

Abstract
Although increased synchrony of the neural activity in the basal ganglia may underlie the motor deficiencies exhibited in Parkinson's disease (PD), how this synchrony arises, propagates through the basal ganglia, and changes under dopamine replacement remains unknown. Gap junctions could play a major role in modifying this synchrony, because they show functional plasticity under the influence of dopamine and after neural injury. In this study, confocal imaging was used to detect connexin-36, the major neural gap junction protein, in postmortem tissues of PD patients and control subjects in the putamen, subthalamic nucleus (STN), and external and internal globus pallidus (GPe and GPi, respectively). Moreover, we quantified how gap junctions affect synchrony in an existing computational model of the basal ganglia. We detected connexin-36 in the human putamen, GPe, and GPi, but not in the STN. Furthermore, we found that the number of connexin-36 spots in PD tissues increased by 50% in the putamen, 43% in the GPe, and 109% in the GPi compared with controls. In the computational model, gap junctions in the GPe and GPi strongly influenced synchrony. The basal ganglia became especially susceptible to synchronize with input from the cortex when gap junctions were numerous and high in conductance. In conclusion, connexin-36 expression in the human GPe and GPi suggests that gap junctional coupling exists within these nuclei. In PD, neural injury and dopamine depletion could increase this coupling. Therefore, we propose that gap junctions act as a powerful modulator of synchrony in the basal ganglia.
Keywords: Connexin-36, oscillations, globus pallidus, confocal microscopy
In patients with Parkinson's disease (PD) and corresponding animal models, unusually high amounts of synchrony, bursting, and low-frequency oscillations have been recorded.1 These abnormalities are thought to underlie the motor symptoms of PD, but whether they are causal remains unclear.2 Still, the mechanisms for the emergence and transmission of synchrony and oscillations in the basal ganglia remain debated. Experimental and computational studies have shown that interactions between the subthalamic nucleus (STN) and the external segment of the globus pallidus (GPe) are important for the emergence of synchrony.3–6 Other studies highlighted the role of synaptic input from the cortex to the STN.7–11 Changes in the intrinsic properties of the GPe can also lead to synchrony.12–15 Whereas the healthy GPe shows almost no correlations between pairs of neurons despite the presence of local axon collaterals and correlated inputs,16,17 synchronization in the β frequency band (13-30 Hz) is prominent after dopamine loss.16,18,19 Therefore, a decorrelation mechanism has been suggested to exist in the healthy GPe.17 Given the pacemaking function of GPe neurons, intra-GPe synaptic coupling may play an important role in synchronization and desynchronization in the GPe. However, few experimental studies have described changes in the GPe after dopamine loss that would explain the clear shifts in network dynamics seen in PD. Furthermore, how these mechanisms may change under dopamine replacement remains unclear.
Pallidal gap junctions (GJs) could provide such an intrinsic mechanism of synchronization. Gap junctional coupling (GJC) in cortex and striatum has already been proposed to contribute to the pathology of PD.20–24 In striatum and cortex, gap junctions between interneurons consist of connexin-36 (Cx36),25,26 which has also been found in the rat globus pallidus (GP), corresponding to the human GPe.27 Various other neurological pathologies are thought to involve altered GJC, including epilepsy, stroke, spreading depression, and ischemia.28–30 All of these pathologies involve neural injury, which is in general associated with remodeling of GJs.31 Fernandez-Suarez et al.32 reported the death of parvalbumin-positive γ-aminobutyric acid (GABA)ergic neurons in the GPe of animal models of PD, raising the possibility of GJ remodeling. Dopamine can also influence gap junctional coupling: for example, GJs in the retina change their conductance in response to variations in the dopamine level.33–35 Most studies on GJs in the brain found a decrease in GJ conductance with increased dopamine levels.21 Dye coupling, an indication for GJC, was increased in the striatum after dopamine loss in rats.36,37 Dopamine modulation of GJC in the striatum also has been associated with stereotypic behavior,38 emphasizing the potential impact of GJC on clinical characteristics. Although the presence of GJs in the human GPe, internal globus pallidus (GPi), and STN would significantly impact information processing in the basal ganglia, whether they exist and how they may remodel after dopamine depletion remains unknown.
We therefore studied Cx36 expression in the putamen, GPe, GPi, and STN of postmortem tissues from PD patients and control subjects. Furthermore, we incorporated GJs into a basic conductance-based computational model of the basal ganglia to examine their potential influence on synchrony. Based on our findings, we hypothesize that GJs exist between GABAergic neurons of the GPe and GPi and suggest that they undergo redistribution because of neural injury in PD and exhibit up-regulated conductances after dopamine depletion. The existence of numerous high-conductance GJs in the GPe may diminish the ability of pallidal neurons to desynchronize correlated input.
Materials and Methods
Human Tissue Preparation
Human tissue was obtained from The Netherlands Brain Bank, Netherlands Institute for Neuroscience, Amsterdam. All material was collected from donors for or from whom written informed consent for a brain autopsy and the use of the material and clinical information for research purposes had been obtained by the Netherlands Brain Bank. One control subject that showed a local bacterial proliferation in the basal ganglia was excluded (not shown in Table1). All patients were men between 71 and 96 years of age. The control and PD patient groups were matched in age (82 ± 11 years) and postmortem delay (5.1 ± 1.4 h). Based on their clinical information, the control subjects did not suffer from any neurological disease. Quantitative scores on the severity of motor symptoms in the PD patients were not available. The formalin-fixed, paraffin-embedded tissues were deparaffinized using xylene and ethanol. Biopsy specimens were taken from putamen (part of the striatum), GPe, and GPi (6 PD patients and 6 control subjects as described in Table1) and STN (2 PD patients and 2 control subjects, partly coinciding with the previous group). Tissues were immersed in 25% sucrose for at least 48 hours before being frozen to prevent tissue damage. Frozen biopsy specimens were then sectioned using a cryostat along the coronal plane at a thickness of 60 µm.
