

Abstract
The generation of functional microvascular networks is critical for the development of advanced in vitro models to replicate pathophysiological conditions. Mural cells provide structural support to blood vessels and secrete biomolecules contributing to vessel stability and functionality. We investigated the role played by two endothelium-related molecules, angiopoietin (Ang-1) and transforming growth factor (TGF-β1), on bone marrow-derived human mesenchymal stem cell (BM-hMSC) phenotypic transition toward a mural cell lineage, both in monoculture and in direct contact with human endothelial cells (ECs), within 3D fibrin gels in microfluidic devices. We demonstrated that the effect of these molecules is dependent on direct heterotypic cell-cell contact. Moreover, we found a significant increase in the amount of α-smooth muscle actin in microvascular networks with added VEGF and TGF-β1 or VEGF and Ang-1 compared to networks with added VEGF alone. However, the addition of TGF-β1 generated a non-interconnected microvasculature, while Ang-1 promoted functional networks, confirmed by microsphere perfusion and permeability measurements. The presence of mural cell-like BM-hMSCs coupled with the addition of Ang-1 increased the number of network branches and reduced mean vessel diameter compared to EC only vasculature. This system has promising applications in the development of advanced in vitro models to study complex biological phenomena involving functional and perfusable microvascular networks.
Introduction
A functional microvascular network is essential to deliver nutrients, oxygen and immune cells to tissues and organs.1 Endothelial cells (ECs) contribute to the maintenance of vascular integrity by developing tight and adherens junctions2 and express a broad spectrum of receptor molecules such as selectins, vascular cell adhesion molecules and intercellular adhesion molecules involved in multiple cell-cell interactions.3–4 However, the generation of a functional vasculature involves the recruitment of mural cells, and the development of organ-specific matrices and elastic laminae surrounding blood vessels.1, 5
There are numerous factors that are involved in vessel development and maturation. A variety of endothelium-specific molecules cooperate to promote the generation of microvascular networks, including five members of the vascular endothelial growth factor (VEGF) family, four molecules belonging to the angiopoietin group and one of the large ephrinfamily.6 Other non-endothelium specific growth factors are also required for blood vessel formation, such as proteins of the transforming growth factor (TGF-β) family.7 The newly formed microvessels are stabilized by recruited mural cells, i.e. pericytes, smooth muscle cells and fibroblasts, which contribute to the deposition of local extracellular matrix (ECM).1 ECs secrete specific proteins, such as platelet derived growth factor (PDGF-B), promoting mural cell recruitment,8 while mural cells secrete multiple factors including angiopoietin (Ang-1), which leads to lower vascular permeability by maximizing the interactions between ECs and surrounding support cells.9 Moreover, it is known that signalling involving sphingosine-1-phosphate-1 (S1P1) expressed by both ECs and mural cells represents a key pathway for mural cell recruitment.10–11 TGF-β1 is a multifunctional cytokine produced by mural cells and ECs which is involved in multiple processes, including ECM production and mesenchymal cell differentiation into mural cells, with both pro- and anti-angiogenic properties depending on concentration and local microenvironment.12–14
The generation of physiological-like microvascular systems is required for the development of both in vivo long-lasting blood vessels15–16 and advanced in vitro models able to better replicate multiple biological phenomena where the interaction between capillaries and organ-specific tissues is critical. Several 2D in vitro models were recently developed to investigate vascular network related phenomena, such as mesenchymal cell differentiation into smooth muscle cells upon co-culture with ECs17–18 or VEGF-induced vessel permeability.19 However, the importance of 3D in vitro models lies in the possibility to mimic physiological cell-cell and cell-matrix interactions within biological or synthetic matrices where cells show morphologies and differentiation abilities considerably different from those observed on 2D surfaces.20–24 3D microfluidic assays were performed to study angiogenesis25–30 and different methods were applied to replicate microvessel structure.31–35 Furthermore, other studies analysed the effect of endothelial secreted factors such as PDGF-B in the regulation of pericyte recruitment36 and the influence these stabilizing cells provide in terms of endothelial basement membrane generation and expression of integrins that recognize the newly deposited matrix.37 Moreover, our group has recently developed an in vitro model to generate microvascular networks by vasculogenesis to study cancer metastases.38–39
The present study is focused on the generation of functional, perfusable 3D human microvascular network, co-culturing ECs and bone marrow-derived human mesenchymal stem cells (BM-hMSCs) within a microfluidic device by vasculogenesis-like process. The ability of stem cells to acquire a mural phenotype is critical in developing physiological microvessels. In this framework, we investigated the effect of Ang-1, a key molecule in vessel stabilization,7 whose role could be related to the recruitment of mesenchymal cells.40–42 Our model represents a significant step forward toward the generation of more physiological microvessels compared to endothelialized microchannels or micronetworks generated within 3D gels or spheroids suspended in standard multiwell plates.31, 35, 37 We demonstrate that the co-culture of ECs and BM-hMSCs leads to the generation of realistic human microvascular networks in which readily-available bone marrow multipotent cells show a phenotypic transition toward a mural cell lineage and co-localize with ECs. Particularly, we show the dual role of Ang-1 as vessel stabilizer and promoter of phenotypic transition toward mural cells. This system could potentially be used to develop advanced in vitro models to study and characterize complex biological phenomena such as intra- and extravasation of circulating tumor cells (CTCs) or immune cells,43 and better clarify pathophysiological shear stress conditions on in vivo-like capillaries.44–45
Experimental
Microfluidic system
A microfluidic device consisting of two lateral media channels and a central gel channel was adopted in the present work. Microfabrication details were previously reported for other systems developed by our group.46–47 Briefly, the microfluidic device was made of PDMS (poly-dimethyl-siloxane; Silgard 184, Dow-Chemical) through soft lithography techniques and SU-8 micropatterned silicon wafers. Inlet and outlet ports were bored by means of disposable biopsy punches and the PDMS structure was bonded to a glass coverslip after 60 s oxygen plasma treatment to create 200 μm deep microchannels. Trapezoidal posts with an inter-post distance of 330 μm were employed to separate the 1,300 μm wide gel channel from the lateral media channels and promote an ideal gel filling. Microfluidic channels were coated with PDL (poly-D-lysine hydrobromide; 1 mg/ml; Sigma-Aldrich) solution to promote matrix adhesion. Next, a thrombin solution, obtained dissolving 20 μl thrombin (100 U/ml) within 500 μl cell culture medium, was used to resuspend cells and 10 μl aliquots were mixed with 10 μl fibrinogen solution (5.0 mg/ml) to generate a fibrin gel. A 10 μl pipette was used to fill the gel channel and microdevices were incubated within humid chambers for 10 min at room temperature to form the hydrogel. Following gelation, cell culture medium was added to the media channels and microfluidic devices were cultured for at least 7 days under static conditions with daily medium replacement.
