

Abstract
Cyclooxygenase (Cox) is a key enzyme in the biosynthetic metabolism of prostaglandins. The inducible isoform of Cox-2 has been implicated in inflammation and its specific inhibition can be used to treat noninfectious inflammatory diseases, such as rheumatoid arthritis. Borrelia burgdorferi, the agent of Lyme disease, can induce joint inflammation. Here we show that B. burgdorferi induced the upregulation of cox-2 gene expression in murine joints at the onset of arthritis in infected mice. The level of mRNA expression correlated with the degree of inflammation. The specific inhibition of Cox-2 diminished the degree of joint inflammation, without affecting B. burgdorferi-specific antibody or cytokine responses. Cox-2 activity is therefore associated with the genesis of infectious arthritis caused by B. burgdorferi.
Keywords: Lyme arthritis, Inflammation, Cyclooxygenase-2, Borrelia burgdorferi
1. Introduction
Lyme disease, caused by Borrelia burgdorferi, is the most common vector-borne illness in the United States [1]. The disease usually begins with a pathognomonic skin rash, erythema migrans, that can be accompanied by constitutional symptoms such as fevers and myalgia [2]. If not treated, the disease can involve the musculoskeletal, cardiovascular and nervous system [2]. Acute Lyme arthritis is one of the most prevalent clinical manifestations of persistent infection in humans [2].
The pathogenesis of acute Lyme arthritis is complex and depends on several factors, including spirochetal virulence and the host response to infection [3–8]. Ultimately, the recruitment of inflammatory cells, predominantly neutrophils, results in joint swelling. The production of proinflammatory cytokines by these cells in response to B. burgdorferi [4–6] and presumably other factors induce the typical joint effusions and underlying arthritis commonly observed in patients, and in experimental models of B. burgdorferi infection. In rare instances, chronic Lyme arthritis can occur in humans. The pathogenesis of this form of arthritis differs from acute arthritis in that it may result from cross-reaction of B. burgdorferi-specific antibodies with self antigens, like LFA-1 [9]. C3H/HeNCr (C3H) mice can be infected with B. burgdorferi, and develop acute arthritis. The murine model partially mimics acute human disease: it is most severe at 2–4 weeks of infection and resolves over several months [4]. Several weeks after challenge with B. burgdorferi, mice develop tibiotarsal swelling with a neutrophilic joint exudate. After 1–2 months the arthritis resolves. However, the mice remain persistently infected. The murine model has been very useful for studying the pathogenesis of Lyme borreliosis.
Cyclooxygenase (Cox) is a key regulator of prostaglandin (PG) synthesis. Cox exists in two isoforms, Cox-1 and Cox-2, encoded by separate genes [10]. Cox-1 is constitutively expressed in most tissues and is responsible for the physiological production of PGs [10]. The inducible isoform of Cox, Cox-2, has been extensively correlated with inflammation [10]. Nonsteroidal anti-inflammatory drugs that target Cox-2 are used for the treatment of noninfectious inflammatory illnesses such as osteoarthritis, rheumatoid arthritis and gout. The role of Cox-2 during infectious inflammatory arthritides, such as Lyme arthritis, has, however, not yet been extensively examined [11]. We investigated the role of Cox-2 in a murine model of Lyme arthritis.
2. Materials and methods
2.1. Mice, B. burgdorferi and infections
Six-week-old C3H mice were purchased from the Frederick Cancer Research Center, Frederick, MD, USA. Mice were housed in filter frame cages and given food and water ad libitum. The animals were euthanized with CO2.
A clonal low passage B. burgdorferi N40 isolate (cN40), with proven infectivity and pathogenicity was used throughout the studies [4]. Spirochetes were grown in modified Barbour–Stoenner–Kelly (BSK) II medium at 33°C. Individual mice were challenged with a dose of 104 spirochetes administered by intradermal injection in the back, as previously described [4]. At sacrifice, blood, spleen, urinary bladder and skin (at the inoculation site) were collected aseptically and cultured in BSK II medium to check for the infection status of the mice. Joints (both knees and tibiotarsi) were fixed in formalin, embedded in paraffin and examined microscopically for evidence of disease [4]. Prevalence of arthritis was assessed by examination of both knees and tibiotarsal joints from each mouse. The severity of the tibiotarsal arthritis was scored on a scale of 0 (no disease) to 3 (severe disease) as described [4]. Active arthritis was graded, tabulated, and distinguished from resolving lesions (no active exudation). Arthritis is characterized by neutrophilic infiltration that may be accompanied by edema, the thickening of the tendon sheath and bone resorption.
