

Abstract
Dendrites and the dendritic spines of neurons play key roles in the connectivity of the brain and have been recognized as the locus of long-term synaptic plasticity, which is correlated with learning and memory. The development of dendrites and spines in the mammalian central nervous system is a complex process that requires specific molecular events over a period of time. It has been shown that specific molecules are needed not only at the spine's point of contact, but also at a distance, providing signals that initiate a cascade of events leading to synapse formation. The specific molecules that act to signal neuronal differentiation, dendritic morphology, and synaptogenesis are tightly regulated by genetic and epigenetic programs. It has been shown that the dendritic spine structure and distribution are altered in many diseases, including many forms of mental retardation (MR), and can also be potentiated by neuronal activities and an enriched environment. Because dendritic spine pathologies are found in many types of MR, it has been proposed that an inability to form normal spines leads to the cognitive and motor deficits that are characteristic of MR. Epigenetic mechanisms, including DNA methylation, chromatin remodeling, and the noncoding RNA-mediated process, have profound regulatory roles in mammalian gene expression. The study of epigenetics focuses on cellular effects that result in a heritable pattern of gene expression without changes to genomic encoding. Despite extensive efforts to understand the molecular regulation of dendrite and spine development, epigenetic mechanisms have only recently been considered. In this review, we will focus on epigenetic mechanisms that regulate the development and maturation of dendrites and spines. We will discuss how epigenetic alterations could result in spine abnormalities that lead to MR, such as is seen in fragile X and Rett syndromes. We will also discuss both general methodology and recent technological advances in the study of neuronal dendrites and spines.
Keywords: epigenetics, neurodevelopment, dendritic spine, synapse, microRNA, methyl-CpG binding protein 2 (MeCP2), mental retardation
1 Introduction: Dendrites and spines in development and diseases
1.1 Introduction to neuronal dendrites and dendritic spines
Santiago Ramon y Cajal, historically the founder of neuroscience and Nobel Prize winner, was the first to propose in the late 19th century that the nervous system is made up of individual neurons that are able to communicate with neighboring cells through long projecting axons and highly branched dendrites. He first described dendritic spines in 1891 as “the tips of charge or points of reception of impulses, their retraction would result in the individualization or disaggregation of neurons. The awake state would correspond to the swelling and lengthening of spines, while the resting state would correspond to the retraction of these appendages” (Cajal, 1891).
Cajal's observations were based on Golgi staining and a light microscope over 100 years ago. To date, significant advances in biology have confirmed that dendritic spines can undergo long-term modifications, such as changes in number and shape, in response to novel experiences, suggesting that dendritic spines are the locus of long-term synaptic plasticity associated with memory storage in the brain (Segal, 2005). In light of advances in genetic and molecular tools, scientists still ponder why dendrites and dendritic spines become grossly impaired in individuals faced with mental retardation and neuropathology.
The brain is composed of a complex network of neurons that communicate with each other through specialized cell junctions called synapses (Fig. 1A, B). Most of these synaptic junctions are chemical synapses, in which a chemical neurotransmitter is released by the axon of the presynaptic neuron. The neurotransmitter can diffuse into the synaptic cleft (space between synaptic contact), where it can act on the corresponding neurotransmitter receptors on the postsynaptic neuron (Fig. 1B). The synapses are typically found on the dendritic shaft, located on stubby spines and filipodia (long, thin dendritic protrusions), and are characterized by the clustering of certain proteins on the pre- and postsynaptic sites of contact. Dendritic spines have been classified into many types based on their shape. The most common types of spines in the central nervous system (CNS), and also the primary focus of pathological studies, are the following: mushroom-like spines with a bulbous head attached to the dendrite by a narrow neck, short and stubby spines with no neck, and long and thin spines with no head enlargement (Fiala et al., 2002). It is believed that long, thin spines are mostly immature spines, and they develop into the more mature mushroom-like spines (Fig. 2). During development, dendrites start out with no spines, and during synaptogenesis and neuronal maturation, filipodia emerge from the dendrites and form nascent synapses with axons (Fiala et al., 1998; Fiala et al., 2002). Mature synapses are gradually formed on mushroom-like spines that have a well-defined head (Harris and Kater, 1994), and the density of mature spines increases along the dendrite as the neuron matures and forms functional connections with the surrounding brain circuitry (Zhao et al., 2006)(Fig. 3). More about the specific molecular machinery found in the mature excitatory synapse will be discussed in the next section.
Fig. 1. Schematic diagram of a mature neuron and synapse.
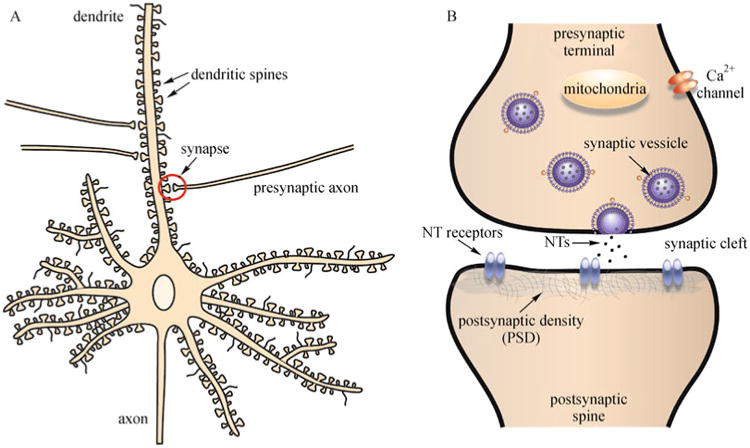
A: A mature neuron has elaborate processes that are composed of multiple dendrites and one axon. Dendrites contain a large number of dendritic spines that form contacts, or synapses, with other neurons. The synapses are typically found on the dendritic shaft, located on stubby spines and filipodia. B: A neuronal synapse is the point of contact between two neurons, a presynaptic neuron and a postsynaptic neuron. It is characterized by specific synaptic proteins located on the pre- and postsynaptic sites.
Fig. 2. Common dendritic spine types in the CNS.
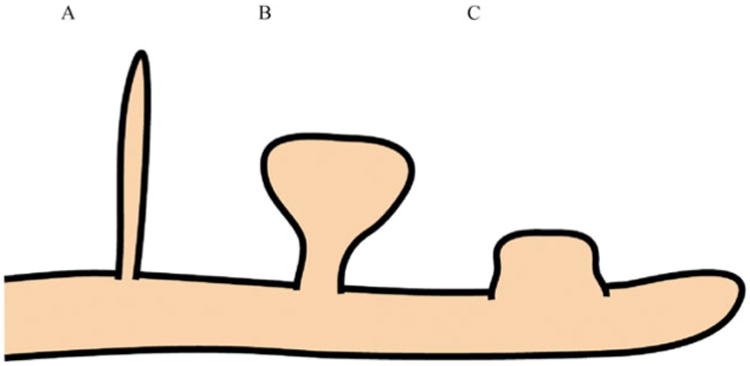
Three types of dendritic spines are commonly found in the CNS. A: the long and thin filipodia spine with no head enlargement; B: the mushroom-like spine with a bulbous head attached to the dendrite by a narrow neck; and C: the short and stubby spine with no neck. It is believed that long, thin spines are mostly immature spines, and they develop into the more mature mushroom-like spines. CNS: central nervous system.
Fig. 3. The stages of morphological development of dendrites and dendritic spines.
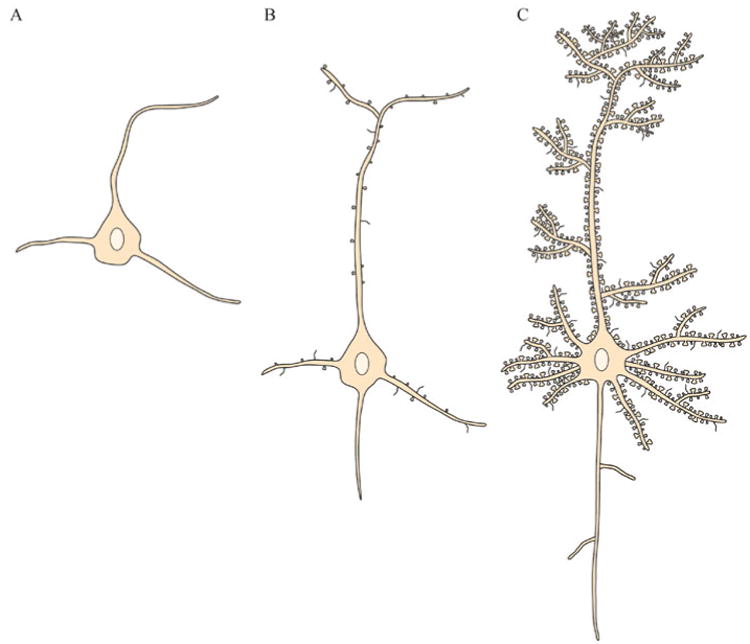
A: Immature neurons have shorter dendrites, and these dendrites have no spines. B: During synaptogenesis and neuronal maturation, neurons develop a more complex dendritic arbor, and these dendrites begin to form spines. Some of these spines start to receive input from the axons of other neurons. C: Mature neurons have elaborate dendritic trees and a high density of mature spines. These neurons form functional connections with other neurons and participate in the brain circuitry.
1.2 Activity-dependent modulation of gene expression controls dendrite and spine development
Glutamatergic neurons in the mammalian brain have elaborate dendrites covered with dendritic spines. These spines function as the primary sites of excitatory synaptic input for the neuron (Alvarez and Sabatini, 2007). The formation of an excitatory synapse in the CNS during development is initiated by the contact between the presynaptic axon and the postsynaptic dendrite, followed by the recruitment of pre- and postsynaptic proteins to the site of contact, and the stabilization of these interactions. Dendritic development and synapse formation are highly influenced by neuronal activities. Spine pruning occurs during early postnatal development, characterized by an experience-dependent loss of spines that selectively maintains spines of active synapses resulting in the appropriate maturation of neuronal circuitry (Grutzendler et al., 2002).
A major area of research in neurodevelopment is how neuronal activity-dependent modulation of gene expression affects dendritic development and synapse formation. It has been shown that dendritic and spine morphogenesis depends on proper neuronal development and activation of glutamate receptors, which maintain appropriate connections between neurons (Parrish et al., 2007b). Activation at the synapse leads to calcium influx into the dendrites of the postsynaptic neuron, which regulates the dendritic outgrowth of postsynaptic neurons. Calcium not only functions locally at the site of entry, but also leads to changes in gene transcription in the nucleus. Calcium influx through the N-methyl-D-aspartate (NMDA) receptor or voltage-sensitive calcium channels (VSCCs) during development can activate many signaling pathways, such as the calcium-sensitive calcium/calmodulin kinases (CaMKs) known to be important for signal transduction in neurons. For example, activated CaMKII regulates the number of α-amino-3-hydroxyl-5-methyl-4-isoxazolepropionate (AMPA) receptors at the synapse and the complexity of neuronal dendrites by influencing actin cytoskeleton (Dillon and Goda, 2005). In cultured neurons, CaMK activity initiates signaling to the nucleus, where activation of cAMP response element binding protein (CREB) leads to activity-dependent gene expression and subsequent dendritic morphological changes (Redmond et al., 2002; Wayman et al., 2006). Thus, intrinsic gene expression programs can be modified by neuronal activity to modulate spine morphogenesis and dendritic development (Cohen and Greenberg, 2008).
1.3 Dendritic spine pathology
Because spines are the key sites of synaptic input, altered spine morphology associated with pathological conditions may have a dramatic impact on the properties of the individual neuron, the neural networks, and mental function as a whole. In fact, dendritic spine distribution and structure is abnormal in many diseases and injuries, as well as many forms of mental retardation (MR) (Fig. 4). Thus, it has been proposed that the cognitive and motor deficits observed in MR may result from altered spine development and function. Understanding the effect of altered spines in pathological conditions will provide researchers with a better understanding of how spines contribute to normal synaptic conditions during development and learning in the adult brain.
Fig. 4. Pathological changes in spine density. Common pathological alterations in dendritic spines are altered spine density and abnormal spine shapes.
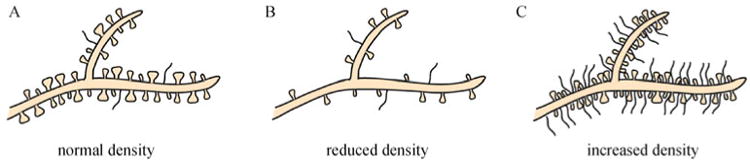
A: Healthy neuronal dendrites contain spines with a typical variation of spine types. B: Reduced spine density is common in many cognitive and developmental disorders, such as Rett syndrome. C: Increased spine density with abnormally increased long and thin types of spines is commonly found in patients with fragile X syndrome.
As summarized in Table 1, morphological dendritic spine abnormalities are found in many types of pathological conditions, including mental retardation such as is seen in autism spectrum disorder (Persico and Bourgeron, 2006), Rett syndrome (Zhou et al., 2006), and fragile X syndrome (Bagni and Greenough, 2005). Spine abnormalities are also found in schizophrenia (Lewis et al., 2003), depression, stress (Pittenger and Duman, 2008), drug addiction (Robinson and Kolb, 2004), epilepsy, and neurodegenerative disorders like Alzheimer's disease, Parkinson's disease (Day et al., 2006), and Huntington's disease (Spires et al., 2004).
