

Abstract
Optogenetics is the use of genetic methods combined with optical technology to achieve gain or loss of function within neuronal circuits. The field of optogenetics has been rapidly expanding in efforts to restore visual function to blinding diseases such as retinitis pigmentosa (RP). Most work in the field includes a group of light-sensitive retinaldehyde-binding proteins known as opsins. Opsins couple photon absorption to molecular signaling chains that control cellular ion currents. Targeting of opsin genes to surviving retinal cells is fundamental to the success of optogenetic therapy. Viral delivery, primarily adeno-associated virus, using intravitreal injection for inner retinal cells and subretinal injection for outer retinal cells, has proven successful in many models. Challenges in bioengineering remain for optogenetics including relative insensitivity of opsins to physiologic light levels of stimulation and difficulty with viral delivery in primate models. However, targeting optogenetic therapy may present an even greater challenge. Neural and glial remodeling seen in advanced stages of RP result in reorganization of remaining neural retina, and optogenetic therapy may not yield functional results. Remodeling also poses a challenge to the selection of cellular targets, with bipolar, amacrine and ganglion cells all playing distinct physiologic roles, and affected by remodeling differently. Although optogenetics has drawn closer to clinical utility, advances in opsin engineering, therapeutic targeting and ultimately in molecular inhibition of remodeling will play critical roles in the continued clinical advancement of optogenetic therapy.
Keywords: Opsins, Optogenetics, Retinitis Pigmentosa
INTRODUCTION
Optogenetics is the use of genetic methods combined with optical technology to achieve gain or loss of function within neuronal circuits.[1,2] The idea of activating selective neuronal circuits or cells with light was introduced in the 1970s with the discovery of bacteriorhodopsin, which functioned as an ion pump activated by visible light.[3,4,5] Since then, advances in discovery of optically responsive proteins and viral delivery of genetic material in animal models have made optogenetics a rapidly evolving field. The validity of optogenetics was widely demonstrated in various organisms, but it was not until 2002 that successful studies were reported in vertebrate models. Zemelman et al have reported the simultaneous expression of Drosophila arrestin-2, photoreversible Drosophila rhodopsin, and the alpha subunit of Drosophila photoreceptor G-protein sensitized cultured hippocampal neurons to light stimulation.[6] However, this approach used a difficult triple transfection and yielded only moderate light-sensitivity.[7] These drawbacks led to exploration for simpler and more effective signaling proteins, and the field of optogenetics has rapidly expanded. One major objective of optogenetics is the restoration of visual function in blinding diseases such as retinitis pigmentosa (RP).
DEFINING OPSINS
Most of optogenetics is based on a group of light-sensitive retinaldehyde-binding proteins known generically as opsins. The strategy of using opsins as retinoid-based light sensors for optogenetics technologies is schematized in Figure 1. The best known opsins are those of vertebrate retinal photoreceptors that couple photon absorption to molecular signaling chains that ultimately control cellular ion currents. Opsins are categorized as Type 1, used by prokaryotes for phototaxis or coupling light energy to cellular functions, and Type 2 found in metazoans involved in higher order processes such as vision and phase setting of circadian rhythms.[8] Types 1 and 2 opsins do not share significant homology,[9] but both use retinaldehyde variants as their chromophores. In this setting, the retinaldehyde chromophore in vertebrate photoreceptors is the light sensor and upon photon absorption, isomerizes from the 11-cis to the all-trans configuration to induce conformational changes in opsin.[10]
Figure 1.
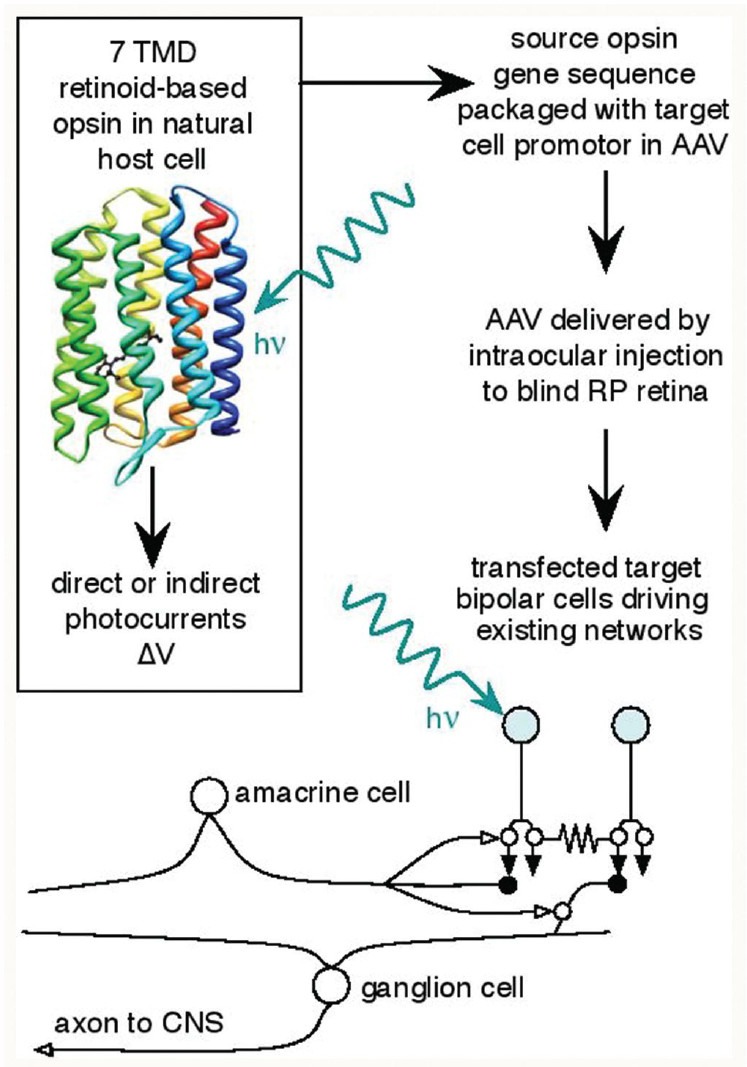
Opsins for retinitis pigmentosa therapy. AAV, adenoassociated virus; TMD, transmembrane domain; ΔV, membrane voltage change; hv, photon; CNS, central nervous system. Rhodopsin image from public domain.