2.2. RNA extraction and RT-PCR
Joint (ankle) tissue was used to extract RNA by the thioisocyanate method [12] using the reagents and protocol of the Micro RNA Isolation kit following the manufacturer’s instructions (Stratagene, La Jolla, CA, USA). 5–10 μg were used to obtain cDNA with an RT-PCR kit (Stratagene) using random primers. cox-2 mRNA was amplified with primers: 5′-TCA GCC AGG CAG CAA ATC CTT G-3′ and 5′-TAG TCT CTC CTA TGA GTA TGA GTC-3′. Conditions of the PCR were: 94°C, 45 s; 55°C, 45 s; and 72°C, 45 s for 35 cycles.
2.3. CD4+ T cell restimulation assays
Splenic CD4+ T cells were purified by negative selection using biotinylated antibodies against CD8a, Ly6G, Mac-1, B220, class II (I-Ak and I-Ek) and pan NK molecules (BD Pharmingen, San Diego, CA, USA), followed by incubation with avidin bound to magnetic microbeads and passed through a magnetic column (Miltenyi Biotec, Auburn, CA, USA). 106 CD4+ T cells per ml were incubated with 106 mitomycin C-treated (50 μg ml−1, 37°C, 40 min) (Sigma Chemical Co., Saint Louis, MO, USA) syngeneic splenocytes per ml, in the presence of B. burgdorferi extract (10 μg ml−1). After 40 h of incubation, the supernatants were recovered and analyzed for the presence of interleukin (IL)-2, and interferon (IFN)γ by capture enzyme-linked immunosorbent assay (ELISA).
2.4. Cytokine ELISA
For the determination of the levels of IL-2 and IFNγ in restimulation supernatants and mouse sera, antibodies specific for murine IL-2 and IFNγ (BD Pharmingen), were used, following the manufacturer’s instructions with some modifications. 96-well ELISA plates (ICN, Costa Mesa, CA, USA) were coated with the capture antibody (2 μg ml−1) for 2 h at 37°C. After blocking with phosphate-buffered saline (PBS) plus 10% fetal calf serum (FCS) (PBS/FCS) overnight at 4°C, samples were applied and incubated 1 h at 37°C. The biotinylated detection antibody (1 μg ml−1) was then added after washing the plates with PBS plus Tween 20 (0.5% v/v) (PBS/Tween). Quantification of cytokine levels was made after incubating the plates with horseradish-conjugated avidin and adding the substrate for the enzyme (TMB, Kirkegaard and Perry Laboratories, Inc., Gaithersburg, MD, USA). Plates were read at 450 nm after stopping the color developing reaction (TMB 1 Component Stop Solution, Kirkegaard and Perry Laboratories, Inc.). The values indicated were calculated by extrapolation from a dose–response curve generated using standard concentrations of recombinant mouse IL-2 and IFNγ (BD Pharmingen).
2.5. Determination of B. burgdorferi-specific antibody titers
B. burgdorferi-specific IgM and IgG levels in the sera from the infected animals were determined by ELISA using biotinylated rat monoclonal anti-mouse-specific antibodies to mouse IgM and IgG (BD Pharmingen) as follows: an ELISA plate (ICN) was coated overnight at 4°C with 0.5 μg ml−1 of B. burgdorferi lysate in coating buffer (bicarbonate buffer, pH 9.6), and blocked for 2 h at room temperature with PBS/FCS. After two washes with PBS/Tween 20, sera (1/100 dilution) were applied and incubated for 1 h at room temperature. Sera from uninfected mice were used as controls. The wells were washed and the specific antisera were applied and incubated for 1 h at room temperature. After four washes, horseradish peroxidase-conjugated streptavidin was applied and incubated for 1 h at room temperature. Plates were read at 450 nm after adding peroxidase substrate (TMB, Kirkegaard and Perry Laboratories, Inc.) and stopping the reaction with TMB 1 Component Stop Solution (Kirkegaard and Perry Laboratories, Inc.).
3. Results
3.1. Infection with B. burgdorferi induces the upregulation of cox-2 in the joints of the infected mice
We examined joint tissue from mice experimentally infected with B. burgdorferi to determine whether the presence of the spirochete would be associated with the upregulation of the cox-2 gene. At 14 and 60 days of infection, joints from B. burgdorferi-infected and control (uninfected) mice were used to obtain RNA. These time points represent the periods in which acute arthritis (14 days) and the resolution of inflammation (60 days) are consistently observed in the mouse model of Lyme borreliosis [4]. RT-PCR analysis revealed a prominent band at 14 days of infection (Fig. 1). At 60 days, the level of cox-2 mRNA was lower but still detectable (Fig. 1). cox-2 mRNA was not detected in control mice (Fig. 1; time 0). These data showed that cox-2 gene expression was induced in the joints of mice experimentally infected with B. burgdorferi.
