

Abstract
MLL1, located on human chromosome 11, is disrupted in distinct recurrent chromosomal translocations in several leukemia subsets. Studying the MLL1 gene and its oncogenic variants has provided a paradigm for understanding cancer initiation and maintenance through aberrant epigenetic gene regulation. Here we review the historical development of model systems to recapitulate oncogenic MLL1-rearrangement (MLL-r) alleles encoding mixed-lineage leukemia fusion proteins (MLL-FPs) or internal gene rearrangement products. These largely mouse and human cell/xenograft systems have been generated and used to understand how MLL-r alleles affect diverse pathways to result in a highly penetrant, drug-resistant leukemia. The particular features of the animal models influenced the conclusions of mechanisms of transformation. We discuss significant downstream enablers, inhibitors, effectors, and collaborators of MLL-r leukemia, including molecules that directly interact with MLL-FPs and endogenous mixed-lineage leukemia protein, direct target genes of MLL-FPs, and other pathways that have proven to be influential in supporting or suppressing the leukemogenic activity of MLL-FPs. The use of animal models has been complemented with patient sample, genome-wide analyses to delineate the important genomic and epigenomic changes that occur in distinct subsets of MLL-r leukemia. Collectively, these studies have resulted in rapid progress toward developing new strategies for targeting MLL-r leukemia and general cell-biological principles that may broadly inform targeting aberrant epigenetic regulators in other cancers.
With the identification of the Philadelphia chromosome in 1960 [1] and its subsequent molecular characterization [2–4], a number of parallel studies identified genes at recurrent chromosomal breakpoints in a variety of hematologic neoplasms. Several of these studies identified a gene disrupted by 11q23 translocations associated with childhood acute leukemia, named HRTX, HRX, ALL-1, MLL, or MLL1. The similarity of mixed-lineage leukemia protein (MLL1) to the Drosophila positional identity regulator trithorax [5–8] led immediately to two major hypotheses: (1) MLL1 fusion proteins (MLL-FPs) likely transform cells through altering gene expression, and (2) HOX genes are likely direct downstream targets of MLL1 and may therefore play a role in leukemia initiated by MLL1-rearrangement (MLL-r) alleles. MLL1 translocations occur at the highest frequency in very young childhood leukemia patients, where they exhibit predominantly pro-B/pre-B-cell immunophenotypes with very poor response to therapy. MLL1 translocations in adults more commonly produce acute myelogenous leukemia (AML), suggesting that the developmental history or cell of origin influences the lineage transformed [9,10].
Here, we review two major aspects of work aiming to understand the molecular mechanisms by which MLL-r alleles result in leukemia: (1) efforts to accurately model MLL-r leukemia and (2) the use of these models to understand pathways that are critical for or modulate features of MLL-r leukemia. Ultimately these pathway studies are aimed at discovering strategies to therapeutically target MLL-r leukemia, but many of the findings are relevant to other leukemia-initiating oncogenes or other cancers, and thus the principles learned are likely to be more broadly relevant for cancers in which epigenetic deregulation plays a major role in pathogenesis.
Successes and limitations in modeling MLL-r leukemia
Mixed-lineage leukemia fusion proteins are largely hematopoietic-specific oncogenes, illustrated by the fact that ubiquitous expression results exclusively in hematologic malignancies [11]. To develop animal models of MLL-r leukemia and to develop structure-function assays, retroviral transduction strategies [12] and mouse knock-in alleles [13] were pursued. Both approaches were used to demonstrate the sufficiency and tissue selectivity of MLL-FPs in generating leukemia, but neither approach perfectly recapitulated all features of the corresponding human disease. Nonetheless, these systems served as the first structure-function assays and in vivo models, and they have been used extensively to reveal a multitude of important mechanistic information relating to MLL-FP–initiated leukemogenesis.
The use of murine retroviruses to express MLL-FPs first demonstrated the sufficiency of MLL-eleven;nineteen leukemia (ENL) to transform mouse bone marrow cells [12,14]. Several other MLL-FPs were tested shortly thereafter, and the requirement for specific sequences within the fusion partner illustrated its active participation in the transformation process. Finding specific sequences that were required in the fusion partner argued against the possibility that loss of the C-terminus contributed to MLL-FP–mediated leukemia. Significantly, the retroviral transformation assay allowed the rapid identification of critical functional motifs in both the MLL1 N terminus and the FP, and it launched the identification of important interactors and molecular mechanisms, described below. However, concerns regarding the high level of retrovirally expressed MLL-FP and the nonphysiologic presence of two remaining wild-type (WT) copies of endogenous MLL1 prompted the creation of additional animal models.
Development of knock-in mouse models of MLL-FP leukemia went through several rounds of refinement. One of the first important observations using this approach was that expression of an MLL-ALL1 fused gene, chromosome 9 (AF9) protein knocked into the mouse MLL1 locus was sufficient for the development of a long-latency AML [13]. Surprisingly, a similar knock-in encoding a transcriptionally inert lacZ fusion also produced AML (although with very long latency) [15]. This finding led to the hypothesis that stabilizing the MLL1 N terminus from proteolytic turnover or oligomerization (two attributes conferred by lacZ) contributed to leukemogenesis, a concept supported by subsequent molecular studies (reviewed in [16]). This first germline knock-in strategy resulted in developmental defects and overt phenotypes in nonhematopoietic tissues, limiting the utility of this model [11,17].
Subsequently, Cre-loxP strategies were pursued to express MLL-FPs in a manner that mimicked the human translocation and avoided ubiquitous expression of the MLL-FP. In a first generation of conditional strategies, an invertor mouse was generated in which the inverted AF4 cDNA was inserted downstream of the breakpoint homology region in the murine MLL1 gene [18]. Upon tissue-specific Cre expression, the MLL-AF4 transcript is produced exclusively in that tissue. However, continuous expression of Cre recombinase results in the reversal of the inversion reaction and restoration of the locus back to WT, reducing the level of MLL-AF4 expression. Nonetheless, this animal model allowed for tissue-specific MLL-AF4 expression. Remarkably, restricting the expression of MLL-AF4 to lymphoid cells using Lck-Cre, Rag1-Cre, or CD19-Cre still resulted in either AML or diffuse large B-cell leukemia, not the B-cell acute lymphocytic leukemia (ALL) phenotype observed most commonly in childhood MLL-r leukemia [18]. Finally, translocator mice were engineered such that MLL-ENL or MLL-AF9 would be expressed upon interchromosomal translocation in a manner most similar to human chromosomal translocations [19]. In this case, limiting the recombination event to hematopoietic stem cells (HSCs) or T-cell progenitors resulted again in predominantly AML. Cumulatively, few strategies have been successful in recapitulating the young age or B-cell phenotype characteristic of human MLL-r childhood leukemia in mouse model systems, although examples of lymphoid or mixed-lineage leukemia have been reported using retroviral strategies [20,21] and MLL-AF4 conditional knock-in, as discussed below [22].
An exception to the myeloid propensity discussed above was a conditional knock-in mouse model similar to those described above, except that expression of MLL-AF4 occurs upon excision of a stop cassette before the AF4 cDNA insertion. Using the inducible Mx1-Cre transgene or infecting bone marrow cells with a self-inactivating Cre virus, both AML and B-ALL were observed in induced animals. Furthermore, genomic analysis of the resulting B-ALL compared with normal pre-B cells was remarkably informative for identifying consistent epigenomic abnormalities induced by MLL-FPs [22]. These studies in particular allowed direct comparisons between murine and human MLL-r B-ALL using genome-wide methods (discussed below).
In contrast to the difficulty transforming a relevant B-cell population in the mouse model systems, human umbilical cord blood (UCB) can yield high frequencies of B-ALL [23,24]. By infecting lineage-depleted or CD34+ UCB cells with MLL-FPs, both the Dick and Mulloy groups observed 50%–90% of xenografted animals succumbing to B-ALL, depending on the particular immuno-compromised recipient used. This occurred despite the use of either AF9 or ENL fusion partners, not typically associated with B-ALL [9]. The cell lines generated by this method exhibited high similarity to MLL-r patient lymphoblasts at the transcriptome level [24]. However, other groups expressing the more B-lymphoid-associated MLL-AF4 protein observed only enhancement of multiline-age engraftment in xenografts, but not full transformation of UCB CD34+ cells or embryonic stem (ES) cell-derived hematopoietic progenitors [25]. In contrast, MLL-extra eleven-nineteen leukemia fusion (EEN, also called SH3GL1/SH3D2B) was capable of transforming ES-derived hematopoietic progenitors [26]. The reasons for the distinct findings regarding MLL-AF4 transformation of human cells are not yet clear.
One important but mechanistically distinct paradigm for MLL-r leukemogenesis is represented by a unique internal rearrangement found in approximately 10% of adult AML. This rearrangement does not involve fusion to a partner gene, but, rather, an internal sequence duplication results in a repeated N-terminal region, termed MLL partial tandem duplication (PTD). The molecular consequences of the PTD in chromatin targeting or modifying are not well understood, but the duplicated region generally harbors DNA recognition, nuclear localization, and CXXC regions [27], suggesting enhanced or altered target gene localization. An allele recreating the PTD from the endogenous MLL1 locus results in AML only when crossed to other mutations that occur together with this particular MLL-r allele [28–30]. The observation of elevated HOXA gene expression in hematopoietic cells of MLL-PTD knock-in mice supported the hypothesis that this molecule also functions through a gain-of-function mechanism [31]. Given that no ectopic activation domain is present in the MLL-PTD molecule, the transcriptional effector mechanisms and target genes altered in this disease are likely distinct from those altered by MLL-FPs.
Despite the limitations of the diverse models of MLL-r leukemia, significant conceptual advances have been made regarding the mechanisms of gene deregulation, role of direct protein interactors, and requisite downstream pathways that modulate the penetrance and phenotype of MLL-r leukemia. An important caveat is that the vast majority of structure-function and pathway information was derived using the retroviral expression of MLL-FPs, which predominantly produces AML. As a consequence, most conclusions regarding MLL-r leukemia pathways may relate to AML but not necessarily to ALL.
Constitutive protein interactors
Menin–lens epithelial-derived growth factor interaction with MLL1 and MLL-FPs
An early approach in understanding MLL-FP–driven leukemia was to identify functionally significant interacting proteins. One of the best characterized protein interactions occurs between the MLL1 N terminus and two proteins, termed Menin and lens epithelial-derived growth factor (LEDGF), which are involved in targeting MLL1 or MLL-FPs to chromatin [32,33]. The human MEN1 gene is a tumor-suppressor gene discovered in familial multiple endocrine neoplasia type 1 [34]. The Menin protein encoded by the MEN1 gene was identified as a constitutive component of the native MLL1 and related MLL2 complexes in several cell lines [35,36], where it bridges interaction with LEDGF [32]. LEDGF contains a PWWP domain, which binds to histone H3 trimethyl-lysine 36 (H3K36me3), particularly in nucleosomes [37]; this modification is associated with actively transcribed gene bodies. The recruitment of MLL-FPs via the PWWP domain of LEDGF, presumably through H3K36me3 binding, is necessary and can mediate transformation by MLL-ENL, circumventing the need for Menin [32]. Furthermore, when the region in LEDGF interacting with the Menin/MLL N terminus is overexpressed, this peptide suppressed colony growth of MLL-FP–transformed mouse and human cells, suppressed MLL-FP–dependent target genes, and significantly delayed leukemogenesis in a mouse model of MLL-AF9–induced AML [38]. A similar interfering peptide from the MLL1 N terminus also inhibits MLL-AF9–driven leukemogenesis [33]. These data collectively indicate that the chromatin-recruiting activity of this N-terminal complex is critical for leukemogenesis by MLL-FPs.
The Menin-MLL1 interaction domain structures have been solved [39,40], and significant progress has been made developing and improving small molecule inhibitors to disrupt this protein-protein interaction [41,42]. One concern is that the endogenous WT MLL1-Menin interaction is equally well disrupted by targeting this particular protein interaction. However, genetic deletion of Menin demonstrated that MLL1-Menin interaction is not necessary for many physiologic MLL1-dependent functions during hematopoiesis in vivo [43]. Furthermore, genomic studies support the concept that only a subset of genomic sites are regulated and bound by Menin and MLL1/MLL-FPs together [43,44]. Thus, MLL-FPs may be more sensitive to drugs disrupting the MLL1-Menin interface, owing to the particular genes depending more strongly on Menin-based recruitment (Fig. 1).
Figure 1.