Table 1.
List of patients from whom tissue of putamen, GPe and GPi has been analyzed.
| Patient | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PD | No | No | No | No | No | No | 11 years | 8 years | 4 years | 3 years | 21 years | 10 years |
| Cause of death | Heart failure | Bronchus carcinoma | Myocardial infarction | Sepsis | Cardiac asthma | Urinary tract infection | Metabolic de-regulation | Heart failure | Respiratory insufficiency | Respiratory insufficiency | De-hydration | Euthanasia |
| Age | 96 | 74 | 87 | 71 | 85 | 96 | 87 | 84 | 83 | 87 | 74 | 81 |
| Post-mortem delay | 6 h 30 min | 5 h | 4 h | 7 h 40 min | 4 h 15 min | 4 h 10 min | 4 h 45 min | 5 h 10 min | 5 h 45 min | 7 h 20 min | 2 h 50 min | 4 h 30 min |
| Tremor | No | No | No | No | No | No | Yes | Yes | No | Yes | Yes | Yes |
| Stiffness | No | No | No | No | No | No | Yes | No | Yes | Yes | No | Yes |
| Brady-/ akinesa | No | No | No | No | No | No | Yes | Yes | Yes | Yes | Yes | Yes |
| l-Dopa medication | No | No | No | No | No | No | Not during last year | Yes | Yes | Yes | No | Yes |
| Cx36 Putamen (spots/mm2) | 1770.11 | 830.06 | 1266.14 | 1171.14 | 1187.16 | 788.27 | 2331.67 | 2158.69 | 1722.85 | 865.00 | 1475.43 | 1938.93 |
| Cx36 Putamen (spot area/%) | 0.02217 | 0.01780 | 0.02812 | 0.01850 | 0.02625 | 0.01717 | 0.03941 | 0.02427 | 0.02589 | 0.01636 | 0.03772 | 0.03738 |
| Cx36 GPe (spots/mm2) | 1266.48 | 902.24 | 1001.31 | 370.92 | 919.42 | 1082.69 | 1299.22 | 1462.38 | 1579.71 | 710.65 | 1254.11 | 1610.66 |
| Cx36 GPe (spot area/%) | 0.01832 | 0.01339 | 0.02149 | 0.00750 | 0.01881 | 0.02450 | 0.02531 | 0.01658 | 0.02556 | 0.01110 | 0.02942 | 0.03765 |
| Cx36 GPi (spots/mm2) | 1058.63 | 556.38 | 712.29 | 771.33 | 709.13 | 778.60 | 2165.37 | 2204.04 | 1109.00 | 1954.85 | 940.23 | 1219.69 |
| Cx36 GPi (spot area/%) | 0.01559 | 0.00847 | 0.01307 | 0.01235 | 0.01775 | 0.01539 | 0.03772 | 0.02505 | 0.01718 | 0.02467 | 0.02264 | 0.03335 |
We show clinical information and detected Cx36 levels for individual subjects and patients. Occurrence of the PD symptoms tremor, stiffness and brady- or akinesia were described based on the clinical reports. The symptoms were assumed to be absent if not mentioned in the report.
Fluorescent Labeling and Confocal Imaging for Cx36 Detection
We used triple labeling to image Cx36 on GABAergic neurons. Free-floating sections were first permeabilized and blocked with phosphate buffered saline containing 0.5% Triton-X-100 and 10% goat serum. Next, sections were incubated in primary and secondary antibodies for approximately 24 hours and 8 hours, respectively. Dilutions (1:300) of mouse monoclonal immunoglobulin IgG1 anti-Cx36 (Invitrogen, Carlsbad, USA) and rabbit polyclonal immunoglobulin IgG anti-GAD-65/67 (Sigma, St. Louis, USA) were used to detect GJs and GABAergic neurons, respectively. 4',6-Diamidino-2-phenylindole was applied in a 1:500 dilution to label the cell nuclei. The secondary antibodies were conjugated to Alexa Fluor 488 and 633 (Invitrogen). To reduce autofluorescence, we applied an autofluorescence eliminator reagent (Millipore, Billerica, USA) for 10 min. The samples were then rinsed in ethanol and mounted on glass slides with Fluoromont-G (Electron Microscopy Sciences, Hatfield, USA). Imaging was performed on a Nikon A1 confocal microscope with a 100× oil lens. To avoid bleed-through, we sequentially scanned the specimens with individual lasers. A minimum of 20 images per tissue group was taken, with a resolution of 1024 × 1024 pixels or 0.124 µm in both directions. Our tissue selection process was not blinded. However, we tried to minimize the effects of unblinded sampling by selecting tissue areas solely based on the 4',6-diamidino-2-phenylindole signal.
Quantification of the Cx36 Signal
Confocal micrographs were analyzed using ImageJ.39 We used a threshold segmentation approach to quantitatively estimate the level of Cx36 expression: assuming that the image intensity histogram is a bimodal distribution, the threshold was defined as the arithmetic mean of p1, the peak intensity of the background noise, and p2, the highest signal intensity:
| 1 |
Images without a bimodal intensity distribution or with bright unspecific staining were rejected. In the segmented image, only spots with an area between 4 and 40 pixels were considered. In this way, segmentation of noise and unspecific labeling was suppressed.