Fig. 1.
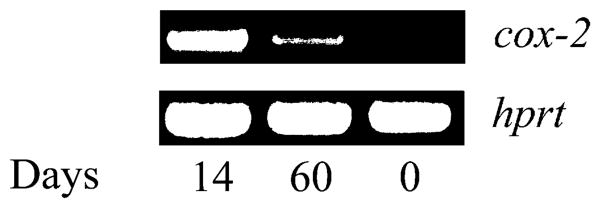
cox-2 gene expression is upregulated in the joints upon infection with B. burgdorferi. C3H mice were infected with B. burgdorferi and at 14 and 60 days of infection, joint RNA was tested for the presence of cox-2 mRNA by RT-PCR. Equal loads of RNA were ensured by amplifying hprt mRNA. Control uninfected mice showed no expression of cox-2 mRNA (day 0).
3.2. Inhibition of Cox-2 activity during experimental Lyme borreliosis modulates arthritis severity
We then tested the role of Cox-2 activity in the genesis of acute murine Lyme arthritis. Several specific inhibitors of the inducible isoform of Cox have been described [13]. The compound 3-(3,4 difluorophenyl)-4-(4-(methylsulfonyl)phenyl)-2-(5H)-furanone (MF-Tricyclic) is a potent Cox-2 inhibitor that shows high specificity over Cox-1 inhibition [14]. Mice were infected with 104 spirochetes and fed chow in which MF-Tricyclic was included (27 mg kg−1) until sacrifice. As a control, normal chow from the same manufacturer was used (Merck Frost Canada, Inc.). The mean concentration of MF-Tricyclic in the plasma of the infected, treated mice was 1.3 ± 0.2 μg ml−1. At 14 days of infection, mice treated with the Cox-2-inhibitor showed a significant reduction in arthritis severity, compared to controls (average degree of arthritis ± S.E.M.; control: 1.2 ± 0.2; treated: 0.5 ± 0.1; Student’s t-test, P <0.01).
3.3. Cox-2 inhibition does not prevent an efficient T and B cell response to B. burgdorferi
Recent reports have shown that Cox-2 activity is involved in T cell activation. The specific inhibition of Cox-2 activity during T cell activation resulted in lower levels of IL-2 in Jurkat and human T cells stimulated in vitro by TCR/CD3 or PMA [15]. To test the effect of specific Cox-2 inhibition on T cell activity during murine Lyme arthritis, we analyzed recall responses of purified CD4+ T lymphocytes to B. burgdorferi antigens. The level of IL-2 produced by CD4+ T cells in response to spirochetal antigens was equivalent in MF-Tricyclic-treated and control mice, indicating that the treatment did not affect the ability of these cells to respond to the bacteria (Fig. 2A). Similarly, IFNγ levels were also equivalent in both groups of mice, indicating that Cox-2 inhibition had no effect on CD4+ T cell differentiation during infection with the spirochete (Fig. 2B). Furthermore, levels of IFNγ in the sera of the infected animals were not affected by the treatment (Fig. 2C).
Fig. 2.

Immune responses to B. burgdorferi are not affected by the in vivo inhibition of Cox-2. Recall immune responses to B. burgdorferi antigens were performed in 2-week-infected C3H mice, using purified CD4+ T cells and syngeneic antigen presenting cells. After 40 h, the supernatants were analyzed for IL-2 (A) and IFNγ (B) levels by cytokine ELISA. The sera levels of IFNγ were also determined in the infected mice (C). Data shown corresponds to one of three representative experiments.
We also measured B. burgdorferi-specific IgM and IgG levels in the sera of the B. burgdorferi-infected animals, and B. burgdorferi-infected mice treated with the Cox-2 inhibitor, at 2 weeks. No differences were observed in the levels of B. burgdorferi-specific antibodies (Fig. 3). These data indicate that the Cox-2 inhibitor did not impair the development of T and B cell responses to B. burgdorferi.
Fig. 3.