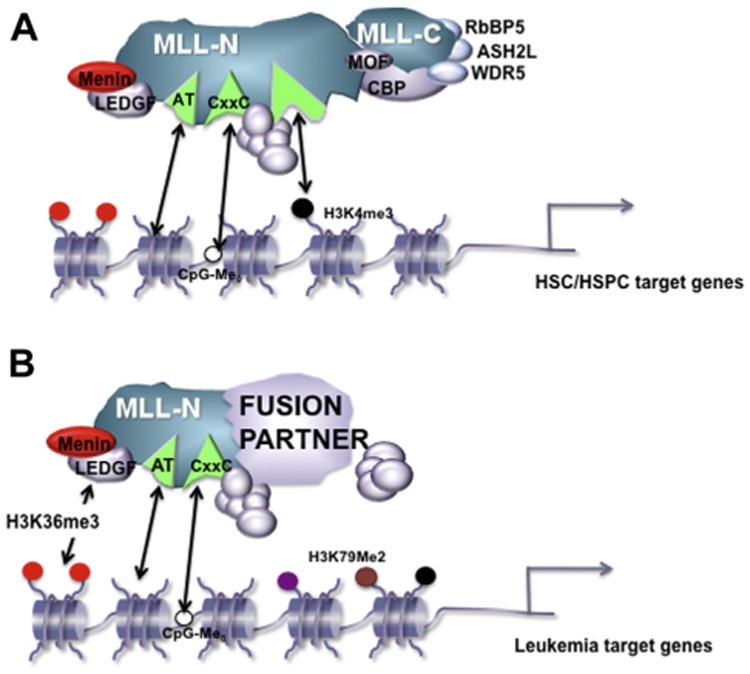
Proposed differential use of chromatin-targeting modules at (A) target genes regulated by the WT MLL1 complex during steady-state hematopoiesis or (B) target genes critical for leukemogenesis regulated by MLL-FPs. The LEDGF protein is shown recruiting MLL-FP complexes to H3K36 methylated histones, whereas the WT MLL1 complex may simply rely more on other chromatin/DNA-binding activities of the complex. Although the identity of HSC/HSPC versus leukemia target genes certainly overlap, the diagram indicates a stronger degree of dependence on Menin/LEDGF for those targets for which high level expression is required to transform hematopoietic cells. HSPC=Hematopoietic stem and progenitor cell.
CpG binding motif and polymerase associated factor complex binding (CXXC-region interactors)
Structure-function assays also identified a region within the MLL1 N terminus encompassing a domain termed ‘CXXC’ or ‘DNA methyltransferase (DNMT) homology region’ as critical for MLL-ENL–mediated transformation [45]. This region was also shown to contain two independent repression domains (RDs), identified by transient transfection assays [46]. Domain-swapping experiments illustrated that only the CXXC domain of MLL1, and not those of related CXXC-containing proteins, could support leukemogenic activity of MLL-ENL or MLL-AF9 [47,48]. Further structural studies guided the development of precise site-directed mutants and demonstrated that this domain protects CpGs at the HOXA9 locus (a direct MLL1 and MLL-FP target gene) from DNA methylation. This CpG protection activity was specifically implicated in MLL-AF9 transformation capability [49].
In a distinct line of investigation, two groups reported that the polymerase associated factor complex (PAFc) interacted directly with a region interdigitated with the CXXC and RD domains [50,51]. The PAFc in mammals is composed of multiple proteins, including PAF1, CDC73, CTR9, LEO1, WDR61/SKI8 and RTF1. This complex was originally identified in S. cerevisiae as a polymerase II (Pol II)-associated complex and is required for maintaining H3K4me3 levels, Pol II elongation efficiency, and 3′ end processing at certain genes [52]. In mammals, the PAFc has been implicated in the control of specific genes depending on the tissue examined, but a particular sensitivity of HOXA gene expression to depletion of PAFc components had been noted [53]. The PAFc is connected to histone modifications through its association with a histone 2B, lysine 120 (H2BK120) ubiquitinating complex containing ring finger proteins 20/40 (RNF20/40) and ubiquitin-conjugating enzyme E2 E1 (UBCH6) subcomplexes, which act as an E3 ubiquitin ligase and E2 conjugating enzyme, respectively, specific for H2BK120 (equivalent to H2BK123 in S. cerevisiae). Therefore, PAFc brings to a subset of genes coordinated epigenetic modifications and enhancement of Pol II elongation and processivity.
Due to the direct interaction between MLL-FPs and PAF1 and the role of the PAFc in HOX gene expression, collaboration of this complex with MLL-AF9 was examined. Coexpression of five PAFc components augments MLL1-WT or MLL-AF9–mediated transcriptional activation of luciferase reporters driven by the HOXA9 promoter. Furthermore, knockdown of CDC73 or CTR9 reduces leukemia colonies upon replating in vitro, transcriptional activation of HOXA9, and recruitment of MLL-AF9 to endogenous target loci [51]. Alternatively, conditional knockout of CDC73 or expression of a PAF1-dominant negative peptide demonstrated that the MLL1 N terminus–PAF1 interaction is critical for MLL-AF9–driven gene expression and leukemogenesis [54]. Furthermore, knockdown of PAFc components or mutation of the PAF1-interacting region results in the release of MLL-FPs from target genes; thus, these interactions may help target MLL-FPs to their target genes [50,51]. Surprisingly, normal hematopoiesis is minimally affected by disruption of the MLL1-PAF1 interaction using the dominant negative peptide, although PAF1 also interacts with the endogenous WT MLL1 protein. These findings suggested that the MLL-FP–PAF1 interaction could be a therapeutic target [54].
In a related study, the H2BK120 ubiquitin ligase RNF20 was identified in a high-throughput shRNA screen for genes that facilitate MLL-AF9–mediated leukemogenesis. Upon depletion of RNF20, MLL-AF9/N-RASG12D-initiated leukemia progressed more slowly in vivo, leukemogenic target genes were down-regulated, and histone H3, dimethyl lysine 79 (H3K79me2) enrichment at these target genes was reduced [55]. Leukemia cells transformed by MLL-FP are more dependent on RNF20 to maintain their oncogenic transcriptional program than normal hematopoietic cells, which may relate to the altered transcriptional effector activity brought by the MLL-FP [55]. In addition to influencing HOX gene expression, RNF20 is required to mark genes in mitosis for sustained expression in interphase [56] and for genomic stability in other settings [57]. Overall, the PAFc and RNF20 appear to function to ensure high-level expression of genes critical for MLL-FP–initiated leukemogenesis.
Sequence-specific interacting transcription factors
Mixed-lineage leukemia protein does not harbor sequence-specific DNA-binding activity. Thus, an attractive hypothesis to explain the target gene selectivity of MLL1 is that sequence-specific transcriptional regulators, singly or in combination, recruit MLL1 to particular enhancers or promoter regions. The Drosophila ortholog trithorax can act through definable genetic response elements (trithorax/polycomb response elements [TRE/PRE]) in conjunction with polycomb proteins. Polycomb response elements can be bound by sequence-specific proteins that are thought to recruit polycomb and trithorax proteins [58], so it is plausible that sequence-specific proteins in mammals help recruit MLL1 and MLL-FPs to particular target sequences. Several sequence-specific transcriptional regulators have been implicated in MLL-FP leukemogenic pathways; below, we describe the best functionally characterized interactions. Importantly, none of these proteins are constitutive components of MLL1 complexes, but may interact transiently with MLL1/MLL-FPs.
c-MYB: multitude of mixed-lineage leukemia protein pathway interactions
Cell line studies established that Myeloblastoma virus homolog (c-MYB) depletion can rate-limit MLL-ENL–driven leukemia, and that forced HOXA9/MEIS1 expression can maintain high c-MYB levels when MLL-ENL is withdrawn [59]. The Hess group later showed that c-MYB regulatory elements were cobound directly by HOXA9/MEIS1, suggesting that c-MYB is a second-tier, indirect target of MLL-ENL [60]. In addition, there is some evidence that MLL-FPs bind directly to the c-Myb promoter (Fig. 2), and shRNA depletion of c-Myb demonstrates that it is required to sustain MLL-AF9/N-RASG12D-driven AML in vivo [61].
Figure 2.
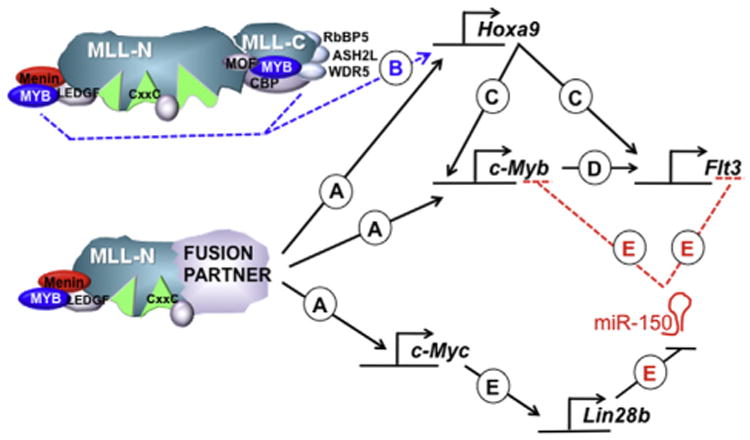
Regulatory relationships involving c-MYB in MLL-FP–mediated leukemogenesis. (A) MLL-FPs bind and directly activate the HOXA9, c-MYB, and c-MYC regulatory elements. (B) c-MYB binds directly to endogenous MLL1 potentially recruiting the complex to HOXA9 regulatory elements through N- or C-terminal interactions. (C) HOXA9 directly activates c-MYB and FLT3. (D) c-MYB directly activates the IL7R (not shown) and FLT3 genes, and (E) activation of c-MYC results in activation of Lin28b, which then inhibits processing of miR-150, resulting in increased steady state c-MYB and FLT3 expression. See main text for more detailed explanations and references.
Two other physical interactions between MLL1 and c-MYB also connect these pathways (Fig. 2). First, c-MYB can cooperatively bind with the WT MLL1 C terminus, together with CREB-binding protein (CBP)/p300 proteins [62,63]. However, this region is not maintained in MLL1 fusion oncoproteins, and therefore this interaction could only be pertinent to leukemogenesis if it acts through assisting the endogenous MLL1 protein (discussed below). Second, a distinct MLL1-N-terminal–c-MYB interaction was reported to occur indirectly through Menin, the MLL-FP binding partner [64]. In this study, direct interaction between c-MYB and Menin recruits MLL1 and results in H3K4me3 enrichment in human leukemia cells at the HOXA9 promoter and enhancement of total HOXA9 levels (Fig. 2). Thus, the multiplicity of interactions between MLL1/MLL-FPs and c-MYB may cumulatively result in the dependency of MLL-FPs on this particular pathway. In fact, a positive autoregulatory loop has been proposed connecting HOXA9/MEIS1 and c-MYB as important downstream targets of MLL-FPs [64].
Additionally, c-MYB has been implicated in MLL-FP–initiated leukemia through complex microRNA (miR)-mediated networks (Fig. 2E). Several groups noted the relative underexpression of miR-150 in MLL-r leukemia relative to matched normal cells or similar non-MLL-r leukemia. Two studies in particular investigated further the role of miR-150 in MLL-r leukemia and showed that MLL-FPs must downregulate miR-150 to efficiently transform cells [65,66]. Interestingly, Jiang et al. found that, although MLL-FPs appear to directly activate transcription of the miR-150 gene, expression of MLL-AF9 resulted in consistently less mature miR-150 [65]. These authors identified a regulatory circuit initiated by MLL-AF9 transactivation of the c-MYC gene, which in turn increases the expression of LIN28B, which then binds the pre-miR-150 species and prevents its maturation. In addition, c-MYB and FLT3 were identified as major targets of miR-150 in MLL-AF9 transformed cells, and additional targets were identified by integrating transcriptome data and in silico target predictions. Collectively, reduced expression of these targets accounts for the observation that ectopic expression of miR-150 in MLL-AF9–transduced cells severely delayed [65] or blocked [66] leukemogenesis.
cAMP-response element binding/CBP/p300 interactions
A sequence-specific transcriptional regulator that can recruit the MLL1 C-terminus is the cAMP-response element binding (CREB) transactivator together with its coactivators CBP/p300 [62]. The phosphorylation-dependent interaction between CREB and CBP exhibits cooperative binding with a specific transactivation domain of MLL1, similar to that described above for c-MYB [63]. However, the corresponding domain on CBP/p300 can either interact with c-MYB-MLL1 or CREB-MLL1 [63]. While these intriguing structural data have not yet found an in vivo relevance, CREB itself has been implicated in AML [67] and has even been shown to accelerate MLL-AF6-driven leukemia using a retroviral model [68]. These authors also found that inhibition of could limit MLL-FP–driven leukemia growth in vitro and in vivo. Glycogen synthase kinase 3–dependent phosphorylation of CREB promotes HOXA9/MEIS1 association and transcriptional activity, thus enhancing the ability of this complex to activate downstream leukemogenic target genes [68]. Thus, the potential role of CREB and endogenous MLL1 (discussed below) to enhance MLL-FP–driven leukemia could occur at several levels, as described for c-MYB. Intriguingly, fusion of MLL1 to EP300 or CREBBP (encoding CBP or p300) occurs in treatment-related AML [69], demonstrating that permanent recruitment of these proteins by covalent fusion is leukemogenic [70,71].