Rhodopsin is the visual pigment of rod photoreceptor cells in marine fishes and terrestrial vertebrates and is based on the 11-cis isomer of retinaldehyde. The polyene chain 11-cis retinaldehyde is covalently linked at C15 to the ε-amino N of lysine 296 in the opsin chain. The insertion of retinaldehyde into the binding pocket shifts the absorption band of 11-cis retinaldehyde from the ultraviolet (UV) to the visible blue-green spectrum. In mammals, the cone opsins expressed by the short-wave system 1 (SWS1) gene generate blue-shifted pigments, whereas the long-wave system (LWS) gene generates a green shifted pigment in nonprimates, and both green and red shifted pigments in primates.
Visual opsins are all G-protein coupled receptors (GPCRs) characterized by 7 transmembrane domains (TMDs). The bound 11-cis retinaldehyde acts as an inverse agonist that prevents activation of its cognate G-protein transducin in the dark state.[11,12] In rods and cones, photon capture activates a multiprotein transduction cascade and effects a drop in outer segment cyclic guanosine monophosphate (cGMP), which leads to the closure of cyclic nucleotide-gated cation channels as cGMP dissociates. This depresses an ongoing inward “dark current” and is observed macroscopically as a hyperpolarization of the photoreceptor. Like excitatory neurons in the brain, photoreceptors use glutamic acid as their neurotransmitter[13] via Ca2+-dependent vesicle fusion, releasing high rates of glutamate when they are depolarized (dark) and reducing glutamate release upon hyperpolarization (light).
As an optogenetics tool, rhodopsin is moderately fast and very sensitive, but it requires at least four transduction proteins to couple photon capture to membrane potential, as well as a ready supply of 11-cis retinaldehyde, adenosine triphosphate and guanosine triphosphate to run transduction reactions. Further, rhodopsin opens channels in the dark and closes them in the light, which poses energetic problems in keeping channels closed without depleting these small molecules. It is still possible that the signaling cascade could be re-engineered to exploit rhodopsin's high extinction coefficient and amplification.
Melanopsin is an opsin expressed in a subset of mammalian ganglion cells known as intrinsically photosensitive ganglion cells (ipGCs). Signaling from ipGCs appears to influence circadian phase setting, pupillary constriction at high brightness, and sleep regulation.[14] ipGCs may also trigger photophobia in migraine.[15] ipGCs also have input from rod and cone pathways, and hence the differential role of melanopsin in their full range of functions remains unclear. Melanopsin also uses 11-cis retinaldehyde as a primary chromophore, absorbs blue-green light,[16] and acts as a GPCR to drive cell signaling likely through a Gq/11G-protein.[17] Unlike rhodopsin, photoactivated melanopsin triggers depolarization of ganglion cell transient receptor potential-like cation channels, probably through phospholipase C signaling, although the exact mechanism remains a matter of debate.[18] Thus, melanopsin is appropriate for optogenetics by virtue of its polarity. The drawbacks to using this opsin for neural signaling include its very slow speed and requirement for native transducers. Even so, melanopsin has successfully been exploited to restore light responsivity in rd1 mice,[19] a model of human autosomal recessive RP. Thus ectopic expression of melanopsin in any ganglion cell appears effective in activating light responses, implying that all retinal neurons may have intrinsic access to 11-cis retinaldehyde and possess the essential transduction chain used by this opsin. However, melanopsin triggers extremely sustained, persistent excitations making it ineffective for signaling at speeds necessary for simple tasks like walking.
Halorhodopsin, first described by Matsuno-Yagi and Mukohata in 1977, is a 7 TMD retinaldehyde-binding protein with a λmax of 570 nm that acts as a light-gated anion-selective pump in a group of the Archaea known as halobacteria.[20] It is not a GPCR. A variant from Natronomonas phoraonis (NpHR) has been of particular interest due to its fast reactivity.[21] It has successfully been expressed in mouse retinal ganglion cells[22,23] and mouse and human cones ex vivo.[24] Limits to application of NpHR, include its lack of sensitivity, inability to excite cells and tendency to inactivate in mammalian cells.
Channelrhodopsins (ChRs) are a family of cation-channel forming algal 7 TMD opsins that bind all-trans retinaldehyde via a Schiff's-base linkage, as do vertebrate visual opsins. ChRs absorb blue-green light (λmax 470 nm) and use photoisomerization of all-trans retinaldehyde to the 13-cis bent form to gate nonselective cation channels generating a depolarizing inward current in most cells. The rhodopsin-like functions of ChR were initially discovered in a range of phototactic motile unicellular organisms.[25] And ultimately ChR1 and ChR2 (similar proteins with different activation behaviors) were described in Chlamydomonas reinhardtii[26] and expressed in nonneuronal mammalian cell lines by Nagel et al.[27] Later experiments reported activation of spike potentials in response to blue light stimulation in mammalian neurons expressing ChR2.[28,29] Similarly, in mouse models, control of action potential firing in response to blue light was achieved in hippocampal slices following stereotactic injection of ChR2-encoding viruses.[30] Several successful studies have now exploited ChR2 expression in retinal bipolar cells to drive upstream signaling in mouse models of RP.[31,32] While ChR2 and several engineered variants represent strong proof of principle for optogenetics therapies, they lack sufficient photon capture efficacy to be used under normal lighting conditions.