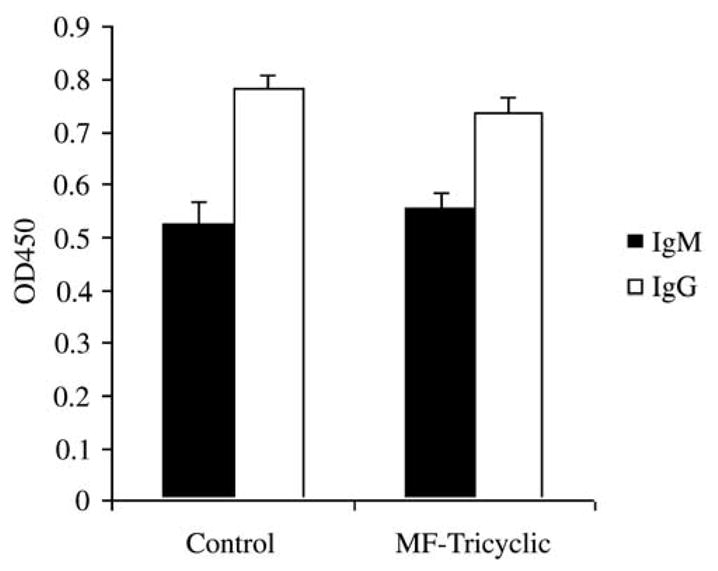
Antibody responses to B. burgdorferi are not affected by the in vivo inhibition of Cox-2 during murine infection. B. burgdorferi-specific IgM and IgG levels were determined in the sera of individual mice at 2 weeks of infection by ELISA. Results represent the average ± S.E.M. of three independent experiments.
4. Discussion
Arthritis is one of the most common manifestations of human Lyme disease. The exact mechanism by which B. burgdorferi induces inflammation is not completely understood, although the recruitment of proinflammatory cells has been implicated in the development of arthritis. These cells, predominantly PMNs, can partially control the number of spirochetes in the joints especially in conditions where an acquired immune response is absent [16]. Nevertheless, these cells also produce, and respond to, mediators of inflammation that can severely damage joint tissue. Pro-inflammatory cytokines and other signals have been shown to upregulate cox-2 expression especially in noninfectious inflammatory processes [17,18].
Our results show that inducible Cox activity could be involved in the genesis of murine Lyme arthritis. The gene was upregulated in the joints of experimentally infected mice but not uninfected mice. Balb/c mice, that developed only mild inflammation when infected with the spirochete, had lower levels of cox-2 mRNA in their joints (not shown) suggesting that levels of Cox-2 expression correlate with the degree of inflammation. Furthermore, the inhibition in vivo of Cox-2 activity during experimental infection of the mice reduced the degree of acute inflammation. These data suggest that Cox-2 activity may be implicated in the recruitment and/or activation of neutrophils. The development of murine Lyme arthritis is therefore the result of coordinated action of several activities associated with neutrophils that ultimately damage the joint tissue.
Reports have suggested that Cox-2 may affect early events occurring upon T cell activation, such as IL-2 and CD25 upregulation [15]. Our results indicate that the Cox-2 inhibitor did not affect T cell activation by B. burgdorferi, since comparable levels of IL-2 and IFNγ were found in both groups of mice upon restimulation of CD4+ T cells in vitro. B. burgdorferi-specific antibody production was also similar in experimental and control animals. As humoral and cellular responses can influence the bacteria burden, these data are suggestive of effective T–B cell cooperation and B cell function.
Cox-2 and Cox-2 inhibitors play important roles in modulating inflammation, particularly inflammatory responses due to autoimmune diseases. The role of Cox-2 in infectious diseases has not yet been fully explored. These data demonstrate that B. burgdorferi induced cox-2 expression in mice and that Cox-2 activity may be important for the joint inflammation that results in murine Lyme arthritis. Studies in patients with Lyme arthritis would be required to correlate these experimental results with human disease because the mechanisms underlying Lyme arthritis in the mouse and man may not be identical.
Acknowledgments
We thank Debbie Beck for her excellent technical assistance and Pauline Luk, Merck-Frosst, Montreal, for performing the MF-Tricyclic assays. This work was supported by grants from the Medical Schools Grant Program (Merck and Co., Inc) and the National Institutes of Health. E.F. is the recipient of a Clinical-Scientist Award in Translational Research from the Burroughs Wellcome Fund.