AML1/RUNX1 family member pathway interactions
AML1/RUNX1 encodes a transcription factor of the Runt-related (RUNX) family which is critical for the developmental emergence of HSCs and is disrupted by mutation or translocation in leukemia [72,73]. The significant association of RUNX1 loss-of-function mutations with the MLL-PTD rearrangement [74,75] prompted investigators to address whether RUNX1 loss and MLL-r alleles cooperate in the development of leukemia. Nishimoto et al. found that RUNX1-excised bone marrow can be transformed with MLL-ENL to produce AML with more accelerated kinetics compared with MLL-ENL–transduced WT cells. Although the number of leukemia-initiating cells (LICs) was not affected by loss of RUNX1, proliferation of leukemia cells was enhanced. In MLL-ENL;RUNX1-/- cells, reduced expression of the RUNX1 transcriptional target Cdkn2a (encoding p16INK4A and p19ARF) was observed, and Cdkn2a knockdown phenocopied RUNX1 loss (accelerated leukemogenesis) [76]. However, in a distinct study, RUNX1 knockdown actually inhibited MLL-AF9–driven proliferation using human UCB CD34+ cells and primary MLL-r patient lymphoblasts [77]. This group showed, using murine conditional knockout alleles, that loss of all RUNX protein function after transformation by MLL-AF9 (through coknockout of the critical cofactor CBFβ) resulted in reduced colony self-renewal in vitro and delayed leukemia kinetics in vivo. The authors suggested that, although RUNX1 may act as a tumor suppressor in MLL-ENL–initiated AML, some RUNX-regulated genes must also be required for MLL-r leukemia [77]. Thus, therapeutic targeting of this pathway may require subtle intervention. Furthermore, RUNX1 mutations do not typically occur in MLL-FP leukemia, but are found in a fraction of MLL-PTD AMLs and collaborate in the mouse model of MLL-PTD discussed earlier [29,75]. Mutations of RUNX1 that occur in leukemia are not usually complete loss-of-function alleles [73]. Also, RUNX2 is directly bound and upregulated by MLL-FPs in some murine and human leukemia cells [22,78,79]. Collectively these studies suggest that therapeutic strategies to manipulate RUNX protein levels or function may benefit from a more in-depth molecular characterization of RUNX proteins and activity in distinct leukemia models.
The above findings assess the role of RUNX proteins as modulators of MLL-FP or MLL-r disease. Distinct lines of investigation have shown that RUNX1 is a direct target gene of MLL-AF4 in B-ALL and that RUNX1 is required to maintain growth of two MLL-AF4 cell lines but not other MLL-FP cell lines [80]. In addition, several groups have obtained evidence that MLL1 interacts directly with RUNX1, suggesting that it could recruit MLL1 to target genes [80–82]. Using a chromatin immunoprecipitation followed by high-throughput DNA sequencing (ChIP-Seq) approach, Wilkinson et al. identified a subset of genes bound by both MLL-AF4 and RUNX1, suggesting coordinate regulation of some proleukemia target genes [80]. Therefore, depending on the cell type (ALL vs. AML) and particular MLL-r allele in question, several distinct mechanisms may underlie collaboration between RUNX1 and different oncogenic MLL-FP or MLL-PTD proteins; further studies will clarify the generality of these mechanisms.
Transcriptional processivity-related mechanisms
Since the initial structure-function assays identifying critical regions within the fusion partner [14], it has been clear that ectopic or deregulated transcriptional activity is a common feature brought by many FPs. However, it took several years and many distinct approaches to arrive at the observation that many common MLL-FPs act through gain-of-recruitment of protein complexes that influence gene expression through regulating the efficiency of Pol II elongation during transcription [83]. One approach that contributed to this conclusion was the purification of complexes containing AF4 and AF5Q31 using multiple common MLL-FPs [84,85]. The finding of multiple translocation partners in a complex, as well as components of a known Pol II activating complex, positive transcription elongation factor-b (P-TEFb), led to a hypothesis that many, if not all, MLL-FPs would recruit the P-TEFb complex to MLL1 target genes in a constitutive manner. The P-TEFb complex is composed of the cyclin/cyclin-dependent kinase pair cyclin T1/CDK9, which phosphorylates the RNA Polymerase II C terminus, as well as other protein complexes that limit active elongation of RNA Polymerase II (NELF and DSIF, reviewed in [86]). P-TEFb activity enhances the transition of promoter-restrained Pol II to an actively elongating state, thus increasing expression of the target gene. Interaction of AF4 with the P-TEFb complex stimulates its kinase activity, and several MLL-FPs exhibit increased complex recruitment to their target genes [84,87].
The other recruited enzymatic activity that influences transcriptional elongation contains the unique Dot1-like (DOT1L) histone methyltransferase. Using a biochemical approach, one of the FPs, AF10, was shown to interact with DOT1L and depend on the histone methyltransferase activity of this enzyme for transformation [88]. The enzyme DOT1L catalyzes H3K79 methylation, with H3K79me2 typically found enriched just downstream of the transcription start site of actively transcribed genes (reviewed in [89]). Genome-wide studies illustrated the generality of H3K79me2-enrichment at MLL-FP target genes, including in mouse models expressing diverse MLL-FPs and in leukemia patient samples (reviewed in [10]). This observation is mechanistically related to the transcriptional processivity-related processes described above; DOT1L complexes associate with P-TEFb complexes through the shared component ENL [83]. Both ENL and AF9 interact directly with DOT1L, but in a manner that is mutually exclusive with other elongation complex interactions [84,90]. Recently, drugs that inhibit DOT1L enzymatic activity have been developed and were shown to be effective at blocking leukemogenesis by MLL-AF4, MLL-ENL, MLL-AF9, and, surprisingly, MLL-PTD and MLL-AF6, which do not directly recruit DOT1L [91-93].
An additional MLL-FP–recruited protein that influences histone methylation is protein arginine methyltransferase 1 (PRMT1), which methylates histone H4 arginine 3 (H4R3), as well as arginine residues on nonhistone proteins [94]. The SRC Homology 3 (SH3) domain of EEN, a less common MLL-FP, interacts with a complex that recruits PRMT1 [95]. Evidence for the relevance of this association came from experiments in which fusion of the MLL1 N terminus to PRMT1 directly enhanced the self-renewal of transduced hematopoietic cells, HOX gene expression, and enrichment of H4R3me at the HOXA9 promoter. All of these activities depended upon the catalytic activity of PRMT1. Conversely, specific knockdown of PRMT1 suppressed MLL-EEN-mediated transformation and leukemogenesis [95].
These studies provide several examples of what is undoubtedly a much deeper list of transcriptional effector complexes that function in parallel or downstream of MLL-FPs to enable transformation. Although many of these factors seem nonspecific, since they are ubiquitously expressed general transcriptional effectors or remodeling proteins, specific requirements for MLL-FP–mediated leukemogenesis has been shown in several cases (e.g., [91]). Therefore, the specific composition of the general transcription elongation complexes in different tissues, particularly hematopoietic tissues, may underlie this observed specificity. The precise molecular mechanisms by which these enabling pathways act in MLL-FP-transformed cells versus normal tissues will be critical for determining the therapeutic promise of targeting these particular pathways in MLL-r leukemia.
Direct transcriptional targets
HOXA9 and its cofactors
The very first MLL1 target genes to be implicated as important deregulated targets of MLL-FPs in leukemia were the HOXA cluster genes, particularly HOXA9, owing to its consistently high expression in patient AML in samples and murine MLL-FP–initiated AML [96]. Therefore, the first epistasis experiments addressed the role of HOXA9 as a critical MLL-FP target using germline HOXA9 knockout cells. Such studies utilized either a knock-in MLL-AF9 mouse model crossed to a germline HOXA9 mutant or retroviral MLL-ENL introduced into HOXA9 knockout bone marrow cells. Although the MLL-ENL retroviral model exhibited an absolute dependence on HOXA9 or HOXA7 for in vitro transformation and in vivo AML [97], the knock-in model exhibited no delay in AML onset or penetrance in MLL-AF9;HOXA9-/- animals, but phenotypic differences in the resulting AML were observed [98]. Using the MLL-GAS7 FP, no absolute requirement for HOXA7 or HOXA9 was found, although loss of either HOX protein could delay leukemogenesis [99]. The use of germline HOXA9 and HOXA7 mutants in these experiments meant that the initial progenitor population was already altered [99], and thus the delay in AML progression could be attributed either to the difference in the initial transformed population or, alternatively, to the failure to maintain a full leukemogenic gene expression program in the MLL-GAS7;HOXA9-/- leukemia. In addition, redundancies between HOX genes [100] suggests that the true importance of a HOXA9-driven pathway may be attenuated by compensatory upregulation of other HOX genes in these model systems.
Proteins of the HOX family gain specificity and affinity for DNA through interaction with cofactors, particularly the PBX and MEIS proteins [101]. Interestingly, several of these cofactors or cofactor partner genes are also direct targets of MLL-FPs and contribute to leukemogenesis, such as MEIS1, PBX2/3, DACH1, EYA1 and SIX1. For example, using shRNA-mediated knockdown or fetal MEIS1-/- hematopoietic progenitors, MEIS1 was shown to be required for transformation by a variety of MLL-FPs in human and mouse cells [102,103]. The lethality of MLL-FP–initiated leukemia correlates experimentally with the ability of diverse MLL-FPs to induce high levels of MEIS1, and ectopically expressing MEIS1 accelerates further leukemia initiated by MLL-FPs. The ability to interact with DNA, transcriptional effectors, and PBX proteins were all required for MEIS1 to promote in vitro transformation by MLL-AF9 [103]. These authors also implicated PBX2 and PBX3 collectively through demonstrating that PBX2-/-;PBX3-knockdown progenitors exhibited reduced in vitro transformation by MLL-FPs. However, deeper investigation illustrated that PBX3 is more consistently up-regulated in human AML and is also functionally required for in vitro transformation by MLL-AF9 [104]. In contrast, PBX1 expression is strongly dependent on endogenous MLL1 in HSCs [105], suggesting that PBX1 differs functionally from PBX2/3 in leukemogenic versus normal hematopoietic activities.
DACH1, EYA1, and SIX1 comprise an interacting transcription factor network that regulates organogenesis across diverse species [106]. Interestingly, all three of these proteins can be induced by MLL-FPs, and DACH1 also can directly interact with HOXA9 to enhance the activation of HOXA9 target genes, including MEIS1 [107]. Thus, MLL-FPs (at least MLL-AF9) coordinately activate two interacting pathways (HOXA9-PBX1-MEIS1 and EYA1-SIX1-DACH1), effectively ensuring that no interacting partner becomes rate-limiting for transformation. Thus, these pathways could collectively be critical targets in MLL-FP–mediated leukemogenesis.
MicroRNAs that affect the HOX/MEIS interconnected pathways also have an impact on MLL-FP–driven leukemogenesis. In particularly, miR-196b has a complex relationship to MLL1 and is located between HOXA9 and HOXA10. Like the adjacent HOX genes, miR-196b is regulated by endogenous MLL1 and is also upregulated in murine and human MLL-r leukemia [108,109]. Not only do MLL1 and MLL-FPs enhance the expression of miR-196b, but HOXA9 does as well, although this may be cell-type specific [110]. In addition, HOXA9 expression can be attenuated by this microRNA, since HOXA9, along with the cooperating MEIS1 gene, are both miR-196b direct targets [111]. Thus, a self-limiting, autoregulatory loop may be part of miR-196b's function; however, its specific role in leukemia is more complex. Antagomir treatment to deplete miR-196b significantly delayed or blocked MLL-FP–mediated leukemogenesis [108,112]. However, ectopic expression of miR-196b also delayed retroviral MLL-FP leukemia in primary recipients but accelerated leukemogenesis upon secondary transplantation [111]. The apparent contradiction in the role of miR196b in MLL-FP leukemogenesis suggests the existence of an autoregulatory loop that cells must circumvent through upregulation of other pathways.
Upstream regulators of HOX genes have also been implicated in MLL-r leukemia. For example, caudal-related homeodomain-containing proteins CDX1, 2, and 4 are redundantly required for embryonic hematopoiesis [113,114], and CDX2 is frequently overexpressed in AML [115]. These divergent homeodomain proteins activate, among other targets, a subset of HOXA and HOXB genes in hematopoietic cells, and were therefore considered potential collaborators or parallel regulators in MLL-FP–initiated leukemia. Although CDX4 knockout in adult hematopoietic cells had no overt consequence on hematopoiesis, retroviral MLL-AF9–initiated AML in this genetic background was slightly delayed. MLL-AF9;CDX4-/- leukemia cells managed to upregulate HOXA genes similarly to WT cells [116]. However, shRNA-mediated depletion of CDX2 in human MLL-r AML lines reduced proliferation and colony frequency [115]. Thus, members of this family of HOX regulators likely act redundantly with each other and in parallel with MLL-FPs to support high levels of HOX gene expression and leukemogenesis. However, a pan-CDX inhibitor or reagent has not been developed to test the collective importance of CDX family members in MLL-FP–driven leukemia cells.