Gap Junctional Coupling in a Basic Model of the Basal Ganglia
Depending on their architecture and strength, GJs can be both synchronizing or desynchronizing40–42 and can interact in a nonlinear way with inhibitory synapses.43 Computational modeling can be used to study how a correlated input from cortex to STN affects synchronization, and how synchrony is spread to other nuclei. We implemented the network model proposed by Rubin and Terman,44 including 16 cells to represent each of STN, GPe, and GPi, using MATLAB.45 As shown in Figure 1A, the STN received excitatory input from the cortex, and both GPe and GPi received inhibitory input from the striatum. We added GJs between pairs of neurons inside the GPe and GPi. The neural dynamics in the GPe and GPi were thus governed by:
| 2 |
where Cm is the membrane capacity, V the transmembrane voltage, and Iionic, Isyn, IGJ, and Iapp the ionic, synaptic, GJ, and applied currents, respectively. GJs were modeled as ohmic resistors:
| 3 |
with GJ conductance gGJ. The ΔV represents the difference in transmembrane voltage between the connecting cells. We chose two different GJC architectures (Fig. 1B) to estimate the effect of newly synthesized GJ channels: (1) sparse coupling with an average of 0.5 GJs per cell and (2) numerous coupling with an average of 1 GJ per cell. To simulate dopamine modulation of the GJ strength, the GJ conductance in the GPe (gGPe) and GPi (gGPi) was varied between 0 and 0.25 mS/cm2, a realistic range for neural GJs46 but low compared with the conductances of chemical synapses. The STN received excitatory input from the cortex in the form of white noise, either completely correlated or completely uncorrelated. The inhibitory input from the striatum to the GPe and GPi was uncorrelated white noise. To quantify synchrony, we performed principal component analysis on spike activity as described in Lourens.47 In short, we calculated the number of principal components (PCs) needed to describe 95% of the information contained in the spike times for all 16 cells in each nucleus. High synchronization is associated with a small number of PCs, indicating that little variation is needed to describe the network state.
Figure 1.
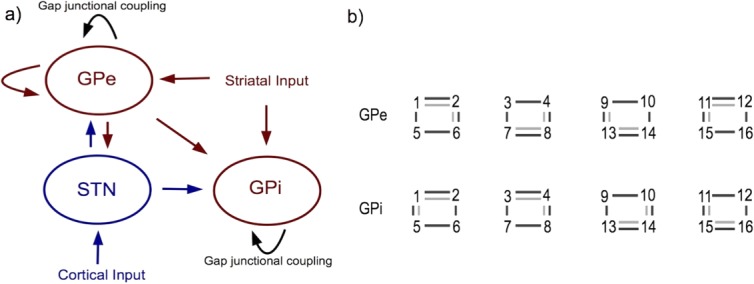
Placement of GJs added to the Rubin-Terman model [41]. (a) General setup of STN, GPe, GPi and inputs from striatum and cortex. Red indicates inhibitory connections, blue excitatory connections, black GJC. (b) GJ architectures in the GPe and GPi. Numbers represent the 16 cells in both nuclei, connected in groups of four via GJs and in the GPe also via inhibitory synapses (not shown). Light grey lines indicate the architecture for sparse GJC, dark grey lines the architecture for numerous GJC. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Results
Cx36 Is Present in the Human Putamen, GPe and GPi, But Not in the STN
The STN tissues from neither PD patients nor control subjects showed significant levels of Cx36 (data not shown) and were thus excluded from further analysis. However, we found a clear punctuate pattern of Cx36 labeling in the putamen, GPe, and GPi of all subjects (Fig. 2), which was absent in a negative control without the primary antibody (data not shown). Table1 summarizes the clinical background of all subjects and the results of the Cx36 quantification. An average of 18, but at least 12 images per tissue group could be included for image analysis. The GJC in the putamen is well described and thought to be increased after dopamine depletion.36,37 We therefore used Cx36 expression in the putamen as a reference for numerous GJCs. Control subjects showed the most Cx36 expression in the putamen; less Cx36 was found in their GPe and GPi. Compared with controls, the number of Cx36 spots in PD patients increased by 50% in the putamen, 43% in the GPe and 109% in the GPi (Fig. 3A). Furthermore, the cumulative area of the Cx36 spots increased significantly in the putamen and GPi of PD patients, but no significant increase was detected in the GPe (Fig. 3B).
Figure 2.
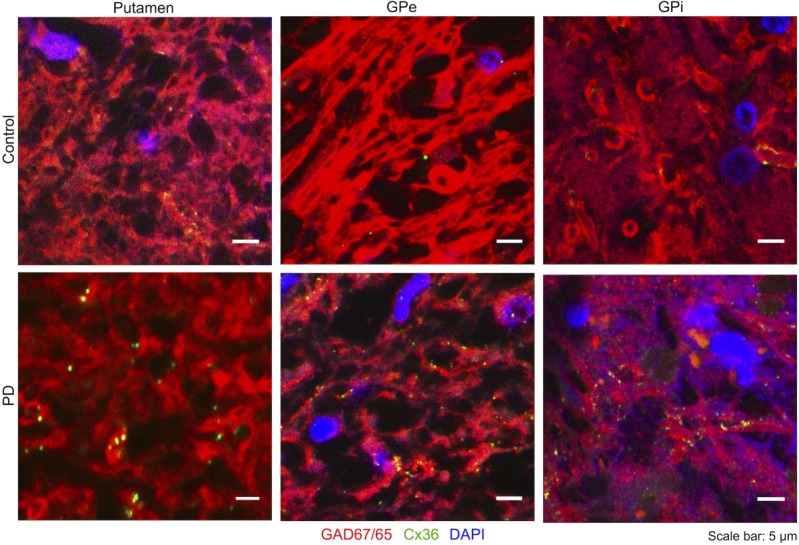
Cx36 in the human basal ganglia: Small high-resolution outtakes from confocal images. Cell nuclei are labelled by DAPI (blue), GABAergic neurons by anti-GAD65/67 (red) and Cx36 by anti-Cx36 (green). Some Cx36 is present in the putamen, GPe and GPi of control subjects, while an increase of Cx36 can be seen in the PD patients. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 3.