Table 1. Spine pathology related to genetic mutations and mental retardation.
| disorder | genetic abnormality | spine pathology | reference |
|---|---|---|---|
| Rett syndrome | MECP2 mutation | reduced spine density, reduced dendritic complexity and maturation | Belichenko et al., 1994; Amir et al., 1999; Moretti et al., 2006; Smrt et al., 2007 |
| fragile X syndrome | FMR1 protein deficiency | increased spine density, abnormal spine morphology | Hinton et al., 1991; Wisniewski et al., 1991; Comery et al., 1997; Irwin et al., 2001 |
| Angelman syndrome | chromosome 15q11–q13, UBE3A | reduced spine density, abnormal spines, small RNA pathway | Miura et al., 2002; Lalande and Calciano, 2007; Ding et al., 2008 |
| Prader–Willi syndrome | chromosome 15q11–q13 | autism, reduced cognitive ability | Battaglia, 2005; Sahoo et al., 2008 |
| Down's syndrome | chromosome 21 trisomy | reduced spine density, reduced dendritic complexity | Marin-Padilla, 1972, 1976; Suetsugu and Mehraein, 1980; Takashima et al., 1981; Ferrer and Gullotta, 1990; Takashima et al., 1994 |
| Lafora disease | EPM2 mutation | reduced spine density | Busardz et al., 1987 |
| Patau syndrome | chromosome 13 trisomy | abnormal spine morphology | Marin-Padilla, 1972 |
| tuberous sclerosis | mutations of TSC1 or TCS2 genes | reduced spine density, abnormal spine morphology | Machado-Salas, 1984 |
| Niemann–Pick disease | deficiency of sphingomyelinase | reduced spine density, reduced dendritic complexity | Walkley and Baker, 1984; Higashi et al., 1993; Sarna et al., 2003 |
| Potocki–Lupski syndrome | duplications of 17p11.2 | autism | Potocki et al., 2007 |
| Smith–Magenis syndrome | chromosome 17p11.2, RA11 mutations | autism, abnormal brain anatomy | Slager et al., 2003; Elsea and Girirajan, 2008 |
| Williams–Beuren syndrome | 7q11.23 (CYLN2, LIMK1, FZD9) | reduced brain volume, altered spine morphology | Hoogenraad et al., 2002; Meng et al., 2002; Zhao et al., 2005; Berg et al., 2007; Lim et al., 2007 |
| 22q11.2 deletion syndrome and DiGeorge syndrome | deletion of 22q11.2, DGCR8 | miRNA pathway, autism, smaller spines, smaller dendrites | Lee and Lupski, 2006; Gothelf et al., 2007; Kobrynski and Sullivan, 2007; Stark et al., 2008 |
Aberrant dendritic spine development includes a broad range of changes in spine morphology and structure, such as increases or decreases in spine density, altered spine size or shape, dendritic beading with subsequent loss of spines, and ectopic spines in abnormal locations (Fiala et al., 2002). Since the shape and structure of a spine are closely associated with its function, the presence of abnormal spine morphology in many of these diseases suggests that the resulting cognitive phenotype is a result of dysfunctional spines. Understanding the effect of altered spines in pathological conditions will provide researchers with a better understanding of how spines contribute to normal synaptic conditions during development and learning in the adult brain.
The spine pathogenesis among various types of mental retardation is strikingly similar. These observations suggest that various genetic and epigenetic deficits related to mental retardation may be the result of abnormal dendrites and spines leading to the disruption of neural homeostasis and the ability of the brain to return to a set point following perturbation (Ramocki and Zoghbi, 2008). Interestingly, many of the mental retardation susceptibility genes encode proteins that regulate neuronal dendrite and spine development. This suggests that the molecular factors involved in behavioral and cognitive processes function in a tightly regulated homeostatic fashion. More specifically, single genes or noncoding RNAs do not encode specific cognitive processes, but instead encode biological processes. It is when a particular biological process, such as synaptic transmission, is disrupted during development that we can appreciate the resulting numerous neurological phenotypes.
Despite the fact that several protein pathways have been identified as critical players in spine development and pathology, the molecular pathogenesis of aberrant spine morphology in these diseases has yet to be clearly and comprehensively elucidated. It is apparent that complex programs of gene expression work to shape the developing nervous system. In this review, we will discuss the roles of epigenetic mechanisms in this important process. Revealing the roles of signaling factors and epigenetic gene regulators in dendritic spine pathologies will provide us with a better understanding of dendritic and spine disease and offer new approaches to treating neurodevelopmental disorders.
2 Epigenetic regulations are critical for neuronal dendritic development
2.1 Introduction to epigenetic regulation
In eukaryotic cells, genomic DNA exists in the form of chromatin and is tightly associated with histones and other chromatin proteins. Epigenetic regulation is defined as heritable changes in gene expression that are not coded within the DNA sequence itself. Epigenetic modulation of the genome involves three interacting systems, DNA methylation, histone modification (Egger et al., 2004; Li and Zhao, 2008), and noncoding RNA-mediated processes (Li and Zhao, 2008) (see Table 2). Recent literature has demonstrated that the phenotype of the cell is not only dependent on the genotype, but also the epigenotype. For example, DNA methylation within promoter regions (e.g., CpG islands) can result in heritable silencing of gene expression and has likely evolved as a host defense mechanism against viral sequences (Bestor and Tycko, 1996; Yoder et al., 1997). This type of epigenetic modulation can imprint dynamic environmental changes on a fixed genome, resulting in a stably transmitted alteration of phenotype, and has long been proposed to be an epigenetic silencing mechanism of fundamental importance (Holliday and Pugh, 1975; Riggs, 1975). The epigenotype shows far greater plasticity than the genotype during normal development, and disruption of these systems can lead to inappropriate expression or silencing of genes, resulting in “epigenetic diseases.” This section will review the epigenetic mechanisms that regulate dendrite and spine development and related diseases.
Table 2. Summary of epigenetic modifications and their direct effects on gene expression.
| epigenetic modification | target | direct effect on gene expression |
|---|---|---|
| DNA methylation | CpG dinucleotides | repression |
| H3 (K4, K36, K79) | activation | |
| histone methylation | H3 (K9, K27), H4 (K20) | repression |
| H3 (R17, R23), H4 (R3) | activation | |
| histone acetylation | H3 (K9, K14, K18, K56), H4 (K5, K8, K13, K16) | activation |
| microRNA | mRNA | repression |
2.1.1 DNA methylation
DNA methylation involves covalent modification of cytosine at position C5 in CpG dinucleotides. Over 70% of CpG dinucleotides in the mammalian genome are methylated, and most DNA methylation occurs at CpG dinucleotides. The exception is CpG islands, defined as more than 500 base pairs of sequence composed of 55% GC content. CpG islands are found in the promoters and the first exon of about 40% of the mammalian genes, and they are normally kept free of DNA methylation by mechanisms that are still unclear (Takai and Jones, 2002). Methylation of CpG islands is associated with stable heritable transcriptional silencing (Jones and Baylin, 2002). The methylation of CpG dinucleotides is catalyzed by several DNA methyltransferases (DNMTs). The de novo establishment of DNA methylation relies on DNMT3a and 3b and cofactor DNMT3L, whereas the maintenance of DNA methylation depends on DNMT1, which specifically recognizes semi-methylated DNA and methylates the remaining strand (Bestor, 2000; Robertson et al., 2000; Jaenisch and Bird, 2003). Mammalian DNA methylation has been implicated in a diverse range of cellular functions, including tissue-specific gene expression, cell differentiation, cell fate determination, genomic imprinting, and X chromosome inactivation (Bird, 2002). Here we focus on the function of DNA methylation in neurodevelopment.
Deletion of DNMT1 in neural progenitor cells leads to hypomethylation in neurons and subsequent abnormalities in synaptic maturation and function (Golshani et al., 2005; Hutnick et al., 2009). Although deletion of DNMT1 in neuronal precursors at E9-E10 leads to normal development through birth, it adversely affects neuronal survival after birth, resulting in death of the mice (Fan et al., 2001). Recently, Feng et al. have shown that DNMT1 and DNMT3a regulate synaptic function in neurons (Feng et al., 2010). Feng's group made use of conditional mutant mice that lacked Dnmt1, Dmnt3a, or both specifically in post-mitotic forebrain neurons of the postnatal mice. Their findings suggest that DNMT1 and DNMT3a are required for synaptic plasticity and learning and memory, likely due to their functions in maintaining DNA methylation and controlling gene expression in post-mitotic neurons. Although these studies provide sufficient evidence that DNMTs and DNA methylation are critical for neuronal development, the function of DNA methylation in postmitotic neurons of the adult brain is still for the most part unknown. DNMT1 is highly expressed in neurons, and the deletion of DNMT1 from post-mitotic neurons of postnatal brains leads to neuronal death (Fan et al., 2001), suggesting that DNMT1 may have other roles in addition to its methyltransferase activity. In fact, DNMT1 has been shown to form a complex with histone deacetylases (HDACs) and participates in transcriptional repression (Robertson et al., 2000; Jaenisch and Bird, 2003), suggesting that DNMTs may have complex roles in transcriptional regulation. Researchers have yet to identify specific genes affected by Dnmt deficiency, which impacts synaptic function, as well as learning and memory.
Despite the magnitude of literature on DNMTs and DNA methylation, much less is known about DNA demethylation. It has been proposed that the steady-state methylation of a particular gene is a dynamic equilibrium between methylase and demethylase activities. It has been found that methyl-CpG binding protein 2 (MBD2) has DNA demethylase activity (Bhattacharya et al., 1999; Detich et al., 2002). It has also been proposed that Gadd45a erases DNA methylation marks by DNA repair-mediated DNA demethylation (Barreto et al., 2007). More recently, Gadd45b was found to be important for the activity-induced demethylation of promoters and the expression of corresponding genes critical for adult neurogenesis, such as Bdnf and Fgf-1 (Ma et al., 2009). It was also shown that demethylation can take place in the absence of DNMT1 and DNMT3a in vivo (Feng et al., 2010). Although researchers have yet to elucidate the mechanisms of active DNA demethylation in the brain, it has been proposed that DNA oxidation and repair are possible mechanisms underlying this process.
DNA methylation represses gene transcription either through directly blocking the access of transcription factors to their binding sites or through indirectly recruiting MBDs. The MBD protein family consists of a growing number of DNA-binding proteins with the ability to recognize methylated CpGs in the genome. The MBD family includes at least MBD1, MBD2, MBD3, MBD4, MECP2, and Kaiso (Klose and Bird, 2006). In a sense, MBDs translate genomic CpG methylation into gene expression changes; therefore, these proteins are the central components of the DNA methylation pathway. DNA methylation is important in mammalian brain development (Chahrour and Zoghbi, 2007). One of the best examples of this occurs in Rett syndrome (RTT), an X-linked dominant pervasive neurodevelopmental disorder caused by de novo mutations in methyl-CpG binding protein 2 (MeCP2) (Amir et al., 1999). MeCP2 is thought to be involved in the structural conformation of chromatin. Once MeCP2 is bound to methylated DNA, MeCP2 recruits a complex of chromatin-remolding enzymes that help to condense the DNA surrounding the MeCP2 binding site, and silence transcription (Bird, 2002; Chahrour and Zoghbi, 2007). Mutations in MeCP2 lead to Rett syndrome, a severe neurodevelopmental disorder. As further discussed below, extensive evidence suggest that Mecp2 plays an important role in dendritic development and neuronal maturation.
2.1.2 Histone code
Histones are abundant nuclear proteins. Eukaryotic genomic DNA wraps around histones, which form the basic unit of chromatin, the nucleosome that consists of ∼147 base pairs of DNA wrapped around a core histone octamer (∼1.65 turns). Each histone octomer includes two copies of H2A, H2B, H3, and H4 histones, and all of them can undergo different types of post-translational modifications, such as acetylation, methylation, and phosphorylation (Lachner et al., 2003). The types and sites of histone modifications, the so-called “histone code,” have a significant impact on chromatin structure and gene expression.
Chromatin is present in two states: heterochromatin and euchromatin. Heterochromatin is in a condensed state that is repressive for gene transcription. When the chromatin is in an open state, called euchromatin, genes can be transcribed. Chromatin remodeling is a mechanism that alters the chromatin structure and functions to modulate DNA-protein interactions and gene activity without changing genomic DNA sequences. The chromatin structure and maintenance are not only critical for gene expression, but also for many cellular processes, such as chromosome segregation during mitosis and X-chromosome inactivation (Grewal and Elgin, 2007).
Recent genome-wide studies have demonstrated that histone modifications, as well as recruitment of other chromatin proteins, can be used as markers for gene expression state. Among histone modifications, lysine acetylation and methylation are the most characterized markers. For example, methylation of a histone at lysine 4 (H3K4), H3K36, or H3K79 is correlated with open chromatin and generally active gene transcription, whereas methylation of H3K9, H3K27, or H4K20 is correlated with condensed chromatin and gene inactivation (Santos-Rosa et al., 2002; Krogan et al., 2003; Schubeler et al., 2004). Additionally, mono-, di-, and trimethylation at the same lysine residues lead to different levels of gene activation or repression and are involved in distinct cellular pathways (Barski et al., 2007). Histone acetylation leads to less condensed chromatin structure and can be used to mark transcriptionally active regions. On the other hand, histone hypoacetylation is associated with more condensed heterochromatin and is used to mark transcriptionally inactive regions (Grewal and Elgin, 2007). Additionally, histones are subject to a number of other modifications, including phosphorylation, ubiqutinylation, etc. (Lachner et al., 2003). Thus, post-translational modifications of histones demonstrate a high degree of diversity and complexity and reflect the importance of the “histone code” in gene expression regulation.
The enzymes that catalyze histone modifications are critical components of the epigenome and play important roles during neurodevelopment. For example, studies of histone acetylation have focused on two opposing enzymes, histone acetyltransferases (HAT) that catalyze acetylation, and histone deacetylases (HDAC) that catalyze deacetylation. Transcription activators can either recruit HATs or utilize their own internal HAT domains (for example CREB binding protein, CBP) to catalyze histone acetylation to promote active chromatin structure. Conversely, transcription repressors can recruit HDACs that lead to histone deacetylation and gene repression. The opposing activities of HATs and HDACs are important for gene transcription regulation and therefore are tightly regulated during development. Alteration of these processes leads to developmental disorders, such as Rubinstein-Taybi syndrome, which results from heterozygote mutations of CBP. It is thus not surprising that HDAC inhibitors, such as valproic acid (VPA), have been developed and used to treat diseases, including psychiatric disorders (Phiel et al., 2001). VPA has been shown to increase neuronal differentiation in hippocampal neural progenitors (Hsieh et al., 2004). More recently, VPA has been shown to alter the morphology of motor cortex neurons in a rat model of autism (Snow et al., 2008) and affect neurite outgrowth in mouse neuroblastoma cells (Yamauchi et al., 2008; Yamauchi et al., 2009). Another HDAC inhibitor, trichostatin A (TSA), has also been shown to increase neuronal differentiation of neural stem cells (NSCs) and enhance the dendritic length and complexity of NSC-derived neurons (Balasubramaniyan et al., 2006). More recently, it was found that the neuronal abundant HDAC2 suppresses synapse formation and dendritic spine development of hippocampal neurons through its negative regulation of multiple neuronal genes. The authors demonstrated that HDAC2 functions to suppress synaptic plasticity and memory formation (Guan et al., 2009).