Evi1 and the Mecom locus
Several nonhomeodomain transcriptional regulators that are also direct targets of MLL1 or in MLL-FP–driven leukemia have been assessed as significant downstream effectors in MLL-r leukemia. The MDS-EVI1 complex locus (Mecom) encodes the Myelodysplasia syndrome 1-ecotropic virus integration site-1 (MDS-EVI1) and EVI1 proteins through the use of two alternative transcriptional start sites (reviewed in [117]). These two major isoforms differ by the presence of a PR-domain (subclass of histone methyltransferase domain) in the longer MDS-EVI1 protein. Deregulation of EVI1 can occur through translocation or other cytogenetic abnormalities, but nearly 50% of adult AML with MLL-r alleles could be categorized as Mecom-high [118]. The Mecom-high group tended to exhibit mono-blastic morphology and defined a higher risk category [118,119]. Mecom transcripts were highly expressed in HSCs, rapidly downregulated upon differentiation, and very strongly MII1- and Men1-dependent [105]. The longer, PR-domain containing isoform produced from this locus was critical for HSC function, in part through maintaining expression of the cyclin-dependent kinase inhibitor p57KIP2 [120]. Interestingly, transformation of HSCs with MLL-AF9 or MLL-ENL resulted in sustained Mecom expression whereas transformation of myeloid progenitors could not reactivate this gene [119,121]. Loss of both isoforms [122] or just the MDS-EVI1 isoform [123] profoundly limited MLL-FP leukemia, either before or after leukemia initiation, suggesting a central role in determining the penetrance and/or aggressive phenotypic characteristics. These observations, together with poor prognosis of the EVI1-high AML subgroup, suggested that MLL-r alleles that arise in HSCs may be capable of sustaining a distinct gene expression program corresponding to a more aggressive leukemia, as compared with translocations that occur in committed progenitors [124].
MEF2C in leukemia-initiating cells
Myocyte enhancer factor-2C (MEF2C) regulatory elements are bound by MLL-AF4 based on ChIP-Seq data and MEF2C was found to be overexpressed in LIC-enriched populations in murine MLL-AF9 leukemia cells [79,125,126]. However, consistent upregulation of MEF2C in pediatric AML or ALL is not apparent using unfractionated human leukemia samples (Li et al., unpublished data). Nonetheless, shRNA-mediated knockdown of MEF2C using the MLL-AF9 retroviral mouse model reduced colony growth and slowed disease progression in vivo [126]. In addition, MEF2C was identified independently in a screen for genes that rate-limited the progression of MLL-AF9–initiated AML [127]. Genetic deletion of MEF2C resulted in only a slight delay in primary or secondary transplanted MLL-ENL AML cells, or no delay at all if cells are injected directly into the peritoneum. The delay was attributed to a deficiency in homing and migration based on the reduced expression of homing and adhesion molecules including matrix metalloproteinases, chemokines, and their receptors [125].
c-MYC downstream of mixed-lineage leukemia fusion proteins
Myelocytomatosis oncogene cellular homolog (c-MYC) has been implicated as an important downstream effector and likely direct target [128] based on correlative and functional data. Multiple ChIP-Seq experiments identified the c-Myc gene as a direct transcriptional target of MLL1 or MLL-FPs, even in mitotic chromatin [44,78,129,130]. Recent attention has been refocused on c-MYC in leukemia generally, based on unexpected mechanisms by which it can be targeted. Two studies in 2011 identified inhibition of Bromodomain-containing 4 (BRD4) binding to chromatin as an effective c-MYC attenuator and an effective strategy for blocking growth of MLL-r leukemia [130,131]. One of the high-throughput shRNA screens discussed above identified BRD4 as a collaborator for MLL-AF9/N-RASG12D-driven AML [131], and the other approach utilized a proteomic discovery approach to arrive at the connection between BRD4 and MLL-FP complexes [130]. Both studies demonstrated that pharmacologic inhibition of BRD4-chromatin binding was highly effective at downregulating c-MYC expression and blocking MLL-AF9–initiated AML in mouse and human cells. The dual acetyl-lysine binding protein BRD4 plays a role in transcription elongation through P-TEFb interaction [86]. Thus, the mechanism by which the BRD4-displacing compounds act is through reduced PAFc and super elongation complex recruitment to genes that are rate-limited by transcriptional elongation [130]. Why c-MYC and other MLL-FP target genes are particularly sensitive to BRD4 removal from chromatin is an area of ongoing investigation and significant clinical significance (reviewed in [132]).
Non-target pathways that interact with MLL-r alleles
FLT3 signaling pathway interactions
Fms related tyrosine kinase 3 (FLT3) is a type III receptor tyrosine kinase and is the most common mutated gene in adult AML. Not only is FLT3 overexpression characteristic of MLL-r childhood leukemia [133], but activating mutations in FLT3 also occur in ∼ 10%–30% of MLL-r adult AML [75]. In mouse models, an activated FLT3 allele can result in fully transformed AML when crossed to the MLL-PTD model described earlier [28]. When activated FLT3 alleles are introduced together with MLL-AF9 using a retroviral transduction model, AML progression is accelerated [134]. The role of FLT3 in accelerating leukemia progression occurs without affecting the frequency of LICs within the resulting leukemia, arguing for enhanced survival of bulk leukemia due to FLT3 expression or hyperactivity. FLT3 is not likely a direct transcriptional target of MLL1 itself, but is more likely regulated by HOXA9, based on genetic and knockdown data in B-cell progenitors [135], and direct binding of HOXA9 to FLT3 regulatory elements in murine leukemia cells [60]. An ongoing phase III trial is currently testing the value of the FLT3 inhibitor lestaurtinib in conjunction with chemotherapy in infant MLL-r leukemia.
The concept of targeting activated FLT3 in MLL-r leukemia has been challenged by several publications using FLT3 loss-of-function animals [136,137]. Although genetic deletion of FLT3 in MLL-ENL–initiated leukemia delayed leukemia progression, FLT3 deletion before MLL-AF9 introduction had no effect on latency [137]. Normal development of AML in the absence of FLT3 was also observed using both MLL-ENL and MLL-CBP-induced AML [136]. Thus, if targeting this pathway in MLL-r leukemia is to be beneficial, it will likely require coordinated inhibition of compensatory pathways.
RAS/MEK/ERK pathways
Gain-of-function mutations in K-RAS and N-RAS have been detected in MLL-r ALL and AML, in both childhood and adult cases, ranging from 10%–30% depending on the population sampled ([138] and references therein). Furthermore, the presence of RAS mutant alleles were a strong independent predictor of poor outcome in MLL-r infant patients [138]. Mutant alleles are often subclonal, and it is not clear whether they are involved in the initiation of disease or evolve later as MLL-FP–expressing cells expand and compete with WT cells. Thus, collaboration between activated RAS alleles and MLL-FPs has been tested in several distinct animal models. Mice harboring a knock-in allele of MLL-AF9 exhibit slightly accelerated AML when crossed to N-RasG12V transgenic animals [139], and retrovirally-expressed MLL-SEPT6 or MLL-ENL plus retroviral N-RASG12V [140], as well as MLL-AF9 coexpressed with K-RASG12D [61], exhibit shorter survival times and increased leukemia burden. Using a unique transgenic MLL-AF4 mouse crossed to a K-RasG12D allele, Tamai et al. observed a leukemia/lymphoma of the B-cell lineage with the mean survival time cut in half relative to the MLL-AF4 transgenic animals [141]. Although the transgenic MLL-AF4 system illustrated that activated RAS was required for maintenance of the AML in vivo, the mechanisms leading to the acceleration of leukemia in most cases have not been identified.
Several indirect pathway interactions between MLL-FPs and RAS have also been reported. For example, expression of MLL-AF4 and related FPs in HeLa cells activates RAS activity as measured by phosphorylation of downstream effectors and RAS-responsive reporter genes [142]. Furthermore, EphrinA7 overexpression induced by MLL-AF4 or MLL-AF9 resulted in ERK phosphorylation in a K562 cell line system [143]. A more direct mechanism of RAS activation has recently been shown for a particular MLL-FP, MLL-AF6. The AF6 fusion partner homodimerizes and is also a RAS-GTP interacting, cytoplasmic protein; thus, the ability of MLL-AF6 to interfere with normal AF6 function was tested. Using human MLL-AF6 cells, the fusion protein was shown to redistribute endogenous AF6 to the nucleus and reduce its ability to inhibit RAS signaling [144]. Therefore, two types of epigenetic regulation occur with such fusion proteins: both direct effects of the MLL-FP on expression of target genes and indirect transcriptional effects through modulating RAS signaling in this case. These results demonstrate that events resulting in RAS/MEK/ERK activation may lead to increased penetrance of MLL-r leukemia and resistance to therapy. Combination therapies blocking activation of this pathway may provide a benefit for a subset of MLL-FPs-initiated leukemia.
Integrin β3/SYK pathway
As discussed previously, multiple shRNA-based modifier screens have been performed with the goal of identifying genes that are essential for contributing to MLL-AF9–initiated leukemia. One such approach focused on the integrin receptor gene, ITGB3, which was identified in a high-throughput shRNA screen for collaborators of MLL-AF9–mediated leukemogenesis. Using shRNA knockdown, genetic knockout, and inhibitor studies, the authors found that limiting or abolishing ITGB3 expression severely rate-limited MLL-AF9–driven leukemogenesis in a murine retroviral model [127]. ITGB3 ablation impaired MLL-AF9–transformed cell homing, resulted in the downregulation of LIC-specific transcriptional programs, and induced differentiation. These effects were mediated by the integrin-regulated kinase, spleen tyrosine kinase (SYK). Because SYK inhibitors are currently in clinical use for lymphoma, it may be feasible to test the combination of SYK inhibition together with other therapies in MLL-r leukemia.
Wnt/β-catenin pathway in parallel to MLL-FP function
The Wnt/β-catenin pathway was implicated in the maintenance of HSCs in the hematopoietic niche, as well as in LICs through multiple gain- and loss-of-function approaches. Two studies in 2010 demonstrated the importance of (β-catenin (encoded by the Ctnnb1 gene) activation in MLL-FP–initiated leukemia. Using retroviral MLL-AF9 or retroviral HOXA9/MEIS1 coexpression, Wang et al. used a comparative strategy to identify genes enriched/reactivated in LICs [145]. Analysis of the resulting data suggested that prostaglandin signaling and (β-catenin activation may be selectively activated in LICs in both MLL-AF9 and HOXA9/MEIS1-transformed cells. Nuclear (active) β-catenin was abundant in MLL-AF9 and HOXA9/MEIS1 LIC-enriched cells but in not the bulk of leukemia cells. Functionally, Ctnnb1-deficient HOXA9/MEIS1 - or MLL-AF9–transformed cells are significantly reduced in leukemia-initiating activity in vivo, suggesting that active β-catenin is required to sustain LIC production downstream or in parallel to the HOXA9/MEIS1 transcriptional program, and thus, indirectly, MLL-FPs [145]. Yeung et al. also illustrated the dependence of MLL-FPs on β-catenin signaling using mouse models and human AML cells [146]. These authors found that β-catenin loss early in the establishment of MLL-ENL–driven leukemia also reduced growth in vitro, survival in limiting cytokines, and persistence of transformed cells in engrafted animals, suggesting a broader role for β-catenin beyond the LIC in sustaining leukemia. Given the mild defects of Ctnnb1 deletion in normal bone marrow cells, targeting this pathway may offer unexpected leukemia specificity [147]. The first Wnt pathway inhibitors are now reaching clinical trials, and novel inhibitors are being developed [148,149].
NFκB in collaboration with mixed-lineage leukemia fusion proteins
In recent studies, the nuclear factor (NF)-κB pathway has been implicated in parallel with MLL-FPs in leukemogenesis. Using transformed MLL1-/- murine embryo fibroblasts (MEFs), Wang et al. demonstrated that tumor necrosis factor (TNF)α failed to induce properly RelA/p65-dependent genes, which were also shown by ChIP polymerase chain reaction (PCR) to be bound by (and presumably regulated by) MLL1. These authors also invoke other H3K4 methyl-transferases through the observation that knockdown of WDR82, a component of the SET1A/B complexes also impairs TNFα-induced gene expression [150]. MLL2-deficient macrophages also exhibit impaired induction of RelA/p65-dependent gene induction, but this was attributed to an indirect signaling defect rather than a chromatin accessibility defect [151]. These studies illustrated a role for MLL1 and the related MLL2 in the proper induction of NFκB-depen-dent genes, some of which could contribute to leukemogenesis. In a more direct approach, Kuo et al. demonstrated through drug screening and data mining approaches that inhibition of RelA/p65 limits MLL-FP–driven leukemia [152]. Although direct interaction between MLL1 or MLL-FPs and RelA/p65 was not shown, the authors demonstrate that the binding of MLL-AF10 to HOXA9 and MEIS1 regulatory elements is reduced upon NFκB inhibition, thus implicating this sequence specific transcriptional regulator in facilitating access to these genes [152]
CEBPα and PU.1 as permissive factors
Using similar approaches, both CAAT enhancer binding protein (CEBP)α and PU.1 have been implicated as parallel regulators of critical leukemogenesis pathways in MLL-FP–driven AML [153,154]. In the case of CEBPα, established MLL-ENL leukemia is not sensitive to loss of this transcriptional regulator, however deletion of CEBPα even in purified granulocyte macrophage progenitors renders cells resistant to MLL-ENL–mediated leukemogenesis and also incapable of HOXA9 induction. In addition to enabling HOXA9 overexpression, C/EBPα must regulate other important leukemogenic targets downstream of the HOXA9/MEIS1 pathway, since HOXA9/MEIS1 coexpression also cannot transform CEBPA-/- cells [153]. Similarly, MLL-AF9–transformed PU.1 hypomorphic bone marrow exhibits delayed leukemia development, and conditional deletion of PU.1 in MLL-AF9 leukemia completely blocks leukemogenesis. PU.1 likely acts in parallel to MLL-FPs, in that it directly activates a subset of HOXA9/MEIS1 regulated genes such as MEIS1 itself, PBX3, c-Kit, and FLT3, where its binding site is frequently found together with MEIS1 sites at enhancers [154]. These transcriptional regulators also play very important roles in myelopoiesis, so targeting these pathways would have to involve very sophisticated approaches to achieve selectivity toward leukemia cells. Nonetheless, understanding the relationship between parallel pathways critical for the initiation or maintenance of MLL-r leukemia will broaden our understanding of the leukemogenic pathways and how they differ from physiologic myelopoiesis/granulopoiesis.