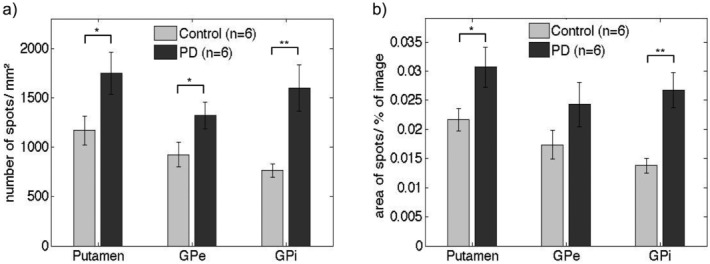
Average expression of Cx36: (a) number of spots per mm2; (b) total area of the spots as a fraction of the total image. Cx36 spots are significantly (p < 0.05) increased in all three nuclei. The increase in cumulative area of detected Cx36 spots is significant only in putamen and GPi. The standard errors of the mean are presented as thin bars. *p < 0.05; **p < 0.01 (two sample t-test).
Gap Junctional Coupling Controls Synchrony in a Basic Model of the Basal Ganglia
Based on our experimental findings of Cx36 in the human GPe and GPi, we used a small network model to demonstrate the effect of GJC in these nuclei. Figure 4 shows the results of our principal component analysis for different GJ densities, GJ conductances gGPe and gGPi, and cortical input. We compared the number of PCs as we increased gGPe and gGPe to model modulation of the GJ conductance by dopamine. For sparse GJC and uncorrelated cortical input, the increase in GJ conductance induced moderate synchronization as indicated by a decrease in the number of PCs (Fig. 4A). Similar results were achieved with correlated cortical input to the STN (Fig. 4B). In contrast, cortical inputs impacted synchronization when numerous GJs were present. When cortical input was uncorrelated, higher GJC conductances in the sparse architecture led to partial synchronization, generating a minimum of 4 PCs (Fig. 4C). Under the influence of correlated cortical input, a minimum of 2 PCs could be achieved, indicating almost complete synchronization (Fig. 4D). Thus, in our computational model, synchrony in the basal ganglia depended on pallidal GJC as well as the cortical input to STN. Although the STN itself did not contain any GJs, pallidal GJs could influence its synchrony. Furthermore, reducing the number of GJs to on average 0.25 per cell led to desynchronization at medium GJ conductances (data not shown).
Figure 4.

Results of the computational model. Principal component analysis of neural activity in the STN (first column), GPe (second column) and GPi (third column) at sparse (a and b) and numerous (c and d) GJC in GPe and GPi as well as with uncorrelated (a and c) and uncorrelated (b and d) inputs from the cortex. Bars show the number of principal components dependent on GJ conductance in the GPe (gGPe) and in the GPi (gGPi). Points with GJ conductance zero indicate the reference without GJC. If GJC is sparse, a rise of GJ conductance leads to only moderate synchronization. Correlated input from the cortex is not able to completely synchronize activity in the basal ganglia (a and b). However, if GJC is numerous, the basal ganglia are vulnerable to synchronize with increased gap junction conductances and also with correlated input from the cortex (c and d). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Discussion and Conclusions
We detected Cx36 in the human putamen, GPe, and GPi, suggesting GJC in these nuclei. In contrast, no Cx36 was found in the human STN. A Cx36 remodeling was seen in the GPe and GPi of the PD patients. In a small network model of the basal ganglia, we demonstrated that numerous high-conductance GJs rendered the GPe more susceptible to synchronize with cortical inputs transmitted via the STN. Cells of the STN also showed synchrony dependent on pallidal GJs, although they were not coupled via GJs themselves. We suggest pallidal GJC as a mechanism for the transmission and reinforcement of neural synchrony in the dopamine-depleted basal ganglia, which can be tested in vivo or in vitro in an animal model of PD. We predict that the application of a GJ blocker directly on the GPe and GPi will decrease neural synchronization in the dopamine-depleted basal ganglia. Should this prove true, seeing how GJ blocking affects the motor signs of the animal would be interesting. A direct involvement of GJC in the pathophysiology of PD would open up novel treatment possibilities, including pharmacological modulation of GJs.
Our findings of Cx36 in the human GPe and GPi are novel. Cx36 has already been detected in healthy rat GP.27 As with our human tissue, the level of Cx36 in the rat GP was low compared with Cx36 in the rat striatum. Kita48 also described single GJs in the rat GP using electron microscopy. Although the messenger ribonucleic acid of Cx36 has been found in rat STN and GP,49 no GJs were found in the rat STN using electron microscopy.50 We also did not find significant levels of Cx36 in the human STN. Gao et al.51 showed up-regulated expression of Cx36 in the motor cortex and striatum of a rat model of PD. In the striatum, they found an increase of 38% using immunohistochemistry and 15% with immunoblotting. Similarly, our immunohistochemistry of the putamen yielded a 50% increase in the number of Cx36 spots and a 42% increase in their area.
The use of human tissue imposed limitations on our study. First, the total number of subjects was low, but all of them showed Cx36 in GPe and GPi. Furthermore, the tissue had postmortem delays between 3 and 8 hours. Although this is a short time for processing human tissue, some proteins may have already degenerated. Moreover, the tissue has been fixed in formalin, which restricts the labeling quality and hinders the possibility of immunoblotting. The quality of the human tissue is therefore not comparable to directly fixed animal tissue. Additionally, most PD patients in this study were treated with L-3,4-dihydroxyphenylalanine (l-dopa), which also may have effects on Cx36 expression. However, PD patients # 7 and # 11 did not receive l-dopa the year before their death, and their Cx36 levels were not lower than those of the other PD patients. In general, postmortem studies do not allow a definite statement on whether the observed changes are causal, compensatory, or epiphenomenal for pathology. Studies with animal models of PD will be appropriate to address these questions in further detail.
