

SUMMARY
High-grade gliomas (HGGs) are extremely lethal tumors. Survival has not changed significantly in the past decades. The only known prognostic factors in pediatric HGGs (pHGGs) are extent of resection and histologic grade. Treatment has historically been based on adult trials because of the rarity of pHGGs and the lack of genomic tools to explore their unique molecular characteristics. The recent advances in molecular biological data helped divide these tumors into distinct subgroups. In this review, the authors focus on major molecular targets of alterations in pHGGs: histone H3.3, telomeres, PDGFRA, IDH, BRAF V600E, ACVR1 and NTRK and briefly highlight the difference with the adult counterpart.
KEYWORDS : epigenetics, high-grade glioma, histone, molecular biology, pediatric
Practice points.
Pediatric high-grade gliomas (pHGGs) are rare and highly lethal.
Treatment options for pHGGs are limited and largely ineffective.
Diagnosis of pHGGs is based mostly on histology.
pHGGs and adult high-grade gliomas are histologically similar.
At the molecular level, pHGGs and adult high-grade gliomas are distinct entities.
Genetic and epigenetic alterations, such as DNA methylation and histone modifications, play important roles in pHGGs.
Effective and feasible diagnostic tools based on genetic and epigenetic modifications are currently under investigation.
Future classification and treatment of pediatric high-grade gliomas (pHGGs) must be based on both histology and molecular characteristics of the tumors.
High-grade gliomas (HGGs), including glioblastomas (GBMs), anaplastic astrocytomas and diffuse intrinsic pontine gliomas (DIPGs), are the most devastating and lethal brain tumors in adults and children [1,2]. HGG comprise 8–12% of all pediatric CNS tumors [3,4], with the incidence of HGGs in children up to 19 years of age being approximately 0.85 per 100,000 [5]. After the age of 19 years, the incidence of HGGs increases significantly. On average, 7.2 per 100,000 are affected, and the number increases to 14.6 per 100,000 between the ages of 75 and 84 years [6].
Unfortunately, the survival of children with HGGs has not changed significantly in the past decades despite improvements in the fields of chemotherapy, radiation therapy delivery and neurosurgical techniques that enhance gross total resection. The 2-year survival rates for children with supratentorial HGGs are 10–30% [7,8], and those for children with DIPGs are less than 10% [7,9,10]. Furthermore, treating young children with HGGs is a special challenge because of the irreversible effect of radiation therapy on neurocognition and quality of life [11]. The only known prognostic factors that significantly affect event-free and overall survival rates of children with HGGs are the extent of tumor resection and histologic grade. More than 90% of tumor resection and anaplastic astrocytoma subtype confer relatively better prognosis [3,12–15]. Unfortunately, achieving maximum tumor resection is difficult in most cases because of high tumor vascularity, invasiveness and tumor location. Tumors invading vital structures, tumors located in the midline and tumors involving brainstem are not amenable to complete resection [13]. In the pediatric population, robust correlation of molecular markers with the prognosis in HGGs has not been established as opposed to adult HGGs (aHGGs). In the adult population, molecular markers such as MGMT status, are starting to be a part of prognosis of HGGs. Adult patients with MGMT overexpression were shown to confer better prognosis when treated concomitantly with the alkylating agent temozolomide and radiation [16], this correlation failed to be replicated in pHGGs [12].
The terminology, classification and treatment of pHGGs have traditionally been based on data from adult studies due to the rarity of these tumors in the pediatric population. This explains the fact that only few large randomized clinical trials have been conducted on pHGGs [13,15]. This approach has proven ineffective, as evidenced by the disappointing results of earlier studies on pHGGs (CCG 943, CCG 945 and ACNS0126) [12,14].
In an effort to decipher the molecular pathogenesis and broaden our knowledge about the underlying genetic abnormalities of HGGs and consequently improve treatment approaches and outcome, HGGs were the first cancer type to be studied by the Cancer Genome Atlas Research Network. Initial studies included adult samples only [17,18]. Assumptions were made that the results can be extrapolated to pHGGs. However, the growing body of molecular and genetic information in recent years has demonstrated that pediatric and aHGGs are distinct entities that only marginally overlap [6,19–21]. HGGs have been subclassified across all age spectrums into six subgroups on the basis of characteristic DNA methylation patterns, gene-expression profiles and mutation profiles: IDH, K27, G34, RTKI (PDGFRA amplification/overexpression), RTK II (classic); and mesenchymal (very few copy number alterations [CNAs] or point mutations) [20]. Age- and location-specific groups have been identified by using this classification system [20,22]. aHGGs and pHGGs harbored few overlapping genetic alterations; however, in large part, most of the mutations were specific for a separate age group. aHGGs mainly harbored alterations in EGFR, PTEN, CDKN2A and CDKN2B, TERT, IDH1 and FGFR genes enriched mainly in the classical (RTKII), mesenchymal and proneural subtypes [6,18,23]. On the other hand, pHGGs mostly had mutations in PDGFRA, H3F3A, HISTH3B/C, ATRX, SETD2 and ACVR1 genes. RTKI, K27, G34 and proneural subtypes enrichment were associated with pHGGs [6,8,20]. Additionally, CNAs were distinct between adult and pHGGs. pHGGs showed frequent gain of chromosome 1 (19–29 vs 9%), and lower frequency of chromosome 7 gain (13 vs 74%) [6,8]. The difference in the biology, cell signaling and consequential response to treatment in adults and children requires more detailed and individualized approach to these patients in order to provide the best diagnostic and treatment strategies [20,22].