Cell culture
BM-hMSCs were harvested from patients undergoing hip arthroplasty and selected by plastic adherence according to a previously optimized protocol.48 Cells of passage 6 or lower were cultured in standard alpha-minimum essential medium (αMEM; Invitrogen)containing non-essential aminoacids, sodium pyruvate and L-glutamine, supplemented with 10% fetal bovine serum (FBS; Invitrogen), biological buffer and antibiotics. Red fluorescent protein (RFP)-transfected human umbilical vein endothelial cells (HUVECs) were commercially obtained (Angio-Proteomie) and cultured in endothelial growth medium (EGM-2MV; Lonza) with full supplements (EGM-2MV bullet kit; Lonza), which was considered as standard endothelial growth medium. All the experiments were conducted using HUVECs of passage 6. HUVECs were suspended at 12×106 cells/ml in standard endothelial growth medium+thrombin and mixed with BM-hMSCs (6×106 cells/ml suspension in BM-hMSC growth medium+thrombin). The obtained suspension was mixed with fibrinogen solution (1:1 ratio) and injected into the gel channel. After gelation, lateral media channels were filled with standard endothelial growth medium supplemented with 50 ng/ml VEGF (Peprotech). The medium was replaced every 24 hours. All cultures were kept in a humidified incubator maintained at 37°C and 5% CO2. After day 1, selected microdevices were cultured with standard endothelial growth medium supplemented with 50 ng/ml VEGF and 100 ng/ml Ang-1 (Peprotech) or 1 ng/ml TGF-β1 (Peprotech) to analyse the effect of these molecules on microvascular network generation and BM-hMSC phenotypic transition toward a mural cell lineage. In order to promote the generation of perfusable microvascular networks, HUVECs were seeded at day 2 within lateral media channels of the identified optimal configuration, introducing 40 μl cell suspension at 1×106 cells/ml cell density. Non adhered cells were washed away flowing fresh medium after 1 h. A schematic representing the microfluidic device and the cell culture model is shown in Fig. 1. Control experiments were performed seeding HUVECs without BM-hMSCs at the same cell density applied in co-culture assays and the vasculatures generated were analysed in the same manner. Furthermore, BM-hMSC phenotypic transition control experiments were conducted seeding mesenchymal stem cells alone at the same cell density set for co-cultures. The influence of the direct contact between ECs and BM-hMSCs was assessed seeding mesenchymal stem cells into the gel region and a HUVEC monolayer on the interface between gel and media channel.
Fig. 1.
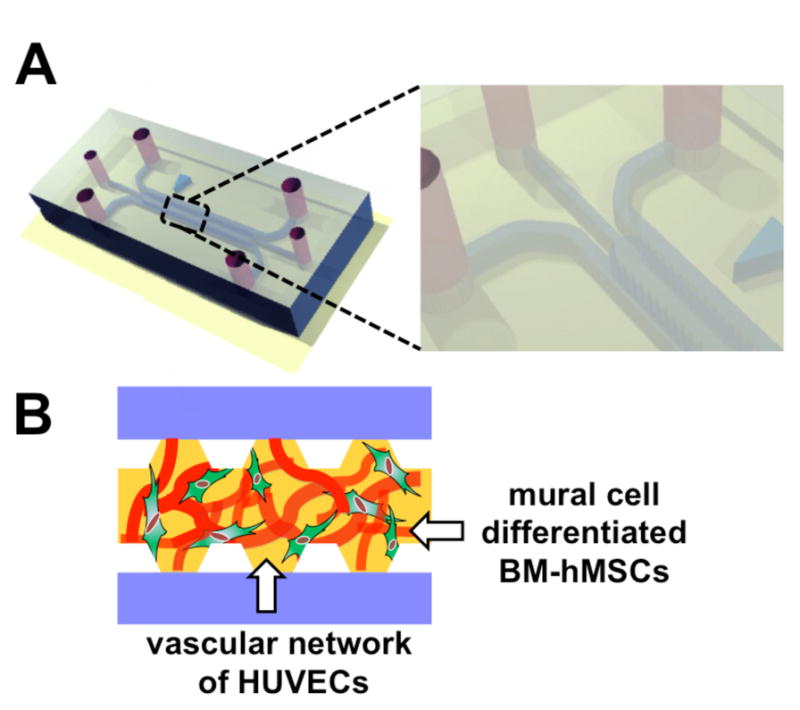
Schematic of the microfluidic system. Microfluidic device composed of two lateral media channels and one interposed 1,300 μm wide gel channel embedding HUVECs and BM-hMSCs in a 2:1 ratio (A). A vasculogenesis-like approach allows the generation of functional, perfusable microvascular networks with ECs (red) surrounded by BM-hMSCs (green) characteristics of a mural cell lineage in a fibrin matrix (B).
Immunofluorescent staining
Samples were washed with phosphate buffered saline (PBS; Invitrogen) and fixed with 4% paraformaldehyde (PFA) for 15 min at room temperature. Next, cells were washed twice with PBS and incubated with 0.1% Triton-X 100 solution for 5 min at room temperature. After washing twice with PBS, cells were blocked with 5% bovine serum albumin (BSA) + 3% goat serum solution for at least 3h at 4°C. Alpha-smooth muscle actin (α-SMA) was labeled with mouse monoclonal antibody (abcam7817) at 1:100 dilution, SM22α was labelled with rabbit polyclonal antibody (abcam14106) at 1:1000 dilution, vascular endothelial-cadherin (VE-cadherin) and laminin were labeled with rabbit polyclonal antibody (abcam33168 and abcam30320, respectively) at 1:100 dilution, zonula occludens-1 (ZO-1) was labeled with mouse polyclonal antibody (Invitrogen; 339100) at 1:100 dilution, and NG2 was labelled with rabbit polyclonal antibody (abcam83178) at 1:200 dilution. Fluorescently labeled secondary antibodies (Invitrogen) were used at 1:200 dilution. Cell nuclei were stained with 4′6-Diamidino-2-Phenylindole (DAPI;5 mg/ml; Invitrogen)at 1:500 dilution while F-actin filaments were stained with AlexaFluor633 phalloidin (Invitrogen) at 1:100 dilution. If not differently specified, all the images were captured using a confocal microscope (Olympus IX81) and processed with Imaris software (Bitplane Scientific Software).
Data analysis
Microvascular network morphology
Microvascular networks were analysed using Fiji software (http://fiji.sc/Fiji). The endothelial cell RFP signal was used to compute projected 2D areas of the network and the Fiji 2D skeletonize plugin was applied to determine number of branches, average branch length and total network length. Projected 3D stacks representing each region of interest (ROI, 533x426 μm2) were pre-processed with Fiji software to enhance contrast (10%), filter noise (application of “despeckle” algorithm and “gaussian blur” filter) and convert the images to a binary format by applying the “triangle” threshold method.49 In addition, the Fiji 3D skeletonize plugin was used to further analyse the number of branches in the longest structure detected within each ROI, according to the abovementioned protocol. Moreover, 3D skeletonize data were noise filtered applying a 25 μm threshold value to remove image artefacts. The threshold value was calculated by averaging multiple measurements and comparing 3D confocal images to3D skeleton reconstructions.