We also considered limitations related to GJ functionality and their role in the basal ganglia. First, detection of Cx36 on cell membranes does not directly imply GJC, but also can indicate hemi-channels or nonfunctional GJs. Fukuda et al.52 estimated that 2% to 5% of Cx36 spots indicate functional GJs in the open configuration. In pathological states such as PD, the average function of the GJs may change. Nevertheless, the occurrence and the detected levels of Cx36 give indications of GJC. We neglected potential GJC between different nuclei, because GJs typically occur between adjacent cells53 of the same type.54–57 However, we cannot exclude the possibility of GJC, for example, to and from the striatum, because GJC between different nuclei is potentially not explored yet. Second, we did not investigate the expression of Cxs other than Cx36. GJC between GABAergic neurons mainly involves Cx36,23,52,58 but in particular glutamatergic neurons may express other Cxs such as Cx45.59 Thus, we cannot provide a definite statement on the absence of GJs in the human STN. Third, for GJC to occur, we need to assume that anatomical contacts exist between cells. Although local axon collaterals are extensively present in the human GPe,60 they seem to be low in number in the GPi.61 Finally, the computational model demonstrated synchronizing roles of pallidal GJs depending on their strength and architecture, but may not realistically reproduce the neural dynamics in the whole basal ganglia. Specifically, it cannot replicate oscillations within different frequency ranges. To achieve this, the model would need to include more cells and account for heterogeneity within the nuclei.
Based on our findings of Cx36 in the human GPe and GPi, we hypothesize GJC in these nuclei. Furthermore, we suggest a remodeling of GJs after neural injury and an increase of GJ conductance after dopamine depletion in PD. In PD patients, dopamine loss also occurs in the GPe (−82%) and GPi (−51%).62 Application of dopamine to the GP of rat models of PD improved the rats' motor behavior.63 Dye-coupling in the striatum of 6-hydroxydopamine rat models of PD, indicative of GJC, can increase up to fourfold.36 A similar increase in GJC in the GPe and GPi after dopamine depletion would be sufficient to trigger synchronization in the basal ganglia. In our computational model, small changes in GJ conductance of approximately 0.05 mS/cm2 could shift the basal ganglia to a synchronized state. We stress that the pathology of PD also may involve altered GJC in the cortex, striatum, substantia nigra, and retina. Using a computational model, Damodaran et al. [24] described how altered GJC between fast-spiking interneurons of the striatum can lead to an imbalance of the direct and indirect pathways in PD. GJs in the cortex could be involved in the generation of β oscillations.64 Modulation of the GJs between dopaminergic cells of the substantia nigra pars compacta65 may contribute to cell death,66 and similar processes may occur in the retina.
Gap junction remodeling also may depend on factors other than neural injury and the dopamine level. Palacios-Prado et al.67 demonstrated an increase in GJC with decreasing intracellular magnesium concentration. Given that the intracellular magnesium concentration is decreased in the resting brain of patients with PD and other neurological diseases,68 GJC may indeed increase in PD. In contrast, Sung et al.66 showed that overexpression of α-synuclein, as seen in PD brains, can decrease GJC. The role of GJs in health and disease is far from understood, and their interplay with chemical synapses can sculpt neural network dynamics in various ways.
Acknowledgments
We thank the Netherlands Brain Bank for providing human tissue and related support, Dr. Anne- Marie van Cappellen van Walsum for help in identifying different brain nuclei, and Dr. Gerco Hassink for his invaluable technical support.
Author Roles
1. Research Project: A. Conception, B. Organization, C. Execution; 2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript Preparation: A. Writing the First Draft, B. Review and Critique.
B.C.S.: 1A, 1C, 2A, 2B, 3A
T.H.: 1A, 2C, 3B
Y. Z.: 2C, 3B
S.A.v.G.: 1A, 2C, 3B
R.J.A.v.W.: 1A, 2C, 3B
Financial Disclosures
We acknowledge funding of the Netherlands Organization for Scientific Research (NWO) on the NDNS+-project “Desynchronization of Parkinsonian Oscillations in the Subthalamic Nucleus” and from MIRA, Institute for Biomedical Technology and Technical Medicine, University of Twente. B. C. Schwab is further supported by the German National Academic Foundation. B. C. Schwab receives funding of the Netherlands Organization for Scientific Research (NWO) on the NDNS+-project “Desynchronization of Parkinsonian oscillations in the Subthalamic Nucleus” and from MIRA, Institute for Biomedical Technology and Technical Medicine, University of Twente. She receives human tissue from the Netherlands Brain Bank, is supported by the German National Academic Foundation, and got travel grants from the International Basal Ganglia Society, the Organization for Computational Neuroscience, NDNS+, and the International Brain Organization. T. Heida is employed at the University of Twente, Netherlands, and further receives funding of the BrainGain project, the Technology Foundation STW, and the TWIN Dutch Institute for Neuromodulation. Y. Zhao is supported by the BrainGain SmartMix Program, the Whitaker International Program, and the NutsOhraFund. S. A. van Gils is employed at the University of Twente, Netherlands, and receives funding from the NWO, MIRA Institute, and Twente Graduate School. R. J. A. van Wezel is employed at the Radboud University Nijmegen, Netherlands, and receives funding from the NutsOhra Fund, the European Marie-Curie project, and MIRA.