This review provides an insight into the common molecular, genetic and epigenetic alterations in pHGGs and their promising role in the diagnosis and treatment of these tumors, in addition to highlighting the major differences between adult and pediatric patients with HGGs.
Role of genetics & epigenetics in pHGGs
Epigenetics is the study of changes in gene expression occurring without corresponding alterations in the DNA sequence and has been studied extensively in the past 15 years [24,25]. It also involves the analysis of stable long-term alterations in the transcriptional potential of cells that are not necessarily heritable. Thus, epigenetic regulation is essential for the regulation of all DNA-based processes, including transcription, DNA repair, replication and the development and differentiation of cells; consequently, its disruption plays a role in the development of cancer in humans [24]. Two important mechanisms of epigenetic regulation are DNA methylation and histone modifications (Figure 1) [25].
Figure 1. . Role of epigenetic changes in gene expression.

(A) Addition or removal of methyl groups by methyltransferases leads to hyper- or hypo-methylation of DNA, leading to gene repression or activation, respectively. (B) Chromosome segment composed of compact DNA wrapped around octamers of core histones (nucleosomes) in which the DNA material is inaccessible and gene functions can not be modified. (C) Epigenetic modifications in histone tails lead to loosening of DNA, rendering genes accessible for modifications of gene activity (i.e., activation and repression). (D) Common post-translational modifications occurring in the histone H3.3 tail that regulate gene transcription. (E) Acetylation at specific amino acid residues leads to transcription activation, whereas methylation leads to transcription inactivation.
Reproduced with permission from [26] © American Association for Cancer Research.
• DNA methylation
DNA methylation is a postreplication process catalyzed by DNA methyltransferases in which a methyl group is added to the DNA. In mammals, DNA methylation occurs almost exclusively in CpG sites, converting cytosine to methyl-cytosine [25,27]. Alterations in DNA methylation occur in many human diseases: in cancers, these changes mainly occur through the hypermethylation of promoter regions of tumor suppressor genes, leading to their inactivation [25,27–29]. However, DNA hypomethylation has recently been found to play a major role in driving HGGs via transcriptional activation of oncogenes [30].
• Histone modifications
Histones are key components in chromosomes that play both structural and functional roles. They wrap the long strands of DNA around an octamer composed of the core histones (H2A, H2B, H3 and H4) and group these DNA-wrapped octamers into structural units called nucleosomes to assemble the highly compacted chromatin complex, a structural state that affects both DNA replication and gene expression (Figure 1B) [31,32]. Histone H3.3 is a variant of histone H3 that can act either as part of the core nucleosome (classical or canonical form) that functions mainly in the S-phase of the cell cycle or as part of the nonclassical (noncanonical) form that has a wider range of roles in different processes, including replacement of displaced nucleosomes, DNA repair, meiotic recombination, chromosome segregation and transcription initiation and termination [33]. This noncanonical form is deposited in transcriptionally active regions of the DNA regardless of the phase of the cell cycle [33,34]. Histone H3.3 can also affect telomere lengthening, a major contributor to oncogenesis [35], as it is incorporated into pericentromeric and telomeric regions [34]. This process is chaperoned by ATRX and the death-associated protein complex (DAXX) [34,36,37].
Until recently, histone genes were thought to be highly preserved in eukaryotes, and no specific disease entities were associated with mutations in histone genes [2,36]. However, recent studies have reported recurrent somatic mutations in regulatory genes H3F3A (which encodes histone H3.3), and HIST1H3B and HIST1H3C (which encode histone H3.1) in DIPGs, thereby shedding light on the biology of gliomagenesis in children and young adults with HGGs [2,6,17,20,30,34,36–38]. Gain-of-function mutations in H3F3A lead to single-codon changes within histone H3.3 at the N-terminal site of the histone tail, which is enriched in residues that are targets for post-translational modifications. These alterations affect not only the transcriptional state of cells but also all the processes that depend on DNA, such as replication, repair, regulation of gene expression, and maintenance of telomeres and centromeres [2,6,30,34,36].
Two unique changes occur at the histone tail of histone H3.3 (and, less frequently, H3.1) in 33–50% of pHGGs [30,34,36,37], and although tumors that harbor these mutations are histologically indistinguishable, they have different methylation patterns [30,39]. First, the substitution of methionine at lysine 27 in the encoded protein H3.3K27M leads to global DNA hypomethylation and a dramatic decrease in the repressive histone mark trimethylated H3K27 (H3K27me3). This modification is mediated via the inactivation of the histone methyltransferase EZH2 (part of PRC2), leading to the transcriptional de-repression of certain genes that may be oncogenic in nature (Figure 2A) [2,6,34,36]. Second, substitution of arginine or valine at glycine 34 (G34R/V) promotes gliomagenesis via a more complicated mechanism that involves decreased K36me3 on the nearby nucleosomes and eventual upregulation of the MYCN oncogene (Figure 2B) [20,34,36,38]. Additionally, a recent study reported loss-of-function mutations in SETD2, an H3K36 methyltransferase, in 15% of pHGGs [40], and these mutations were mutually exclusive from G34R/V mutations [6]. SETD2 mutations partially overlapped with IDH1 mutations and were not identified in DIPGs [21]. SETD2 mutations are hypothesized to play a role in gliomagenesis via depletion of H36me3 (Figure 2C) [6,40].