Another histone modification enzyme in the spotlight is enhancer of zeste homolog 2 (Ezh2), an H3-K27 histone methyltransferase and a part of the Polycomb group (PcG) protein complexes. PcG proteins and Ezh2 are important for neurogenesis and stem cell function in the brain (O'Carroll et al., 2001; Lee et al., 2006). PcG proteins are known to function in maintaining the bivalent chromatin state in stem cells (Boyer et al., 2006; Lee et al., 2006). For example, genes in embryonic stem cells related to differentiation contain both activating (methylated-H3K4) and repressing (trimethylated-H3K27) chromatin markers (Azuara et al., 2006; Bernstein et al., 2006). This bivalent chromatin state is believed to “prime” genes for expression, but “hold them in check” at the same time, therefore controlling the balance between proliferation and differentiation in embryonic stem cells (Bernstein et al., 2007). Recently PcG proteins have been shown to regulate dendritic arborization in Drosophila sensory neurons in a cell-autonomous manner (Parrish et al., 2007a, 2007b). We have recently found that Ezh2 expression is controlled by the crosstalk between MeCP2 and microRNAs (Szulwach et al., 2010), suggesting that aberrant regulation of histone methyltransferase could be involved in the mammalian neuronal development and pathogenesis of neurodevelopmental disorders.
Together, these studies provide evidence that histone modification and chromatin structure modulation play significant roles in the regulation of neuronal development and the formation of dendrites and spines.
2.1.3 Noncoding RNAs
The human genome contains only about 2% coding genes, whereas more than 80% of the genome is transcribed into RNA. Mounting evidence points to important roles for noncoding RNAs in gene expression regulation and cell phenotype determination (Li and Zhao, 2008). Micro-RNAs (miRNAs) are a class of small noncoding RNAs that are transcribed from the genome. Experimental evidence indicates that miRNAs function to modulate gene expression at the post-translational level by partial base-paring with the seed sequence located in the 3′ untranslated region (UTR) of the protein-coding mRNAs, leading to repression of translation efficiency or cleavage of the target mRNA (Filipowicz et al., 2008). miRNAs are expressed in many different tissues, particularly in the brain. The expression levels and patterns of miRNAs in the brain are dynamically regulated, suggesting that they play important roles in neuronal development (Bushati and Cohen, 2007; Barbato et al., 2008). One of the most studied brain specific miRNAs is miR-9. This miRNA has been of particular interest because it plays an important role in neurogenesis (Krichevsky et al., 2006), zebrafish brain patterning (Leucht et al., 2008), and its expression levels increase as neuronal precursors differentiate into the neurons. It was shown that miR-9 and miR-124 repress BAF53a, a process important for NSC proliferation and post-mitotic dendritic outgrowth (Yoo et al., 2009). Additionally, miR-9 regulates the expression of orphan nuclear receptor (Tlx), a gene important for self renewal of NSCs (Shi et al., 2004). Additionally, Tlx also regulates expression of miR-9, demonstrating a regulatory loop that functions to regulate neurogenesis (Zhao et al., 2009). Tlx is also targeted by Let-7b (Zhao et al., 2010), a member of the let-7 miRNA family. The let-7 miRNA family was one of the first families of miRNAs found to regulate stem cell function (Rybak et al., 2008; Liu and Zhao, 2009; Schwamborn et al., 2009). Additionally, let-7 is known to affect self-renewal and differentiation, and it has been shown to interact with the 3′UTR of high mobility group A2 (HMGA2), a known target of let-7. Together, let-7 and HMGA2 regulate the expression of p16Ink4a partially responsible for aged-dependent decline in self-renewal ability of brain NSCs (Nishino et al., 2008).
It has been shown previously that miRNAs are important for a number of cellular processes, such as differentiation, apoptosis, metabolism, and dendritic development (Brennecke et al., 2003; Johnston and Hobert, 2003; Chang et al., 2004; Chen et al., 2004; Poy et al., 2004). Loss of important components of the miRNA pathway, such as Dicer and DGCR8, can alter the proliferation and differentiation of stem cells (Kanellopoulou et al., 2005; Wang et al., 2007). Additionally, miRNAs have been demonstrated to play a role in the modulation of proliferation and differentiation of different stem cell types (Ivey et al., 2008; Szulwach et al., 2010).
One of the most exciting recent discoveries in neuroscience is that miRNAs have a role in synaptic plasticity. Learning and memory in the brain requires the synapse to undergo long-term modifications. It is believed that these changes require the local translation of factors important for synaptic function (Sutton and Schuman, 2005). Local protein synthesis at the dendritic spine involves transport of mRNAs to the dendritic compartment; however, we know little about how translation at the synaptic compartment is regulated. In Drosophila melanogaster, it has been shown that mRNAs important for synaptic plasticity are targets of the miRNA pathway (Ashraf et al., 2006). The authors have analyzed the brains of RISC mutants and demonstrated that protein translation of neuronal CaMKII is increased in dicer-, armitage-, or aubergine-mutant brains. At the same time, Schratt et al. were able to directly link a specific miRNA to dendrite and spine development in mammalian neurons (Schratt et al., 2006). First, they showed that miR-134 is localized in the dendrites of cultured mouse hippocampal neurons and is in dendritic spines that are apposed to synapsin-positive presynaptic terminals. Next, they found that overexpression of miR-134 leads to decreased spine size, whereas inhibition of endogenous miR-134 leads to increased spine size. Therefore miR-134 may act as a negative regulator of dendritic spine maturation. Recently, the same group found that miR-138 is important for dendritic patterning and spine morphogenesis. miR-138 is highly enriched in the brain and is localized in the dendrites. The authors revealed that miR-138 negatively regulates dendritic spine size in rat hippocampal neurons by controlling the protein translation of APT1 (Siegel et al., 2009). In addition, brain-specific miR-124 is localized at presynaptic terminals of Aplysia and regulates synaptic plasticity by regulating the transcription factor CREB (Rajasethupathy et al., 2009). Recently, a neuronal activity-dependent miRNA, miR-132, was shown to regulate dendritic development by targeting a Rho family GTPase-activating protein, p250GAP (Wayman et al., 2008). We have demonstrated that neuron-enriched miR-137 has a significant impact in vivo and in vitro on the maturation and dendritic morphogenesis of young hippocampal neurons by regulating the translation of Mib1, a ubiquitin ligase known to be important for neurogenesis and neurodevelopment (Smrt et al., 2010). Overall, these reports demonstrate that miRNAs have the capacity to modulate the expression of mRNAs important for dendrite and spine development (Fig. 5). The identification of these specific miRNAs that are important for dendritic development and synaptic plasticity are critical for our understanding of miRNAs in the CNS. New methods for detecting and quantifying miRNA at dendritic spines and synapses will help untangle the role of miRNAs in dendritic spine development and synaptic plasticity (see review Schratt, 2009).
Fig. 5. Epigenetic regulation of biogenesis and functions of miRNA in the neuron.
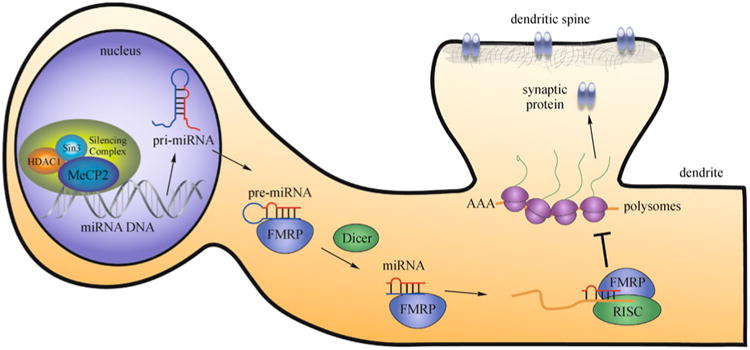
Transcription of miRNA, which can be regulated by epigenetic machinery, leads to the production of miRNA transcripts (pri-miRNAs), which are cleaved in the nucleus and exported into the cytoplasm as pre-miRNAs. The pre-miRNA is processed by the RNase Dicer and is likely associated with other accessory proteins, such as fragile X mental retardation protein (FMRP), to form an intermediate miRNA duplex. The leading strand of the miRNA duplex can then associate with the miRNA-induced silencing complex (miRISC), and then pair with sequences located on the 3′UTR of target mRNAs. This leads to translational repression of the target mRNA. Recent evidence suggests that RNA-binding proteins like FMRP interact with miRNAs to modulate mRNA translation. This example illustrates the effect of miRNA-mediated translational repression of proteins important for dendritic spine function.
2.2 Epigenetic diseases that affect dendritic spine development
Cognitive functions like learning and memory are extremely complex and require a precise balance of epigenetic and regulatory mechanisms. Extensive data support the link between epigenetic dysregulation of gene expression and neurodevelopmental disorders (Table 1). In this section, we will focus on two of the best-characterized epigenetic disorders with known dendritic pathology, Rett syndrome and fragile X syndrome.
2.2.1 Rett syndrome
Rett syndrome (RTT) is a neurodevelopmental disorder and an autism spectrum disorder. RTT is one of the most common forms of mental retardation in young females. RTT patients develop normally until 6–18 months of age, and thereafter experience a myriad of neurological deficits, including seizures, ataxia, and stereotypical hand movements. RTT is caused by loss-of-function mutations in MECP2, an X-linked gene encoding the MeCP2 protein, one of the MBDs. MeCP2 is a central player in epigenetic regulation by binding methylated DNA and recruiting factors, such as histone deacetylases, leading to the repression of gene expression (Amir et al., 1999; Bird, 2002) (Fig. 5).
Extensive evidence supports the role of MeCP2 in neuronal maturation (Hagberg et al., 1983; Smrt et al., 2007). The neurological symptoms of RTT appear after a seemingly normal embryonic development, suggesting that MeCP2 is not required for early developmental neurogenesis. However, MeCP2 expression coincides with a period of synaptogenesis (Akbarian et al., 2001; Shahbazian and Zoghbi, 2002; Zoghbi, 2003), suggesting that Mecp2-dependent epigenetic modulation of gene expression is important for the maturation and maintenance of neurons during brain development. Several mouse models have contributed to our understanding of the function of Mecp2 in neuronal and dendritic spine development (Bienvenu and Chelly, 2006; Chahrour and Zoghbi, 2007; Smrt et al., 2007). Expression studies in these animal models indicate that MeCP2 expression steadily increases during neuronal maturation, and is low or absent in immature neurons (Kishi and Macklis, 2004). Both RTT patients and Mecp2 mutant mice have excess immature neurons in the olfactory epithelium and reduced transition of immature neurons into mature neurons (Matarazzo et al., 2004; Smrt et al., 2007). Additionally, human postmortem tissues show less complex dendritic arborization, smaller soma size, decreased dendritic spine density (Fig. 4B), and lowered levels of the dendritic cytoskeletal protein microtubule-associated protein 2 (MAP2) in RTT brains (Kaufmann and Moser, 2000; Armstrong, 2002). On the other hand, exogenous Mecp2 expression leads to increased neurite complexity in cultured neurons (Jugloff et al., 2005; Smrt et al., 2007). Consistent with human pathology, adult Mecp2 mutant mice have smaller soma sizes and less complex dendrites in layer II/III pyramidal neurons in the cortex (Kishi and Macklis, 2004). Using retroviral labeling of new cells, we have shown that Mecp2-deficient mice have reduced dendritic spine density and exhibit delayed maturation of newborn dentate granule neurons (Smrt et al., 2007). On the other hand, both we and others have shown that exogenous MeCP2 expression can lead to increased neurite complexity in cultured neurons (Jugloff et al., 2005; Smrt et al., 2010), further suggesting an important role for MeCP2 in dendritic development.
Extensive efforts have been devoted to the identification of downstream effectors of MeCP2, but only a handful of them have been verified, including BDNF, DLX5, and DLX6 (Tudor et al., 2002; Chahrour and Zoghbi, 2007). Since these known targets cannot fully explain the deficits associated with MeCP2 deficiency, the identification of additional MeCP2 effector genes is needed to paint a full picture of the pathogenesis of RTT. Recently, epigenetic regulation of noncoding RNAs by MeCP2 has been suggested as a novel mechanism for understanding the pathogenesis of Rett syndrome. It was found that the miR-184 transcript is imprinted and exclusively expressed on the paternal allele and that MeCP2 binds upstream of miR-184 before neuronal depolarization, and releases after depolarization in an activity-dependent manner (Nomura et al., 2008). However, the miR-184 expression level is decreased in Mecp2 KO brains, which contradicts the authors' finding. It is possible that miR-184 is indirectly regulated by MeCP2. In fact, we have found that miR-184 is directly regulated by MBD1 (Liu et al., 2010). We have shown that MeCP2 can directly regulate the expression of a subset of miRNAs in primary neural progenitor cells isolated from postnatal brains. One of these miRNAs, miR-137, modulates neural progenitor cell (NPC) proliferation and differentiation by regulating protein translation of Ezh2, a histone H3K27 methyltransferease (Szulwach et al., 2010). In developing neurons, miR-137 represses dendrite and spine morphogenesis by targeting another neurodevelopmental factor, Mib-1 (Smrt et al., 2010). Therefore, it is conceivable that MeCP2 regulates neuronal dendrite and spine development through noncoding RNAs. Functional crosstalk between the DNA methylation pathway and small regulatory RNA could be an important and novel mechanism regulating mammalian neurodevelopment (Fig. 5).
2.2.2 Fragile X syndrome
Fragile X syndrome (FXS) is the most common cause of inherited mental retardation and is attributed to mutations in the X-linked FMR1 gene. Epigenetic mechanisms have been implicated in the molecular mechanisms of the disease (Graff and Mansuy, 2009). Typically, fragile X syndrome is caused by the expansion of a polymorphic CGG repeat in the 5′ UTR of the gene. If the CGG expansion reaches more than 200 repeats in female carriers, it is considered a full mutation. As a result, the repeat, the upstream CpG island, and the surrounding sequence become hypermethylated, and the gene is silenced (Oberle et al., 1991). One distinct characteristic of patients with FXS is that they have many more dendritic spines than normal individuals (Fig. 4C), and their spines are longer and thinner, resembling immature spines (Hinton et al., 1991; Irwin et al., 2001). It has been suggested this is due to misregulated development and elimination. This phenotype is also seen in the mouse model of FXS (Hinton et al., 1991; Comery et al., 1997; Greenough et al., 2001; Irwin et al., 2001; Nimchinsky et al., 2001). During dendritic spine development, synapses may be formed in excess numbers. Therefore, maturation and pruning are needed to establish the final synaptic pattern. In FXS, the activity-dependent events that lead to removal of excess or inappropriately placed synapses do not occur (Galvez et al., 2003).