References
- Wichmann T. Basal ganglia discharge abnormalities in Parkinson's disease. Parkinson's Dis Rel Disord. 2006:21–25. doi: 10.1007/978-3-211-45295-0_5. http://link.springer.com/chapter/10.1007/978-3-211-45295-0_5. [DOI] [PubMed] [Google Scholar]
- Quiroga-Varela A, Walters JA, Brazhnik E, Marin C, Obeso JA. What basal ganglia changes underlie the parkinsonian state? The significance of neuronal oscillatory activity. Neurobiol Dis. 2013 doi: 10.1016/j.nbd.2013.05.010. , doi: 10.1016/j.nbd.2013.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plenz D, Kital ST. A basal ganglia pacemaker formed by the subthalamic nucleus and external globus pallidus. Nature. 1999;400:677–682. doi: 10.1038/23281. [DOI] [PubMed] [Google Scholar]
- Terman D, Rubin JE, Yew AC, Wilson CY. Activity patterns in a model for the subthalamopallidal network of the basal ganglia. J Neurosci. 2002;22:2963–2976. doi: 10.1523/JNEUROSCI.22-07-02963.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan MD, Magill PJ, Terman D, Bolam JP, Wilson CJ. Move to the rhythm: oscillations in the subthalamic nucleus-external globus pallidus network. Trends Neurosci. 2002;25:525–531. doi: 10.1016/s0166-2236(02)02235-x. [DOI] [PubMed] [Google Scholar]
- Fan KY, Baufreton J, Surmeier JD, Chan CS, Bevan MD. Proliferation of external globus pallidus-subthalamic nucleus synapses following degeneration of midbrain dopamine neurons. J Neurosci. 2013;32:13718–13728. doi: 10.1523/JNEUROSCI.5750-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magill PJ, Bolam JP, Bevan MD. 2001. Dopamine regulates the impact of the cerebral cortex on the subthalamic nucleus–globus pallidus network. Neuroscience. 2001;106:313–330. doi: 10.1016/s0306-4522(01)00281-0. [DOI] [PubMed] [Google Scholar]
- Brown P. Oscillatory nature of human basal ganglia activity: relationship to the pathophysiology of Parkinson's disease. Mov Disord. 2003;18:357–363. doi: 10.1002/mds.10358. [DOI] [PubMed] [Google Scholar]
- Sharott A, Magill PJ, Harnack D, Kupsch A, Meissner W, Brown P. Dopamine depletion increases the power and coherence of f3-oscillations in the cerebral cortex and subthalamic nucleus of the awake rat. Eur J Neurosci. 2005;21:1413–1422. doi: 10.1111/j.1460-9568.2005.03973.x. [DOI] [PubMed] [Google Scholar]
- Hahn PJ, McIntyre CC. Modeling shifts in the rate and pattern of subthalamopallidal network activity during deep brain stimulation. J Comput Neurosci. 2010;28:425–441. doi: 10.1007/s10827-010-0225-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana Y, Iwamuro H, Kita H, Takada M, Nambu A. Subthalamo-pallidal interactions underlying parkinsonian neuronal oscillations in the primate basal ganglia. Eur J Neurosci. 2011;34:1470–1484. doi: 10.1111/j.1460-9568.2011.07865.x. [DOI] [PubMed] [Google Scholar]
- Chan CS, Glajch KE, Gertler TS. 2011. HCN channelopathy in external globus pallidus neurons in models of Parkinson's disease. Nat Neurosci. 2011;14:85–92. doi: 10.1038/nn.2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miguelez C, Morin S, Martinez A, Goillandeau M, Bezard E, Bioulac B, Baufreton J. Altered pallido-pallidal synaptic transmission leads to aberrant firing of globus pallidus neurons in a rat model of Parkinson's disease. J Physiol. 2012;590:5861–5875. doi: 10.1113/jphysiol.2012.241331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CJ. Active decorrelation in the basal ganglia. Neuroscience. 2013;250:467–482. doi: 10.1016/j.neuroscience.2013.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab BC, Heida T, Zhao Y, Marani E, van Gils SA, van Wezel RJA. Synchrony in Parkinson's disease: importance of intrinsic properties of the external pallidal segment. Front Syst Neurosci. 2013;7:60. doi: 10.3389/fnsys.2013.00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nini A, Feingold A, Slovin H, Bergman H. Neurons in the globus pallidus do not show correlated activity in the normal monkey, but phase-locked oscillations appear in the MPTP model of parkinsonism. J Neurophysiol. 1995;74:1800–1805. doi: 10.1152/jn.1995.74.4.1800. [DOI] [PubMed] [Google Scholar]
- Bar-Gad I, Heimer G, Ritov Y, Bergman H. Functional correlations between neighboring neurons in the primate globus pallidus are weak or nonexistent. J Neurosci. 2003;23:4012–4016. doi: 10.1523/JNEUROSCI.23-10-04012.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz A, Vaadia E, Bergman H. Firing patterns and correlations of spontaneous discharge of pallidal neurons in the normal and the tremulous 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine vervet model of parkinsonism. J Neurosci. 2000;20:8559–8571. doi: 10.1523/JNEUROSCI.20-22-08559.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimer G, Bar-Gad I, Goldberg JA, Bergman H. Dopamine replacement therapy reverses abnormal synchronization of pallidal neurons in the 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine primate model of parkinsonism. J Neurosci. 2002;22:7850–7855. doi: 10.1523/JNEUROSCI.22-18-07850.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamawaki N, Stanford IM, Hall SD, Woodhall GL. Pharmacologically induced and stimulus evoked rhythmic neuronal oscillatory activity in the primary motor cortex in vitro. Neuroscience. 2008;151:386–395. doi: 10.1016/j.neuroscience.2007.10.021. [DOI] [PubMed] [Google Scholar]