Note that to the extent that vessels cross at different z-planes, this could lead to errors in estimating the number of branches and vessel segments. To quantify this effect, 2D and 3D skeletons were compared as described in the “Generation of microvascular networks” section.
Finally, average vessel diameter was quantified with Imaris software. The temporal evolution of the network was assessed through daily monitoring by phase-contrast and fluorescence microscopy (Nikon Eclipse Ti).
Quantification of BM-hMSC phenotypic transition
BM-hMSC phenotypic transition was quantified through the α-SMA signal in terms of percentage of active pixels, mean signal intensity and total signal intensity (normalized by the minimum value among the three conditions, i.e. VEGF, VEGF+Ang-1 and VEGF+TGF-β1) within each ROI. All intensity values were obtained after subtracting the background signal.
Microvascular network perfusion and permeability quantification
Vessel permeability was quantified according to a previously described method.50 Briefly, the medium in all reservoirs was aspirated and two reservoirs of the opposite media channels were injected with 40μl of fluorescent dextran (70kDa,green, Invitrogen) diluted with endothelial growth medium for a final concentration of 25 μg/ml. Concentrations were then determined by confocal imaging every 1 min for 5 min once equilibrium was established (i.e. constant intensity within the vessels, 5–10 min). Permeability was quantified by obtaining the average intensity at the initial and final time points considering a region of interest including both the vessel and the surrounding ECM. Permeability was computed from vessel segments in the central region of the gel channel to avoid border effects according to the following formula:50
where Ii, If and Ib represent the initial, final and background average intensities, respectively, Δt is the time interval between two captured images and d is the average diameter of the vessel.
Finally, 40 μl of medium containing 10 μm diameter fluorescent microspheres were introduced within a single reservoir (medium was aspirated from the other reservoirs) of a live sample to demonstrate the presence of a perfusable microvascular network connecting lateral media channels.
Statistics
Microvascular network and differentiation data are averages of measurements of 6(min) to 9(max) regions from 3 independent devices. Results are shown as mean ± standard error of the mean (SEM). The comparisons between groups were assessed using unpaired Student’s t-test. Statistical significance was assumed for p<0.05. All tests were performed with SigmaPlot12.
Results and discussion
BM-hMSCs phenotypic transition toward mural cells
The ability of BM-hMSCs to get a transition toward mural cells was increased in the presence of HUVECs (Fig. 2). Phenotypic transition was visualized by staining with mural cell marker, α-SMA (Fig. 2A–C, green). Addition of Ang-1 or TGF-β1 to VEGF rich standard endothelial growth medium further increased phenotypic transition, as quantified by both percentage area covered and signal intensity of α-SMA marker. α-SMA+ BM-hMSCs covered 4.63 ± 0.77% of the entire area imaged on devices supplemented with VEGF only while devices with added VEGF + Ang-1and VEGF + TGF-β1 showed significantly higher values: 14.2± 1.52% and14.3 ± 1.13%, respectively (Fig. 2D). The mean intensity of α-SMA marker was 2.11 ± 0.32 fold higher with VEGF + Ang-1 devices and 2.36 ± 0.32 fold higher with VEGF + TGF-β1 than VEGF only devices (Fig. 2E). The total sum of α-SMA signal was 4.72 ± 0.99 fold higher with VEGF+Ang-1 whereas VEGF + TGF-β1 devices showed values5.06 ± 0.79 fold higher when normalized to VEGF only devices (Fig. 2F).
Fig. 2.

BM-hMSCs phenotypic transition in presence of HUVECs. When co-cultured with HUVECs, BM-hMSCs undergo a transition toward mural cells. Phenotypic transition can be visualized by staining with mural cell marker, α-smooth muscle actin (green). Cell nuclei were stained with DAPI (blue) (A–C). Addition of Ang-1 or TGF-β1 in VEGF supplemented standard endothelial growth medium induces even more phenotypic transition, as quantified by both percent area covered and αSMA intensity (D–F). Scale bars represent 100 μm.
α-SMA + BM-hMSCs co-localized with HUVECs and wrapped around the newly formed microvascular network (Fig. 3A, α-SMA labelled in green). Culturing BM-hMSCs in VEGF supplemented standard endothelial growth medium did not induce phenotypic transition nor did the addition of VEGF + Ang-1 or VEGF + TGF-β1 without HUVECs (Fig. 3B–D, α-SMA labelled in green, F-actin stained with phalloidin, yellow). The generation of HUVEC monolayers on the interface between gel and media channel showed that only those BM-hMSCs that reached ECs were α-SMA+ (Fig. 3E, α-SMA labelled in green; F-actin stained with phalloidin, yellow), thus demonstrating that direct contact is required to induce a phenotypic transition toward a mural cell lineage.
Fig. 3.
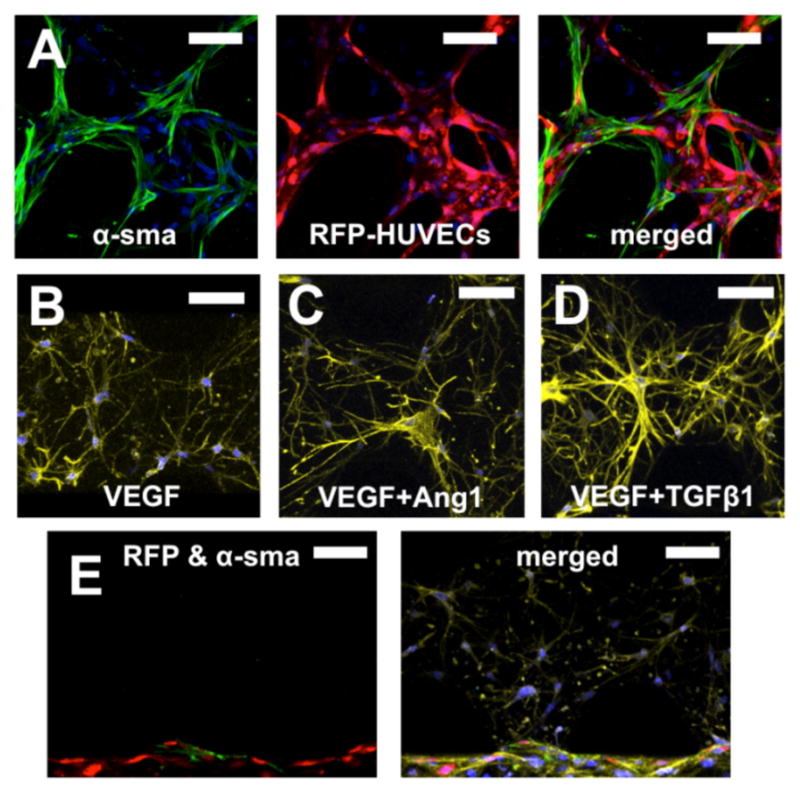
Co-localization of HUVECs and BM-hMSCs enabled BM-hMSCs phenotypic transition toward mural cells. Most of α-SMA+ BM-hMSCs (green) co-localized with vasculature formed by RFP-transfected HUVECs (red) in fibrin gel (A). Culturing BM-hMSCs in VEGF supplemented standard endothelial growth medium did not induce phenotypic transition, nor did addition of VEGF+Ang-1 or VEGF+TGF-β1 without HUVECs (α-SMA green, F-actin stained with phalloidin, yellow) (B–D). When HUVECs (red) were cultured within a lateral media channel and BM-hMSCs in the fibrin gel, only those BM-hMSCs that reached the HUVEC monolayer showed signs of phenotypic transition (α-SMA, green; F-actin, yellow) (E). Cell nuclei were stained with DAPI (blue). Scale bars =100 μm.