Fragile X mental retardation protein (FMRP) is an RNA-binding protein, part of the heterogeneous nuclear ribonucleoproteins (hnRNPs), which function in many aspects of mRNA metabolism and biology, including nuclear export of mRNA and subcellular localization (Van de Bor and Davis, 2004). FMRP is part of a large protein complex that is involved in the transportation and translation of mRNA in neurons. Studies have shown that abnormal spines in FXS are associated with impaired neuronal plasticity; therefore, it has been suggested that FMRP may function to transport coding and noncoding RNA to the synapse and participate in local protein synthesis in dendrites (Fig. 5). Local protein synthesis in dendrites modulated by FMRP can potentially influence biochemical pathways or signaling cascades involved in spine morphogenesis, such as Rac1, MAP1B, CamKII, calbindin, and cadherins (Grossman et al., 2006; Penagarikano et al., 2007). The mechanism by which FMRP regulates translation of synaptic factors at the dendritic spine is still under investigation. It has also been shown that the bidirectional transport of the FMRP-mRNA complex between the soma to the dendrites and spines is driven by neuronal activity (Antar et al., 2004; Ling et al., 2004; Antar et al., 2005), indicative of multiple roles for FMRP in the activity-dependent regulation of gene expression at the dendritic spine.
It has been suggested that FMRP can associate with miRNAs and regulate the expression of a subset of target synaptic mRNAs (Jin et al., 2004; Vanderklish and Edelman, 2005; Penagarikano et al., 2007; Fiore et al., 2008) (Fig. 5). Recently, it was shown that a number of microRNAs, including miR125b and miR-132, can associate with FMRP in the mouse brain. While these miRNAs have opposing effects on dendritic spine morphology, knockdown of FMRP ameliorates the effects of overexpressing these microRNAs on spine morphology. The FMRP-associated miRNA 125b was found to target glutamate receptor subunit NR2A, suggesting FMRP-associated miRNAs may have a profound impact on synaptic plasticity, as well as on the pathophysiology of FXS (Edbauer et al., 2010). Although there is supporting evidence that FMRP collaborates with miRNAs to suppress the expression of genes important for dendritic spine morphology and synaptic plasticity, the details of how miRNA and FMRP interact are unclear. One proposed interaction is that miRNA and mRNA could act as the “kissing complex” RNA structure that has previously been proposed to bind the KH2 domain of FMRP (Bassell and Warren, 2008).
3 Methods for studying the epigenetic regulation of dendrite and spine development
3.1 Genetic and molecular methods
Significant advances in genetics and molecular biology have made a profound impact on our understanding of neurodevelopment and disease. Here we will summarize a few important methods.
3.1.1 Drosophila genetics
Drosophila neurogenetics was born in Seymour Benzer's lab at Caltech in the mid-1960s, when he proposed that genes can control behaviors in the fruit fly (Benzer, 1967). Benzer's trainees went on to isolate the shaker (sk) gene, which is a member of the transient receptor potential (trp) ion channel family (Papazian et al., 1987). A number of classical studies of brain functional genetics involved the use of loss-of-function approaches in Drosophila (Hotta and Benzer, 1970; Lin et al., 1998; Lush et al., 1998; Min and Benzer, 1999).
Many neurologic as well as other human diseases can be modeled using Drosophila to characterize genetic, epigenetic, and cellular pathways that lead to the disease state. Drosophila genetics are appealing to biologists because the tools of this trade are relatively simple and behavioral paradigms are well characterized, providing an extremely powerful method to go from mutant phenotypes to genotypes (forward genetics). Thus, the use of Drosophila models of human disease has grown rapidly and significantly contributed to our understanding of the human pathogenesis. The molecular and cellular pathways in Drosophila are generally considered to be highly conserved with vertebrates; approximately 75% of human genes known to be associated with disease correspond to a Drosophila ortholog (Reiter et al., 2001). Drosophila can be used to conduct assays ranging from genetic screens to drug target validation. For example, genetic modifier screens can identify proteins and genes that interact with pathways associated with pathology (Egger et al., 2004). Additionally, flies are excellent subjects for high-throughput drug screening libraries and are responsible for the identification of mGluR5 as a drug target for treating fragile X syndrome (Marsh and Thompson, 2006; Chang et al., 2008).
Drosophila has been used to understand the genetic pathways implicated in fragile X syndrome. The Drosophila genome contains the gene Drosophila fragile X-related (dfxr or dfmr1), which is homologous to FMR1 (Wan et al., 2000) and contains all the functional motifs related to FMRP. It was found that Drosophila genetics could be used to show dFXR plays a role in neuronal development, including synaptic formation, axonal growth, and dendritic development (Zhang et al., 2001; Dockendorff et al., 2002; Morales et al., 2002; Lee et al., 2003; Michel et al., 2004).
Drosophila has been used for studying epigenetic regulation related to histones and miRNAs. For example, dFMR1 is important in the miRNA pathway (Caudy et al., 2002; Ishizuka et al., 2002; Yang et al., 2007) and has been shown to modulate miR-124a levels, which are important for dendritic branching in Drosophila (Xu et al., 2008). However, CpG methylation is essentially absent in Drosophila, making it an invalid model for studying mammalian DNA methylation.
3.1.2 Genetic mouse models
Genetic manipulation of the mouse genome along with the characterization of mutant phenotypes has made a profound impact on our understanding of the pathophysiology of many human diseases. Mice and humans share 99% of their genes and have similar physiological and biochemical features, making genetic mouse models a powerful tool to study human disease (Rosenthal and Brown, 2007). This section will briefly describe the most common mouse models used to study Rett syndrome and fragile X syndrome, both implicated in abnormal spine development.
3.1.2.1 The MeCP2 mouse
To model Rett syndrome and related disorders in mice, three mouse models were generated with different Mecp2 genetic alterations. One of these models is the Mecp2 conditional KO mouse, which lacks either exon 3 or both exons 3 and 4 of the Mecp2 gene (Chen et al., 2001; Guy et al., 2001). These conditional knockout mice develop normally during the first 3–6 weeks of life, and thereafter develop motor dysfunction, hind limb clasping, and breathing irregularities. Mutant brains show reduced brain weight and more densely packed neurons, but do not show neuroanatomical abnormalities. The female mice, which are heterozygous for the Mecp2 mutation (Mecp2 +/−), show behavioral phenotypes that are less severe and with a later onset.
Another mouse model was generated by truncating Mecp2 at amino acid 308. This truncation resulted in a hypomorphic allele that contains a truncated C-terminal region, reminiscent of Rett syndrome patients found with C-terminal deletions (Shahbazian et al., 2002). These mice have many similarities with the conditional knockout model described above, but have a less severe phenotype that leads to longer longevity.
The third mouse model involves a two-fold over-expression of human MeCP2 that leads to an initial increase in synaptic plasticity and contextual learning; however, by 20 weeks of age the mice develop progressive neurological phenotypes, including motor abnormalities, hind limb clasping, and seizures, and they die by 1 year of age (Collins et al., 2004). This is also seen in the clinic, where duplication of MECP2 in human male patients causes severe mental retardation. Moreover, selective overexpression of MeCP2 in post-mitotic neurons, under the tau promoter, leads to a progressive neurological phenotype in mice (Luikenhuis et al., 2004).
In yet another mouse model, deletion of Mecp2 specifically in post-mitotic neurons using a CaMKII Cre transgene results in a phenotype resembling the Mecp2−/y knockout. This suggests that dysfunction of brain-specific MeCP2 leads to the neurological phenotype observed in Rett syndrome (Chen et al., 2001; Gemelli et al., 2006). Additionally, when Mecp2 is expressed in post-mitotic neurons under the tau promoter in null mice, the Rett phenotype is rescued, further supporting this idea (Luikenhuis et al., 2004). Taken together, these studies demonstrate that perturbations in the homeostatic balance of MeCP2 expression can result in aberrant neurological functioning.
3.1.2.2 The fragile X mouse
FMR1 is highly conserved between humans and mice (Ashley et al., 1993). The FMR1 knockout mouse was generated by disrupting exon 5 of the FMR1 gene by homologous recombination. This resulted in the absence of normal FMRP protein (Bakker et al., 1994). These mice show cognitive and anatomical impairments that are related to those seen in human patients, making the fragile X knockout mouse an important model to study the function of FMRP. The fragile X knockout mouse displays distinct phenotypes, such as hyperactivity, special learning deficits, macroorchidism, and altered dendritic spines (Bakker et al., 1994; Kooy, 2003). Among the similarities between the human pathology and the mouse model is cognitive function. It has been found that FMR1 KO mice have special learning deficits when challenged with the Morris water maze and radial arm maze (Bakker et al., 1994; Kooy et al., 1996; Mineur et al., 2002; Kooy, 2003), two tests generally considered to reflect problems in hippocampal functioning (Morris et al., 1982; Logue et al., 1997). We found that FMR1 KO mice exhibit impaired hippocampal neurogenesis, which may contribute to the deficits in hippocampus-dependent learning (Luo et al., 2010). Among structural abnormalities in the mouse model, FMR1 KO mice show macroorchidism due to increased Sertoli cell proliferation (Slegtenhorst-Eegdeman et al., 1998). No gross neuroanatomical differences are observed in human patients or fragile X mouse brains (Bakker et al., 1994); however, it is well appreciated that patients and mice have long, thin immature spines with increased spine density in dendrites of the cortex (Comery et al., 1997; Irwin et al., 2001). FRM1 KO mice also show a propensity for epileptic seizures. Seizures occur in fragile X patients (Hull and Hagerman, 1993) and can be elicited by auditory stimuli in knockout mice (Chen and Toth, 2001). Epileptic seizures in fragile X are likely due to dendritic spine abnormalities in mice and patients. Fragile X mice have been used for developing therapeutic treatments, though the results are still controversial. Pharmacological approaches have been proposed to compensate for the loss of FMRP (Kooy, 2003; Penagarikano et al., 2007). More work is required for a better understanding of the mechanism of how FMRP regulates mRNA translation, and how FMRP functions in activity-dependent local protein synthesis in the dendrite.
3.1.3 Isolation and RNA content analysis of synaptoneurosomes
Synaptoneurosomes are small vesicle structures that are prepared by subcellular fractionation of brain homogenate. These structures are known to be enriched in dendritic spines (Lugli et al., 2008). It has been shown that polyribosomes, as well as many mRNAs, are present in dendrites and are recruited into dendritic spines (Ostroff et al., 2002; Bourne et al., 2007). Gene chip analysis of postsynaptic density (PSD) fraction-associated mRNAs shows that mRNAs encoding many postsynaptic proteins are highly concentrated in PSD fractions (Suzuki et al., 2007). In addition, synaptoneurosomes also contain a large number of miRNAs. To characterize microRNA expression at the synapse, Lugli and collaborators isolated synaptic fractions from the mouse forebrain and analyzed microRNA expression using microarrays; enriched microRNAs were subsequently confirmed by quantitative RT-PCR (qRT-PCR) (Lugli et al., 2008). Lugli et al. found that a number of microRNAs were highly enriched in synaptoneurosomes and are predominately associated with PSDs. Their study further supports the role of miRNAs in protein synthesis at the synapse. It is likely that synaptosomes contain other types of RNAs and perhaps even DNA. With the development of next-generation sequencing, more synaptosome RNAs will be revealed, providing a better picture of the regulatory network governing dendrite and spine development.
3.2 Histological methods
3.2.1 The Golgi method
The Golgi method was discovered by Camillo Golgi and published in 1873 (Golgi, 1873); however, it was popularized by Ramon e Cajal, who modified the method to study a myriad of species and tissues during many developmental periods, including the brain (De Carlos and Borrell, 2007). Since then, the application of the Golgi technique in neurosciences has been used to study morphological abnormalities in dendrites that are typical in diseases associated with mental retardation. The pioneers of Golgi impregnations to study dendrites in MR were Huttenlocher (Huttenlocher, 1970, 1974) and Purpura (Purpura, 1975), who relied on post-mortem and biopsy material. Using the Golgi method, Parpura found cortical pyramidal neurons to have shorter and less complex dendritic branches in individuals with unclassified MR (Purpura, 1974). This dendritic “dysgenesis” was also observed in individuals with chromosomopathies and genetic disorders associated with MR (Marin-Padilla, 1972; Ferrer et al., 1984) (see Fig. 4). Golgi impregnation in Rett syndrome post-mortem tissue showed a reduction in the dendritic arborization of cortical neurons throughout life in RTT patients (Armstrong et al., 1995). Similar results were obtained using this method in an animal model of RTT lacking MeCP2 (Kishi and Macklis, 2004). In fragile X syndrome, Golgi preparations showed long dendritic spines (Hinton et al., 1991), and the FMR1 knockout mouse also shows spine dysgenesis (Comery et al., 1997).
3.2.2 Lipophilic dye
Lipophilic dyes are fluorescent substances that can be microinjected into a single cell and visualized using a confocal microscope to study neuronal morphology in both animals and humans (Belichenko et al., 1994). This method offers advantages over the Golgi method because that experimenters can select the neuron(s) that are to be infused with the dye and perform digital 3-D reconstruction of dendritic morphology with laser confocal imaging, instead of the traditional light microscope with Camera-Lucida method. Belichenco's group used the dye Lucifer Yellow and confocal microscopy to reconstruct the 3-D morphology of neurons in the prefrontal, motor, and temporal areas of RTT brain tissue. They showed reduced apical dendritic morphology and spine density, as well as dendritic segments lacking spines, which they called “naked dendrites,” in pyramidal neurons of the frontal cortex (Belichenko et al., 1994; Belichenko et al., 1997). Belichenko and his colleagues used this method to study the morphology of neurons in a number of diseases and injuries associated with spine pathology (Belichenko et al., 1994; Belichenko et al., 1997; Johansson and Belichenko, 2002; Belichenko et al., 2004).
3.2.3 The single-cell genetic approach
The use of retrovirus-mediated gene delivery, also known as the single-cell genetic approach, has made it possible to study the morphology and functional properties of newborn neurons throughout their lifetime (Gage, 2002; van Praag et al., 2002). Newborn neurons in the adult hippocampus have been used as a model to study neuronal development, because they recapitulate many features of embryonic hippocampal development (Song et al., 2005). When using the retroviral approach, newborn cells in the hippocampus are infected with retrovirus, causing them to express a live reporter, such as green fluorescent protein (GFP), throughout the cell, including the dendritic processes spines. This allows researchers to conduct a temporal analysis of the dendrite and spine development of newborn neurons by direct visualization of living newborn cells (Zhao et al., 2006). We used the single-cell genetic approach in an animal model of RTT to investigate the role of Mecp2 in developing neurons of the adult hippocampus (Smrt et al., 2007). We found that four-week-old new neurons in adult Mecp2 knockout animals showed reduced dendritic spine density, a characteristic feature of immature neurons suggesting that newborn dentate granule neurons lacking Mecp2 have impaired maturation. Using a similar approach, we demonstrate that miR-137 overexpression inhibits dendritic morphogenesis (Smrt et al., 2010). These data are consistent with similar findings using other methods described in this section to study dendritic spines under pathological conditions and support the idea that abnormal dendrite and spine development is the common point of vulnerability leading to the neurological deficits in diseases associated with mental retardation. The retrovirus-labeled GFP+ neurons can be further analyzed using immuno-electron microscopy to reveal synapse formation and structure (Duan et al., 2007; Toni et al., 2008).