- Traub RD, Whittington MA. Cortical Oscillations in Health and Disease. Oxford University Press; 2010. [Google Scholar]
- Weinberger M, Dostrovsky JO. A basis for the pathological oscillations in basal ganglia: the crucial role of dopamine. Neuroreport. 2011;22:151. doi: 10.1097/WNR.0b013e328342ba50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dere E. Gap Junctions in the Brain. Elsevier; 2012. [Google Scholar]
- Damodaran S, Evans RC, Blackwell KT. Synchronized firing of fast-spiking interneurons is critical to maintain balanced firing between direct and indirect pathway neurons of the striatum. J Neurophysiol. 2014;111:836–848. doi: 10.1152/jn.00382.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galarreta M, Hestrin S. Electrical synapses between GABA-releasing interneurons. Nat Rev Neurosci. 2001;2:425–433. doi: 10.1038/35077566. [DOI] [PubMed] [Google Scholar]
- Fukuda T. Network architecture of gap junction-coupled neuronal linkage in the striatum. J Neurosci. 29:1235–1243. doi: 10.1523/JNEUROSCI.4418-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rash JE, Staines WA, Yasumura T, Patel D, Furman CS, Stelmack GL, Nagy JI. Immunogold evidence that neuronal gap junctions in adult rat brain and spinal cord contain connexin-36 but not connexin-32 or connexin-43. Proc Natl Acad Sci U S A. 2000;97:7573–7578. doi: 10.1073/pnas.97.13.7573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemani VM, Binder DK. Emerging role of gap junctions in epilepsy. Histol Histopathol. 2005;20:253–259. doi: 10.14670/HH-20.253. [DOI] [PubMed] [Google Scholar]
- Wang Y, Denisova JV, Kang KS, Fontes JD, Zhu BT. Belousov AB. Neuronal gap junctions are required for NMDA receptor-mediated excitotoxicity: implications in ischemic stroke. J Neurophysiol. 2010;104:3551–3556. doi: 10.1152/jn.00656.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargiotas P, Muhammad S, Rahman M. Connexin 36 promotes cortical spreading depolarization and ischemic brain damage. Brain Res. 2012;1479:80–85. doi: 10.1016/j.brainres.2012.08.046. [DOI] [PubMed] [Google Scholar]
- Belousov AB. Novel model for the mechanisms of glutamate-dependent excitotoxicity: role of neuronal gap junctions. Brain Res. 2012;1487:123–130. doi: 10.1016/j.brainres.2012.05.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Suarez D, Celorrio M, Lanciego JL, Franco R, Aymerich MS. Loss of parvalbumin-positive neurons from the globus pallidus in animal models of Parkinson disease. J Neuropathol Exp Neurol. 2012;71:973–982. doi: 10.1097/NEN.0b013e3182717cba. [DOI] [PubMed] [Google Scholar]
- McHahon DG, Knapp AG, Dowling JE. Horizontal cell gap junctions: single-channel conductance and modulation by dopamine. Proc Natl Acad Sci U S A. 1989;86:7639–7643. doi: 10.1073/pnas.86.19.7639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampson ECGM, Weiler R, Vaney DI. Regulation of gap-junction permeability between A-type horizontal cells in rabbit retina-effects of ph and dopamine. Invest Ophthalmol. 1992;33:1406–1406. [Google Scholar]
- Li H, Zhang Z, Blackburn MR, Wang SW, Ribelayga CP, O'Brien J. Adenosine and dopamine receptors coregulate photoreceptor coupling via gap junction phosphorylation in mouse retina. J Neurosci. 2013;33:3135–3150. doi: 10.1523/JNEUROSCI.2807-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Walsh JP, Hull CD, Howard SG, Buchwald NA, Levine MS. Dye-coupling in the neostriatum of the rat: I. Modulation by dopamine-depleting lesions. Synapse. 1989;4:229–237. doi: 10.1002/syn.890040308. [DOI] [PubMed] [Google Scholar]
- Onn SP, Grace AA. Alterations in electrophysiological activity and dye coupling of striatal spiny and aspiny neurons in dopamine-denervated rat striatum recorded in vivo. Synapse. 1999;33:1–15. doi: 10.1002/(SICI)1098-2396(199907)33:1<1::AID-SYN1>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Moore H, Grace AA. A role for electrotonic coupling in the striatum in the expression of dopamine receptor-mediated stereotypies. Neuropsychopharmacology. 2002;27:980–992. doi: 10.1016/S0893-133X(02)00383-4. [DOI] [PubMed] [Google Scholar]
- Rasband WS . Image J. Bethesda, MD: U.S. National Institutes of Health. Available from: http://imagej.nih.gov/ij/
- Chow CC, Kopell N. Dynamics of spiking neurons with electrical coupling. Neural Comput. 2000;12:1643–1678. doi: 10.1162/089976600300015295. [DOI] [PubMed] [Google Scholar]
- Lewis TJ, Rinzel J. Dynamics of spiking neurons connected by both inhibitory and electrical coupling. J Comput Neurosci. 2003;14:283–309. doi: 10.1023/a:1023265027714. [DOI] [PubMed] [Google Scholar]
- Vervaeke K, Lorincz A, Gleeson P, Farinella M, Nusser Z, Silver RA. Rapid desynchronization of an electrically coupled interneuron network with sparse excitatory synaptic input. Neuron. 2010;67:435–451. doi: 10.1016/j.neuron.2010.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeuty B. The combined effects of inhibitory and electrical synapses in synchrony. Neural Comput. 2005;17:633–670. doi: 10.1162/0899766053019917. [DOI] [PubMed] [Google Scholar]
- Rubin JE, Terman D. High frequency stimulation of the subthalamic nucleus eliminates pathological thalamic rhythmicity in a computational model. J Comput Neurosci. 2004;16:211–235. doi: 10.1023/B:JCNS.0000025686.47117.67. [DOI] [PubMed] [Google Scholar]