Several studies investigated mesenchymal cell differentiation, testing the necessity for direct contact with endothelial cells,51 the effect of mechanical stress52 and the addition of different molecules, including TGF-β153 and sphingosylphosphorylcholine (SPC).54 However, these previous studies were not conducted in a 3D microenvironment, which plays a critical role.22
In our study we examined the influence of direct EC contact and the effect of additive molecules, i.e. Ang-1 and TGF-β1, on BM-hMSC phenotypic transition in a more physiological-like 3D microenvironment compared to previously reported 2D assays.17–18 Although the role of Ang-1 in vessel stabilization is well known,6 only a few studies investigated its effect on smooth muscle-like cell recruitment. However, they did not analyse the role of Ang-1 on mesenchymal stem cell phenotypic transition toward a mural lineage.40–42 In this framework, we have shown addition of Ang-1 (100 ng/ml) not only recruits but also induces BM-hMSC phenotypic transition toward mural cells when mediated by EC co-culture. Furthermore, our results show that the addition of TGF-β1 (1 ng/ml) in the absence of ECs does not exert a significant influence on BM-hMSC commitment toward a mural cell lineage, in agreement with results shown by Au and coauthors,15 which used even higher concentrations (10 ng/ml). However, other studies highlighted the effect of this molecule on the expression of mural cell markers,12, 55 thus the role of this multifunctional cytokine is not entirely clear and could promote different responses in different conditions (e.g. concentrations, cell type, cell microenvironment).1 Addition of TGF-β1 exerted a strong influence on BM-hMSC phenotypic transition only when BM-hMSCs were in direct contact with ECs, emphasizing the need for close interaction with ECs.
Generation of microvascular networks
As we found that HUVECs had crucial role in BM-hMSCs phenotypic transition toward mural cells, we investigated, in reverse, the role of α-SMA+ BM-hMSCs in the vasculogenesis-like process. Formation of microvascular networks was monitored over time during 6 days of culture in microfluidic system (Fig. 4). HUVECs and BM-hMSCs were uniformly distributed in fibrin gel at the time of gel filling (12×106 cells/ml and 6×106 cells/ml, respectively), and remained mixed. RFP-transfected HUVECs enabled to distinguish between the two cell types throughout the culture and to monitor the vasculogenesis-like process. HUVECs did not generate cell sheets when suspended in gel, and connected to each other to form pieces of vessel structures and gradually formed anastomoses of vessels so that most microvasculature was composed by either one or two continuous microvascular networks by day 6 of culture. The presence of BM-hMSCs did not result in gel contraction problems.
Fig. 4.
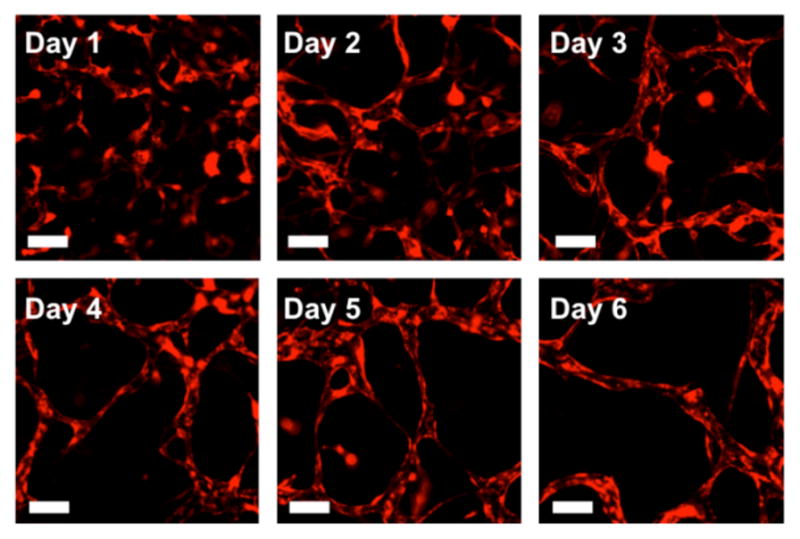
Formation of microvascular networks by vasculogenic-like process. HUVECs as well as BM-hMSCs are uniformly distributed within the gel at the time of gel filling and remain mixed by day 1 (RFP-HUVECs, red). HUVECs gradually interconnect to form a mostly continuous microvascular network by day 6 of culture. Scale bars =100μm.
The effect of mural cell-like BM-hMSCs on vasculogenesis-like processes was investigated by first comparing the networks generated with and without BM-hMSCs in standard endothelial growth medium supplemented with 50 ng/mlVEGF. Networks formed with BM-hMSCs under VEGF only, VEGF+Ang-1, and VEGF+TGF-β1 conditions were compared as done in BM-hMSC phenotypic transition experiments, since each condition gave rise to a different degree of transition. Clear differences were observed in the morphology of vascular networks for different conditions (Fig. 5A–D). HUVEC only devices exhibited interconnected and perfusable microvessels that had the largest diameter, whereas all networks generated in presence of BM-hMSCs retained smaller vessel diameters, while still forming a network. Differences in the size of the microvascular networks were quantified by measuring the percent area that HUVECs occupied. This value varied from 51.9 ± 1.1%for HUVEC only to 37.1 ± 0.97% for HUVECs with BM-hMSCs in VEGF only, and 35.5 ± 0.81% and 26.1 ± 0.72% for VEGF+Ang-1 and VEGF+TGF-β1 (Fig. 5E). Average vessel diameters were also measured (Fig. 5F): networks formed by HUVEC only had an average diameter (84.2 ± 2.7 μm) that was more than two fold larger than the networks that were co-cultured with BM-hMSCs, regardless of added factors (40.8 ± 1.5 μm VEGF only; 36.2 ± 1.6 μm VEGF+Ang-1; 21.5 ± 0.68 μm VEGF+TGF-β1). This is possibly due to the presence of BM-hMSCs in the gel which restricted the 3D volume that HUVECs could occupy, therefore limiting vessel size and network area and mimicking a more physiological microvasculature.1, 15 Interestingly, VEGF+Ang-1 and VEGF+TGF-β1 supplemented systems showed significantly smaller diameters compared to VEGF devices but the area covered with the addition of VEGF+TGF-β1 was statistically lower compared to the other HUVEC+BM-hMSC conditions.