Figure 2. . Schematic presentation of mutations in H3F3A gene leading to gliomagenesis.

(A) substitution of methionine at lysine 27 in the histone tail leads to a decrease in the repressive histone mark H3K27me3 via the inactivation of the histone methyltransferase EZH2 (part of the PRC2), leading to the transcriptional activation of oncogenes. (B) substitution of arginine or valine at glycine 34 leads to a decrease in K36me3 on the same or nearby nucleosomes and upregulation of the MYCN oncogene. (C) SETD2 (H3K36 methyltransferase), hypothesized to play a role in gliomagenesis via depletion of H3K36 and consequent upregulation of the MYCN oncogene. SETD2 mutations can cause temozolomide resistance and MSI, leading to defective MMR.
MMR: Mismatch repair; MSI: Microsatellite instability.
• Alternate telomere lengthening
Telomeres are regions of repetitive nucleotides (TTAGGG) at the end of chromatids. They are essential in multiple biologic functions including cell stability and replication. They guard the chromosome endings from recombination and fusion that can lead to genetic instability [41]. Normally, in somatic cells, they facilitate the organization of chromosomes within the nucleus, gene expression and, consequently, control the replicative capacity of human cells. Telomeres become shortened with each cell division cycle, and eventually this leads to arrest of cell division and senescence. This shortening occurs via p53-dependent or -independent mechanisms. Telomerase, a reverse trascriptase protein encoded by the TERT gene, replenishes the shortened replication-dependent telomeres by adding nucleotides (telomere lengthening) [41–43]. Cells that acquire alternative mechanisms of telomere lengthening independently from telomerase (alternative lengthening of telomeres [ALT]) become immortal, genetically instable and, eventually, cancerous. This mechanism occurs in approximately 5–10% of human cancers, including HGGs [43].
H3.3-ATRX/DAXX-TP53 pathway
The tumor suppressor gene TP53 encodes the p53 protein and plays a major role in regulating the cell cycle and preventing the proliferation of genetically abnormal cells. Mutations in p53 are pivotal for gliomagenesis, cell survival, and invasion. Mutations in TP53 gene are mostly missense mutations that lead to overexpression of the p53 protein in the cells, which can be easily measured to verify the TP53 status. However, tumors with nonsense TP53 mutations lack p53 overexpression, which limits the use of p53 as a sensor of these mutations [44]. The frequency of TP53 mutation is directly proportional to age [7,11]. In a recent analysis of the long-term survival of children with HGGs treated on the CCG-945 Phase III trial, TP53 mutations were found at significantly lower rates in children younger than 3 years at diagnosis than in children aged 3–6 years at diagnosis (28 vs 70%) [11]. This finding might explain why patients younger than 3 years have better treatment outcomes despite the fact that the majority of these children do not receive radiation therapy [7,11]. Moreover, TP53 mutations may play a role in ALT [42], as discussed above. Concurrent mutations in the H3.3-ATRX/DAXX pathway occur in a third of pHGGs and in all tumors harboring the G34R/V mutation. In addition, somatic TP53 mutations occur in approximately half of all human cancers and in most tumors having mutations in H3F3A, ATRX or both [6,20,34,36,45]. Disruption of the H3.3-ATRX/DAXX chromatin-remodeling pathway and TP53 can lead to ALT and genomic instability [21,34,36,42].
PDGFRA (RTK I)
PDGF stimulates growth, migration and survival of several cell types through its receptors (PDGFRs). Focal amplification and overstimulation of these receptors can cause tumorigenesis [23,46]. Dysregulation of the PDGFR pathway occurs in approximately 14% of pHGGs and 36–50% of DIPGs [6,8,10]. However, the prevalence of PDGFRA amplification in DIPGs may be overestimated because most of the studies used autopsy samples of patients pretreated with radiation. However, PDGFRA amplification was detected in a number of pretreatment samples [20]. Strong overexpression of PDGFRA receptors occurs even in tumors lacking the PDGFRA amplification [47], highlighting the significance of this pathway in the development of pHGGs.
IDH
IDH is a mitochondrial enzyme that plays a central role in energy production and metabolism of cells [21]. Mutations in IDH1 and IDH2 are gain-of-function mutations that most commonly result in amino acid substitutions at arginine 132 (R132) [30]. Theses substitutions lead to accumulation of the oncometabolite 2-hydroxyglutarate (2-HG) and disruption of the methylome of the mutated GBM by extensive DNA hypermethylation at several loci, which results in the glioma CpG island methylator phenotype [6,48]. Methylation of K27 and K36 is also disrupted by elevated levels of the onco-metabolite 2-HG [20]. Mutations in IDH1 overlap with those in ATRX, TP53 and SETD2, suggesting that these pathways might have a complex and coordinated mechanism of action [20,40,45,49]. IDH mutations probably play an important role in malignant transformation of gliomas, as they are mainly found in grade 2 gliomas and secondary HGGs and only rarely in primary HGG in adults, suggesting a distinct pathway of tumorigenesis of primary HGGs [50–52]. In the pediatric population, IDH mutations are usually encountered in adolescents and young adults (>14 years of age) [6,51].
