharmacogenet Genomics. 2009;19:113–20. doi: 10.1097/FPC.0b013e32831bd976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Patel D, Moonis M. Clinical implications of aspirin resistance. Expert Rev Cardiovasc Ther. 2007;5:969–75. doi: 10.1586/14779072.5.5.969. [DOI] [PubMed] [Google Scholar]
- 81.Bierend A, Rau T, Maas R, Schwedhelm E, Boger RH. P2Y12 polymorphisms and antiplatelet effects of aspirin in patients with coronary artery disease. Br J Clin Pharmacol. 2008;65:540–7. doi: 10.1111/j.1365-2125.2007.03044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee CR, North KE, Bray MS, Couper DJ, Heiss G, Zeldin DC. Cyclooxygenase polymorphisms and risk of cardiovascular events: the Atherosclerosis Risk in Communities (ARIC) study. Clin Pharmacol Ther. 2008;83:52–60. doi: 10.1038/sj.clpt.6100221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Montes R, Nantes O, Alonso A, Zozaya JM, Hermida J. The influence of polymorphisms of VKORC1 and CYP2C9 on major gastrointestinal bleeding risk in anticoagulated patients. Br J Haematol. 2008;143:727–33. doi: 10.1111/j.1365-2141.2008.07414.x. [DOI] [PubMed] [Google Scholar]