- MATLAB version 7.14. Natick, Massachusetts. USA: The MathWorks Inc; 2012. [Google Scholar]
- Fortier PA. Detecting and estimating rectification of gap junction conductance based on simulations of dual-cell recordings from a pair and a network of coupled cells. J Theor Biol. 2010;265:104–114. doi: 10.1016/j.jtbi.2010.03.048. [DOI] [PubMed] [Google Scholar]
- Lourens MAJ. Neural network dynamics in Parkinson's disease. University of Twente; 2013. [Google Scholar]
- Kita H. Parvalbumin-immunopositive neurons in rat globus pallidus: a light and electron microscopic study. Brain Res. 1994;657:31–41. doi: 10.1016/0006-8993(94)90950-4. [DOI] [PubMed] [Google Scholar]
- Vandecasteele M, Deniau JM, Glowinski J, Venance L. Electrical synapses in basal ganglia. Rev Neurosci. 2007;18:15–36. doi: 10.1515/revneuro.2007.18.1.15. [DOI] [PubMed] [Google Scholar]
- Chang HT, Kita H, Kitai ST. The fine structure of the rat subthalamic nucleus: an electron microscopic study. J Comp Neurol. 1983;221:113–123. doi: 10.1002/cne.902210110. [DOI] [PubMed] [Google Scholar]
- Gao M, Wang HL, Wang LC, Chen XW. Modulation of connexin 36 expression in basal ganglia and motor cortex in rat model of Parkinson's disease. Chinese Journal of Contemporary Neurology and Neurosurgery. 2013;13:697–703. [Google Scholar]
- Fukuda T, Kosaka T, Singer W, Galuske RA. Gap junctions among dendrites of cortical GABAergic neurons establish a dense and widespread intercolumnar network. J Neurosci. 2006;26:3434–3443. doi: 10.1523/JNEUROSCI.4076-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soehl G, Maxeiner S, Willecke K. Expression and functions of neuronal gap junctions. Nat Rev Neurosci. 2005;6:191–200. doi: 10.1038/nrn1627. [DOI] [PubMed] [Google Scholar]
- Galarreta M, Hestrin S. A network of fast-spiking cells in the neocortex connected by electrical synapses. Nature. 1999;402:72–75. doi: 10.1038/47029. [DOI] [PubMed] [Google Scholar]
- Gibson JR, Beierlein M. Connors BW. Two networks of electrically coupled inhibitory neurons in neocortex. Nature. 1999;402:75–79. doi: 10.1038/47035. [DOI] [PubMed] [Google Scholar]
- Tamas G, Buhl EH, Lorincz A, Somogyi P. Proximally targeted GABAergic synapses and gap junctions synchronize cortical interneurons. Nat Neurosci. 2000;3:366–371. doi: 10.1038/73936. [DOI] [PubMed] [Google Scholar]
- Hestrin S, Galarreta M. Electrical synapses define networks of neocortical GABAergic neurons. Trends Neurosci. 2005;28:304–309. doi: 10.1016/j.tins.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Connors BW, Long MA. Electrical synapses in the mammalian brain. Annu Rev Neurosci. 2004;27:393–418. doi: 10.1146/annurev.neuro.26.041002.131128. [DOI] [PubMed] [Google Scholar]
- Ma Y, Hioki H, Konno M. Expression of gap junction protein connexin36 in multiple subtypes of GABAergic neurons in adult rat somatosensory cortex. Cerebral Cortex. 2011;21:2639–2649. doi: 10.1093/cercor/bhr051. [DOI] [PubMed] [Google Scholar]
- François C, Percheron G, Yelnik J, Heyner S. A Golgi analysis of the primate globus pallidus. I. Inconstant processes of large neurons, other neuronal types, and afferent axons. J Comp Neurol. 1984;227:182–199. doi: 10.1002/cne.902270205. [DOI] [PubMed] [Google Scholar]
- Parent M, Parent A. The pallidofugal motor fiber system in primates. Parkinsonism Rel Disord. 2004;10:203–211. doi: 10.1016/j.parkreldis.2004.02.007. [DOI] [PubMed] [Google Scholar]
- Rajput AH, Sitte HH, Rajput A, Fenton ME, Pifl C, Hornykiewicz O. Globus pallidus dopamine and Parkinson motor subtypes. Neurology. 2008;70:1403–1410. doi: 10.1212/01.wnl.0000285082.18969.3a. [DOI] [PubMed] [Google Scholar]
- Galvan A, Floran B, Erlij D, Aceves J. Intrapallidal dopamine restores motor deficits induced by 6-hydroxydopamine in the rat. J Neural Transm. 2001;108:153–166. doi: 10.1007/s007020170085. [DOI] [PubMed] [Google Scholar]
- Traub RD, Bibbig A, Fisahn A, LeBeau FE, Whittington MA, Buhl EH. A model of gamma frequency network oscillations induced in the rat CA3 region by carbachol in vitro. Eur J Neurosci. 2000;12:4093–4106. doi: 10.1046/j.1460-9568.2000.00300.x. [DOI] [PubMed] [Google Scholar]
- Vandecasteele M, Glowinski J, Venance L. Electrical synapses between dopaminergic neurons of the substantia nigra pars compacta. J Neurosc. 2005;25:291–298. doi: 10.1523/JNEUROSCI.4167-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung JY, Lee HJ, Jeong EI, Oh Y, Park J, Kang KS, Chung KC. Alpha-synuclein overexpression reduces gap junctional intercellular communication in dopaminergic neuroblastoma cells. Neurosci Lett. 2007;416:289–293. doi: 10.1016/j.neulet.2007.02.025. [DOI] [PubMed] [Google Scholar]
- Palacios-Prado N, Hoge G, Marandykina A. Intracellular magnesium-dependent modulation of gap junction channels formed by neuronal connexin36. J Neurosci. 2013;33:4741–4753. doi: 10.1523/JNEUROSCI.2825-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbiroli B, Iotti S, Cortelli P, Martinelli P, Lodi R, Carelli V, Montagna P. Low brain intracellular free magnesium in mitochondrial cytopathies. J Cereb Blood Flow Metabol. 1999;19:528–532. doi: 10.1097/00004647-199905000-00007. [DOI] [PubMed] [Google Scholar]