Finally, a novel fusion of photochemistry and genetics removes the necessity of using opsins altogether and instead exploits naturally expressed, high-conductance ion channels charged with photosensitive azobenzenes. Caporale et al created a modified glutamate receptor LiGluR (based on ionotropic glutamate receptor iGluR6) modified with an exposed cysteine for cross linking to a maleimide-azobenzene-glutamate (MAG) adduct.[33] After expression through adeno-associated virus (AAV) delivery, intravitreal injection of MAG activates a photosensitive glutamate receptor. Photoactivation at 380 nm allowed the glutamate end-link to occupy the binding site in LiGluR and activate the cation channel, whereas photoactivation at 500 nm withdrew the link. Like ChR2, expression of this protein after intravitreal injection of AAV-LiGluR followed MAG linking restored visual responsiveness as assayed by a number of physiological techniques. However, again, the world is not a set of UV-green events, and some modification of this scheme is necessary to render it practical for human vision.
VIRAL DELIVERY
Optogenetic therapy for degenerative retinal diseases hinges on successful targeting of opsin or other genes to surviving retinal cells. Viral vectors are the most widely validated method for opsin delivery to neuronal or other tissue cells, with vector administration via subretinal injection targeting outer retinal cells and intravitreal injection targeting inner retinal cells.[34] Over thirty papers in the past 5 years from the Hauswirth group have demonstrated the efficacy of AAV-based gene delivery. Lentiviral and adenoviral models have been shown to express opsins in mouse, rat, and primate tissues.[35] The advantages to using viral vectors (as opposed to nonbiologic methods such as nanoparticles or gene-gun approaches) include rapid delivery, high infectivity, and potential cell-specificity based on pseudotyped AAV variants with different cell tropisms. The drawback to AAV is that promoter sequences must be relatively small and cell-specific, as they must drive strong protein expression.[36] However, growing understanding of pseudotyped AAV efficacies[37] and coupling a wider-range of cell-specific transcriptional markers to high-efficiency expression systems are rapidly easing these concerns.[38]
A technical approach to overcome some of these challenges and optimize opsin performance in model systems includes viral delivery of opsins into Cre recombinase transgenic mice, allowing tighter control of gene expression in the research setting.[35,36] Indeed, there has been an explosion of studies that prove the efficacy of Cre-driver lines for cell-specific targeting.[39] Advances in viral delivery have allowed for light-induced changes and activation of axons distant to cell bodies which have been virally infected to express opsin proteins. This method of distant activation has been described as projection-based control.[40,41] However, these approaches are not viable clinical interventions, and direct AAV targeting still remains the best proof-of-principle approach.
Delivery of and signaling by opsins in primate models have proven to be more challenging. Lentiviral delivery of ChR2 to cortical neurons of macaques without observed behavioral response has previously been reported,[42,43] but recently Lee et al successfully delivered ChR2 to parvalbulin + inhibitory neurons in mouse V1 cortex and directly modulated behavior, suggesting that primate brain targeting is feasible.[44] Delivery of opsins with confirmed function has also been reported in primate stem cell neurons[45] and ex vivo retina.[24] However, targeting primate retina in vivo remains a challenge for AAV-based methods, as intravitreal delivery is compromised by poor transport through the inner limiting membrane.[46] The cost of scaling viral production to deliver the large particle numbers necessary for the primate eye has also been part of the problem. While recent advances in animal models have proven valuable in understanding the role and function of opsins within neuronal circuits, challenges remain in clinical application of optogenetic therapy in humans.
CHALLENGES IN CLINICAL APPLICATION OF OPTOGENTICS: REMODELING
As noted above, characteristics of the currently identified opsins make their use in clinical settings a challenge. ChRs, halorhodopsins and azobenzene modified targets remain so insensitive that they require extreme, nonphysiological stimulus fluxes, and the dependence of some signaling regimes on UV stimulation is clearly problematic. Melanopsin is similar to invertebrate rhodopsins in chomophore associations and use of Gq/11-protein signaling, but its response is so sustained as to prevent signaling of environmental cues. The use of rhodopsin itself is complicated by the requirement of intrinsic transduction pathways (ON bipolar cells), but new breakthroughs in this system are in the offing. In addition to these challenging properties of opsins, AAV delivery to primates has proven difficult.[47]
These are largely bioengineering challenges that should be rapidly soluble by designing higher sensitivity opsins, improving AAV targeting and designing optimized expression systems. Given this, does anything prevent rapidly deploying optogenetics in human retinal degenerations such a RP or even atrophic age-related macular degeneration? The answer is yes: Retinal remodeling.