BRAF V600E
The serine/threonine kinase BRAF is a member of the RAS/RAF/MEK/ERK kinase pathway, which plays a key role in cellular division, survival and metabolism [53]. A missense mutation at amino acid position 600 of BRAF (BRAF V600E) leads to overactivation of the gene and, consequently, uncontrolled cell proliferation. BRAF V600E was initially associated with melanoma and medullary thyroid carcinoma but later found in several types of low- and high-grade glial brain tumors [53–55]. Among the low-grade gliomas in children, BRAF V600E was most frequently detected in pleomorphic xanthoastrocytomas (66%), followed by gangliogliomas (18%) and pilocytic astrocytoma (9%) [55]. This mutation was also found in a small subset of HGGs [55,56], and a recent study showed high occurrence of this mutation specifically in the epithelioid subtype of GBMs [54].
ACVR1 mutation
The ACVR1 gene (also known as ALK2), encodes a protein that is a member of the bone morphogenetic protein type I receptor family [57]. Mutations in ACVR1 were initially described in patients with fibrodysplasia ossificans progressiva [22,57–59], who are not predisposed to cancer, suggesting that the ACVR1 mutation is not a driver mutation and requires additional mutations to cause gliomagenesis [60]. However, the ACVR1 receptor plays an important role in development of the mouse embryo and in early left–right patterning [58]. Furthermore, gain-of-function mutations in ACVR1 encode several amino acid substitutions that activate the ACVR1 pathway and lead to increased levels of the phosphorylated proteins SMAD1, SMAD5 and SMAD8, which consequently cause overexpression of the downstream activin signaling target proteins ID1, ID2 and ID3 [22,58,59]. Interestingly, reports of recent studies document ACVR1 mutations in 20–22% of DIPGs [22,58,59], and these tumors also harbored H3F3A mutations, mostly H3.1 [58,59].
NTRK fusion
Neurotrophins are a family of proteins that are important for neuronal cell survival, differentiation, and death and exert their effects through the neurotrophic tyrosine receptor kinase (NTRK) family [61,62]. Fusions in NTRK genes occur in papillary thyroid carcinomas, pediatric low-grade gliomas and aHGGs [62,63]. A recent study identified five fusions in NTRK1, NTRK2 and NTRK3 in 4% of DIPGs and 10% of pHGGs [60]: two of these (TPM3-NTRK1 and ETV6-NTRK3) were oncogenic in different cancer types [60,64–67]. A striking finding in this study was the presence of these fusions in 40% of nonbrainstem pHGGs in children younger than 3 years of age [60].
Chromosomal alterations
HGGs in children are characterized by an incidence of chromosomal aberrations lower than that in adults, and approximately 15% of pHGGs lack any aberrations [6,8,20,68,69]. The most frequent chromosomal imbalances found in children are gain of chromosome 1q (20%), loss of 16q (18%), and loss of 4q (15%) [69]. Amplifications of MYC or MYCN were reported to occur in 8% of children with HGGs [20,68,69]. Tumors with mutations in the H3F3A-ATRX/DAXX-TP53 pathway have high CNAs [36]. One study reported that pHGGs with few or no CNAs were associated with a better prognosis [68].
Secondary glioblastoma multiforme in children
In contrast to the situation in adults, HGGs in children rarely arise from lower-grade tumors. Data on the molecular characteristics of secondary HGGs in children are scarce. However, a study of 11 secondary HGGs in patients with a median age of 13.6 years revealed molecular signatures similar to those seen in adult primary and secondary GBMs, including overexpression of TP53, deletion of RB1 and/or CDKN2A, and abnormalities in the PTEN pathway [70].
From the laboratory to the clinic: connecting the dots
Some alterations in the genetic and epigenetic machinery of pHGGs correlate well with clinical parameters such as tumor location and patient age [6,20,30,34,39] as shown in Table 1. K27M tumors confer poorer prognosis than do G34R/V tumors [6,20,30]. As discussed earlier, IDH mutations in adults are almost exclusively encountered in secondary GBM that serves as a strong diagnostic marker. In children, secondary GBM is rare. Similar to adults, pHGGs with IDH1 mutations have a better clinical outcome than do those with IDH wild-type [50,51]. Pollack et al. showed in their study one-year overall survival of 100% in patients with mutations versus 81% in those without mutations [51]. One study found a correlation between SETD2 mutations and defective DNA mismatch repair through microsatellite instability, which might confer temozolomide-resistance to these tumors (Figure 2C) [71,72]. The median age of patients having tumors with PDGFRA alterations is 11.4 years [6]. PDGFRA amplification is more commonly observed in postmortem samples which might be related to the exposure of the tumor to radiation therapy [6,37]. Clinical significance of PDGFRA amplification in pHGG is controversial, one study reported improved survival in these patients [73]. While another study did not show any prognostic significance of PDGFRA amplification in pediatric patients, it showed an adverse outcome of this amplification in IDH mutant primary GBM [74]. BRAF V600E point mutations can be restricted to the cerebral hemispheres in young patients [6,75]. The recently reported mutations in ACVR1 were mostly identified in pretreated samples [6,22,58,59]. This highlights the possible important role of this alteration in DIPGs. A study reported that children with HGGs harboring ACVR1 mutations were predominantly girls and had a longer overall survival than those without such mutations [59]. However, because of the rarity of pHGGs, more large-scale studies are needed to draw firm conclusions.