Histone-modifying complexes
Since the realization that mammalian trithorax-group (Trx-G) and polycomb-group (Pc-G) homologs participate in cancer, the paradigm of their mutual antagonism has informed the discovery of pathways that could restore normal gene expression programs to neoplastic cells in which one side of the Trx-G/Pc-G balance is disrupted [155,156]. Two Pc-G complexes (PRC1 and PRC2) function in gene repression nucleated by H2AK119 and H3K27 modifications, respectively. In the case of leukemia driven by MLL-FPs, the role of several PRC1 and PRC2 complex components as parallel/downstream pathways has been delineated through independent approaches.
Enhancer of Zeste homolog 2 (EZH2) is the enzymatic component of PRC2, catalyzing H3K27me3, associated with gene repression. Tanaka et al. showed that MLL-AF9 transformation of EZH2-/- bone marrow cells reduces cell growth and maintenance of the undifferentiated state in vitro and reduces LIC frequency in vivo [157]. Deletion of EZH2 in an established AML only delayed leukemia onset by two weeks, resulting in an altered phenotype that more closely resembled chronic myelomonocytic leukemia. EGR1 was identified as a derepressed gene responsible for some of the differentiation/growth inhibition phenotypes, although derepression of CDKN2A, among hundreds of other genes, was also observed. Genome-wide studies and Western blots demonstrated redundancy with the related EZH1, which maintains some H3K27me3 upon EZH2 deletion [157]. Similarly, Neff et al. observed a delay in MLL-AF9–driven leukemia progression in EZH2-/- hematopoietic progenitors only upon secondary transplant, but, in contrast, deletion of EED, a distinct PRC2 component, resulted in significant (> threefold) delay in leukemia onset, and animals that developed leukemia exhibited incomplete deletion of the EED gene [158]. This delay was attributed to downregulation of a suite of c-MYC-regulated genes. EED is critical for all canonical PRC2 activities and also regulates prosurvival genes in HSCs [158,159]. In addition, shRNA-knockdown screens [160] identified PRC2 members EED and SUZ12, as well as KDM1A, as potent MLL-AF9/N-RASG12D collaborators. Further studies demonstrated that knockdown of EED, SUZ12, EZH2, or EZH1 slowed leukemia progression and in vitro growth, and the authors recapitulated the in vitro growth suppression by individually expressing the derepressed target genes ID2, ZMAT3, PERP, and CDKN1A. Importantly, all three studies illustrate the importance of PRC2 components in sustaining MLL-AF9–initiated leukemia involving parallel pathways. Furthermore, these studies suggest that blocking PRC2 function can dominantly enhance differentiation of MLL-AF9 leukemia, illustrating that this principle may be particularly effective in combination with other therapies in the treatment MLL-r leukemia. In fact, a drug-like stabilized α-helix (SAH) of EZH2 (SAH-EZH2) selectively inhibits H3K27me3. This is thought to occur by disrupting the EZH2 complex with embryonic ectoderm cevelopment (EED) and reducing EZH2 protein levels, resulting in growth arrest and differentiation of MLL-AF9 leukemic cells [161].
Two components of the PRC1 complex, BMI1 and chromobox homolog 8 (CBX8), have also been studied in detail downstream of MLL-FPs. Germline Bmi1-/- granulocyte macrophage progenitors transduced with MLL-AF9 exhibited reduced colonies upon replating, enhanced differentiation, and delayed latency in vivo [162], although the latency difference was not significant in another study [163]. Interestingly, reintroduction of Bmi1 into Bmi1-/-;MLL-AF9 leukemia was not capable of restoring normal growth or gene expression, suggesting an irreversible epigenetic commitment resulting in derepression of some important leukemia-inhibiting targets such as TBX15 and CDKN1A [162].
Through distinct mechanisms, the PRC1 component CBX8 was also required for MLL-ENL and MLL-AF9–induced leukemogenesis [164,165]. CBX8 directly interacts with AF9, and this interaction is necessary for transformation. Rather than derepressing HOX gene activation, CBX8-/- cells or human MLL-FP cell lines expressing CBX8 knockdown shRNAs exhibited reduced HOXA9 expression. The role of CBX8 as an enabler of MLL-AF9 transformation was not due to its participation in the PRC1 complex, but rather recruitment of the histone acetyltranferase TAT-interacting protein 60 (TIP60) and positive effect on gene expression at MLL-AF9 regulated genes [164]. However, Maethner et al. present evidence for a distinct mechanism by which MLL-ENL neutralizes the repressive activity of CBX8 as part of PRC1, thus allowing transformation [165]. All of these approaches illustrate that the initial hypothesis that Trx-G proteins, such as MLL1, and Pc-G proteins oppose each other's functions epigenetically at HOX genes is far too simplistic to account for the diverse effects of knocking out individual PRC components in hematopoiesis or MLL-FP–mediated leukemo-genesis. Undoubtedly, the distinct PRC components interact genetically with MLL-FPs through complex and diverse pathways still to be fully unraveled.
The KDM1A/LSD1 enzyme catalyzes lysine-specific demethylation of mono- and dimethyl-H3K4 or H3K9, depending on associated proteins. The presence of LSD1 in the native MLL1 protein complex [166] and its repressive capability prompted an assessment of its role in MLL-FP initiated leukemia. Loss of in vitro self-renewal was observed upon knockdown of KDM1A in MLL-AF9–transformed cells, which was accompanied by delayed AML latency in vivo. Interestingly, KDM1A knockdown in leukemia cells exhibited derepression of PRC2-repressed genes broadly based on genomic analyses, as well as downregulation of genes associated with the MLL-FP transformation program. Pharmacologic inhibition of KDM1A using tranylcypromine analogs phenocopied KDM1A knockdown in both murine and primary human MLL-r AML cells [167]. On the other hand, deletion of KDM1A during normal hematopoiesis revealed a failure of HSC differentiation and derepression of HSC-signature genes in more mature populations (including MLL-FP upregulated genes such as c-MYB, HOXA9, and MEIS1 [168]). This observation suggests that KDM1A loss would collaborate with MLL-FPs to enhance the undifferentiated state of leukemia cells. However, KDM1A-/- HSCs were defective in engraftment and persistence in chimeric animals, defects attributed to a failure to self-renew. The paradoxical role of KDM1A in normal hematopoiesis versus MLL-FP–driven leukemia underscores the need to determine the precise molecular pathways involved in promoting leukemogenesis versus those required for enabling differentiation of hematopoietic cells.
Finally, evidence supporting the role of endogenous MLL1 itself in MLL-FP–mediated leukemia comes from two studies. One study used knockdown and MLL1 deletion approaches to illustrate the requirement for endogenous MLL1 to promote MLL-AF9–driven leukemogenesis [169]. The second approach demonstrated that the localization (by ChIP) of MLL-AF9 to the HOXA9 promoter region in MEFs required endogenous MLL1, suggesting that chromatin modifications performed by MLL1 may prepare the locus for binding by MLL-AF9, perhaps through its H3K4 methylation activity, since this is absent from fusion proteins [50]. However, a recent study illustrates that cells lacking the H3K4 methylation domain of MLL1 fully support leukemogenesis by MLL-AF9, so any requirement for endogenous MLL1 may be through indirect means [170].
Chromatin remodeling complexes
ATP-dependent chromatin remodeling complexes have also been implicated in enabling MLL-FP leukemogenesis. RuvB-like AAA ATPase 2 (RUVBL2), a helicase with chromatin remodeling activity (in the NUA4/INO80 complex), is upregulated by MLL-AF9 and is important for maintaining survival and proliferation of a conditionally-transformed MLL-AF9 cell line, as well as the human THP-1 (MLL-AF9 expressing) cell line. The connection between MLL-AF9 and RUVBL2 pathways is not yet clear, although proliferation and survival are both compromised upon RUVBL2 knockdown [171]. Similarly, the BRM/SWI2 related gene-1 (BRG1) helicase, a component of the ATP-dependent switch/sucrose non-fermenting (SWI/SNF) chromatin-remodeling complex, was also shown to facilitate MLL-AF9/N-RASG12D-driven AML based on knockdown studies. In this case, the mechanism is likely to involve regulating transcription factor access to a critical c-MYC downstream enhancer [172].
DNA methylation: correlations and relationships with mixed-lineage leukemia fusion protein leukemogenesis
Several groups have noted that MLL-r leukemia exhibits overall hypermethylated DNA, extending to both promoter regions and repetitive elements [173,174]. This observation suggests a mechanism by which MLL-FPs generally alter the efficiency of CpG methylation. Evidence for such a mechanism has come from an MLL-PTD/FLT3-ITD mouse model, in which increased levels of DNMT1, DNMT3A, and DNMT3A protein were observed [175]. Furthermore, a LIC population within retrovirally transformed MLL-AF9 leukemia exhibited increased DNMT1 transcript levels, and even loss of one allele of DNMT1 significantly delayed self-renewal in vitro and leukemia development in vivo [176,177]. In addition to DNMT enzymes, ten-eleven translocation methylcytosine dioxygenase (TET) proteins also influence global CpG methylation levels through converting 5-methylcytoxine to 5-hydroxymethylcytosine, which then can be converted to thymidine through base-excision repair [178]. Researchers identified TET1 as upregulated in a subset of MLL-r AML, as well as MLL-AF9–transduced mouse bone marrow [179]. These authors also showed that TET1 knockdown or knockout in MLL-AF9 cells exhibited slower disease progression relative to MLL-AF9 transformed WT cells. Together with the observations of increased global methylation and DNMT expression described above, these studies suggest that MLL-r leukemia exhibits more epigenetic plasticity (both increased DNA methylation and demethylation potential), facilitating the selection of leukemogenic clones with optimal gene expression levels for the maintenance of the transformed state, or possibly evading drug targeting strategies.
Summary
Although a significant number of genes have been identified whose activities are either required for or play a significant role in either initiating or sustaining MLL-r–dependent transformation of hematopoietic cells, the work covered in this review (Table 1) likely represents the tip of a very large iceberg. None of the shRNA-mediated discovery approaches have been designed to achieve genome-wide saturation, and genetic confirmation experiments require significant time to complete. As technologies to edit the genome become more routinely employed, the complete collection of genes that are required to generate and maintain an MLL-FP–dependent AML may be within reach. However, since the retroviral transduction strategy is the most amenable to use of shRNA or genome editing techniques, the complete compendium of genes required for MLL-r leukemia is likely to come from this model system. Acute myelogenous leukemia driven by other MLL-FPs, AML initiated by the MLL-PTD, and ALL models still remain the least well understood with respect to critical downstream/parallel pathways required for leukemogenesis. Many fewer epistasis experiments have been performed using the knock-in models of MLL-r leukemia, but discordant results comparing requirement for HOXA9 in the retroviral and knock-in models of MLL-AF9–initiated leukemia [97,98] demonstrate how important it is to determine the accuracy of each model in predicting the relevance of particular genetic networks in the corresponding human disease.