Retinal remodeling is by far the most serious barrier to treating RP and other retinal degenerations with optogenetics, prosthetics, cell therapies or late-stage gene therapies.[48,49,50,51,52] RP is a collection of progressive diseases that continue to alter the neural retina long after photoreceptors have died. Every form of RP transitions through three phases that strongly influence the function of the surviving neural retina.[48]
In Phase I, stressed photoreceptor cells rapidly change their synaptic associations with downstream retinal bipolar cells. In Phase II, photoreceptor cell death peaks, leading to the decimation of the outer nuclear layer truncation of bipolar cell dendrites, and the formation of the distal glial seal that completely separates the remnant neural retina from the surviving RP and choroid. This glial seal is formed by remnant Müller cell microvilli cross-linked by homocellular intermediate junctions and is not a glial scar like those formed by GFAP + astrocytes in damaged neuropil of the central nervous system. In cone sparing forms of RP near the macula, surviving rod bipolar cells reprogram their signaling from their classical mGluR6-based ON polarity to an iGluR-based OFF polarity.[50] Phase III is a persistent state of neural and glial remodeling, with progressive cell death, aberrant neuritogenesis, rewiring of all classes of neurons including the formation of pathologic synaptic microneuromas, pathologic neuronal migration of ganglion and amacrine cells to distal retina, and bipolar cell and amacrine cell migration to proximal retina. Further, retinal pigment epithelial cells begin to invade the neural retina, often descending completely to the inner limiting membrane. This generates the appearance of sharply delineated bone spicules. When this presentation is seen in ophthalmoscopy [Figure 2], it means that the retina is already in a state of advanced Phase III remodeling [Figure 3], and optogenetics or any other interventions will be largely ineffective. Figure 3b shows the effect of remodeling in a human retina, including absence of ganglion cells, virtually complete depletion of bipolar cells, and severe reorganization of the remnant neural retina through neuronal migration and glial hypertrophy. Figure 3c demonstrates rod bipolar cell reprogramming in human cone-sparing RP. Importantly, the spared cones often lack opsin expression, but retain some synaptic connectivity. Finally, Phase III Müller cells begin to chaotically reprogram into varied states of altered glutamine metabolism, implying that glial support of the essential glutamate cycle may be compromised.[53]
Figure 2.
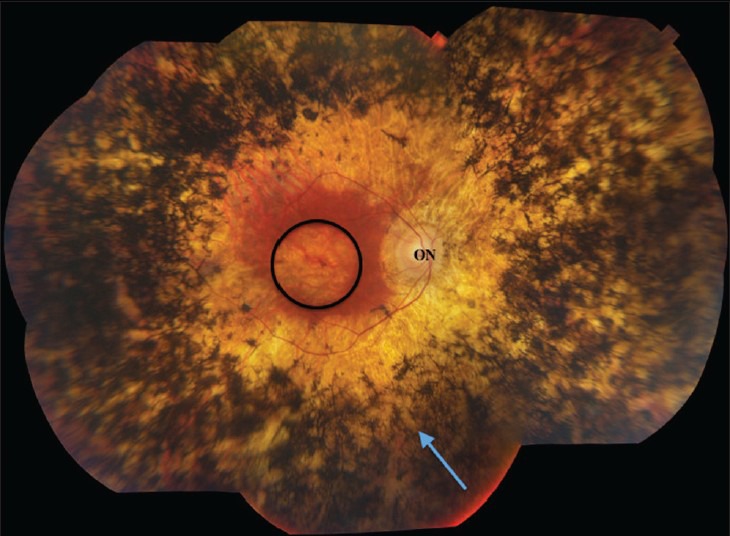
Fundus imaging of advanced human retinitis pigmentosa. Bone spicule pigment from invading retinal pigment epithelial cells (arrow) dominates the retinal periphery. Retinal thinning is extensive, with relative macular preservation. ON, optic nerve head; circle, macula.
Figure 3.
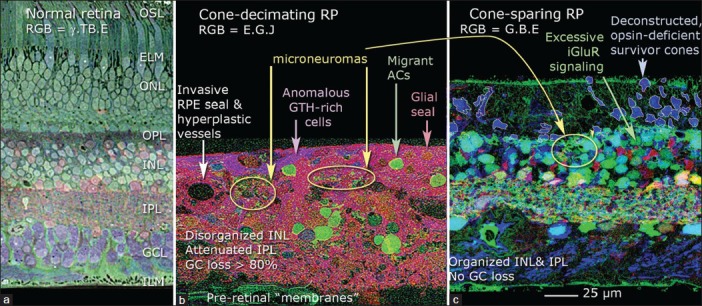
Retinal remodeling in human retinitis pigmentosa (RP). (a) Normal primate retina visualized with computational molecular phenotyping (Marc and Jones, 2002) using RGB = γ. TB.E mapping (gamma-aminobtyric acid; TB, toluidine blue; E, glutamate). OSL, outer segment layer; ELM, external limiting membrane; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer; ILM, inner limiting membrane. (b) Cone-decimated advanced RP (Foundation Fighting Blindness Accession no. 133, 67-year-old female, no light perception, advanced simplex RP, 2.5 hours postmortem). RGB = E.G.J mapping (E, glutamate; G, glycine; J, glutathione). From Marc et al, 2003, by permission of the authors. RPE, retinal pigment epithelium; AC, amacrine cell; GC, ganglion cell. (c) Cone-sparing RP (University of Utah Lions Eye Bank, 21-year-old male, central vision only, 2 hours postmortem). Excitation mapping using 1-amino-4-guanidobutane (AGB) and 25 μM kainate stimulation (Marc and Jones, 2002) reveals aberrant iGluR (ionotropic glutamate receptor) excitation in surviving rod bipolar cells. RGB = G.B.E. mapping (G, glycine; B, AGB; E, glutamate). From Marc et al, 2007, by permission of the authors.
A major defect of virtually all animal research models on optogenetics is the use of early stages in retinal degeneration before significant remodeling begins. For example, Lagali et al show successful ChR2 expression in ON bipolar cells that still have dendrites.[31] This occurs only in Phase I animals. Ultimately, the expression of mGluR6 in ON bipolar cells in rd1 mice is strongly depressed in Phase III,[48] and Phase III interventions based on that promoter may be ineffective. The key question in the rd1 mouse is whether bipolar cell expression of ChR2 can be achieved in animals that are 200 postnatal day (PND) of age or older; or over 300 PND in P23H rhodopsin mutant models; or 600 PND in the RCS rat, as these are faithful mimics of human Phase III RP. No one has yet achieved visual function restoration in a Phase III retina.