Fig. 5.
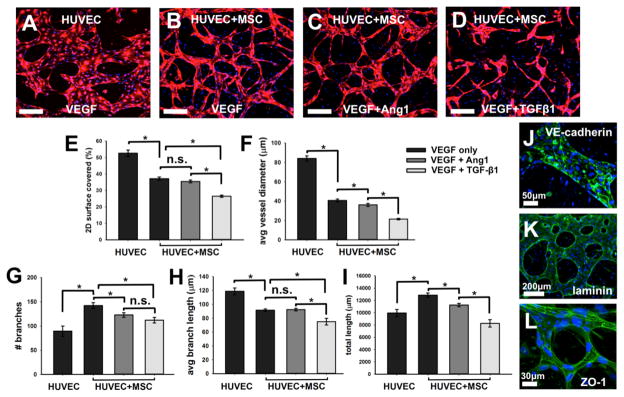
Generation of microvascular networks in the presence of BM-hMSC and different biomolecules. Initially uniformly dispersed HUVECs form perfusable networks by vasculogenesis-like process in VEGF supplemented standard endothelial growth medium with and without BM-hMSCs or with VEGF+Ang-1 and BM-hMSCs (A–D). Microvascular networks under various conditions are quantified E–I). Functionality of microvessel sare confirmed by staining with VE-cadherin (J, green) for adherens junctions, laminin (K, green) for matrix secretion, and ZO-1 (L, green)for tight junctions. Cell nuclei were stained with DAPI (blue). Scale bars = 200μm, unless noted.
We further compared networks by quantifying them using different metrics. The HUVEC only condition had 93.8 ± 9.4 branches, while HUVEC+BM-hMSC microfluidic devices resulted in nearly a 1.5 fold increase (142.2 ± 5.9) with VEGF, 122.0±4.6 branches with VEGF+Ang-1 and 112.2 ± 9.1 branches with VEGF+TGF-β1 (Fig. 5G). When we used the3D skeletonize plugin we obtained on average a 7.7% increase in the number of branches compared to the 2D flat projection method. This difference was due both to the presence of small vessel protrusions, which were not detected with the 2D plugin, and the increased impact of background noise in 3D imaging, the effect of which was attenuated by applying the 2D protocol. However, the general trends resulting in a decrease of the number of branches comparing VEGF, VEGF+Ang-1 and VEGF+TGF-β1conditions were confirmed (Fig. S4). The length of all branches was also measured, resulting in an average branch length of 114.3 ± 7.9 μm for HUVEC only and lower values for co-culture conditions, with 91.6± 1.8 μm for VEGF, 92.5 ± 1.9 μm for VEGF+Ang-1 and 75.1 ± 4.8 μm for VEGF+TGF-β1 (Fig. 5H), accrued in a total length of 10.13 ± 0.39 mm, 12.86 ± 0.35 mm, 11.26 ± 0.28 mm, and 8.28 ± 0.60 mm for HUVEC only and co-cultured with BM-hMSC in VEGF only, VEGF+Ang-1 and VEGF+TGF-β1, respectively (Fig. 5I).
While the addition of TGF-β1 during culture enhanced BM-hMSCs phenotypic transition toward a mural cell lineage, there seemed no advantage in terms of generating a microvascular network. Systems with added TGF-β1 generally resulted in fragmented networks with unconnected vessel segments throughout the duration of the experiment (Fig. 5D), showing that the presence of α-SMA+ BM-hMSC does not necessarily lead to the generation of a well-formed, perfusable vasculature. However, as previously reported,13 TGF-β1 was shown to behave as a pro- or anti-angiogenic molecule depending on concentration and microenvironment. Moreover, it was demonstrated to be a positive regulator of endothelial cell migration and proliferation or vessel maturation.56 Based on these observations and according to our results, we hypothesize TGF-β1 played a critical and negative role on the generation of the microvascular network, compromising any potential stabilizing effect of αSMA+ BM-hMSCs.
The addition of VEGF+Ang-1 in co-culture promoted the generation of microvascular networks characterized by total lengths comparable to HUVEC monoculture while VEGF conditioned co-cultures attained even greater total lengths. However, the sub-structure of the network showed more branches and reduced average branch lengths for VEGF+Ang-1 and VEGF only co-culture conditions, compared to HUVEC only. This seems to be related to the formation of cell aggregates resembling small islands connected by microvessels with a reduced number of sprouts in the HUVEC only condition, which contributes to the generation of a less organized and less physiological structure.57 Furthermore, we found significant differences comparing some network parameters, including average vessel diameter and number of branches within VEGF and VEGF+Ang-1 conditions. We speculate the reduced number of branches within VEGF+Ang-1 devices could be due to the stabilizing role of Ang-1,58 while the presence of BM-hMSCs with a differentiation toward a mural cell lineage could promote lower diameters, as previously showed with pericytes by Stratman and colleagues36. However, we would also like to highlight data reported by Richardson and co-authors, who did not correlate the presence of α-SMA+ cells with vessel cross-sectional area59.
Interestingly, VEGF+Ang-1 added systems not only promoted formation of microvascular networks with small vessel diameters, but also strongly induced phenotypic transition of BM-hMSCs toward mural cells, at a level comparable to the VEGF+TGF-β1 condition. For these reasons the addition of VEGF+Ang-1 was considered the optimal condition, leading to interconnected microvessels surrounded by α-SMA+ BM-hMSCs (Fig. S3). However, this condition alone did not allow perfusion, because while vessel network had formed inside the fibrin gel, there were no openings into the media channels. To compensate, EC monolayers were grown within the lateral channels to create anastomoses with the fibrin gel embedded network; this eventually allowed for perfusion of 10μm diameter fluorescent microbeads into the vascular network (Movie S1). Upon encountering an opening of the network in the gel, microbeads flowed from the media channel into the vessel, travelled through the vascular network and exited from the other side of the gel into the opposite media channel. These experiments confirmed that even the narrowest network generated with VEGF+Ang-1 was continuous and perfusable. Perfusion of the microvascular network with 70 kDa dextran (Fig. S5) confirmed the presence of lumens and showed a diffusive permeability of 6.7×10−7± 2.74×10−7 cm/s at day 6 after seeding (n=5 within 2 independent devices). Our values are in the range of other previously reported in vitro data31, 35, 60–61 but closer to in vivo venular vessel permeability.62
Finally, the optimized VEGF+Ang-1 added model was characterized through immunofluorescence assays to demonstrate the presence of a functional microvasculature. Endothelial adherens and tight junctions were stained with anti-VE-cadherin antibody and anti-ZO-1 antibody, respectively, showing that the initially dispersed single cell mixture of HUVECs elongated and migrated to form microvessels in which endothelial cells are tightly adhered(Fig. 5J and L and Movie S2).Furthermore, functionality of the microvessels was confirmed by staining for laminin (Fig. 5K), which represents one of the major ECM protein secreted by vasculature.63 The presence of laminin, surrounding the entire network, is a clear indication of a functional microvasculature.