4 Conclusions
In this review, we have discussed the role of epigenetics in dendritic and spine morphogenesis. Since Cajal's epic contributions to neuroscience in the late 19th and early 20th centuries, contemporary science and advances in genetic and molecular tools have enabled researchers to look deeper into the underlying mechanisms of dendritic and synaptic development. The advent of epigenetic research has provided much new knowledge and also opened up more questions regarding the role of gene expression regulation in neurodevelopment. Our focus as researchers is to use our growing knowledge of molecular biology and genetics to understand how dendrites and dendritic spines become grossly impaired in individuals faced with mental retardation and neuropathology.
Recent studies have shown that mutations in the epigenetic machinery lead to many forms of mental retardation disorders. Synaptic plasticity, as well as learning and memory, is believed to depend on proper neuron-neuron communication, and identification of the genes involved in synaptic development is crucial to understanding these human disorders.
The important challenge for both scientists and clinicians is to identify the molecular basis for cognitive and behavioral symptoms in order to design adequate pharmacological treatments. A number of epigenetic-based therapeutic methods have been proposed as treatments for epigenetic-associated diseases. Many pharmacological agents have been discovered that alter methylation patterns on DNA and histones, many of which are being extensively tested in vitro and in clinical trials (Egger et al., 2004). However, epigenetic reagents, such as HDAC or DNA methylation inhibitors, have met with concerns about the specificity of the drug's action. MicroRNAs have recently been identified as promising candidates for the treatment of diseases, such as heart disease and cancers. The challenge is to identify the specific miRNA(s) that could be considered treatment targets and to deliver that miRNA or miRNA inhibitor precisely to the target cells with high efficiency and without side effects.
Acknowledgments
We would like to thank Ms C. T. Strauss for editing the manuscript. This work is supported by grants from the International Rett Syndrome Foundation (IRSF) and the NIH (Nos: MH080434 and MH078972). R.D.S. is supported by a Minority Supplement to NIH grant (No. MH080434).
References
- Akbarian S, Chen RZ, Gribnau J, Rasmussen TP, Fong H, Jaenisch R, Jones EG. Expression pattern of the Rett syndrome gene MeCP2 in primate prefrontal cortex. Neurobiol Dis. 2001;8(5):784–791. doi: 10.1006/nbdi.2001.0420. [DOI] [PubMed] [Google Scholar]
- Alvarez VA, Sabatini BL. Anatomical and physiological plasticity of dendritic spines. Annu Rev Neurosci. 2007;30:79–97. doi: 10.1146/annurev.neuro.30.051606.094222. [DOI] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23(2):185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Antar LN, Afroz R, Dictenberg JB, Carroll RC, Bassell GJ. Metabotropic glutamate receptor activation regulates fragile x mental retardation protein and FMR1 mRNA localization differentially in dendrites and at synapses. J Neurosci. 2004;24(11):2648–2655. doi: 10.1523/JNEUROSCI.0099-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antar LN, Dictenberg JB, Plociniak M, Afroz R, Bassell GJ. Localization of FMRP-associated mRNA granules and requirement of microtubules for activity-dependent trafficking in hippocampal neurons. Genes Brain Behav. 2005;4(6):350–359. doi: 10.1111/j.1601-183X.2005.00128.x. [DOI] [PubMed] [Google Scholar]
- Armstrong D, Dunn JK, Antalffy B, Trivedi R. Selective dendritic alterations in the cortex of Rett syndrome. J Neuropathol Exp Neurol. 1995;54(2):195–201. doi: 10.1097/00005072-199503000-00006. [DOI] [PubMed] [Google Scholar]
- Armstrong DD. Neuropathology of Rett syndrome. Ment Retard Dev Disabil Res Rev. 2002;8(2):72–76. doi: 10.1002/mrdd.10027. [DOI] [PubMed] [Google Scholar]
- Ashley CT, Sutcliffe JS, Kunst CB, Leiner HA, Eichler EE, Nelson DL, Warren ST. Human and murine FMR-1: alternative splicing and translational initiation downstream of the CGG-repeat. Nat Genet. 1993;4(3):244–251. doi: 10.1038/ng0793-244. [DOI] [PubMed] [Google Scholar]
- Ashraf SI, McLoon AL, Sclarsic SM, Kunes S. Synaptic protein synthesis associated with memory is regulated by the RISC pathway in Drosophila. Cell. 2006;124(1):191–205. doi: 10.1016/j.cell.2005.12.017. [DOI] [PubMed] [Google Scholar]
- Azuara V, Perry P, Sauer S, Spivakov M, Jørgensen HF, John RM, Gouti M, Casanova M, Warnes G, Merkenschlager M, Fisher AG. Chromatin signatures of pluripotent cell lines. Nat Cell Biol. 2006;8(5):532–538. doi: 10.1038/ncb1403. [DOI] [PubMed] [Google Scholar]
- Bagni C, Greenough WT. From mRNP trafficking to spine dysmorphogenesis: the roots of fragile X syndrome. Nat Rev Neurosci. 2005;6(5):376–387. doi: 10.1038/nrn1667. [DOI] [PubMed] [Google Scholar]
- Bakker CE, Verheij C, Willemsen R, Helm R, Oerlemans F, Vermey M, Bygrave A, Hoogeveen A, Oostr BA, Reyniers E, De Boule K, D'Hooge R, Cras P, van Velzen D, Nagels G, Martin JJ, De Deyn PP, Darby JK, Willems PJ. Fmr1 knockout mice: a model to study fragile X mental retardation. Cell. 1994;78(1):23–33. [PubMed] [Google Scholar]
- Balasubramaniyan V, Boddeke E, Bakels R, Küst B, Kooistra S, Veneman A, Copray S. Effects of histone deacetylation inhibition on neuronal differentiation of embryonic mouse neural stem cells. Neuroscience. 2006;143(4):939–951. doi: 10.1016/j.neuroscience.2006.08.082. [DOI] [PubMed] [Google Scholar]
- Barbato C, Giorgi C, Catalanotto C, Cogoni C. Thinking about RNA? MicroRNAs in the brain. Mamm Genome. 2008;19(7–8):541–551. doi: 10.1007/s00335-008-9129-6. [DOI] [PubMed] [Google Scholar]
- Barreto G, Schäfer A, Marhold J, Stach D, Swaminathan SK, Handa V, Döderlein G, Maltry N, Wu W, Lyko F, Niehrs C. Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature. 2007;445(7128):671–675. doi: 10.1038/nature05515. [DOI] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129(4):823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Bassell GJ, Warren ST. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron. 2008;60(2):201–214. doi: 10.1016/j.neuron.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia A. The inv dup(15) or idic(15) syndrome: a clinically recognisable neurogenetic disorder. Brain Dev. 2005;27(5):365–369. doi: 10.1016/j.braindev.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Belichenko PV, Hagberg B, Dahlström A. Morphological study of neocortical areas in Rett syndrome. Acta Neuropathol. 1997;93(1):50–61. doi: 10.1007/s004010050582. [DOI] [PubMed] [Google Scholar]
- Belichenko PV, Masliah E, Kleschevnikov AM, Villar AJ, Epstein CJ, Salehi A, Mobley WC. Synaptic structural abnormalities in the Ts65Dn mouse model of Down Syndrome. J Comp Neurol. 2004;480(3):281–298. doi: 10.1002/cne.20337. [DOI] [PubMed] [Google Scholar]
- Belichenko PV, Oldfors A, Hagberg B, Dahlström A. Rett syndrome: 3-D confocal microscopy of cortical pyramidal dendrites and afferents. Neuroreport. 1994;5(12):1509–1513. [PubMed] [Google Scholar]
- Benzer S. Behavioral mutants of drosophila isolated by countercurrent distribution. Proc Natl Acad Sci U S A. 1967;58(3):1112–1119. doi: 10.1073/pnas.58.3.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg JS, Brunetti-Pierri N, Peters SU, Kang SH, Fong CT, Salamone J, Freedenberg D, Hannig VL, Prock LA, Miller DT, Raffalli P, Harris DJ, Erickson RP, Cunniff C, Clark GD, Blazo MA, Peiffer DA, Gunderson KL, Sahoo T, Patel A, Lupski JR, Beaudet AL, Cheung SW. Speech delay and autism spectrum behaviors are frequently associated with duplication of the 7q11.23 Williams-Beuren syndrome region. Genet Med. 2007;9(7):427–441. doi: 10.1097/gim.0b013e3180986192. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128(4):669–681. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, Jaenisch R, Wagschal A, Feil R, Schreiber SL, Lander ES. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125(2):315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- Bestor TH. The DNA methyltransferases of mammals. Hum Mol Genet. 2000;9(16):2395–2402. doi: 10.1093/hmg/9.16.2395. [DOI] [PubMed] [Google Scholar]
- Bestor TH, Tycko B. Creation of genomic methylation patterns. Nat Genet. 1996;12(4):363–367. doi: 10.1038/ng0496-363. [DOI] [PubMed] [Google Scholar]
- Bhattacharya SK, Ramchandani S, Cervoni N, Szyf M. A mammalian protein with specific demethylase activity for mCpG DNA. Nature. 1999;397(6720):579–583. doi: 10.1038/17533. [DOI] [PubMed] [Google Scholar]
- Bienvenu T, Chelly J. Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nat Rev Genet. 2006;7(6):415–426. doi: 10.1038/nrg1878. [DOI] [PubMed] [Google Scholar]
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16(1):6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- Bourne JN, Sorra KE, Hurlburt J, Harris KM. Polyribosomes are increased in spines of CA1 dendrites 2 h after the induction of LTP in mature rat hippocampal slices. Hippocampus. 2007;17(1):1–4. doi: 10.1002/hipo.20238. [DOI] [PubMed] [Google Scholar]
- Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, Bell GW, Otte AP, Vidal M, Gifford DK, Young RA, Jaenisch R. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441(7091):349–353. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- Brennecke J, Hipfner DR, Stark A, Russell RB, Cohen SM. bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell. 2003;113(1):25–36. doi: 10.1016/s0092-8674(03)00231-9. [DOI] [PubMed] [Google Scholar]
- Busard HL, Renier WO, Gabreëls FJ, Jaspar HH, Slooff JL, Janssen AJ, Van Haelst UJ. Lafora disease: a quantitative morphological and biochemical study of the cerebral cortex. Clin Neuropathol. 1987;6(1):1–6. [PubMed] [Google Scholar]
- Bushati N, Cohen SM. microRNA functions. Annu Rev Cell Dev Biol. 2007;23:175–205. doi: 10.1146/annurev.cellbio.23.090506.123406. [DOI] [PubMed] [Google Scholar]
- Cajal SR. Sur la structure de l'ecorce cérébrale de quelques mammifères. Cellule. 1891;7:125–176. [Google Scholar]
- Caudy AA, Myers M, Hannon GJ, Hammond SM. Fragile X-related protein and VIG associate with the RNA interference machinery. Genes Dev. 2002;16(19):2491–2496. doi: 10.1101/gad.1025202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56(3):422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Chang S, Bray SM, Li Z, Zarnescu DC, He C, Jin P, Warren ST. Identification of small molecules rescuing fragile X syndrome phenotypes in Drosophila. Nat Chem Biol. 2008;4(4):256–263. doi: 10.1038/nchembio.78. [DOI] [PubMed] [Google Scholar]
- Chang S, Johnston RJ, Jr, Frøkjaer-Jensen C, Lockery S, Hobert O. MicroRNAs act sequentially and asymmetrically to control chemosensory laterality in the nematode. Nature. 2004;430(7001):785–789. doi: 10.1038/nature02752. [DOI] [PubMed] [Google Scholar]
- Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303(5654):83–86. doi: 10.1126/science.1091903. [DOI] [PubMed] [Google Scholar]
- Chen L, Toth M. Fragile X mice develop sensory hyperreactivity to auditory stimuli. Neuroscience. 2001;103(4):1043–1050. doi: 10.1016/s0306-4522(01)00036-7. [DOI] [PubMed] [Google Scholar]
- Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27(3):327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- Cohen S, Greenberg ME. Communication between the synapse and the nucleus in neuronal development, plasticity, and disease. Annu Rev Cell Dev Biol. 2008;24:183–209. doi: 10.1146/annurev.cellbio.24.110707.175235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins AL, Levenson JM, Vilaythong AP, Richman R, Armstrong DL, Noebels JL, David Sweatt J, Zoghbi HY. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum Mol Genet. 2004;13(21):2679–2689. doi: 10.1093/hmg/ddh282. [DOI] [PubMed] [Google Scholar]
- Comery TA, Harris JB, Willems PJ, Oostra BA, Irwin SA, Weiler IJ, Greenough WT. Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc Natl Acad Sci U S A. 1997;94(10):5401–5404. doi: 10.1073/pnas.94.10.5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day M, Wang Z, Ding J, An X, Ingham CA, Shering AF, Wokosin D, Ilijic E, Sun Z, Sampson AR, Mugnaini E, Deutch AY, Sesack SR, Arbuthnott GW, Surmeier DJ. Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat Neurosci. 2006;9(2):251–259. doi: 10.1038/nn1632. [DOI] [PubMed] [Google Scholar]
- De Carlos JA, Borrell J. A historical reflection of the contributions of Cajal and Golgi to the foundations of neuroscience. Brain Res Rev. 2007;55(1):8–16. doi: 10.1016/j.brainresrev.2007.03.010. [DOI] [PubMed] [Google Scholar]
- Detich N, Theberge J, Szyf M. Promoter-specific activation and demethylation by MBD2/demethylase. J Biol Chem. 2002;277(39):35791–35794. doi: 10.1074/jbc.C200408200. [DOI] [PubMed] [Google Scholar]
- Dillon C, Goda Y. The actin cytoskeleton: integrating form and function at the synapse. Annu Rev Neurosci. 2005;28:25–55. doi: 10.1146/annurev.neuro.28.061604.135757. [DOI] [PubMed] [Google Scholar]