Finally, identifying proper cellular targets has also been challenging. Should light sensing genes be inserted into bipolar cells, amacrine cells, or ganglion cells, and if so, which ones? Figure 4 illustrates the global organization of a normal mammalian retina. The flow of signals in the retina can be thought of as a four-layer model with rods, SWS1-cones (blue cones) and LWS cones (red or green cones) as layer 1; bipolar cells as layer 2; amacrine cell networks that aggregate and tune bipolar cell signals as layer 3; and ganglion cells that collect the amacrine cell and bipolar cell signals as layer 4. Finally, Type AII amacrine cells are unique to mammals and form a hub that aggregates and distributes signals for all types of ganglion cells except primate midget pathways. We refer to this AII amacrine cell hub function as mimicking layer “2.5” as it is interspersed between bipolar cells (BCs) and all other neurons. In RP [Figure 5] the photoreceptor layer 1 is ablated. Assuming that all networks in the neural retina remain normal (which is not true), picking a target network is difficult. If we choose to target ON bipolar cells [Figure 5a] through mGluR6 promoter or enhancer sequences, we must successfully target multiple varieties of ON cells because they have the best chance of appropriately driving retinal ganglion cells. The same is true of the AII amacrine cell, except it would be simpler to target if we knew the right promoter, which we do not. Targeting bipolar cells may be difficult in late-stage because they remodel rapidly and die slowly in most retinal degenerations. While ganglion cells survive the longest, along with some gamma-aminobutyric-acid-ergic (GABA-ergic) amacrine cells,[48,49] they require networks to generate appropriate visual signals and drive central pathways with appropriate timing. Thus, the expression of the same opsin in every ganglion cell may create a corrupted visual experience [Figure 5b] even if it restores crude light sensibility. If we can discover transcriptional controls for human foveal midget ganglion cells, it may be possible to target them specifically. Finally, the reality of remodeling is that many target neurons may die, and the remaining networks will become scrambled in ways that may render functional vision via optogenetics unlikely [Figure 6]. It appears that late-stage remodeling is not slowed by interventions such as subretinal transplantation of normal fetal retina into transgenic rats early in life with advanced retinal degenerations.[54] Thus, it is unlikely that even optogenetics signaling will attenuate remodeling, since processes like pathologic de novo neuritogenesis in retinal degenerations depend on retinoic acid signaling rather than network activity.[55]
Figure 4.
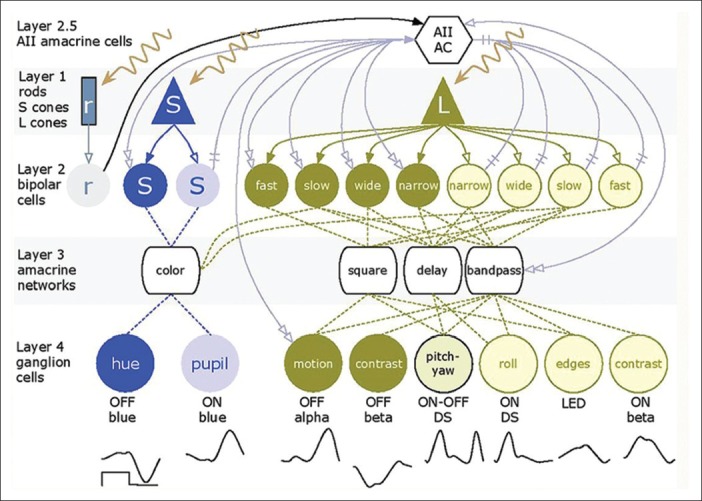
The basic organization of the mammalian retina. Rods (r), short-wave system 1 (S) and long-wave system (L) cones form layer 1 and drive cognate sets of layer 2 ON (light color) and OFF (dark) bipolar cells via sign-conserving (solid single arrows) and sign-inverting (single open arrows) glutamate synapses. Bipolar cells drive a large array (over 30 classes) of specialized layer 3 amacrine cells and these mixed bipolar-amacrine networks drive different layer 4 ganglion cells that transmit different visual information to the brain. The AII amacrine cell (layer 2.5) aggregates bipolar cell inputs into a single hub and distributes that information for both rod and cone vision. The waveforms under layer 4 represent the different analog voltage response profiles of ganglion cells to the onset and offset of a bright long-wave length flash.
Figure 5.
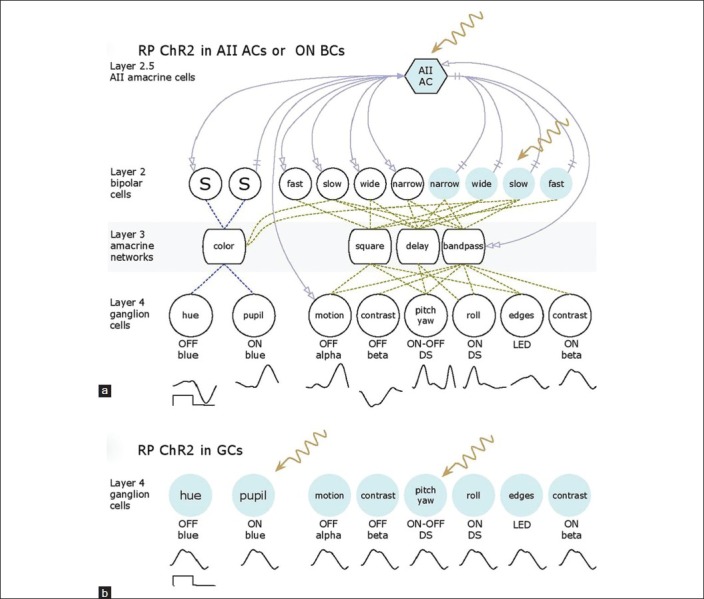
Optogenetics schemes using ChR2 in the retinitis pigmentosa retina after loss of layer 1 photoreceptors. (a) Targeting either ON bipolar cells or AII amacrine cells with ChR2 (color) can potentially drive all ganglion cell classes with correct polarities and waveforms. (b) Targeting ganglion cells generates the same waveform in all ganglion cell classes, which may compromise useful vision.