Table 1. . Summary of most frequent molecular and clinical characteristics of pediatric high-grade gliomas.
| Mutations/cytogenetics | H3F3AmutK27 | H3F3AmutG34 | RTK I (PDGFRA ampl.) | SETD2 | ACVR1 | BRAFV600E | NTRK |
|---|---|---|---|---|---|---|---|
| Age distribution (years) | 5–29 | Median: 20 | Median: 11.4 | >12 | 5–9 | 6.5–20 | <3 |
| Tumor location |
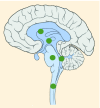 23–43% |
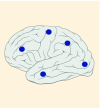 12–14% |
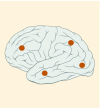 8–39% |
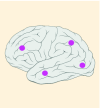 15% |
 20–22% |
 10% |
 40% |
| Therapeutic Significance | None | None | PDGFR inhibitors | Temozolomide resistance | Potential (ALK inhibitors) | BRAF inhibitors | Potential (NTRK inhibitors) |
K27M tumors are found in the midline areas (thalamus, pons, brainstem and spine), they tend to occur at a median age of 10.5 years. G34R/V tumors are found in the cerebral hemispheres, usually occur in adolescents (median age: 18 years) [6,18,20,36,37]. Focal amplifications in PDGFRA are mainly associated with diffuse intrinsic pontine gliomas, the incidence might be overestimated because of the suspected overexpression of PDGFRA in radiation-exposed tumors [6,8,20,68], and with a subset of hemispheric tumors. The median age of patients having tumors with PDGFRA alterations is 11.4 years [6]. Mutations in SETD2 occur exclusively in the cerebral hemispheres in patients older than 12 years [6,40,71]. A recent study found an association between SETD2 mutations and defective DNA mismatch repair through microsatellite instability [41]. Mutations in ACVR1 are reported exclusively in diffuse intrinsic pontine gliomas and are found in 20–22% of these patients, most of which are in pretreated samples [22,58–60]. BRAF V600E point mutations in pediatric high-grade gliomas occur in young adults and most of the tumors are located in the cerebral hemispheres [6]. NTRK fusions in NTRK1, NTRK2 and NTRK3 genes are found in the cerebral hemispheres of patients younger than 3 years of age [60].
In addition to being promising clinical and prognostic indicators, data about genetic alterations may also play an important role in the diagnosis and treatment of pHGGs. Tests such as whole-genome sequencing and those involving PCR are accurate; however, they are complicated, expensive and not universally available. Simple tests such as immunohistochemistry (IHC) assays are currently being assessed as a more practical and feasible alternative for clinicians to use to diagnose and subclassify pHGGs [20]. Sturm et al. successfully applied IHC to identify six epigenetic GBM subgroups by using commercially available antibodies to detect OLIG2, FOX1 (universal markers for diffuse gliomas and ventral telencephalic marker, respectively), IDH1 R132H mutation, and ALT in samples from pediatric patients [20]. BRAF V600E mutations can also be detected by IHC, and, although not frequently detected in HGGs, the existence of targeted agents against this mutation makes it a promising marker [21]. Mismatch repair deficiency and microsatellite instability can also be detected by IHC. This is important to guide treatment in patients with SETD2 mutations who develop resistance to temozolomide [21,71,76,77].
Conclusion & future perspective
The dissection of pHGGs at the molecular level and the increasing knowledge about the associated pathways that underlie disease pathogenesis highlights the difference with the adult counterpart and the idea of age-related pathways in gliomagenesis. This provides grounds for physicians to investigate future tailored, targeted therapy to improve the dismal outcome of children with HGGs. Although multiple possible targets such as BRAF V600E, PDGFRA, IDH, PRC2, ACVR1, NTRKs and ALT have been identified, the complexity of signaling pathways in gliomagenesis, cell survival and tumor invasion poses a serious challenge for treatment success. Several previous studies targeting specific molecular pathways in HGG failed to produce survival benefit. This could be related to multiple factors including the use of single pathway inhibitors in tumors where multiple pathways are involved in tumorigenesis. Other possible factors include resistance of glioma cells to targeting agents and difficulty of delivering the medications through the blood–brain barrier to the primary tumors. Future treatment regimens of pHGGs should assess the incorporation of targeted agents based on individualized tumor characteristics and patients’ age, and be built into prospective clinical research protocols designed on the basis of histologic and molecular characteristics of tumors. Because of the rarity of HGGs in the pediatric population, collaborative multicenter studies recruiting as many patients as possible are warranted to gain further insights into the molecular pathology of HGGs and improve the outcome and quality of life of patients with this deadly and incurable disease. The latest WHO classification of tumors of the central nervous system (2007 WHO) does not include the molecular characteristics of brain tumors [1]. This is related to the fact that this is a newly evolving field, and the emerging molecular data is abundant, and challenging to interpret. In addition, prior to integrating the molecular information in the diagnosis of brain tumors, standardized testing methods and consistent results must be obtained from multiple independent studies. In their latest meeting, the International Society of Neuropathology addressed the upcoming WHO classification of CNS tumors. The group suggested a layered approach to the diagnosis, which includes four parts: integrated diagnosis, histological classification, WHO grade and molecular information [78].