To reach a complex level of organization, microvascular networks should mature both at the vessel wall and at the network level. Maturation of the vessels includes the recruitment of mural cells and the development of surrounding matrix, while maturation of the network involves an optimal patterning through branching and pruning.1 Although our data on average vessel diameters do not show the existence of a hierarchical organization characterized by different vessel sizes, the presence of vascular endothelial adherens and tight junctions (Fig. 5J and L), BM-hMSC phenotypic transition toward a mural cell lineage confirmed by positive SM22α and NG-2 stainings (Fig. 3A, S2A and S2B), laminin deposition (Fig. 5K) and vessel permeability measurements close to in vivo values suggest a maturing microvascular network, at least at the vessel wall level. Particularly, NG-2+ cells represent a clear indication of a specific phenotypic transition toward pericytic cells.
Overall these results demonstrate the ability to generate perfusable microvascular networks combining ECs and BM-hMSC phenotypically transitioning toward a mural cell lineage, overcoming limitations of previously reported models such as the lack of connections between microvessels and perfusion channels in the presence of supportive cells (e.g. fibroblasts)60 or the sparse association of pericytes with ECs within templated vasculatures.35 Particularly, the absence of high density fibroblast-containing regions within the microfluidic device might enhance the use of this model to study complex biological phenomena within vascularized systems with greater control over the activation of specific signalling pathways.
Conclusions
We developed 3D functional, perfusable microvascular networks composed of human ECs and BM-hMSCs phenotypically transitioning toward mural cells by vasculogenesis-like approach. The role of several critical factors, i.e. Ang-1 and TGF-β1, in BM-hMSC transition toward a mural cell lineage was investigated, showing their effect is influenced by co-culture with ECs. TGF-β1 was found to exert a significant effect on BM-hMSC phenotypic transition, but its presence does not allow the generation of functional microvascular networks. On the other hand, Ang-1 supplemented systems formed interconnected and perfusable microvessels, surrounded by α-SMA+ BM-hMSCs and a laminin rich ECM. This approach can be used to develop advanced in vitro models where the interactions between a functional vasculature characterized by mature vessel walls and tissue parenchyma is critical to mimic pathophysiological processes (intra- and extravasation processes involving leukocytes and cancer cells) and test diffusion and effects of therapeutics in complex microenvironments.
Supplementary Material
Video S1 Perfusion of 10 μm fluorescent microbeads within a microvascular network composed of HUVECs and mural cell differentiated BM-hMSCs. A pressure drop was applied by aspirating the media from all the reservoirs of the microfluidic device and adding 40 μl microbead suspension within a single reservoir.
Fig. S1 Live/Dead assay showing cell viability at day 1. Cells were stained with 1 μl fluorescein diacetate (FDA, green; Sigma-Aldrich) and 3.5 μl propidium iodide (PI, red; Sigma-Aldrich) diluted in 1 ml phosphate buffered saline (PBS). Samples were incubated for 5 min, washed with PBS and observed through confocal microscopy.
Fig. S2 SM22α immunofluorescent staining highlighting BM-hMSC differentiation toward a mural cell lineage. Representative image showing red fluorescent protein (RFP)-transfected human umbilical vein endothelial cells (HUVECs) organized in a microvessel structure wrapped by differentiated BM-hMSCs (SM22α, green). Cell nuclei were stained with 4′6-Diamidino-2-Phenylindole (DAPI, blue).
Fig. S3 Confocal microscopy image representing mural cell differentiated BM-hMSCs (α-smooth muscle actin, green) co-localization with ECs (red). Capillary lumens are indicated by white arrowheads.
Fig. S4 Microvascular network analysis: number of branches. The 3D skeletonize plugin of the Fiji software was applied to compute the number of branches of the longest connected structure within each region of interest (ROI, 533x426 μm2). A 25 μm threshold was applied to filter 3D skeleton data (main text). Representative images of a confocal 3D reconstruction (A), a 2D skeleton obtained with the 2D skeletonize plugin (B) and a 3D volumetric skeleton (C). 3D data for the three different experimental conditions (addition of VEGF, VEGF+Ang-1 and VEGF+TGF-β1). Average values were obtained for a minimum of n=8 regions within 2 or 3 independent devices per condition (D). VEGF: vascular endothelial growth factor; Ang-1: angiopoietin-1; TGF-β1: transforming growth factor-β1.
Fig. S5 Vessel perfusion with 70 kDa fluorescent dextran revealing patent lumen and absence of focal leaks. Representative picture of a microvascular network composed by HUVECs and mural cell differentiated BM-hMSCs treated with VEGF and Ang-1.
3D confocal reconstruction of a representative microvessel stained with anti-VE-cadherin antibody (green). ECs (red) organized in a patent capillary appear tightly connected through a network of vascular adherens junctions. Cell nuclei were stained with DAPI (blue).
Acknowledgments
Support from the National Cancer Institute (R33 CA174550–01 and R21 CA140096) and the Italian Ministry of Health, fellowship support to S. Bersini provided by the Fondazione Fratelli Agostino and Enrico Rocca through the Progetto Rocca Doctoral Fellowship and support to J.S. Jeon provided by Repligen Fellowship in Cancer Research and Draper Fellowship are gratefully acknowledged.