Table 1. Genes and pathways that support MLL-r leukemogenesis.
| Category | Molecule/domain | Relationship to MLL-FP | References |
|---|---|---|---|
| Constitutive protein interactors | MENIN/LEDGF | Direct interaction | [32,33,35,38] |
| CpG/PAF/CXXC | Direct interaction | [45,49–51,54,55] | |
| Sequence-specific interactors | c-MYB (& miR-150) | Direct interaction/Downstream | [59–61,64–66] |
| CREB/CBP/p300 | Direct interaction/Parallel | [68] | |
| AML1/RUNX1 family | Parallel/Downstream | [76,77,80–82] | |
| Transcriptional processivity-related | P-TEFb | Direct interaction | [84,85] |
| DOT1L | Direct interaction | [84,88,90–93] | |
| PRMT1 | Direct interaction | [95] | |
| Direction transcriptional target | HOXA7/HOXA9/CDXs | Downstream/Parallel | [97–99,115,116] |
| MEIS1/PBX2/PBX3 | Downstream | [102,103] | |
| EYA1/SIX1/DACH1 | Downstream | [107] | |
| miR-196b | Downstream | [108,111,112] | |
| MDS-EVI1, EVI1 | Downstream | [122,123] | |
| MEF2C | Downstream | [125–127] | |
| c-MYC | Downstream | [130,131] | |
| Signaling pathway | FLT3 | Parallel | [28,134,136,137] |
| RAS/MEK/ERK | Parallel | [61,139–141] | |
| Integrin β3/SYK | Parallel | [127] | |
| Wnt/β-catenin | Parallel | [145,146] | |
| NFκB | Parallel | [152] | |
| CEBPα/PU.1 | Parallel | [153,154] | |
| Histone modifying complex | EZH2/EED/SUZ12 | Parallel | [157,158] |
| BMI1 | Parallel | [162,163] | |
| CBX8 | Parallel | [164,165] | |
| KDM1A/LSD1 | Parallel | [167] | |
| MLL1 | Parallel | [169] | |
| Chromatin remodeling complex | RUVBL2 | Parallel | [171] |
| BRG1 | Parallel | [172] | |
| DNA methylation | DNMT1/3A/3B | Parallel | [175–177] |
| TET1 | Parallel | [179] |
The category of interaction with MLL-r alleles; name of the molecule, domain, or pathway; and reference numbers are shown. Genes are categorized as they are in the text into general pathways based on physical or functional data.
Although the high-throughput knockdown screens have been performed with mouse cells, in principle these approaches are possible using human hematopoietic progenitors and xenotransplantation [23,24]. If murine models fail to accurately represent pediatric B-ALL, for example, it may be possible to assess gene regulatory pathways using the UCB transformation system. There are certainly differences between MLL-r AML versus ALL that will correspond to differences in important downstream pathways. Thus, pathways that could be targeted in MLL-r pediatric ALL, with which MLL-r alleles are frequently associated, may not benefit from all of the molecular targets identified in the existing preclinical models, which tend to yield AML.
Despite the limitations of existing animal models, significant insight into the pathways deregulated by MLL-r alleles has been achieved. The genes required for MLL-FP transformation can be categorized into coherent pathways (Table 1). For example, structure-function assays identified key motifs within the MLL N terminus and within the fusion partner, which in turn illuminated the role of ectopic recruitment of P-TEFb and DOT1L complexes and Pol II elongation as a major deregulated mechanism in leukemogenesis. However, unanswered questions still remain. For example, without a fusion partner, does the MLL-PTD molecule transform cells though a distinct mechanism? If so, why is it also sensitive to DOT1L inhibitors?
The ability to transfer leukemia through secondary transplantation and to introduce MLL-FPs into distinct hematopoietic populations has also yielded insight into leukemia development. This feature of the retroviral models allowed investigators to define LIC-enriched populations, which in turn revealed some distinct molecular pathways regulated by MLL-FPs as compared with those regulated in the majority of leukemia cells. Defining a small LIC-enriched population enabled the identification of pathways regulated by MLL-FPs specifically involved in leukemia self-renewal, resulting in the definition of a LIC-enriched signature that had prognostic significance [124]. Furthermore, the introduction of MLL-FPs into purified HSCs versus other progenitors illustrated that the cell-of-origin has a permanent impact on gene expression in the resulting leukemia [122] and could be selectively targeted utilizing the LIC-specific pathway information [145].
Ultimately, the translational challenges that remain center on assessment of the animal models and biochemical data to arrive at effective therapeutic strategies to treat MLL-r leukemia. A complete compendium of all the downstream/parallel pathways that are required for MLL-r leukemogenesis is not necessarily useful if these same pathways are also critical for normal hematopoietic function. Therefore, a deeper understanding of why MLL-FPs rely on some pathways more than the equivalent normal hematopoietic population is warranted. Many of the genetic studies illustrate that inhibition of a single pathway or inhibition of a particular MLL-FP–interacting protein is insufficient to completely block leukemia progression. Furthermore, extending the knowledge gained from genetic analysis and identifying pharmacologic targets with minimal effects on normal tissue development and homeostasis will be challenging but of critical importance, especially for young patients. The pace at which novel therapeutic strategies have been assessed at a proof-of-principle level has accelerated recently, as has the development of novel targeted therapeutics. As many of these therapeutic strategies reach preclinical and clinical trials, targeting this particular epigenetic oncoprotein will likely continue to serve as a paradigm for targeting epigenetics in cancer.
Acknowledgments
We thank lab members R. Mako Saito and L. Castillia for commenting on the manuscript. We thank our many colleagues for discussions and sharing unpublished information and viewpoints, and we apologize for work that was omitted for relative brevity. This work was supported by grants from the American Cancer Society (no. RSG-10-242-LIB) and National Institutes of Health (no. HL090036).
Footnotes
Conflict of interest disclosure: No financial interest/relationships with financial interest relating to the topic of this article have been declared.
References
- 1.Nowell PC, Hungerford D. A minute chromosome in human chronic granulocytic leukemia. Science. 1960;132:1497. doi: 10.1126/science.144.3623.1229. [DOI] [PubMed] [Google Scholar]
- 2.Groffen J, Stephenson JR, Heisterkamp N, de Klein A, Bartram CR, Grosveld G. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell. 1984;36:93–99. doi: 10.1016/0092-8674(84)90077-1. [DOI] [PubMed] [Google Scholar]
- 3.Rowley JD. Ph1-positive leukaemia, including chronic myelogenous leukaemia. Clin Haematol. 1980;9:55–86. [PubMed] [Google Scholar]
- 4.Lugo TG, Pendergast AM, Muller AJ, Witte ON. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science. 1990;247:1079–1082. doi: 10.1126/science.2408149. [DOI] [PubMed] [Google Scholar]
- 5.Tkachuk DC, Kohler S, Cleary ML. Involvement of a homolog of Drosophila trithorax by 11q23 chromosomal translocations in acute leukemias. Cell. 1992;71:691–700. doi: 10.1016/0092-8674(92)90602-9. [DOI] [PubMed] [Google Scholar]
- 6.Gu Y, Nakamura T, Alder H, et al. The t(4;11) chromosome translocation of human acute leukemias fuses the ALL-1 gene, related to Drosophila trithorax, to the AF-4 gene. Cell. 1992;71:701–708. doi: 10.1016/0092-8674(92)90603-a. [DOI] [PubMed] [Google Scholar]
- 7.Djabali M, Selleri L, Parry P, Bower M, Young BD, Evans GA. A trithorax-like gene is interrupted by chromosome 11q23 translocations in acute leukaemias. Nat Genet. 1992;2:113–118. doi: 10.1038/ng1092-113. [DOI] [PubMed] [Google Scholar]
- 8.Domer PH, Fakharzadeh SS, Chen CS, et al. Acute mixed-lineage leukemia t(4;11)(q21;q23) generates an MLL-AF4 fusion product. Proc Natl Acad Sci U S A. 1993;90:7884–7888. doi: 10.1073/pnas.90.16.7884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jansen MW, Corral L, van der Velden VH, et al. Immunobiological diversity in infant acute lymphoblastic leukemia is related to the occurrence and type of MLL gene rearrangement. Leukemia. 2007;21:633–641. doi: 10.1038/sj.leu.2404578. [DOI] [PubMed] [Google Scholar]
- 10.Neff T, Armstrong SA. Recent progress toward epigenetic therapies: the example of mixed lineage leukemia. Blood. 2013;121:4847–4853. doi: 10.1182/blood-2013-02-474833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dobson CL, Warren AJ, Pannell R, et al. The mll-AF9 gene fusion in mice controls myeloproliferation and specifies acute myeloid leukaemogenesis. EMBO J. 1999;18:3564–3574. doi: 10.1093/emboj/18.13.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lavau C, Szilvassy SJ, Slany R, Cleary ML. Immortalization and leukemic transformation of a myelomonocytic precursor by retrovirally transduced HRX-ENL. EMBO J. 1997;16:4226–4237. doi: 10.1093/emboj/16.14.4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corral J, Lavenir I, Impey H, et al. An MLL-AF9 fusion gene made by homologous recombination causes acute leukemia in chimeric mice: a method to create fusion oncogenes. Cell. 1996;85:853–861. doi: 10.1016/s0092-8674(00)81269-6. [DOI] [PubMed] [Google Scholar]
- 14.Slany RK, Lavau C, Cleary ML. The oncogenic capacity of HRX-ENL requires the transcriptional transactivation activity of ENL and the DNA binding motifs of HRX. Mol Cell Biol. 1998;18:122–129. doi: 10.1128/mcb.18.1.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dobson CL, Warren AJ, Pannell R, Forster A, Rabbitts TH. Tumorigenesis in mice with a fusion of the leukaemia oncogene MLL and the bacterial lacZ gene. EMBO J. 2000;19:843–851. doi: 10.1093/emboj/19.5.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muntean AG, Hess JL. The pathogenesis of mixed-lineage leukemia. Annu Rev Pathol. 2012;7:283–301. doi: 10.1146/annurev-pathol-011811-132434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen W, Li Q, Hudson WA, Kumar A, Kirchhof N, Kersey JH. A murine MLL-AF4 knock-in model results in lymphoid and myeloid deregulation and hematologic malignancy. Blood. 2006;108:669–677. doi: 10.1182/blood-2005-08-3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Metzler M, Forster A, Pannell R, et al. A conditional model of MLL-AF4 B-cell tumourigenesis using invertor technology. Oncogene. 2006;25:3093–3103. doi: 10.1038/sj.onc.1209636. [DOI] [PubMed] [Google Scholar]
- 19.Drynan LF, Pannell R, Forster A, et al. MLL fusions generated by Cre-loxP-mediated de novo translocations can induce lineage reassignment in tumorigenesis. EMBO J. 2005;24:3136–3146. doi: 10.1038/sj.emboj.7600760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.So CW, Karsunky H, Passegue E, Cozzio A, Weissman IL, Cleary ML. MLL-GAS7 transforms multipotent hematopoietic progenitors and induces mixed lineage leukemias in mice. Cancer Cell. 2003;3:161–171. doi: 10.1016/s1535-6108(03)00019-9. [DOI] [PubMed] [Google Scholar]
- 21.Zeisig BB, Garcia-Cuellar MP, Winkler TH, Slany RK. The oncoprotein MLL-ENL disturbs hematopoietic lineage determination and transforms a biphenotypic lymphoid/myeloid cell. Oncogene. 2003;22:1629–1637. doi: 10.1038/sj.onc.1206104. [DOI] [PubMed] [Google Scholar]
- 22.Krivtsov AV, Feng Z, Lemieux ME, et al. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell. 2008;14:355–368. doi: 10.1016/j.ccr.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barabe F, Kennedy JA, Hope KJ, Dick JE. Modeling the initiation and progression of human acute leukemia in mice. Science. 2007;316:600–604. doi: 10.1126/science.1139851. [DOI] [PubMed] [Google Scholar]
- 24.Wei J, Wunderlich M, Fox C, et al. Microenvironment determines lineage fate in a human model of MLL-AF9 leukemia. Cancer Cell. 2008;13:483–495. doi: 10.1016/j.ccr.2008.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Montes R, Ayllon V, Gutierrez-Aranda I, et al. Enforced expression of MLL-AF4 fusion in cord blood CD34+ cells enhances the hematopoietic repopulating cell function and clonogenic potential but is not sufficient to initiate leukemia. Blood. 2011;117:4746–4758. doi: 10.1182/blood-2010-12-322230. [DOI] [PubMed] [Google Scholar]
- 26.Kong CT, Sham MH, So CW, Cheah KS, Chen SJ, Chan LC. The MLL-Een knockin fusion gene enhances proliferation of myeloid progenitors derived from mouse embryonic stem cells and causes myeloid leukaemia in chimeric mice. Leukemia. 2006;20:1829–1839. doi: 10.1038/sj.leu.2404342. [DOI] [PubMed] [Google Scholar]
- 27.Schichman SA, Caligiuri MA, Gu Y, et al. ALL-1 partial duplication in acute leukemia. Proc Natl Acad Sci U S A. 1994;91:6236–6239. doi: 10.1073/pnas.91.13.6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zorko NA, Bernot KM, Whitman SP, et al. MLL partial tandem duplication and Flt3 internal tandem duplication in a double knock-in mouse recapitulates features of counterpart human acute myeloid leukemias. Blood. 2012;120:1130–1136. doi: 10.1182/blood-2012-03-415067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y, Yan X, Chen A, Sashida G, Xiao Z, Huang G. Modeling and targeting MLL-PTD/RUNX1 related MDS/AML in mouse. Blood Abstract. 2013;122 [Google Scholar]