Footnotes
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
References
- 1.Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chan KM, Fang D, Gan H, et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013;27:985–990. doi: 10.1101/gad.217778.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Broniscer A. Past, present, and future strategies in the treatment of high-grade glioma in children. Cancer Invest. 2006;24:77–81. doi: 10.1080/07357900500449702. [DOI] [PubMed] [Google Scholar]
- 4.Gielen GH, Gessi M, Hammes J, Kramm CM, Waha A, Pietsch T. H3F3A K27M mutation in pediatric CNS tumors: a marker for diffuse high-grade astrocytomas. Am. J. Clin. Pathol. 2013;139:345–349. doi: 10.1309/AJCPABOHBC33FVMO. [DOI] [PubMed] [Google Scholar]
- 5.Wu G, Broniscer A, McEachron TA, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012;44:251–253. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sturm D, Bender S, Jones DT, et al. Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nat. Rev. Cancer. 2014;14:92–107. doi: 10.1038/nrc3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Broniscer A, Gajjar A. Supratentorial high-grade astrocytoma and diffuse brainstem glioma: two challenges for the pediatric oncologist. Oncologist. 2004;9:197–206. doi: 10.1634/theoncologist.9-2-197. [DOI] [PubMed] [Google Scholar]
- 8.Paugh BS, Qu C, Jones C, et al. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J. Clin. Oncol. 2010;28:3061–3068. doi: 10.1200/JCO.2009.26.7252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schroeder KM, Hoeman CM, Becher OJ. Children are not just little adults: recent advances in understanding of diffuse intrinsic pontine glioma biology. Pediatr. Res. 2014;75:205–209. doi: 10.1038/pr.2013.194. [DOI] [PubMed] [Google Scholar]
- 10.Zarghooni M, Bartels U, Lee E, et al. Whole-genome profiling of pediatric diffuse intrinsic pontine gliomas highlights platelet-derived growth factor receptor alpha and poly (ADP-ribose) polymerase as potential therapeutic targets. J. Clin. Oncol. 2010;28:1337–1344. doi: 10.1200/JCO.2009.25.5463. [DOI] [PubMed] [Google Scholar]
- 11.Batra V, Sands SA, Holmes E, et al. Long-term survival of children less than six years of age enrolled on the CCG-945 Phase III trial for newly-diagnosed high-grade glioma: a report from the Children's Oncology Group. Pediatr. Blood Cancer. 2014;61:151–157. doi: 10.1002/pbc.24718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohen KJ, Pollack IF, Zhou T, et al. Temozolomide in the treatment of high-grade gliomas in children: a report from the Children's Oncology Group. Neuro Oncol. 2011;13:317–323. doi: 10.1093/neuonc/noq191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fangusaro J. Pediatric high grade glioma: a review and update on tumor clinical characteristics and biology. Front. Oncol. 2012;2:105. doi: 10.3389/fonc.2012.00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Finlay JL, Zacharoulis S. The treatment of high grade gliomas and diffuse intrinsic pontine tumors of childhood and adolescence: a historical – and futuristic – perspective. J. Neurooncol. 2005;75:253–266. doi: 10.1007/s11060-005-6747-7. [DOI] [PubMed] [Google Scholar]
- 15.MacDonald TJ, Aguilera D, Kramm CM. Treatment of high-grade glioma in children and adolescents. Neuro Oncol. 2011;13:1049–1058. doi: 10.1093/neuonc/nor092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hegi ME, Diserens A-C, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 17.Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brennan CW, Verhaak RG, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 20.Sturm D, Witt H, Hovestadt V, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22:425–437. doi: 10.1016/j.ccr.2012.08.024. [DOI] [PubMed] [Google Scholar]
- 21.Rakopoulos P, Hawkins C. Genetic alterations in paediatric high grade astrocytomas. Diagn. Histopathol. 2014;20:84–90. [Google Scholar]
- 22.Buczkowicz P, Hoeman C, Rakopoulos P, et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat. Genet. 2014;46(5):451–456. doi: 10.1038/ng.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1 . Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 25.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl.):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 26.Gajjar A, Pfister SM, Taylor MD, Gilbertson RJ. Molecular insights into pediatric brain tumors have the potential to transform therapy. Clin. Cancer Res. 2014;20(22):5630–5640. doi: 10.1158/1078-0432.CCR-14-0833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 28.Laird PW. Principles and challenges of genome-wide DNA methylation analysis. Nat. Rev. Genet. 2010;11:191–203. doi: 10.1038/nrg2732. [DOI] [PubMed] [Google Scholar]
- 29.Weber M, Hellmann I, Stadler MB, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007;39:457–466. doi: 10.1038/ng1990. [DOI] [PubMed] [Google Scholar]