Footnotes
Electronic Supplementary Information (ESI) available. See DOI: 10.1039/b000000x/
References
- 1.Jain RK. Nat Med. 2003;9:685–93. doi: 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- 2.Dejana E, Orsenigo F. J Cell Sci. 2013;126(Pt 12):2545–9. doi: 10.1242/jcs.124529. [DOI] [PubMed] [Google Scholar]
- 3.Fukuda M, Hiraoka N, Yeh JC. J Cell Biol. 1999;147:467–70. doi: 10.1083/jcb.147.3.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gout S, Tremblay PL, Huot J. Clin Exp Metastasis. 2008;25:335–44. doi: 10.1007/s10585-007-9096-4. [DOI] [PubMed] [Google Scholar]
- 5.LeBlanc AJ, Krishnan L, Sullivan CJ, Williams SK, Hoying JB. Microcirculation. 2012;19:676–95. doi: 10.1111/j.1549-8719.2012.00207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, Holash J. Nature. 2000;407:242–8. doi: 10.1038/35025215. [DOI] [PubMed] [Google Scholar]
- 7.Carmeliet P. Nat Med. 2000;6:389–95. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- 8.Hellstrom M, Gerhardt H, Kalen M, Li X, Eriksson U, Wolburg H, Betsholtz C. J Cell Biol. 2001;153:543–53. doi: 10.1083/jcb.153.3.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thurston G, Suri C, Smith K, McClain J, Sato TN, Yancopoulos GD, McDonald DM. Science. 1999;286:2511–4. doi: 10.1126/science.286.5449.2511. [DOI] [PubMed] [Google Scholar]
- 10.Armulik A, Abramsson A, Betsholtz C. Circ Res. 2005;97:512–23. doi: 10.1161/01.RES.0000182903.16652.d7. [DOI] [PubMed] [Google Scholar]
- 11.Kluk MJ, Hla T. Biochim Biophys Acta. 2002;1582:72–80. doi: 10.1016/s1388-1981(02)00139-7. [DOI] [PubMed] [Google Scholar]
- 12.Chambers RC, Leoni P, Kaminski N, Laurent GJ, Heller RA. Am J Pathol. 2003;162:533–46. doi: 10.1016/s0002-9440(10)63847-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gohongi T, Fukumura D, Boucher Y, Yun CO, Soff GA, Compton C, Todoroki T, Jain RK. Nat Med. 1999;5:1203–8. doi: 10.1038/13524. [DOI] [PubMed] [Google Scholar]
- 14.Pepper MS. Cytokine Growth Factor Rev. 1997;8:21–43. doi: 10.1016/s1359-6101(96)00048-2. [DOI] [PubMed] [Google Scholar]
- 15.Au P, Tam J, Fukumura D, Jain RK. Blood. 2008;111:4551–8. doi: 10.1182/blood-2007-10-118273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koike N, Fukumura D, Gralla O, Au P, Schechner JS, Jain RK. Nature. 2004;428:138–9. doi: 10.1038/428138a. [DOI] [PubMed] [Google Scholar]
- 17.Goerke SM, Plaha J, Hager S, Strassburg S, Torio-Padron N, Stark GB, Finkenzeller G. Tissue Eng Part A. 2012;18:2395–405. doi: 10.1089/ten.TEA.2012.0147. [DOI] [PubMed] [Google Scholar]
- 18.Guo X, Stice SL, Boyd NL, Chen SY. Am J Physiol Cell Physiol. 2013;304:C289–98. doi: 10.1152/ajpcell.00298.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gavard J, Gutkind JS. Nat Cell Biol. 2006;8:1223–34. doi: 10.1038/ncb1486. [DOI] [PubMed] [Google Scholar]
- 20.Derda R, Laromaine A, Mammoto A, Tang SK, Mammoto T, Ingber DE, Whitesides GM. Proc Natl Acad Sci U S A. 2009;106:18457–62. doi: 10.1073/pnas.0910666106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Even-Ram S, Yamada KM. Curr Opin Cell Biol. 2005;17:524–32. doi: 10.1016/j.ceb.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 22.Griffith LG, Swartz MA. Nat Rev Mol Cell Biol. 2006;7:211–24. doi: 10.1038/nrm1858. [DOI] [PubMed] [Google Scholar]
- 23.Lee J, Cuddihy MJ, Kotov NA. Tissue Eng Part B Rev. 2008;14:61–86. doi: 10.1089/teb.2007.0150. [DOI] [PubMed] [Google Scholar]
- 24.Yamada KM, Cukierman E. Cell. 2007;130:601–10. doi: 10.1016/j.cell.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 25.Bischel LL, Young EW, Mader BR, Beebe DJ. Biomaterials. 2013;34:1471–7. doi: 10.1016/j.biomaterials.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shin Y, Jeon JS, Han S, Jung GS, Shin S, Lee SH, Sudo R, Kamm RD, Chung S. Lab Chip. 2011;11:2175–81. doi: 10.1039/c1lc20039a. [DOI] [PubMed] [Google Scholar]
- 27.Song JW, Bazou D, Munn LL. Integr Biol (Camb) 2012;4:857–62. doi: 10.1039/c2ib20061a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vickerman V, Kamm RD. Integr Biol (Camb) 2012;4:863–74. doi: 10.1039/c2ib00184e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wood LB, Ge R, Kamm RD, Asada HH. Integr Biol (Camb) 2012;4:1081–9. doi: 10.1039/c2ib20054f. [DOI] [PubMed] [Google Scholar]
- 30.Yeon JH, Ryu HR, Chung M, Hu QP, Jeon NL. Lab Chip. 2012;12:2815–22. doi: 10.1039/c2lc40131b. [DOI] [PubMed] [Google Scholar]
- 31.Chrobak KM, Potter DR, Tien J. Microvasc Res. 2006;71:185–96. doi: 10.1016/j.mvr.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 32.Golden AP, Tien J. Lab Chip. 2007;7:720–5. doi: 10.1039/b618409j. [DOI] [PubMed] [Google Scholar]
- 33.Grainger SJ, Putnam AJ. PLoS One. 2011;6:e22086. doi: 10.1371/journal.pone.0022086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hsu YH, Moya ML, Abiri P, Hughes CC, George SC, Lee AP. Lab Chip. 2013;13:81–9. doi: 10.1039/c2lc40787f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng Y, Chen J, Craven M, Choi NW, Totorica S, Diaz-Santana A, Kermani P, Hempstead B, Fischbach-Teschl C, Lopez JA, Stroock AD. Proc Natl Acad Sci U S A. 2012;109:9342–7. doi: 10.1073/pnas.1201240109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stratman AN, Schwindt AE, Malotte KM, Davis GE. Blood. 2010;116:4720–30. doi: 10.1182/blood-2010-05-286872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stratman AN, Malotte KM, Mahan RD, Davis MJ, Davis GE. Blood. 2009;114:5091–101. doi: 10.1182/blood-2009-05-222364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen MB, Whisler JA, Jeon JS, Kamm RD. Integr Biol (Camb) 2013;5:1262–71. doi: 10.1039/c3ib40149a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Whisler JA, Chen M, Kamm R. Tissue Eng Part C Methods. 2013 doi: 10.1089/ten.tec.2013.0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen F, Tan Z, Dong CY, Li X, Xie Y, Wu Y, Chen X, Guo S. Eur J Pharmacol. 2007;568:222–30. doi: 10.1016/j.ejphar.2007.04.047. [DOI] [PubMed] [Google Scholar]