- Ding F, Li HH, Zhang S, Solomon NM, Camper SA, Cohen P, Francke U. SnoRNA Snord116 (Pwcr1/MBII-85) deletion causes growth deficiency and hyperphagia in mice. PLoS One. 2008;3:e1709. doi: 10.1371/journal.pone.0001709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dockendorff TC, Su HS, McBride SM, Yang Z, Choi CH, Siwicki KK, Sehgal A, Jongens TA. Drosophila lacking dfmr1 activity show defects in circadian output and fail to maintain courtship interest. Neuron. 2002;34(6):973–984. doi: 10.1016/s0896-6273(02)00724-9. [DOI] [PubMed] [Google Scholar]
- Duan X, Chang JH, Ge S, Faulkner RL, Kim JY, Kitabatake Y, Liu XB, Yang CH, Jordan JD, Ma DK, Liu CY, Ganesan S, Cheng HJ, Ming GL, Lu B, Song H. Disrupted-In-Schizophrenia 1 regulates integration of newly generated neurons in the adult brain. Cell. 2007;130(6):1146–1158. doi: 10.1016/j.cell.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edbauer D, Neilson JR, Foster KA, Wang CF, Seeburg DP, Batterton MN, Tada T, Dolan BM, Sharp PA, Sheng M. Regulation of synaptic structure and function by FMRP-associated microRNAs miR-125b and miR-132. Neuron. 2010;65(3):373–384. doi: 10.1016/j.neuron.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429(6990):457–463. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- Elsea SH, Girirajan S. Smith-Magenis syndrome. Eur J Hum Genet. 2008;16(4):412–421. doi: 10.1038/sj.ejhg.5202009. [DOI] [PubMed] [Google Scholar]
- Fan G, Beard C, Chen RZ, Csankovszki G, Sun Y, Siniaia M, Biniszkiewicz D, Bates B, Lee PP, Kuhn R, Trumpp A, Poon C, Wilson CB, Jaenisch R. DNA hypomethylation perturbs the function and survival of CNS neurons in postnatal animals. J Neurosci. 2001;21(3):788–797. doi: 10.1523/JNEUROSCI.21-03-00788.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Zhou Y, Campbell SL, Le T, Li E, Sweatt JD, Silva AJ, Fan G. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat Neurosci. 2010;13(4):423–430. doi: 10.1038/nn.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Fabregues I, Coll J, Ribalta T, Rives A. Tuberous sclerosis: a Golgi study of cortical tuber. Clin Neuropathol. 1984;3(2):47–51. [PubMed] [Google Scholar]
- Ferrer I, Gullotta F. Down's syndrome and Alzheimer's disease: dendritic spine counts in the hippocampus. Acta Neuropathol. 1990;79(6):680–685. doi: 10.1007/BF00294247. [DOI] [PubMed] [Google Scholar]
- Fiala JC, Feinberg M, Popov V, Harris KM. Synaptogenesis via dendritic filopodia in developing hippocampal area CA1. J Neurosci. 1998;18(21):8900–8911. doi: 10.1523/JNEUROSCI.18-21-08900.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala JC, Spacek J, Harris KM. Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Brain Res Rev. 2002;39(1):29–54. doi: 10.1016/s0165-0173(02)00158-3. [DOI] [PubMed] [Google Scholar]
- Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008;9(2):102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- Fiore R, Siegel G, Schratt G. MicroRNA function in neuronal development, plasticity and disease. Biochim Biophys Acta. 2008;1779(8):471–478. doi: 10.1016/j.bbagrm.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Gage FH. Neurogenesis in the adult brain. J Neurosci. 2002;22(3):612–613. doi: 10.1523/JNEUROSCI.22-03-00612.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvez R, Gopal AR, Greenough WT. Somatosensory cortical barrel dendritic abnormalities in a mouse model of the fragile X mental retardation syndrome. Brain Res. 2003;971(1):83–89. doi: 10.1016/s0006-8993(03)02363-1. [DOI] [PubMed] [Google Scholar]
- Gemelli T, Berton O, Nelson ED, Perrotti LI, Jaenisch R, Monteggia LM. Postnatal loss of methyl-CpG binding protein 2 in the forebrain is sufficient to mediate behavioral aspects of Rett syndrome in mice. Biol Psychiatry. 2006;59(5):468–476. doi: 10.1016/j.biopsych.2005.07.025. [DOI] [PubMed] [Google Scholar]
- Golshani P, Hutnick L, Schweizer F, Fan G. Conditional Dnmt1 deletion in dorsal forebrain disrupts development of somatosensory barrel cortex and thalamocortical long-term potentiation. Thalamus Relat Syst. 2005;3(3):227–233. doi: 10.1017/S1472928807000222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gothelf D, Feinstein C, Thompson T, Gu E, Penniman L, Van Stone E, Kwon H, Eliez S, Reiss AL. Risk factors for the emergence of psychotic disorders in adolescents with 22q11.2 deletion syndrome. Am J Psychiatry. 2007;164(4):663–669. doi: 10.1176/ajp.2007.164.4.663. [DOI] [PubMed] [Google Scholar]
- Gräff J, Mansuy IM. Epigenetic dysregulation in cognitive disorders. Eur J Neurosci. 2009;30(1):1–8. doi: 10.1111/j.1460-9568.2009.06787.x. [DOI] [PubMed] [Google Scholar]
- Greenough WT, Klintsova AY, Irwin SA, Galvez R, Bates KE, Weiler IJ. Synaptic regulation of protein synthesis and the fragile X protein. Proc Natl Acad Sci U S A. 2001;98(13):7101–7106. doi: 10.1073/pnas.141145998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal SI, Elgin SC. Transcription and RNA interference in the formation of heterochromatin. Nature. 2007;447(7143):399–406. doi: 10.1038/nature05914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman AW, Aldridge GM, Weiler IJ, Greenough WT. Local protein synthesis and spine morphogenesis: Fragile X syndrome and beyond. J Neurosci. 2006;26(27):7151–7155. doi: 10.1523/JNEUROSCI.1790-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grutzendler J, Kasthuri N, Gan WB. Long-term dendritic spine stability in the adult cortex. Nature. 2002;420(6917):812–816. doi: 10.1038/nature01276. [DOI] [PubMed] [Google Scholar]
- Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, Tsai LH. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459(7243):55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27(3):322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- Hagberg B, Aicardi J, Dias K, Ramos O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett's syndrome: report of 35 cases. Ann Neurol. 1983;14(4):471–479. doi: 10.1002/ana.410140412. [DOI] [PubMed] [Google Scholar]
- Harris KM, Kater SB. Dendritic spines: cellular specializations imparting both stability and flexibility to synaptic function. Annu Rev Neurosci. 1994;17:341–371. doi: 10.1146/annurev.ne.17.030194.002013. [DOI] [PubMed] [Google Scholar]
- Higashi Y, Murayama S, Pentchev PG, Suzuki K. Cerebellar degeneration in the Niemann-Pick type C mouse. Acta Neuropathol. 1993;85(2):175–184. doi: 10.1007/BF00227765. [DOI] [PubMed] [Google Scholar]
- Hinton VJ, Brown WT, Wisniewski K, Rudelli RD. Analysis of neocortex in three males with the fragile X syndrome. Am J Med Genet. 1991;41(3):289–294. doi: 10.1002/ajmg.1320410306. [DOI] [PubMed] [Google Scholar]
- Holliday R, Pugh JE. DNA modification mechanisms and gene activity during development. Science. 1975;187(4173):226–232. [PubMed] [Google Scholar]
- Hoogenraad CC, Koekkoek B, Akhmanova A, Krugers H, Dortland B, Miedema M, van Alphen A, Kistler WM, Jaegle M, Koutsourakis M, Van Camp N, Verhoye M, van der Linden A, Kaverina I, Grosveld F, De Zeeuw CI, Galjart N. Targeted mutation of Cyln2 in the Williams syndrome critical region links CLIP-115 haploinsufficiency to neurodevelopmental abnormalities in mice. Nat Genet. 2002;32(1):116–127. doi: 10.1038/ng954. [DOI] [PubMed] [Google Scholar]
- Hotta Y, Benzer S. Genetic dissection of the Drosophila nervous system by means of mosaics. Proc Natl Acad Sci U S A. 1970;67(3):1156–1163. doi: 10.1073/pnas.67.3.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh J, Nakashima K, Kuwabara T, Mejia E, Gage FH. Histone deacetylase inhibition-mediated neuronal differentiation of multipotent adult neural progenitor cells. Proc Natl Acad Sci U S A. 2004;101(47):16659–16664. doi: 10.1073/pnas.0407643101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull C, Hagerman RJ. A study of the physical, behavioral, and medical phenotype, including anthropometric measures, of females with fragile X syndrome. Am J Dis Child. 1993;147(11):1236–1241. doi: 10.1001/archpedi.1993.02160350110017. [DOI] [PubMed] [Google Scholar]
- Hutnick LK, Golshani P, Namihira M, Xue Z, Matynia A, Yang XW, Silva AJ, Schweizer FE, Fan G. DNA hypomethylation restricted to the murine forebrain induces cortical degeneration and impairs postnatal neuronal maturation. Hum Mol Genet. 2009;18(15):2875–2888. doi: 10.1093/hmg/ddp222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenlocher PR. Dendritic development and mental defect. Neurology. 1970;20(4):381. [PubMed] [Google Scholar]
- Huttenlocher PR. Dendritic development in neocortex of children with mental defect and infantile spasms. Neurology. 1974;24(3):203–210. doi: 10.1212/wnl.24.3.203. [DOI] [PubMed] [Google Scholar]
- Irwin SA, Patel B, Idupulapati M, Harris JB, Crisostomo RA, Larsen BP, Kooy F, Willems PJ, Cras P, Kozlowski PB, Swain RA, Weiler IJ, Greenough WT. Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am J Med Genet. 2001;98(2):161–167. doi: 10.1002/1096-8628(20010115)98:2<161::aid-ajmg1025>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Ishizuka A, Siomi MC, Siomi H. A Drosophila fragile X protein interacts with components of RNAi and ribosomal proteins. Genes Dev. 2002;16(19):2497–2508. doi: 10.1101/gad.1022002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivey KN, Muth A, Arnold J, King FW, Yeh RF, Fish JE, Hsiao EC, Schwartz RJ, Conklin BR, Bernstein HS, Srivastava D. MicroRNA regulation of cell lineages in mouse and human embryonic stem cells. Cell Stem Cell. 2008;2(3):219–229. doi: 10.1016/j.stem.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- Jin P, Zarnescu DC, Ceman S, Nakamoto M, Mowrey J, Jongens TA, Nelson DL, Moses K, Warren ST. Biochemical and genetic interaction between the fragile X mental retardation protein and the microRNA pathway. Nat Neurosci. 2004;7(2):113–117. doi: 10.1038/nn1174. [DOI] [PubMed] [Google Scholar]
- Johansson BB, Belichenko PV. Neuronal plasticity and dendritic spines: effect of environmental enrichment on intact and postischemic rat brain. J Cereb Blood Flow Metab. 2002;22(1):89–96. doi: 10.1097/00004647-200201000-00011. [DOI] [PubMed] [Google Scholar]
- Johnston RJ, Hobert O. A microRNA controlling left/right neuronal asymmetry in Caenorhabditis elegans. Nature. 2003;426(6968):845–849. doi: 10.1038/nature02255. [DOI] [PubMed] [Google Scholar]
- Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3(6):415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- Jugloff DG, Jung BP, Purushotham D, Logan R, Eubanks JH. Increased dendritic complexity and axonal length in cultured mouse cortical neurons overexpressing methyl-CpG-binding protein MeCP2. Neurobiol Dis. 2005;19(1–2):18–27. doi: 10.1016/j.nbd.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Kanellopoulou C, Muljo SA, Kung AL, Ganesan S, Drapkin R, Jenuwein T, Livingston DM, Rajewsky K. Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev. 2005;19(4):489–501. doi: 10.1101/gad.1248505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann WE, Moser HW. Dendritic anomalies in disorders associated with mental retardation. Cereb Cortex. 2000;10(10):981–991. doi: 10.1093/cercor/10.10.981. [DOI] [PubMed] [Google Scholar]
- Kishi N, Macklis JD. MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol Cell Neurosci. 2004;27(3):306–321. doi: 10.1016/j.mcn.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31(2):89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Kobrynski LJ, Sullivan KE. Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndromes. Lancet. 2007;370(9596):1443–1452. doi: 10.1016/S0140-6736(07)61601-8. [DOI] [PubMed] [Google Scholar]
- Kooy RF. Of mice and the fragile X syndrome. Trends Genet. 2003;19(3):148–154. doi: 10.1016/s0168-9525(03)00017-9. [DOI] [PubMed] [Google Scholar]
- Kooy RF, D'Hooge R, Reyniers E, Bakker CE, Nagels G, De Boulle K, Storm K, Clincke G, De Deyn PP, Oostra BA, Willems PJ. Transgenic mouse model for the fragile X syndrome. Am J Med Genet. 1996;64(2):241–245. doi: 10.1002/(SICI)1096-8628(19960809)64:2<241::AID-AJMG1>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Krichevsky AM, Sonntag KC, Isacson O, Kosik KS. Specific microRNAs modulate embryonic stem cell-derived neurogenesis. Stem Cells. 2006;24(4):857–864. doi: 10.1634/stemcells.2005-0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogan NJ, Kim M, Tong A, Golshani A, Cagney G, Canadien V, Richards DP, Beattie BK, Emili A, Boone C, Shilatifard A, Buratowski S, Greenblatt J. Methylation of histone H3 by Set2 in Saccharomyces cerevisiae is linked to transcriptional elongation by RNA polymerase II. Mol Cell Biol. 2003;23(12):4207–4218. doi: 10.1128/MCB.23.12.4207-4218.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachner M, O'Sullivan RJ, Jenuwein T. An epigenetic road map for histone lysine methylation. J Cell Sci. 2003;116(Pt 11):2117–2124. doi: 10.1242/jcs.00493. [DOI] [PubMed] [Google Scholar]
- Lalande M, Calciano MA. Molecular epigenetics of Angelman syndrome. Cell Mol Life Sci. 2007;64(7–8):947–960. doi: 10.1007/s00018-007-6460-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JA, Lupski JR. Genomic rearrangements and gene copy-number alterations as a cause of nervous system disorders. Neuron. 2006;52(1):103–121. doi: 10.1016/j.neuron.2006.09.027. [DOI] [PubMed] [Google Scholar]
- Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, Chevalier B, Johnstone SE, Cole MF, Isono K, Koseki H, Fuchikami T, Abe K, Murray HL, Zucker JP, Yuan B, Bell GW, Herbolsheimer E, Hannett NM, Sun K, Odom DT, Otte AP, Volkert TL, Bartel DP, Melton DA, Gifford DK, Jaenisch R, Young RA. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125(2):301–313. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Rådmark O, Kim S, Kim VN. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425(6956):415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- Leucht C, Stigloher C, Wizenmann A, Klafke R, Folchert A, Bally-Cuif L. MicroRNA-9 directs late organizer activity of the midbrain-hindbrain boundary. Nat Neurosci. 2008;11(6):641–648. doi: 10.1038/nn.2115. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Glantz LA, Pierri JN, Sweet RA. Altered cortical glutamate neurotransmission in schizophrenia: evidence from morphological studies of pyramidal neurons. Ann N Y Acad Sci. 2003;1003:102–112. doi: 10.1196/annals.1300.007. [DOI] [PubMed] [Google Scholar]
- Li X, Zhao X. Epigenetic regulation of mammalian stem cells. Stem Cells Dev. 2008;17(6):1043–1052. doi: 10.1089/scd.2008.0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim MK, Kawamura T, Ohsawa Y, Ohtsubo M, Asakawa S, Takayanagi A, Shimizu N. Parkin interacts with LIM Kinase 1 and reduces its cofilin-phosphorylation activity via ubiquitination. Exp Cell Res. 2007;313(13):2858–2874. doi: 10.1016/j.yexcr.2007.04.016. [DOI] [PubMed] [Google Scholar]
- Lin YJ, Seroude L, Benzer S. Extended life-span and stress resistance in the Drosophila mutant methuselah. Science. 1998;282(5390):943–946. doi: 10.1126/science.282.5390.943. [DOI] [PubMed] [Google Scholar]
- Ling SC, Fahrner PS, Greenough WT, Gelfand VI. Transport of Drosophila fragile X mental retardation protein-containing ribonucleoprotein granules by kinesin-1 and cytoplasmic dynein. Proc Natl Acad Sci U S A. 2004;101(50):17428–17433. doi: 10.1073/pnas.0408114101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Teng ZQ, Santistevan NJ, Szulwach KE, Guo W, Jin P, Zhao X. Epigenetic regulation of miR-184 by MBD1 governs neural stem cell proliferation and differentiation. Cell Stem Cell. 2010;6(5):433–444. doi: 10.1016/j.stem.2010.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Zhao X. MicroRNAs in adult and embryonic neurogenesis. Neuromolecular Med. 2009;11(3):141–152. doi: 10.1007/s12017-009-8077-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue SF, Paylor R, Wehner JM. Hippocampal lesions cause learning deficits in inbred mice in the Morris water maze and conditioned-fear task. Behav Neurosci. 1997;111(1):104–113. doi: 10.1037//0735-7044.111.1.104. [DOI] [PubMed] [Google Scholar]
- Lugli G, Torvik VI, Larson J, Smalheiser NR. Expression of microRNAs and their precursors in synaptic fractions of adult mouse forebrain. J Neurochem. 2008;106(2):650–661. doi: 10.1111/j.1471-4159.2008.05413.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikenhuis S, Giacometti E, Beard CF, Jaenisch R. Expression of MeCP2 in postmitotic neurons rescues Rett syndrome in mice. Proc Natl Acad Sci U S A. 2004;101(16):6033–6038. doi: 10.1073/pnas.0401626101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Shan G, Guo W, Smrt RD, Johnson EB, Li X, Pfeiffer RL, Szulwach KE, Duan R, Barkho BZ, Li W, Liu C, Jin P, Zhao X. Fragile x mental retardation protein regulates proliferation and differentiation of adult neural stem/progenitor cells. PLoS Genet. 2010;6(4):e1000898. doi: 10.1371/journal.pgen.1000898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lush MJ, Li Y, Read DJ, Willis AC, Glynn P. Neuropathy target esterase and a homologous Drosophila neurodegeneration-associated mutant protein contain a novel domain conserved from bacteria to man. Biochem J. 1998;332(Pt 1):1–4. doi: 10.1042/bj3320001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma DK, Jang MH, Guo JU, Kitabatake Y, Chang ML, Pow-Anpongkul N, Flavell RA, Lu B, Ming GL, Song H. Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science. 2009;323(5917):1074–1077. doi: 10.1126/science.1166859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado-Salas JP. Abnormal dendritic patterns and aberrant spine development in Bourneville's disease—a Golgi survey. Clin Neuropathol. 1984;3(2):52–58. [PubMed] [Google Scholar]
- Marin-Padilla M. Structural abnormalities of the cerebral cortex in human chromosomal aberrations: a Golgi study. Brain Res. 1972;44(2):625–629. doi: 10.1016/0006-8993(72)90324-1. [DOI] [PubMed] [Google Scholar]
- Marin-Padilla M. Pyramidal cell abnormalities in the motor cortex of a child with Down's syndrome. A Golgi study. J Comp Neurol. 1976;167(1):63–81. doi: 10.1002/cne.901670105. [DOI] [PubMed] [Google Scholar]
- Marsh JL, Thompson LM. Drosophila in the study of neurodegenerative disease. Neuron. 2006;52(1):169–178. doi: 10.1016/j.neuron.2006.09.025. [DOI] [PubMed] [Google Scholar]
- Matarazzo V, Cohen D, Palmer AM, Simpson PJ, Khokhar B, Pan SJ, Ronnett GV. The transcriptional repressor Mecp2 regulates terminal neuronal differentiation. Mol Cell Neurosci. 2004;27(1):44–58. doi: 10.1016/j.mcn.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Meng Y, Zhang Y, Tregoubov V, Janus C, Cruz L, Jackson M, Lu WY, MacDonald JF, Wang JY, Falls DL, Jia Z. Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron. 2002;35(1):121–133. doi: 10.1016/s0896-6273(02)00758-4. [DOI] [PubMed] [Google Scholar]
- Michel CI, Kraft R, Restifo LL. Defective neuronal development in the mushroom bodies of Drosophila fragile X mental retardation 1 mutants. J Neurosci. 2004;24(25):5798–5809. doi: 10.1523/JNEUROSCI.1102-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min KT, Benzer S. Preventing neurodegeneration in the Drosophila mutant bubblegum. Science. 1999;284(5422):1985–1988. doi: 10.1126/science.284.5422.1985. [DOI] [PubMed] [Google Scholar]
- Mineur YS, Sluyter F, de Wit S, Oostra BA, Crusio WE. Behavioral and neuroanatomical characterization of the Fmr1 knockout mouse. Hippocampus. 2002;12(1):39–46. doi: 10.1002/hipo.10005. [DOI] [PubMed] [Google Scholar]
- Miura K, Kishino T, Li E, Webber H, Dikkes P, Holmes GL, Wagstaff J. Neurobehavioral and electroencephalographic abnormalities in Ube3a maternal-deficient mice. Neurobiol Dis. 2002;9(2):149–159. doi: 10.1006/nbdi.2001.0463. [DOI] [PubMed] [Google Scholar]
- Morales J, Hiesinger PR, Schroeder AJ, Kume K, Verstreken P, Jackson FR, Nelson DL, Hassan BA. Drosophila fragile X protein, DFXR, regulates neuronal morphology and function in the brain. Neuron. 2002;34(6):961–972. doi: 10.1016/s0896-6273(02)00731-6. [DOI] [PubMed] [Google Scholar]
- Moretti P, Levenson JM, Battaglia F, Atkinson R, Teague R, Antalffy B, Armstrong D, Arancio O, Sweatt JD, Zoghbi HY. Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J Neurosci. 2006;26(1):319–327. doi: 10.1523/JNEUROSCI.2623-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RG, Garrud P, Rawlins JN, O'Keefe J. Place navigation impaired in rats with hippocampal lesions. Nature. 1982;297(5868):681–683. doi: 10.1038/297681a0. [DOI] [PubMed] [Google Scholar]
- Nimchinsky EA, Oberlander AM, Svoboda K. Abnormal development of dendritic spines in FMR1 knock-out mice. J Neurosci. 2001;21(14):5139–5146. doi: 10.1523/JNEUROSCI.21-14-05139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino J, Kim I, Chada K, Morrison SJ. Hmga2 promotes neural stem cell self-renewal in young but not old mice by reducing p16Ink4a and p19. Arf Expression Cell. 2008;135(2):227–239. doi: 10.1016/j.cell.2008.09.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura T, Kimura M, Horii T, Morita S, Soejima H, Kudo S, Hatada I. MeCP2-dependent repression of an imprinted miR-184 released by depolarization. Hum Mol Genet. 2008;17(8):1192–1199. doi: 10.1093/hmg/ddn011. [DOI] [PubMed] [Google Scholar]
- O'Carroll D, Erhardt S, Pagani M, Barton SC, Surani MA, Jenuwein T. The polycomb-group gene Ezh2 is required for early mouse development. Mol Cell Biol. 2001;21(13):4330–4336. doi: 10.1128/MCB.21.13.4330-4336.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberle I, Rousseau F, Heitz D, Kretz C, Devys D, Hanauer A, Boue J, Bertheas M, Mandel J. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science. 1991;252:1097–1102. doi: 10.1126/science.252.5009.1097. [DOI] [PubMed] [Google Scholar]
- Ostroff LE, Fiala JC, Allwardt B, Harris KM. Polyribosomes redistribute from dendritic shafts into spines with enlarged synapses during LTP in developing rat hippocampal slices. Neuron. 2002;35(3):535–545. doi: 10.1016/s0896-6273(02)00785-7. [DOI] [PubMed] [Google Scholar]
- Papazian DM, Schwarz TL, Tempel BL, Jan YN, Jan LY. Cloning of genomic and complementary DNA from Shaker a putative potassium channel gene from Drosophila. Science. 1987;237(4816):749–753. doi: 10.1126/science.2441470. [DOI] [PubMed] [Google Scholar]
- Parrish JZ, Emoto K, Jan LY, Jan YN. Polycomb genes interact with the tumor suppressor genes hippo and warts in the maintenance of Drosophila sensory neuron dendrites. Genes Dev. 2007a;21(8):956–972. doi: 10.1101/gad.1514507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish JZ, Emoto K, Kim MD, Jan YN. Mechanisms that regulate establishment, maintenance, and remodeling of dendritic fields. Annu Rev Neurosci. 2007b;30:399–423. doi: 10.1146/annurev.neuro.29.051605.112907. [DOI] [PubMed] [Google Scholar]
- Penagarikano O, Mulle JG, Warren ST. The pathophysiology of fragile x syndrome. Annu Rev Genomics Hum Genet. 2007;8:109–129. doi: 10.1146/annurev.genom.8.080706.092249. [DOI] [PubMed] [Google Scholar]
- Persico AM, Bourgeron T. Searching for ways out of the autism maze: genetic, epigenetic and environmental clues. Trends Neurosci. 2006;29(7):349–358. doi: 10.1016/j.tins.2006.05.010. [DOI] [PubMed] [Google Scholar]
- Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem. 2001;276(39):36734–36741. doi: 10.1074/jbc.M101287200. [DOI] [PubMed] [Google Scholar]
- Pittenger C, Duman RS. Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology. 2008;33(1):88–109. doi: 10.1038/sj.npp.1301574. [DOI] [PubMed] [Google Scholar]
- Potocki L, Bi W, Treadwell-Deering D, Carvalho CM, Eifert A, Friedman EM, Glaze D, Krull K, Lee JA, Lewis RA, Mendoza-Londono R, Robbins-Furman P, Shaw C, Shi X, Weissenberger G, Withers M, Yatsenko SA, Zackai EH, Stankiewicz P, Lupski JR. Characterization of Potocki-Lupski syndrome (dup(17) (p11.2p11.2)) and delineation of a dosage-sensitive critical interval that can convey an autism phenotype. Am J Hum Genet. 2007;80(4):633–649. doi: 10.1086/512864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poy MN, Eliasson L, Krutzfeldt J, Kuwajima S, Ma X, Macdonald PE, Pfeffer S, Tuschl T, Rajewsky N, Rorsman P, Stoffel M. A pancreatic islet-specific microRNA regulates insulin secretion. Nature. 2004;432(7014):226–230. doi: 10.1038/nature03076. [DOI] [PubMed] [Google Scholar]
- Purpura DP. Dendritic spine “dysgenesis” and mental retardation. Science. 1974;186(4169):1126–1128. doi: 10.1126/science.186.4169.1126. [DOI] [PubMed] [Google Scholar]
- Purpura DP. Dendritic differentiation in human cerebral cortex: normal and aberrant developmental patterns. Adv Neurol. 1975;12:91–134. [PubMed] [Google Scholar]
- Rajasethupathy P, Fiumara F, Sheridan R, Betel D, Puthanveettil SV, Russo JJ, Sander C, Tuschl T, Kandel E. Characterization of small RNAs in aplysia reveals a role for miR-124 in constraining synaptic plasticity through CREB. Neuron. 2009;63(6):803–817. doi: 10.1016/j.neuron.2009.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramocki MB, Zoghbi HY. Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature. 2008;455(7215):912–918. doi: 10.1038/nature07457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redmond L, Kashani AH, Ghosh A. Calcium regulation of dendritic growth via CaM kinase IV and CREB-mediated transcription. Neuron. 2002;34(6):999–1010. doi: 10.1016/s0896-6273(02)00737-7. [DOI] [PubMed] [Google Scholar]
- Reiter LT, Potocki L, Chien S, Gribskov M, Bier E. A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res. 2001;11(6):1114–1125. doi: 10.1101/gr.169101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs AD. X inactivation, differentiation, and DNA methylation. Cytogenet Cell Genet. 1975;14(1):9–25. doi: 10.1159/000130315. [DOI] [PubMed] [Google Scholar]
- Robertson KD, Ait-Si-Ali S, Yokochi T, Wade PA, Jones PL, Wolffe AP. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat Genet. 2000;25(3):338–342. doi: 10.1038/77124. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Kolb B. Structural plasticity associated with exposure to drugs of abuse. Neuropharmacology. 2004;47(Suppl 1):33–46. doi: 10.1016/j.neuropharm.2004.06.025. [DOI] [PubMed] [Google Scholar]
- Rosenthal N, Brown S. The mouse ascending: perspectives for human-disease models. Nat Cell Biol. 2007;9(9):993–999. doi: 10.1038/ncb437. [DOI] [PubMed] [Google Scholar]