- 30.Bender S, Tang Y, Lindroth AM, et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell. 2013;24:660–672. doi: 10.1016/j.ccr.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 31.Biterge B, Schneider R. Histone variants: key players of chromatin. Cell Tissue Res. 2014;356(3):457–466. doi: 10.1007/s00441-014-1862-4. [DOI] [PubMed] [Google Scholar]
- 32.Ho L, Crabtree GR. Chromatin remodelling during development. Nature. 2010;463:474–484. doi: 10.1038/nature08911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Talbert PB, Henikoff S. Histone variants – ancient wrap artists of the epigenome. Nat. Rev. Mol. Cell Biol. 2010;11:264–275. doi: 10.1038/nrm2861. [DOI] [PubMed] [Google Scholar]
- 34.Yuen BT, Knoepfler PS. Histone H3.3 mutations: a variant path to cancer. Cancer Cell. 2013;24:567–574. doi: 10.1016/j.ccr.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cesare AJ, Reddel RR. Alternative lengthening of telomeres: models, mechanisms and implications. Nat. Rev. Genet. 2010;11:319–330. doi: 10.1038/nrg2763. [DOI] [PubMed] [Google Scholar]
- 36.Schwartzentruber J, Korshunov A, Liu XY, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–231. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 37.Wu G, Broniscer A, McEachron TA, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012;44:251–253. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lewis PW, Muller MM, Koletsky MS, et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 2013;340:857–861. doi: 10.1126/science.1232245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khuong-Quang DA, Buczkowicz P, Rakopoulos P, et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012;124:439–447. doi: 10.1007/s00401-012-0998-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fontebasso AM, Schwartzentruber J, Khuong-Quang DA, et al. Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta Neuropathol. 2013;125:659–669. doi: 10.1007/s00401-013-1095-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.La Torre D, Aguennouz M, Conti A, et al. Potential clinical role of telomere length in human glioblastoma. Translational medicine @ UniSa. 2011;1:243–270. [PMC free article] [PubMed] [Google Scholar]
- 42.Chen YJ, Hakin-Smith V, Teo M, et al. Association of mutant TP53 with alternative lengthening of telomeres and favorable prognosis in glioma. Cancer Res. 2006;66:6473–6476. doi: 10.1158/0008-5472.CAN-06-0910. [DOI] [PubMed] [Google Scholar]
- 43.Henson JD, Neumann AA, Yeager TR, Reddel RR. Alternative lengthening of telomeres in mammalian cells. Oncogene. 2002;21:598–610. doi: 10.1038/sj.onc.1205058. [DOI] [PubMed] [Google Scholar]
- 44.Gillet E, Alentorn A, Doukoure B, et al. TP53 and p53 statuses and their clinical impact in diffuse low grade gliomas. J. Neurooncol. 2014;118(1):131–139. doi: 10.1007/s11060-014-1407-4. [DOI] [PubMed] [Google Scholar]
- 45.Jiao Y, Killela PJ, Reitman ZJ, et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget. 2012;3:709–722. doi: 10.18632/oncotarget.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heldin CH. Targeting the PDGF signaling pathway in tumor treatment. Cell Commun. Signal. 2013;11:97. doi: 10.1186/1478-811X-11-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paugh BS, Zhu X, Qu C, et al. Novel oncogenic PDGFRA mutations in pediatric high-grade gliomas. Cancer Res. 2013;73:6219–6229. doi: 10.1158/0008-5472.CAN-13-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Noushmehr H, Weisenberger DJ, Diefes K, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu XY, Gerges N, Korshunov A, et al. Frequent ATRX mutations and loss of expression in adult diffuse astrocytic tumors carrying IDH1/IDH2 and TP53 mutations. Acta Neuropathol. 2012;124:615–625. doi: 10.1007/s00401-012-1031-3. [DOI] [PubMed] [Google Scholar]
- 50.Turkalp Z, Karamchandani J, Das S. IDH mutation in glioma: new insights and promises for the future. JAMA Neurol. 2014;71(10):1319–1325. doi: 10.1001/jamaneurol.2014.1205. [DOI] [PubMed] [Google Scholar]
- 51.Pollack IF, Hamilton RL, Sobol RW, et al. IDH1 mutations are common in malignant gliomas arising in adolescents: a report from the Children's Oncology Group. Childs Nerv. Syst. 2011;27:87–94. doi: 10.1007/s00381-010-1264-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ducray F, Marie Y, Sanson M. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009;360:2248–2249. author reply 2249. [PubMed] [Google Scholar]
- 53.Dasgupta T, Haas-Kogan DA. The combination of novel targeted molecular agents and radiation in the treatment of pediatric gliomas. Front. Oncol. 2013;3:110. doi: 10.3389/fonc.2013.00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kleinschmidt-DeMasters BK, Aisner DL, Birks DK, Foreman NK. Epithelioid GBMs show a high percentage of BRAF V600E mutation. Am. J. Surg. Pathol. 2013;37:685–698. doi: 10.1097/PAS.0b013e31827f9c5e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schindler G, Capper D, Meyer J, et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol. 2011;121:397–405. doi: 10.1007/s00401-011-0802-6. [DOI] [PubMed] [Google Scholar]