- 41.Saif J, Schwarz TM, Chau DY, Henstock J, Sami P, Leicht SF, Hermann PC, Alcala S, Mulero F, Shakesheff KM, Heeschen C, Aicher A. Arterioscler Thromb Vasc Biol. 2010;30:1897–904. doi: 10.1161/ATVBAHA.110.207928. [DOI] [PubMed] [Google Scholar]
- 42.Zeng H, Li L, Chen JX. PLoS One. 2012;7:e35905. doi: 10.1371/journal.pone.0035905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim D, Haynes CL. Anal Chem. 2013 [Google Scholar]
- 44.Estrada R, Giridharan GA, Nguyen MD, Roussel TJ, Shakeri M, Parichehreh V, Prabhu SD, Sethu P. Anal Chem. 2011;83:3170–7. doi: 10.1021/ac2002998. [DOI] [PubMed] [Google Scholar]
- 45.Song JW, Gu W, Futai N, Warner KA, Nor JE, Takayama S. Anal Chem. 2005;77:3993–9. doi: 10.1021/ac050131o. [DOI] [PubMed] [Google Scholar]
- 46.Chung S, Sudo R, Vickerman V, Zervantonakis IK, Kamm RD. Ann Biomed Eng. 2010;38:1164–77. doi: 10.1007/s10439-010-9899-3. [DOI] [PubMed] [Google Scholar]
- 47.Shin Y, Han S, Jeon JS, Yamamoto K, Zervantonakis IK, Sudo R, Kamm RD, Chung S. Nat Protoc. 2012;7:1247–59. doi: 10.1038/nprot.2012.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lopa S, Mercuri D, Colombini A, De Conti G, Segatti F, Zagra L, Moretti M. J Biomed Mater Res A. 2013 doi: 10.1002/jbm.a.34649. [DOI] [PubMed] [Google Scholar]
- 49.Nilufar S, Morrow AA, Lee JM, Perkins TJ. BMC Syst Biol. 2013;7:66. doi: 10.1186/1752-0509-7-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Curry FE, Huxley VH, Adamson RH. Am J Physiol. 1983;245:H495–505. doi: 10.1152/ajpheart.1983.245.3.H495. [DOI] [PubMed] [Google Scholar]
- 51.Ball SG, Shuttleworth AC, Kielty CM. Int J Biochem Cell Biol. 2004;36:714–27. doi: 10.1016/j.biocel.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 52.Kobayashi N, Yasu T, Ueba H, Sata M, Hashimoto S, Kuroki M, Saito M, Kawakami M. Exp Hematol. 2004;32:1238–45. doi: 10.1016/j.exphem.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 53.Narita Y, Yamawaki A, Kagami H, Ueda M, Ueda Y. Cell Tissue Res. 2008;333:449–59. doi: 10.1007/s00441-008-0654-0. [DOI] [PubMed] [Google Scholar]
- 54.Jeon ES, Moon HJ, Lee MJ, Song HY, Kim YM, Bae YC, Jung JS, Kim JH. J Cell Sci. 2006;119:4994–5005. doi: 10.1242/jcs.03281. [DOI] [PubMed] [Google Scholar]
- 55.Hirschi KK, Rohovsky SA, D’Amore PA. J Cell Biol. 1998;141:805–14. doi: 10.1083/jcb.141.3.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P. EMBO J. 2002;21:1743–53. doi: 10.1093/emboj/21.7.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McDonald DM, Choyke PL. Nat Med. 2003;9:713–25. doi: 10.1038/nm0603-713. [DOI] [PubMed] [Google Scholar]
- 58.Fukuhara S, Sako K, Minami T, Noda K, Kim HZ, Kodama T, Shibuya M, Takakura N, Koh GY, Mochizuki N. Nat Cell Biol. 2008;10:513–26. doi: 10.1038/ncb1714. [DOI] [PubMed] [Google Scholar]
- 59.Richardson TP, Peters MC, Ennett AB, Mooney DJ. Nat Biotechnol. 2001;19:1029–34. doi: 10.1038/nbt1101-1029. [DOI] [PubMed] [Google Scholar]
- 60.Kim S, Lee H, Chung M, Jeon NL. Lab Chip. 2013;13:1489–500. doi: 10.1039/c3lc41320a. [DOI] [PubMed] [Google Scholar]
- 61.Zervantonakis IK, Hughes-Alford SK, Charest JL, Condeelis JS, Gertler FB, Kamm RD. Proc Natl Acad Sci U S A. 2012;109:13515–20. doi: 10.1073/pnas.1210182109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yuan W, Lv Y, Zeng M, Fu BM. Microvasc Res. 2009;77:166–73. doi: 10.1016/j.mvr.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 63.Frantz C, Stewart KM, Weaver VM. J Cell Sci. 2010;123:4195–200. doi: 10.1242/jcs.023820. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1 Perfusion of 10 μm fluorescent microbeads within a microvascular network composed of HUVECs and mural cell differentiated BM-hMSCs. A pressure drop was applied by aspirating the media from all the reservoirs of the microfluidic device and adding 40 μl microbead suspension within a single reservoir.
Fig. S1 Live/Dead assay showing cell viability at day 1. Cells were stained with 1 μl fluorescein diacetate (FDA, green; Sigma-Aldrich) and 3.5 μl propidium iodide (PI, red; Sigma-Aldrich) diluted in 1 ml phosphate buffered saline (PBS). Samples were incubated for 5 min, washed with PBS and observed through confocal microscopy.
Fig. S2 SM22α immunofluorescent staining highlighting BM-hMSC differentiation toward a mural cell lineage. Representative image showing red fluorescent protein (RFP)-transfected human umbilical vein endothelial cells (HUVECs) organized in a microvessel structure wrapped by differentiated BM-hMSCs (SM22α, green). Cell nuclei were stained with 4′6-Diamidino-2-Phenylindole (DAPI, blue).
Fig. S3 Confocal microscopy image representing mural cell differentiated BM-hMSCs (α-smooth muscle actin, green) co-localization with ECs (red). Capillary lumens are indicated by white arrowheads.
Fig. S4 Microvascular network analysis: number of branches. The 3D skeletonize plugin of the Fiji software was applied to compute the number of branches of the longest connected structure within each region of interest (ROI, 533x426 μm2). A 25 μm threshold was applied to filter 3D skeleton data (main text). Representative images of a confocal 3D reconstruction (A), a 2D skeleton obtained with the 2D skeletonize plugin (B) and a 3D volumetric skeleton (C). 3D data for the three different experimental conditions (addition of VEGF, VEGF+Ang-1 and VEGF+TGF-β1). Average values were obtained for a minimum of n=8 regions within 2 or 3 independent devices per condition (D). VEGF: vascular endothelial growth factor; Ang-1: angiopoietin-1; TGF-β1: transforming growth factor-β1.
Fig. S5 Vessel perfusion with 70 kDa fluorescent dextran revealing patent lumen and absence of focal leaks. Representative picture of a microvascular network composed by HUVECs and mural cell differentiated BM-hMSCs treated with VEGF and Ang-1.
3D confocal reconstruction of a representative microvessel stained with anti-VE-cadherin antibody (green). ECs (red) organized in a patent capillary appear tightly connected through a network of vascular adherens junctions. Cell nuclei were stained with DAPI (blue).