- 30.Gaidzik VI, Bullinger L, Schlenk RF, et al. RUNX1 mutations in acute myeloid leukemia: results from a comprehensive genetic and clinical analysis from the AML study group. J Clin Oncol. 2011;29:1364–1372. doi: 10.1200/JCO.2010.30.7926. [DOI] [PubMed] [Google Scholar]
- 31.Dorrance AM, Liu S, Yuan W, et al. MLL partial tandem duplication induces aberrant Hox expression in vivo via specific epigenetic alterations. J Clin Invest. 2006;116:2707–2716. doi: 10.1172/JCI25546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yokoyama A, Cleary ML. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell. 2008;14:36–46. doi: 10.1016/j.ccr.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caslini C, Yang Z, El-Osta M, Milne TA, Slany RK, Hess JL. Interaction of MLL amino terminal sequences with menin is required for transformation. Cancer Res. 2007;67:7275–7283. doi: 10.1158/0008-5472.CAN-06-2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang EH, Ebrahimi SA, Wu AY, Kashefi C, Passaro E, Jr, Sawicki MP. Mutation of the MENIN gene in sporadic pancreatic endocrine tumors. Cancer Res. 1998;58:4417–4420. [PubMed] [Google Scholar]
- 35.Yokoyama A, Somervaille TC, Smith KS, Rozenblatt-Rosen O, Meyerson M, Cleary ML. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell. 2005;123:207–218. doi: 10.1016/j.cell.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 36.Hughes CM, Rozenblatt-Rosen O, Milne TA, et al. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell. 2004;13:587–597. doi: 10.1016/s1097-2765(04)00081-4. [DOI] [PubMed] [Google Scholar]
- 37.Eidahl JO, Crowe BL, North JA, et al. Structural basis for high-affinity binding of LEDGF PWWP to mononucleosomes. Nucleic Acids Res. 2013;41:3924–3936. doi: 10.1093/nar/gkt074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mereau H, De Rijck J, Cermakova K, et al. Impairing MLL-fusion gene-mediated transformation by dissecting critical interactions with the lens epithelium-derived growth factor (LEDGF/p75) Leukemia. 2013;27:1245–1253. doi: 10.1038/leu.2013.10. [DOI] [PubMed] [Google Scholar]
- 39.Murai MJ, Chruszcz M, Reddy G, Grembecka J, Cierpicki T. Crystal structure of menin reveals binding site for mixed lineage leukemia (MLL) protein. J Biol Chem. 2011;286:31742–31748. doi: 10.1074/jbc.M111.258186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang J, Gurung B, Wan B, et al. The same pocket in menin binds both MLL and JUND but has opposite effects on transcription. Nature. 2012;482:542–546. doi: 10.1038/nature10806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grembecka J, He S, Shi A, et al. Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat Chem Biol. 2012;8:277–284. doi: 10.1038/nchembio.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shi A, Murai MJ, He S, et al. Structural insights into inhibition of the bivalent menin-MLL interaction by small molecules in leukemia. Blood. 2012;120:4461–4469. doi: 10.1182/blood-2012-05-429274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li BE, Gan T, Meyerson M, Rabbitts TH, Ernst P. Distinct pathways regulated by menin and by MLL1 in hematopoietic stem cells and developing B cells. Blood. 2013;122:2039–2046. doi: 10.1182/blood-2013-03-486647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang QF, Wu G, Mi S, et al. MLL fusion proteins preferentially regulate a subset of wild-type MLL target genes in the leukemic genome. Blood. 2011;117:6895–6905. doi: 10.1182/blood-2010-12-324699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ayton PM, Chen EH, Cleary ML. Binding to nonmethylated CpG DNA is essential for target recognition, transactivation, and myeloid transformation by an MLL oncoprotein. Mol Cell Biol. 2004;24:10470–10478. doi: 10.1128/MCB.24.23.10470-10478.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xia ZB, Anderson M, Diaz MO, Zeleznik-Le NJ. MLL repression domain interacts with histone deacetylases, the polycomb group proteins HPC2 and BMI-1, and the corepressor C-terminal-binding protein. Proc Natl Acad Sci U S A. 2003;100:8342–8347. doi: 10.1073/pnas.1436338100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bach C, Mueller D, Buhl S, Garcia-Cuellar MP, Slany RK. Alterations of the CxxC domain preclude oncogenic activation of mixed-lineage leukemia 2. Oncogene. 2009;28:815–823. doi: 10.1038/onc.2008.443. [DOI] [PubMed] [Google Scholar]
- 48.Risner LE, Kuntimaddi A, Lokken AA, et al. Functional specificity of CpG DNA-binding CXXC domains in mixed lineage leukemia. J Biol Chem. 2013;288:29901–29910. doi: 10.1074/jbc.M113.474858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cierpicki T, Risner LE, Grembecka J, et al. Structure of the MLL CXXC domain-DNA complex and its functional role in MLL-AF9 leukemia. Nat Struct Mol Biol. 2010;17:62–68. doi: 10.1038/nsmb.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Milne TA, Kim J, Wang GG, et al. Multiple interactions recruit MLL1 and MLL1 fusion proteins to the HOXA9 locus in leukemogenesis. Mol Cell. 2010;38:853–863. doi: 10.1016/j.molcel.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Muntean AG, Tan J, Sitwala K, et al. The PAF complex synergizes with MLL fusion proteins at HOX loci to promote leukemogenesis. Cancer Cell. 2010;17:609–621. doi: 10.1016/j.ccr.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rosonina E, Manley JL. From transcription to mRNA: PAF provides a new path. Mol Cell. 2005;20:167–168. doi: 10.1016/j.molcel.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 53.Zhu B, Zheng Y, Pham AD, et al. Monoubiquitination of human histone H2B: the factors involved and their roles in HOX gene regulation. Mol Cell. 2005;20:601–611. doi: 10.1016/j.molcel.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 54.Muntean AG, Chen W, Jones M, Granowicz EM, Maillard I, Hess JL. MLL fusion protein-driven AML is selectively inhibited by targeted disruption of the MLL-PAFc interaction. Blood. 2013;122:1914–1922. doi: 10.1182/blood-2013-02-486977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang E, Kawaoka S, Yu M, et al. Histone H2B ubiquitin ligase RNF20 is required for MLL-rearranged leukemia. Proc Natl Acad Sci U S A. 2013;110:3901–3906. doi: 10.1073/pnas.1301045110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Arora M, Zhang J, Heine GF, et al. Promoters active in interphase are bookmarked during mitosis by ubiquitination. Nucleic Acids Res. 2012;40:10187–10202. doi: 10.1093/nar/gks820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shiloh Y, Shema E, Moyal L, Oren M. RNF20-RNF40: A ubiquitin-driven link between gene expression and the DNA damage response. FEBS Lett. 2011;585:2795–2802. doi: 10.1016/j.febslet.2011.07.034. [DOI] [PubMed] [Google Scholar]
- 58.Schuettengruber B, Cavalli G. Recruitment of polycomb group complexes and their role in the dynamic regulation of cell fate choice. Development. 2009;136:3531–3542. doi: 10.1242/dev.033902. [DOI] [PubMed] [Google Scholar]
- 59.Hess JL, Bittner CB, Zeisig DT, et al. c-Myb is an essential downstream target for homeobox-mediated transformation of hematopoietic cells. Blood. 2006;108:297–304. doi: 10.1182/blood-2005-12-5014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang Y, Sitwala K, Bronstein J, et al. Identification and characterization of Hoxa9 binding sites in hematopoietic cells. Blood. 2012;119:388–398. doi: 10.1182/blood-2011-03-341081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zuber J, Rappaport AR, Luo W, et al. An integrated approach to dissecting oncogene addiction implicates a Myb-coordinated self-renewal program as essential for leukemia maintenance. Genes Dev. 2011;25:1628–1640. doi: 10.1101/gad.17269211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ernst P, Wang J, Huang M, Goodman RH, Korsmeyer SJ. MLL and CREB bind cooperatively to the nuclear coactivator CREB-binding protein. Mol Cell Biol. 2001;21:2249–2258. doi: 10.1128/MCB.21.7.2249-2258.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.De Guzman RN, Goto NK, Dyson HJ, Wright PE. Structural basis for cooperative transcription factor binding to the CBP coactivator. J Mol Biol. 2006;355:1005–1013. doi: 10.1016/j.jmb.2005.09.059. [DOI] [PubMed] [Google Scholar]
- 64.Jin S, Zhao H, Yi Y, Nakata Y, Kalota A, Gewirtz AM. c-Myb binds MLL through menin in human leukemia cells and is an important driver of MLL-associated leukemogenesis. J Clin Invest. 2010;120:593–606. doi: 10.1172/JCI38030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jiang X, Huang H, Li Z, et al. Blockade of miR-150 maturation by MLL-fusion/MYC/LIN-28 is required for MLL-associated leukemia. Cancer Cell. 2012;22:524–535. doi: 10.1016/j.ccr.2012.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bousquet M, Zhuang G, Meng C, et al. miR-150 blocks MLL-AF9–associated leukemia through oncogene repression. Mol Cancer Res. 2013;11:912–922. doi: 10.1158/1541-7786.MCR-13-0002-T. [DOI] [PubMed] [Google Scholar]
- 67.Cho EC, Mitton B, Sakamoto KM. CREB and leukemogenesis. Crit Rev Oncog. 2011;16:37–46. doi: 10.1615/critrevoncog.v16.i1-2.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang Z, Iwasaki M, Ficara F, et al. GSK-3 promotes conditional association of CREB and its coactivators with MEIS1 to facilitate HOX-mediated transcription and oncogenesis. Cancer Cell. 2010;17:597–608. doi: 10.1016/j.ccr.2010.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rowley JD, Reshmi S, Sobulo O, et al. All patients with the T(11; 16)(q23;p13.3) that involves MLL and CBP have treatment-related hematologic disorders. Blood. 1997;90:535–541. [PubMed] [Google Scholar]
- 70.Lavau C, Du C, Thirman M, Zeleznik-Le N. Chromatin-related properties of CBP fused to MLL generate a myelodysplastic-like syndrome that evolves into myeloid leukemia. EMBO J. 2000;19:4655–4664. doi: 10.1093/emboj/19.17.4655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang J, Iwasaki H, Krivtsov A, et al. Conditional MLL-CBP targets GMP and models therapy-related myeloproliferative disease. EMBO J. 2005;24:368–381. doi: 10.1038/sj.emboj.7600521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Swiers G, de Bruijn M, Speck NA. Hematopoietic stem cell emergence in the conceptus and the role of Runx1. Int J Dev Biol. 2010;54:1151–1163. doi: 10.1387/ijdb.103106gs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mangan JK, Speck NA. RUNX1 mutations in clonal myeloid disorders: from conventional cytogenetics to next generation sequencing, a story 40 years in the making. Crit Rev Oncog. 2011;16:77–91. doi: 10.1615/critrevoncog.v16.i1-2.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tang JL, Hou HA, Chen CY, et al. AML1/RUNX1 mutations in 470 adult patients with de novo acute myeloid leukemia: prognostic implication and interaction with other gene alterations. Blood. 2009;114:5352–5361. doi: 10.1182/blood-2009-05-223784. [DOI] [PubMed] [Google Scholar]
- 75.Network TCGAR. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nishimoto N, Arai S, Ichikawa M, et al. Loss of AML1/Runx1 accelerates the development of MLL-ENL leukemia through down-regulation of p19ARF. Blood. 2011;118:2541–2550. doi: 10.1182/blood-2010-10-315440. [DOI] [PubMed] [Google Scholar]
- 77.Goyama S, Schibler J, Cunningham L, et al. Transcription factor RUNX1 promotes survival of acute myeloid leukemia cells. J Clin Invest. 2013;123:3876–3888. doi: 10.1172/JCI68557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bernt KM, Zhu N, Sinha AU, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20:66–78. doi: 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Guenther MG, Lawton LN, Rozovskaia T, et al. Aberrant chromatin at genes encoding stem cell regulators in human mixed-lineage leukemia. Genes Dev. 2008;22:3403–3408. doi: 10.1101/gad.1741408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wilkinson AC, Ballabio E, Geng H, et al. RUNX1 is a key target in t(4;11) leukemias that contributes to gene activation through an AF4-MLL complex interaction. Cell Rep. 2013;3:116–127. doi: 10.1016/j.celrep.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yu M, Mazor T, Huang H, et al. Direct recruitment of polycomb repressive complex 1 to chromatin by core binding transcription factors. Mol Cell. 2012;45:330–343. doi: 10.1016/j.molcel.2011.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Huang G, Zhao X, Wang L, et al. The ability of MLL to bind RUNX1 and methylate H3K4 at PU.1 regulatory regions is impaired by MDS/AML-associated RUNX1/AML1 mutations. Blood. 2011;118:6544–6552. doi: 10.1182/blood-2010-11-317909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chi P, Allis CD, Wang GG. Covalent histone modifications–miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer. 2010;10:457–469. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yokoyama A, Lin M, Naresh A, Kitabayashi I, Cleary ML. A higher-order complex containing AF4 and ENL family proteins with P-TEFb facilitates oncogenic and physiologic MLL-dependent transcription. Cancer Cell. 2010;17:198–212. doi: 10.1016/j.ccr.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lin C, Smith ER, Takahashi H, et al. AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol Cell. 2010;37:429–437. doi: 10.1016/j.molcel.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Adelman K, Lis JT. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet. 2012;13:720–731. doi: 10.1038/nrg3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bitoun E, Oliver PL, Davies KE. The mixed-lineage leukemia fusion partner AF4 stimulates RNA polymerase II transcriptional elongation and mediates coordinated chromatin remodeling. Hum Mol Genet. 2007;16:92–106. doi: 10.1093/hmg/ddl444. [DOI] [PubMed] [Google Scholar]