Figure 6.
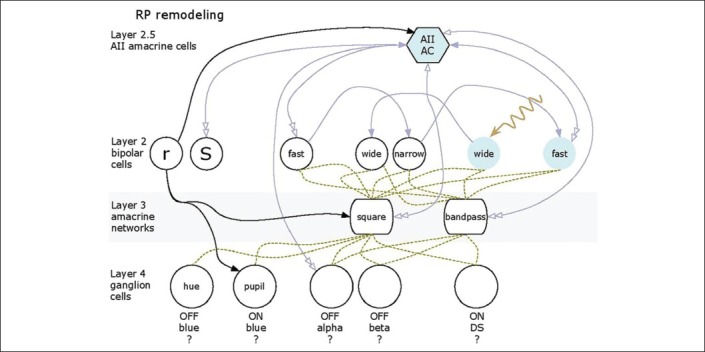
Remodeling severely scrambles networks so that successful ChR2 expression may yield unexpected or fictive responses from surviving retinal cells.
OUTLOOK
The field of optogenetics has grown dramatically from its conception in the 1970s. With the identification of diverse opsins from unicellular organisms, the emergence of largely safe and effective viral vectors for delivery of genetic material and the development of numerous transgenic animal models, optogenetics has grown closer to clinical utility. Independent advances in opsin engineering;[56] viral packing, targeting and expression;[37] and ultimately, molecular control of remodeling[55] are all keys to eventual clinical application of optogenetic therapy for degenerative retinal diseases. It is possible that optogenetics therapy will have to be combined with some form of high-brightness video display to overcome intrinsic insensitivity of existing opsins. Direct comparisons of different cellular targets in large-eyed models of human retinal degenerations[51] and the possible development of nonhuman primate models of RP will also play critical roles in preclinical development of optogenetics. Ultimately, if successful, optogenetics therapies will represent one of the crowning achievements of modern genetics, neurobiology, and photobiology.
ACKNOWLEDGMENTS
This work was supported by an unrestricted grant to the Moran Eye Center, University of Utah School of Medicine by Research to Prevent Blindness.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
REFERENCES
- 1.Yizhar O, Fenno LE, Davidson TJ, Mogri M, Deisseroth K. Optogenetics in neural systems. Neuron. 2011;71:9–34. doi: 10.1016/j.neuron.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 2.Deisseroth K, Feng G, Majewska AK, Miesenböck G, Ting A, Schnitzer MJ. Next-generation optical technologies for illuminating genetically targeted brain circuits. J Neurosci. 2006;26:10380–10386. doi: 10.1523/JNEUROSCI.3863-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crick FH. Thinking about the brain. Sci Am. 1979;241:219–232. doi: 10.1038/scientificamerican0979-219. [DOI] [PubMed] [Google Scholar]
- 4.Oesterhelt D, Stoeckenius W. Rhodopsin-like protein from the purple membrane of Halobacterium halobium. Nat New Biol. 1971;233:149–152. doi: 10.1038/newbio233149a0. [DOI] [PubMed] [Google Scholar]
- 5.Oesterhelt D, Stoeckenius W. Functions of a new photoreceptor membrane. Proc Natl Acad Sci U S A. 1973;70:2853–2857. doi: 10.1073/pnas.70.10.2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zemelman BV, Lee GA, Ng M, Miesenböck G. Selective photostimulation of genetically chARGed neurons. Neuron. 2002;33:15–22. doi: 10.1016/s0896-6273(01)00574-8. [DOI] [PubMed] [Google Scholar]
- 7.Reiner A, Isacoff EY. The Brain Prize 2013: The optogenetics revolution. Trends Neurosci. 2013;36:557–560. doi: 10.1016/j.tins.2013.08.005. [DOI] [PubMed] [Google Scholar]
- 8.Sakmar TP. Structure of rhodopsin and the superfamily of seven-helical receptors: The same and not the same. Curr Opin Cell Biol. 2002;14:189–195. doi: 10.1016/s0955-0674(02)00306-x. [DOI] [PubMed] [Google Scholar]
- 9.Man D, Wang W, Sabehi G, Aravind L, Post AF, Massana R, et al. Diversification and spectral tuning in marine proteorhodopsins. EMBO J. 2003;22:1725–1731. doi: 10.1093/emboj/cdg183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lamb TD. Evolution of phototransduction, vertebrate photoreceptors and retina. Prog Retin Eye Res. 2013;36:52–119. doi: 10.1016/j.preteyeres.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 11.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 12.Frederiksen R, Boyer NP, Nickle B, Chakrabarti KS, Koutalos Y, Crouch RK, et al. Low aqueous solubility of 11-cis-retinal limits the rate of pigment formation and dark adaptation in salamander rods. J Gen Physiol. 2012;139:493–505. doi: 10.1085/jgp.201110685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marc RE. Retinal neurotransmitters. In: Chalupa LM, Werner JS, editors. The Visual Neurosciences. Cambridge, Massachusetts: MIT Press; 2004. pp. 315–330. [Google Scholar]
- 14.Benarroch EE. The melanopsin system: Phototransduction, projections, functions, and clinical implications. Neurology. 2011;76:1422–1427. doi: 10.1212/WNL.0b013e31821671a5. [DOI] [PubMed] [Google Scholar]
- 15.Noseda R, Burstein R. Advances in understanding the mechanisms of migraine-type photophobia. Curr Opin Neurol. 2011;24:197–202. doi: 10.1097/WCO.0b013e3283466c8e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hattar S, Lucas RJ, Mrosovsky N, Thompson S, Douglas RH, Hankins MW, et al. Melanopsin and rod-cone photoreceptive systems account for all major accessory visual functions in mice. Nature. 2003;424:76–81. doi: 10.1038/nature01761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bailes HJ, Lucas RJ. Human melanopsin forms a pigment maximally sensitive to blue light (λmax ≈ 479 nm) supporting activation of G (q/11) and G (i/o) signalling cascades. Proc Biol Sci. 2013;280:20122987. doi: 10.1098/rspb.2012.2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Numata T, Kiyonaka S, Kato K, Takahashi N, Mori Y. Activation of TRP channels in mammalian systems. In: Zhu MX, editor. TRP Channels. Boca Raton, FL: CRC Press; 2011. [PubMed] [Google Scholar]