- 56.Schiffman JD, Hodgson JG, VandenBerg SR, et al. Oncogenic BRAF mutation with CDKN2A inactivation is characteristic of a subset of pediatric malignant astrocytomas. Cancer Res. 2010;70:512–519. doi: 10.1158/0008-5472.CAN-09-1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shore EM, Xu M, Feldman GJ, et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat. Genet. 2006;38:525–527. doi: 10.1038/ng1783. [DOI] [PubMed] [Google Scholar]
- 58.Fontebasso AM, Papillon-Cavanagh S, Schwartzentruber J, et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. 2014;46(5):462–466. doi: 10.1038/ng.2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Taylor KR, Mackay A, Truffaux N, et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat. Genet. 2014;46(5):457–461. doi: 10.1038/ng.2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu G, Diaz AK, Paugh BS, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 2014;46:444–450. doi: 10.1038/ng.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Harel L, Costa B, Fainzilber M. On the death Trk. Dev. Neurobiol. 2010;70:298–303. doi: 10.1002/dneu.20769. [DOI] [PubMed] [Google Scholar]
- 62.Thiele CJ, Li Z, McKee AE. On Trk – the TrkB signal transduction pathway is an increasingly important target in cancer biology. Clin. Cancer Res. 2009;15:5962–5967. doi: 10.1158/1078-0432.CCR-08-0651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Frattini V, Trifonov V, Chan JM, et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat. Genet. 2013;45:1141–1149. doi: 10.1038/ng.2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Butti MG, Bongarzone I, Ferraresi G, Mondellini P, Borrello MG, Pierotti MA. A sequence analysis of the genomic regions involved in the rearrangements between TPM3 and NTRK1 genes producing TRK oncogenes in papillary thyroid carcinomas. Genomics. 1995;28:15–24. doi: 10.1006/geno.1995.1100. [DOI] [PubMed] [Google Scholar]
- 65.Eguchi M, Eguchi-Ishimae M, Tojo A, et al. Fusion of ETV6 to neurotrophin-3 receptor TRKC in acute myeloid leukemia with t(12;15)(p13;q25) Blood. 1999;93:1355–1363. [PubMed] [Google Scholar]
- 66.Lannon CL, Sorensen PH. ETV6-NTRK3: a chimeric protein tyrosine kinase with transformation activity in multiple cell lineages. Semin. Cancer Biol. 2005;15:215–223. doi: 10.1016/j.semcancer.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 67.Li Z, Tognon CE, Godinho FJ, et al. ETV6-NTRK3 fusion oncogene initiates breast cancer from committed mammary progenitors via activation of AP1 complex. Cancer Cell. 2007;12:542–558. doi: 10.1016/j.ccr.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bax DA, Mackay A, Little SE, et al. A distinct spectrum of copy number aberrations in pediatric high-grade gliomas. Clin. Cancer Res. 2010;16:3368–3377. doi: 10.1158/1078-0432.CCR-10-0438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jones C, Perryman L, Hargrave D. Paediatric and adult malignant glioma: close relatives or distant cousins? Nat. Rev. Clin. Oncol. 2012;9:400–413. doi: 10.1038/nrclinonc.2012.87. [DOI] [PubMed] [Google Scholar]
- 70.Broniscer A, Baker SJ, West AN, et al. Clinical and molecular characteristics of malignant transformation of low-grade glioma in children. J. Clin. Oncol. 2007;25:682–689. doi: 10.1200/JCO.2006.06.8213. [DOI] [PubMed] [Google Scholar]
- 71.Li F, Mao G, Tong D, et al. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha. Cell. 2013;153:590–600. doi: 10.1016/j.cell.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schmidt CK, Jackson SP. On your mark, get SET(D2), go! H3K36me3 primes DNA mismatch repair. Cell. 2013;153:513–515. doi: 10.1016/j.cell.2013.04.018. [DOI] [PubMed] [Google Scholar]
- 73.Liang ML, Ma J, Ho M, et al. Tyrosine kinase expression in pediatric high grade astrocytoma. J. Neurooncol. 2008;87:247–253. doi: 10.1007/s11060-007-9513-1. [DOI] [PubMed] [Google Scholar]
- 74.Phillips JJ, Aranda D, Ellison DW, et al. PDGFRA amplification is common in pediatric and adult high-grade astrocytomas and identifies a poor prognostic group in IDH1 mutant glioblastoma. Brain Pathol. 2013;23:565–573. doi: 10.1111/bpa.12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nicolaides TP, Li H, Solomon DA, et al. Targeted therapy for BRAF V600E malignant astrocytoma. Clin. Cancer Res. 2011;17:7595–7604. doi: 10.1158/1078-0432.CCR-11-1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hewish M, Martin SA, Elliott R, Cunningham D, Lord CJ, Ashworth A. Cytosine-based nucleoside analogs are selectively lethal to DNA mismatch repair-deficient tumour cells by enhancing levels of intracellular oxidative stress. Br. J. Cancer. 2013;108:983–992. doi: 10.1038/bjc.2013.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang J, Stevens MF, Bradshaw TD. Temozolomide: mechanisms of action, repair and resistance. Curr. Mol. Pharmacol. 2012;5:102–114. doi: 10.2174/1874467211205010102. [DOI] [PubMed] [Google Scholar]
- 78.Louis DN, Perry A, Burger P, et al. International Society of Neuropathology-Haarlem Consensus Guidelines, for Nervous System Tumor Classification and Grading. Brain Pathol. 2014;24(5):429–35. doi: 10.1111/bpa.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]