References
- 1.Anonymous. Lyme disease – United States, 1996. Morb Mortal Wkly Rep. 1997;46:531–535. [PubMed] [Google Scholar]
- 2.Evans J. Lyme disease. Curr Opin Rheumatol. 1996;8:327–333. doi: 10.1097/00002281-199607000-00009. [DOI] [PubMed] [Google Scholar]
- 3.Yang L, Weis JH, Eichwald E, Kolbert CP, Persing DH, Weis JJ. Heritable susceptibility to severe Borrelia burgdorferi-induced arthritis is dominant and is associated with persistence of large numbers of spirochetes in tissues. Infect Immun. 1994;62:492–500. doi: 10.1128/iai.62.2.492-500.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anguita J, Persing DH, Rinco ¤n M, Barthold SW, Fikrig E. Effect of anti-interleukin 12 treatment on murine Lyme borreliosis. J Clin Invest. 1996;97:1028–1034. doi: 10.1172/JCI118494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keane-Myers A, Nickell SP. Role of IL-4 and IFN-gamma in modulation of immunity to Borrelia burgdorferi in mice. J Immunol. 1995;155:2020–2028. [PubMed] [Google Scholar]
- 6.Matyniak JE, Reiner SL. T helper phenotype and genetic susceptibility in experimental Lyme disease. J Exp Med. 1995;181:1251–1254. doi: 10.1084/jem.181.3.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keane-Myers A, Nickell SP. T cell subset-dependent modulation of immunity to Borrelia burgdorferi in mice. J Immunol. 1995;154:1770–1776. [PubMed] [Google Scholar]
- 8.Lengl-Janssen B, Strauss AF, Steere AC, Kamradt T. The T helper cell response in Lyme arthritis: differential recognition of Borrelia burgdorferi outer surface protein A in patients with treatment-resistant or treatment-responsive Lyme arthritis. J Exp Med. 1994;180:2069–2078. doi: 10.1084/jem.180.6.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gross DM, Forsthuber T, Tary-Lehmann M, Etling C, Ito K, Nagy ZA, Field JA, Steere AC, Huber BT. Identification of LFA-1 as a candidate autoantigen in treatment-resistant Lyme arthritis. Science. 1998;281:703–706. doi: 10.1126/science.281.5377.703. [DOI] [PubMed] [Google Scholar]
- 10.Masferrer JL, Isakson PC, Seibert K. Cyclooxigenase-2 inhibitors. Gastroenterol Clin N Am. 1996;25:363–372. doi: 10.1016/s0889-8553(05)70252-1. [DOI] [PubMed] [Google Scholar]
- 11.Gilroy DW, Tomlinson A, Greenslade K, Seed MP, Willoughby DA. The effects of cyclooxygenase 2 inhibitors on cartilage erosion and bone loss in a model of Mycobacterium tuberculosis-induced monoarticular arthritis in the rat. Inflammation. 1998;22:509–519. doi: 10.1023/a:1022350111213. [DOI] [PubMed] [Google Scholar]
- 12.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 13.Donnelly MT, Hawkey CJ. COX-II inhibitors – a new generation of safer NSAIDs? Aliment Pharmacol Ther. 1997;11:227–236. doi: 10.1046/j.1365-2036.1997.154330000.x. [DOI] [PubMed] [Google Scholar]
- 14.Black SC, Brideau C, Cirino M, Belley M, Bosquet J, Chan CC, Rodger IW. Differential effect of a selective cyclo-oxygenase-2 inhibitor versus indomethacin on renal blood flow in conscious volume-depleted dogs. J Cardiovasc Pharmacol. 1998;32:686–694. doi: 10.1097/00005344-199811000-00002. [DOI] [PubMed] [Google Scholar]
- 15.Iniguez MA, Punzon C, Fresno M. Induction of cyclo-oxygenase-2 on activated T lymphocytes: regulation of T cell activation by cyclooxygenase-2 inhibitors. J Immunol. 1999;163:111–119. [PubMed] [Google Scholar]
- 16.Anguita J, Samanta S, Barthold SW, Fikrig E. Ablation of interleukin-12 exarcebates Lyme arthritis in SCID mice. Infect Immun. 1997;65:4334–4336. doi: 10.1128/iai.65.10.4334-4336.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mark KS, Trickler WJ, Miller DW. Tumor necrosis factor-alpha induces cyclooxygenase-2 expression and prostaglandin release in brain microvessel endothelial cells. J Pharmacol Exp Ther. 2001;297:1051–1058. [PubMed] [Google Scholar]
- 18.Matsuura H, Sakaue M, Subbaramaiah K, Kamitani H, Eling TE, Dannenberg AJ, Tanabe T, Inoue H, Arata J, Jetten AM. Regulation of cyclooxygenase-2 by interferon gamma and transforming growth factor alpha in normal human epidermal keratinocytes and squamous carcinoma cells. Role of mitogen-activated protein kinases. J Biol Chem. 1999;274:29138–29148. doi: 10.1074/jbc.274.41.29138. [DOI] [PubMed] [Google Scholar]