- 19.Lin B, Koizumi A, Tanaka N, Panda S, Masland RH. Restoration of visual function in retinal degeneration mice by ectopic expression of melanopsin. Proc Natl Acad Sci U S A. 2008;105:16009–16014. doi: 10.1073/pnas.0806114105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsuno-Yagi A, Mukohata Y. Two possible roles of bacteriorhodopsin; a comparative study of strains of Halobacterium halobium differing in pigmentation. Biochem Biophys Res Commun. 1977;78:237–243. doi: 10.1016/0006-291x(77)91245-1. [DOI] [PubMed] [Google Scholar]
- 21.Kolbe M, Besir H, Essen LO, Oesterhelt D. Structure of the light-driven chloride pump halorhodopsin at 1.8 a resolution. Science. 2000;288:1390–1396. doi: 10.1126/science.288.5470.1390. [DOI] [PubMed] [Google Scholar]
- 22.Greenberg KP, Pham A, Werblin FS. Differential targeting of optical neuromodulators to ganglion cell soma and dendrites allows dynamic control of center-surround antagonism. Neuron. 2011;69:713–720. doi: 10.1016/j.neuron.2011.01.024. [DOI] [PubMed] [Google Scholar]
- 23.Wu C, Ivanova E, Zhang Y, Pan ZH. rAAV-mediated subcellular targeting of optogenetic tools in retinal ganglion cells in vivo. PLoS One. 2013;8:e66332. doi: 10.1371/journal.pone.0066332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Busskamp V, Duebel J, Balya D, Fradot M, Viney TJ, Siegert S, et al. Genetic reactivation of cone photoreceptors restores visual responses in retinitis pigmentosa. Science. 2010;329:413–417. doi: 10.1126/science.1190897. [DOI] [PubMed] [Google Scholar]
- 25.Foster KW, Smyth RD. Light antennas in phototactic algae. Microbiol Rev. 1980;44:572–630. doi: 10.1128/mr.44.4.572-630.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagel G, Ollig D, Fuhrmann M, Kateriya S, Musti AM, Bamberg E, et al. Channelrhodopsin-1: A light-gated proton channel in green algae. Science. 2002;296:2395–2398. doi: 10.1126/science.1072068. [DOI] [PubMed] [Google Scholar]
- 27.Nagel G, Szellas T, Huhn W, Kateriya S, Adeishvili N, Berthold P, et al. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc Natl Acad Sci U S A. 2003;100:13940–13945. doi: 10.1073/pnas.1936192100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci. 2005;8:1263–1268. doi: 10.1038/nn1525. [DOI] [PubMed] [Google Scholar]
- 29.Li X, Gutierrez DV, Hanson MG, Han J, Mark MD, Chiel H, et al. Fast noninvasive activation and inhibition of neural and network activity by vertebrate rhodopsin and green algae channelrhodopsin. Proc Natl Acad Sci U S A. 2005;102:17816–17821. doi: 10.1073/pnas.0509030102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishizuka T, Kakuda M, Araki R, Yawo H. Kinetic evaluation of photosensitivity in genetically engineered neurons expressing green algae light-gated channels. Neurosci Res. 2006;54:85–94. doi: 10.1016/j.neures.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 31.Lagali PS, Balya D, Awatramani GB, Münch TA, Kim DS, Busskamp V, et al. Light-activated channels targeted to ON bipolar cells restore visual function in retinal degeneration. Nat Neurosci. 2008;11:667–675. doi: 10.1038/nn.2117. [DOI] [PubMed] [Google Scholar]
- 32.Doroudchi MM, Greenberg KP, Liu J, Silka KA, Boyden ES, Lockridge JA, et al. Virally delivered channelrhodopsin-2 safely and effectively restores visual function in multiple mouse models of blindness. Mol Ther. 2011;19:1220–1229. doi: 10.1038/mt.2011.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caporale N, Kolstad KD, Lee T, Tochitsky I, Dalkara D, Trauner D, et al. LiGluR restores visual responses in rodent models of inherited blindness. Mol Ther. 2011;19:1212–1219. doi: 10.1038/mt.2011.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Francis PJ, Mansfield B, Rose S. Proceedings of the First International Optogenetic Therapies for Vision Symposium. Transl Vis Sci Technol. 2013;2:4. doi: 10.1167/tvst.2.7.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang F, Gradinaru V, Adamantidis AR, Durand R, Airan RD, de Lecea L, et al. Optogenetic interrogation of neural circuits: Technology for probing mammalian brain structures. Nat Protoc. 2010;5:439–456. doi: 10.1038/nprot.2009.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fenno L, Yizhar O, Deisseroth K. The development and application of optogenetics. Annu Rev Neurosci. 2011;34:389–412. doi: 10.1146/annurev-neuro-061010-113817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Petrs-Silva H, Dinculescu A, Li Q, Min SH, Chiodo V, Pang JJ, et al. High-efficiency transduction of the mouse retina by tyrosine-mutant AAV serotype vectors. Mol Ther. 2009;17:463–471. doi: 10.1038/mt.2008.