- Rybak A, Fuchs H, Smirnova L, Brandt C, Pohl EE, Nitsch R, Wulczyn FG. A feedback loop comprising lin-28 and let-7 controls pre-let-7 maturation during neural stem-cell commitment. Nat Cell Biol. 2008;10(8):987–993. doi: 10.1038/ncb1759. [DOI] [PubMed] [Google Scholar]
- Sahoo T, del Gaudio D, German JR, Shinawi M, Peters SU, Person RE, Garnica A, Cheung SW, Beaudet AL. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat Genet. 2008;40(6):719–721. doi: 10.1038/ng.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, Schreiber SL, Mellor J, Kouzarides T. Active genes are tri-methylated at K4 of histone H3. Nature. 2002;419(6905):407–411. doi: 10.1038/nature01080. [DOI] [PubMed] [Google Scholar]
- Sarna JR, Larouche M, Marzban H, Sillitoe RV, Rancourt DE, Hawkes R. Patterned Purkinje cell degeneration in mouse models of Niemann-Pick type C disease. J Comp Neurol. 2003;456(3):279–291. doi: 10.1002/cne.10522. [DOI] [PubMed] [Google Scholar]
- Schratt G. microRNAs at the synapse. Nat Rev Neurosci. 2009;10(12):842–849. doi: 10.1038/nrn2763. [DOI] [PubMed] [Google Scholar]
- Schratt GM, Tuebing F, Nigh EA, Kane CG, Sabatini ME, Kiebler M, Greenberg ME. A brain-specific microRNA regulates dendritic spine development. Nature. 2006;439(7074):283–289. doi: 10.1038/nature04367. [DOI] [PubMed] [Google Scholar]
- Schübeler D, MacAlpine DM, Scalzo D, Wirbelauer C, Kooperberg C, van Leeuwen F, Gottschling DE, O'Neill LP, Turner BM, Delrow J, Bell SP, Groudine M. The histone modification pattern of active genes revealed through genome-wide chromatin analysis of a higher eukaryote. Genes Dev. 2004;18(11):1263–1271. doi: 10.1101/gad.1198204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwamborn JC, Berezikov E, Knoblich JA. The TRIM-NHL protein TRIM32 activates microRNAs and prevents self-renewal in mouse neural progenitors. Cell. 2009;136(5):913–925. doi: 10.1016/j.cell.2008.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal M. Dendritic spines and long-term plasticity. Nat Rev Neurosci. 2005;6(4):277–284. doi: 10.1038/nrn1649. [DOI] [PubMed] [Google Scholar]
- Shahbazian M, Young J, Yuva-Paylor L, Spencer C, Antalffy B, Noebels J, Armstrong D, Paylor R, Zoghbi H. Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron. 2002;35(2):243–254. doi: 10.1016/s0896-6273(02)00768-7. [DOI] [PubMed] [Google Scholar]
- Shahbazian MD, Zoghbi HY. Rett syndrome and MeCP2: linking epigenetics and neuronal function. Am J Hum Genet. 2002;71(6):1259–1272. doi: 10.1086/345360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Chichung Lie D, Taupin P, Nakashima K, Ray J, Yu RT, Gage FH, Evans RM. Expression and function of orphan nuclear receptor TLX in adult neural stem cells. Nature. 2004;427(6969):78–83. doi: 10.1038/nature02211. [DOI] [PubMed] [Google Scholar]
- Siegel G, Obernosterer G, Fiore R, Oehmen M, Bicker S, Christensen M, Khudayberdiev S, Leuschner PF, Busch CJ, Kane C, Hübel K, Dekker F, Hedberg C, Rengarajan B, Drepper C, Waldmann H, Kauppinen S, Greenberg ME, Draguhn A, Rehmsmeier M, Martinez J, Schratt GM. A functional screen implicates microRNA-138-dependent regulation of the depalmitoylation enzyme APT1 in dendritic spine morphogenesis. Nat Cell Biol. 2009;11(6):705–716. doi: 10.1038/ncb1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slager RE, Newton TL, Vlangos CN, Finucane B, Elsea SH. Mutations in RAI1 associated with Smith-Magenis syndrome. Nat Genet. 2003;33(4):466–468. doi: 10.1038/ng1126. [DOI] [PubMed] [Google Scholar]
- Slegtenhorst-Eegdeman KE, de Rooij DG, Verhoef-Post M, van de Kant HJ, Bakker CE, Oostra BA, Grootegoed JA, Themmen AP. Macroorchidism in FMR1 knockout mice is caused by increased Sertoli cell proliferation during testicular development. Endocrinology. 1998;139(1):156–162. doi: 10.1210/endo.139.1.5706. [DOI] [PubMed] [Google Scholar]
- Smrt RD, Eaves-Egenes J, Barkho BZ, Santistevan NJ, Zhao C, Aimone JB, Gage FH, Zhao X. Mecp2 deficiency leads to delayed maturation and altered gene expression in hippocampal neurons. Neurobiol Dis. 2007;27(1):77–89. doi: 10.1016/j.nbd.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smrt RD, Szulwach KE, Pfeiffer RL, Li X, Guo W, Pathania M, Teng ZQ, Luo Y, Peng J, Bordey A, Jin P, Zhao X. MicroRNA miR-137 regulates neuronal maturation by targeting ubiquitin ligase mind bomb-1. Stem Cells. 2010;28(6):1060–1070. doi: 10.1002/stem.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow WM, Hartle K, Ivanco TL. Altered morphology of motor cortex neurons in the VPA rat model of autism. Dev Psychobiol. 2008;50(7):633–639. doi: 10.1002/dev.20337. [DOI] [PubMed] [Google Scholar]
- Song H, Kempermann G, Overstreet Wadiche L, Zhao C, Schinder AF, Bischofberger J. New neurons in the adult mammalian brain: synaptogenesis and functional integration. J Neurosci. 2005;25(45):10366–10368. doi: 10.1523/JNEUROSCI.3452-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires TL, Grote HE, Garry S, Cordery PM, Van Dellen A, Blakemore C, Hannan AJ. Dendritic spine pathology and deficits in experience-dependent dendritic plasticity in R6/1 Huntington's disease transgenic mice. Eur J Neurosci. 2004;19(10):2799–2807. doi: 10.1111/j.0953-816X.2004.03374.x. [DOI] [PubMed] [Google Scholar]
- Stark KL, Xu B, Bagchi A, Lai WS, Liu H, Hsu R, Wan X, Pavlidis P, Mills AA, Karayiorgou M, Gogos JA. Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat Genet. 2008;40(6):751–760. doi: 10.1038/ng.138. [DOI] [PubMed] [Google Scholar]
- Suetsugu M, Mehraein P. Spine distribution along the apical dendrites of the pyramidal neurons in Down's syndrome. A quantitative Golgi study. Acta Neuropathol. 1980;50(3):207–210. doi: 10.1007/BF00688755. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Schuman EM. Local translational control in dendrites and its role in long-term synaptic plasticity. J Neurobiol. 2005;64(1):116–131. doi: 10.1002/neu.20152. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Tian QB, Kuromitsu J, Kawai T, Endo S. Characterization of mRNA species that are associated with postsynaptic density fraction by gene chip microarray analysis. Neurosci Res. 2007;57(1):61–85. doi: 10.1016/j.neures.2006.09.009. [DOI] [PubMed] [Google Scholar]
- Szulwach KE, Li X, Smrt RD, Li Y, Luo Y, Lin L, Santistevan NJ, Li W, Zhao X, Jin P. Cross talk between microRNA and epigenetic regulation in adult neurogenesis. J Cell Biol. 2010;189(1):127–141. doi: 10.1083/jcb.200908151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci U S A. 2002;99(6):3740–3745. doi: 10.1073/pnas.052410099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima S, Becker LE, Armstrong DL, Chan F. Abnormal neuronal development in the visual cortex of the human fetus and infant with down's syndrome. A quantitative and qualitative Golgi study. Brain Res. 1981;225(1):1–21. doi: 10.1016/0006-8993(81)90314-0. [DOI] [PubMed] [Google Scholar]
- Takashima S, Iida K, Mito T, Arima M. Dendritic and histochemical development and ageing in patients with Down's syndrome. J Intellect Disabil Res. 1994;38(Pt 3):265–273. doi: 10.1111/j.1365-2788.1994.tb00394.x. [DOI] [PubMed] [Google Scholar]
- Toni N, Laplagne DA, Zhao C, Lombardi G, Ribak CE, Gage FH, Schinder AF. Neurons born in the adult dentate gyrus form functional synapses with target cells. Nat Neurosci. 2008;11(8):901–907. doi: 10.1038/nn.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tudor M, Akbarian S, Chen RZ, Jaenisch R. Transcriptional profiling of a mouse model for Rett syndrome reveals subtle transcriptional changes in the brain. Proc Natl Acad Sci U S A. 2002;99(24):15536–15541. doi: 10.1073/pnas.242566899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Bor V, Davis I. mRNA localisation gets more complex. Curr Opin Cell Biol. 2004;16(3):300–307. doi: 10.1016/j.ceb.2004.03.008. [DOI] [PubMed] [Google Scholar]
- van Praag H, Schinder AF, Christie BR, Toni N, Palmer TD, Gage FH. Functional neurogenesis in the adult hippocampus. Nature. 2002;415(6875):1030–1034. doi: 10.1038/4151030a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderklish PW, Edelman GM. Differential translation and fragile X syndrome. Genes Brain Behav. 2005;4(6):360–384. doi: 10.1111/j.1601-183X.2005.00134.x. [DOI] [PubMed] [Google Scholar]
- Walkley SU, Baker HJ. Sphingomyelin lipidosis in a cat: Golgi studies. Acta Neuropathol. 1984;65(2):138–144. doi: 10.1007/BF00690467. [DOI] [PubMed] [Google Scholar]
- Wan L, Dockendorff TC, Jongens TA, Dreyfuss G. Characterization of dFMR1, a Drosophila melanogaster homolog of the fragile X mental retardation protein. Mol Cell Biol. 2000;20(22):8536–8547. doi: 10.1128/mcb.20.22.8536-8547.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Medvid R, Melton C, Jaenisch R, Blelloch R. DGCR8 is essential for microRNA biogenesis and silencing of embryonic stem cell self-renewal. Nat Genet. 2007;39(3):380–385. doi: 10.1038/ng1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman GA, Davare M, Ando H, Fortin D, Varlamova O, Cheng HY, Marks D, Obrietan K, Soderling TR, Goodman RH, Impey S. An activity-regulated microRNA controls dendritic plasticity by down-regulating p250GAP. Proc Natl Acad Sci U S A. 2008;105(26):9093–9098. doi: 10.1073/pnas.0803072105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman GA, Impey S, Marks D, Saneyoshi T, Grant WF, Derkach V, Soderling TR. Activity-dependent dendritic arborization mediated by CaM-kinase I activation and enhanced CREB-dependent transcription of Wnt-2. Neuron. 2006;50(6):897–909. doi: 10.1016/j.neuron.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Wisniewski KE, Segan SM, Miezejeski CM, Sersen EA, Rudelli RD. The Fra(X) syndrome: neurological, electrophysiological, and neuropathological abnormalities. Am J Med Genet. 1991;38(2–3):476–480. doi: 10.1002/ajmg.1320380267. [DOI] [PubMed] [Google Scholar]
- Xu XL, Li Y, Wang F, Gao FB. The steady-state level of the nervous-system-specific microRNA-124a is regulated by dFMR1 in Drosophila. J Neurosci. 2008;28(46):11883–11889. doi: 10.1523/JNEUROSCI.4114-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi J, Miyamoto Y, Kusakawa S, Torii T, Mizutani R, Sanbe A, Nakajima H, Kiyokawa N, Tanoue A. Neurofibromatosis 2 tumor suppressor, the gene induced by valproic acid, mediates neurite outgrowth through interaction with paxillin. Exp Cell Res. 2008;314(11–12):2279–2288. doi: 10.1016/j.yexcr.2008.03.019. [DOI] [PubMed] [Google Scholar]
- Yamauchi J, Miyamoto Y, Torii T, Mizutani R, Nakamura K, Sanbe A, Koide H, Kusakawa S, Tanoue A. Valproic acid-inducible Arl4D and cytohesin-2/ARNO, acting through the downstream Arf6, regulate neurite outgrowth in N1E-115 cells. Exp Cell Res. 2009;315(12):2043–2052. doi: 10.1016/j.yexcr.2009.03.012. [DOI] [PubMed] [Google Scholar]
- Yang L, Duan R, Chen D, Wang J, Chen D, Jin P. Fragile X mental retardation protein modulates the fate of germline stem cells in Drosophila. Hum Mol Genet. 2007;16(15):1814–1820. doi: 10.1093/hmg/ddm129. [DOI] [PubMed] [Google Scholar]
- Yoder JA, Walsh CP, Bestor TH. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 1997;13(8):335–340. doi: 10.1016/s0168-9525(97)01181-5. [DOI] [PubMed] [Google Scholar]
- Yoo AS, Staahl BT, Chen L, Crabtree GR. MicroRNA-mediated switching of chromatin-remodelling complexes in neural development. Nature. 2009;460(7255):642–646. doi: 10.1038/nature08139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YQ, Bailey AM, Matthies HJ, Renden RB, Smith MA, Speese SD, Rubin GM, Broadie K. Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell. 2001;107(5):591–603. doi: 10.1016/s0092-8674(01)00589-x. [DOI] [PubMed] [Google Scholar]
- Zhao C, Avilés C, Abel RA, Almli CR, McQuillen P, Pleasure SJ. Hippocampal and visuospatial learning defects in mice with a deletion of frizzled 9, a gene in the Williams syndrome deletion interval. Development. 2005;132(12):2917–2927. doi: 10.1242/dev.01871. [DOI] [PubMed] [Google Scholar]
- Zhao C, Sun G, Li S, Lang MF, Yang S, Li W, Shi Y. MicroRNA let-7b regulates neural stem cell proliferation and differentiation by targeting nuclear receptor TLX signaling. Proc Natl Acad Sci U S A. 2010;107(5):1876–1881. doi: 10.1073/pnas.0908750107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Sun G, Li S, Shi Y. A feedback regulatory loop involving microRNA-9 and nuclear receptor TLX in neural stem cell fate determination. Nat Struct Mol Biol. 2009;16(4):365–371. doi: 10.1038/nsmb.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Teng EM, Summers RG, Jr, Ming GL, Gage FH. Distinct morphological stages of dentate granule neuron maturation in the adult mouse hippocampus. J Neurosci. 2006;26(1):3–11. doi: 10.1523/JNEUROSCI.3648-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Hong EJ, Cohen S, Zhao WN, Ho HY, Schmidt L, Chen WG, Lin Y, Savner E, Griffith EC, Hu L, Steen JA, Weitz CJ, Greenberg ME. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. 2006;52(2):255–269. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY. Postnatal neurodevelopmental disorders: meeting at the synapse? Science. 2003;302(5646):826–830. doi: 10.1126/science.1089071. [DOI] [PubMed] [Google Scholar]