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kay JN, De la Huerta I, Kim IJ, Zhang Y, Yamagata M, Chu MW, et al. Retinal ganglion cells with distinct directional preferences differ in molecular identity, structure, and central projections. J Neurosci. 2011;31:7753–7762. doi: 10.1523/JNEUROSCI.0907-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuhlman SJ, Huang ZJ. High-resolution labeling and functional manipulation of specific neuron types in mouse brain by cre-activated viral gene expression. PLoS One. 2008;3:e2005. doi: 10.1371/journal.pone.0002005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petreanu L, Huber D, Sobczyk A, Svoboda K. Channelrhodopsin-2-assisted circuit mapping of long-range callosal projections. Nat Neurosci. 2007;10:663–668. doi: 10.1038/nn1891. [DOI] [PubMed] [Google Scholar]
- 41.Gradinaru V, Thompson KR, Zhang F, Mogri M, Kay K, Schneider MB, et al. Targeting and readout strategies for fast optical neural control in vitro and in vivo. J Neurosci. 2007;27:14231–14238. doi: 10.1523/JNEUROSCI.3578-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Han X, Qian X, Bernstein JG, Zhou HH, Franzesi GT, Stern P, et al. Millisecond-timescale optical control of neural dynamics in the nonhuman primate brain. Neuron. 2009;62:191–198. doi: 10.1016/j.neuron.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Diester I, Kaufman MT, Mogri M, Pashaie R, Goo W, Yizhar O, et al. An optogenetic toolbox designed for primates. Nat Neurosci. 2011;14:387–397. doi: 10.1038/nn.2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee SH, Kwan AC, Zhang S, Phoumthipphavong V, Flannery JG, Masmanidis SC, et al. Activation of specific interneurons improves V1 feature selectivity and visual perception. Nature. 2012;488:379–383. doi: 10.1038/nature11312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weick JP, Johnson MA, Skroch SP, Williams JC, Deisseroth K, Zhang SC. Functional control of transplantable human ESC-derived neurons via optogenetic targeting. Stem Cells. 2010;28:2008–2016. doi: 10.1002/stem.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dalkara D, Kolstad KD, Caporale N, Visel M, Klimczak RR, Schaffer DV, et al. Inner limiting membrane barriers to AAV-mediated retinal transduction from the vitreous. Mol Ther. 2009;17:2096–2102. doi: 10.1038/mt.2009.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gradinaru V, Zhang F, Ramakrishnan C, Mattis J, Prakash R, Diester I, et al. Molecular and cellular approaches for diversifying and extending optogenetics. Cell. 2010;141:154–165. doi: 10.1016/j.cell.2010.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marc RE, Jones BW, Watt CB, Strettoi E. Neural remodeling in retinal degeneration. Prog Retin Eye Res. 2003;22:607–655. doi: 10.1016/s1350-9462(03)00039-9. [DOI] [PubMed] [Google Scholar]
- 49.Jones BW, Watt CB, Frederick JM, Baehr W, Chen CK, Levine EM, et al. Retinal remodeling triggered by photoreceptor degenerations. J Comp Neurol. 2003;464:1–16. doi: 10.1002/cne.10703. [DOI] [PubMed] [Google Scholar]
- 50.Marc RE, Jones BW, Anderson JR, Kinard K, Marshak DW, Wilson JH, et al. Neural reprogramming in retinal degeneration. Invest Ophthalmol Vis Sci. 2007;48:3364–3371. doi: 10.1167/iovs.07-0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jones BW, Kondo M, Terasaki H, Watt CB, Rapp K, Anderson J, et al. Retinal remodeling in the Tg P347L rabbit, a large-eye model of retinal degeneration. J Comp Neurol. 2011;519:2713–2733. doi: 10.1002/cne.22703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jones BW, Kondo M, Terasaki H, Lin Y, McCall M, Marc RE. Retinal remodeling. Jpn J Ophthalmol. 2012;56:289–306. doi: 10.1007/s10384-012-0147-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marc RE, Jones BW, Watt CB, Vazquez-Chona F, Vaughan DK, Organisciak DT. Extreme retinal remodeling triggered by light damage: Implications for age related macular degeneration. Mol Vis. 2008;14:782–806. [PMC free article] [PubMed] [Google Scholar]
- 54.Seiler MJ, Jones BW, Aramant RB, Yang PB, Keirstead HS, Marc RE. Computational molecular phenotyping of retinal sheet transplants to rats with retinal degeneration. Eur J Neurosci. 2012;35:1692–1704. doi: 10.1111/j.1460-9568.2012.08078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin Y, Jones BW, Liu A, Tucker JF, Rapp K, Luo L, et al. Retinoid receptors trigger neuritogenesis in retinal degenerations. FASEB J. 2012;26:81–92. doi: 10.1096/fj.11-192914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang F, Prigge M, Beyrière F, Tsunoda SP, Mattis J, Yizhar O, et al. Red-shifted optogenetic excitation: A tool for fast neural control derived from Volvox carteri. Nat Neurosci. 2008;11:631–633. doi: 10.1038/nn.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]