- 88.Okada Y, Feng Q, Lin Y, et al. hDOT1L links histone methylation to leukemogenesis. Cell. 2005;121:167–178. doi: 10.1016/j.cell.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 89.Nguyen AT, Zhang Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011;25:1345–1358. doi: 10.1101/gad.2057811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.He N, Chan CK, Sobhian B, et al. Human Polymerase-Associated Factor complex (PAFc) connects the Super Elongation Complex (SEC) to RNA polymerase II on chromatin. Proc Natl Acad Sci U S A. 2011;108:E636–645. doi: 10.1073/pnas.1107107108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Daigle SR, Olhava EJ, Therkelsen CA, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011;20:53–65. doi: 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Deshpande AJ, Chen L, Fazio M, et al. Leukemic transformation by the MLL-AF6 fusion oncogene requires the H3K79 methyltransferase Dot1l. Blood. 2013;121:2533–2541. doi: 10.1182/blood-2012-11-465120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kühn MWM, Hadler M, Daigle SR, et al. Myeloid leukemia cells with MLL partial tandem duplication are sensitive to pharmacological inhibition of the H3K79 methyltransferase DOT1L ASH Abstract. 2013 [Google Scholar]
- 94.Wolf SS. The protein arginine methyltransferase family: an update about function, new perspectives and the physiological role in humans. Cell Mol Life Sci. 2009;66:2109–2121. doi: 10.1007/s00018-009-0010-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cheung N, Chan LC, Thompson A, Cleary ML, So CW. Protein arginine-methyltransferase-dependent oncogenesis. Nat Cell Biol. 2007;9:1208–1215. doi: 10.1038/ncb1642. [DOI] [PubMed] [Google Scholar]
- 96.Argiropoulos B, Humphries RK. Hox genes in hematopoiesis and leukemogenesis. Oncogene. 2007;26:6766–6776. doi: 10.1038/sj.onc.1210760. [DOI] [PubMed] [Google Scholar]
- 97.Ayton PM, Cleary ML. Transformation of myeloid progenitors by MLL oncoproteins is dependent on Hoxa7 and Hoxa9. Genes Dev. 2003;17:2298–2307. doi: 10.1101/gad.1111603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kumar AR, Hudson WA, Chen W, Nishiuchi R, Yao Q, Kersey JH. Hoxa9 influences the phenotype but not the incidence of MLL-AF9 fusion gene leukemia. Blood. 2004;103:1823–1828. doi: 10.1182/blood-2003-07-2582. [DOI] [PubMed] [Google Scholar]
- 99.So CW, Karsunky H, Wong P, Weissman IL, Cleary ML. Leukemic transformation of hematopoietic progenitors by MLL-GAS7 in the absence of Hoxa7 or Hoxa9. Blood. 2004;103:3192–3199. doi: 10.1182/blood-2003-10-3722. [DOI] [PubMed] [Google Scholar]
- 100.Bijl J, Thompson A, Ramirez-Solis R, et al. Analysis of HSC activity and compensatory Hox gene expression profile in Hoxb cluster mutant fetal liver cells. Blood. 2006;108:116–122. doi: 10.1182/blood-2005-06-2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Longobardi E, Penkov D, Mateos D, De Florian G, Torres M, Blasi F. Biochemistry of the tale transcription factors PREP, MEIS, and PBX in vertebrates. Dev Dyn. 2014;243:59–75. doi: 10.1002/dvdy.24016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kumar AR, Li Q, Hudson WA, et al. A role for MEIS1 in MLL-fusion gene leukemia. Blood. 2009;113:1756–1758. doi: 10.1182/blood-2008-06-163287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wong P, Iwasaki M, Somervaille TC, So CW, Cleary ML. Meis1 is an essential and rate-limiting regulator of MLL leukemia stem cell potential. Genes Dev. 2007;21:2762–2774. doi: 10.1101/gad.1602107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Li Z, Zhang Z, Li Y, et al. PBX3 is an important cofactor of HOXA9 in leukemogenesis. Blood. 2013;121:1422–1431. doi: 10.1182/blood-2012-07-442004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Artinger EL, Mishra BP, Zaffuto KM, et al. An MLL-dependent network sustains hematopoiesis. Proc Natl Acad Sci U S A. 2013;10:12000–12005. doi: 10.1073/pnas.1301278110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tadjuidje E, Hegde RS. The Eyes Absent proteins in development and disease. Cell Mol Life Sci. 2013;70:1897–1913. doi: 10.1007/s00018-012-1144-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lee JW, Kim HS, Hwang J, et al. Regulation of HOXA9 activity by predominant expression of DACH1 against C/EBPalpha and GATA-1 in myeloid leukemia with MLL-AF9. Biochem Biophys Res Com-mun. 2012;426:299–305. doi: 10.1016/j.bbrc.2012.08.048. [DOI] [PubMed] [Google Scholar]
- 108.Popovic R, Riesbeck LE, Velu CS, et al. Regulation of mir-196b by MLL and its overexpression by MLL fusions contributes to immortalization. Blood. 2009;113:3314–3322. doi: 10.1182/blood-2008-04-154310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Schotte D, Lange-Turenhout EA, Stumpel DJ, et al. Expression of miR-196b is not exclusively MLL-driven but is especially linked to activation of HOXA genes in pediatric acute lymphoblastic leukemia. Haematologica. 2010;95:1675–1682. doi: 10.3324/haematol.2010.023481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Beachy SH, Onozawa M, Silverman D, Chung YJ, Rivera MM, Aplan PD. Isolated Hoxa9 overexpression predisposes to the development of lymphoid but not myeloid leukemia. Exp Hematol. 2013;41:518–529.e515. doi: 10.1016/j.exphem.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li Z, Huang H, Chen P, et al. miR-196b directly targets both HOXA9/MEIS1 oncogenes and FAS tumour suppressor in MLL-rearranged leukaemia. Nat Commun. 2012;3:688. doi: 10.1038/ncomms1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Velu CS, Chaubey A, Phelan JD, et al. Therapeutic antagonists of microRNAs deplete leukemia-initiating cell activity. J Clin Invest. 2014;124:222–236. doi: 10.1172/JCI66005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Davidson AJ, Ernst P, Wang Y, et al. cdx4 mutants fail to specify blood progenitors and can be rescued by multiple hox genes. Nature. 2003;425:300–306. doi: 10.1038/nature01973. [DOI] [PubMed] [Google Scholar]
- 114.Wang Y, Yabuuchi A, McKinney-Freeman S, et al. Cdx gene deficiency compromises embryonic hematopoiesis in the mouse. Proc Natl Acad Sci U S A. 2008;105:7756–7761. doi: 10.1073/pnas.0708951105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Scholl C, Bansal D, Dohner K, et al. The homeobox gene CDX2 is aberrantly expressed in most cases of acute myeloid leukemia and promotes leukemogenesis. J Clin Invest. 2007;117:1037–1048. doi: 10.1172/JCI30182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Koo S, Huntly BJ, Wang Y, et al. Cdx4 is dispensable for murine adult hematopoietic stem cells but promotes MLL-AF9–mediated leukemogenesis. Haematologica. 2010;95:1642–1650. doi: 10.3324/haematol.2010.023168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Metais JY, Dunbar CE. The MDS1-EVI1 gene complex as a retrovirus integration site: impact on behavior of hematopoietic cells and implications for gene therapy. Mol Ther. 2008;16:439–449. doi: 10.1038/sj.mt.6300372. [DOI] [PubMed] [Google Scholar]
- 118.Groschel S, Lugthart S, Schlenk RF, et al. High EVI1 expression predicts outcome in younger adult patients with acute myeloid leukemia and is associated with distinct cytogenetic abnormalities. J Clin Oncol. 2010;28:2101–2107. doi: 10.1200/JCO.2009.26.0646. [DOI] [PubMed] [Google Scholar]
- 119.Bindels EM, Havermans M, Lugthart S, et al. EVI1 is critical for the pathogenesis of a subset of MLL-AF9–rearranged AMLs. Blood. 2012;119:5838–5849. doi: 10.1182/blood-2011-11-393827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhang Y, Stehling-Sun S, Lezon-Geyda K, et al. PR-domain-containing Mds1-Evi1 is critical for long-term hematopoietic stem cell function. Blood. 2011;118:3853–3861. doi: 10.1182/blood-2011-02-334680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Arai S, Yoshimi A, Shimabe M, et al. Evi-1 is a transcriptional target of mixed-lineage leukemia oncoproteins in hematopoietic stem cells. Blood. 2011;117:6304–6314. doi: 10.1182/blood-2009-07-234310. [DOI] [PubMed] [Google Scholar]
- 122.Goyama S, Yamamoto G, Shimabe M, et al. Evi-1 is a critical regulator for hematopoietic stem cells and transformed leukemic cells. Cell Stem Cell. 2008;3:207–220. doi: 10.1016/j.stem.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 123.Zhang Y, Owens K, Hatem L, et al. Essential role of PR-domain protein MDS1-EVI1 in MLL-AF9 leukemia. Blood. 2013;122:2888–2892. doi: 10.1182/blood-2012-08-453662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Krivtsov AV, Figueroa ME, Sinha AU, et al. Cell of origin determines clinically relevant subtypes of MLL-rearranged AML. Leukemia. 2013;27:852–860. doi: 10.1038/leu.2012.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Schwieger M, Schuler A, Forster M, et al. Homing and invasiveness of MLL/ENL leukemic cells is regulated by MEF2C. Blood. 2009;114:2476–2488. doi: 10.1182/blood-2008-05-158196. [DOI] [PubMed] [Google Scholar]
- 126.Krivtsov AV, Twomey D, Feng Z, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442:818–822. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- 127.Miller PG, Al-Shahrour F, Hartwell KA, et al. In Vivo RNAi screening identifies a leukemia-specific dependence on integrin beta 3 signaling. Cancer Cell. 2013;24:45–58. doi: 10.1016/j.ccr.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Schreiner S, Birke M, Garcia-Cuellar MP, Zilles O, Greil J, Slany RK. MLL-ENL causes a reversible and myc-dependent block of myelomonocytic cell differentiation. Cancer Res. 2001;61:6480–6486. [PubMed] [Google Scholar]
- 129.Blobel GA, Kadauke S, Wang E, et al. A reconfigured pattern of MLL occupancy within mitotic chromatin promotes rapid transcriptional reactivation following mitotic exit. Mol Cell. 2009;36:970–983. doi: 10.1016/j.molcel.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dawson MA, Prinjha RK, Dittmann A, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529–533. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zuber J, Shi J, Wang E, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Barbieri I, Cannizzaro E, Dawson MA. Bromodomains as therapeutic targets in cancer. Brief Funct Genomics. 2013;12:219–230. doi: 10.1093/bfgp/elt007. [DOI] [PubMed] [Google Scholar]
- 133.Armstrong SA, Golub TR, Korsmeyer SJ. MLL-rearranged leukemias: insights from gene expression profiling. Semin Hematol. 2003;40:268–273. doi: 10.1016/s0037-1963(03)00196-3. [DOI] [PubMed] [Google Scholar]
- 134.Stubbs MC, Kim YM, Krivtsov AV, et al. MLL-AF9 and FLT3 cooperation in acute myelogenous leukemia: development of a model for rapid therapeutic assessment. Leukemia. 2008;22:66–77. doi: 10.1038/sj.leu.2404951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gwin K, Frank E, Bossou A, Medina KL. Hoxa9 regulates Flt3 in lymphohematopoietic progenitors. J Immunol. 2010;185:6572–6583. doi: 10.4049/jimmunol.0904203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Albouhair S, Morgado E, Lavau C. Flt3 does not play a critical role in murine myeloid leukemias induced by MLL fusion genes. PLoS One. 2013;8:e72261. doi: 10.1371/journal.pone.0072261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kamezaki K, Luchsinger L, Snoeck HW. Differential requirement for wild-type Flt3 in leukemia-initiation among mouse models of human leukemia. Exp Hematol. 2014;42:192–203.e1. doi: 10.1016/j.exphem.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Driessen EM, van Roon EH, Spijkers-Hagelstein JA, et al. Frequencies and prognostic impact of RAS mutations in MLL-rearranged acute lymphoblastic leukemia in infants. Haematologica. 2013;98:937–944. doi: 10.3324/haematol.2012.067983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kim WI, Matise I, Diers MD, Largaespada DA. RAS oncogene suppression induces apoptosis followed by more differentiated and less myelosuppressive disease upon relapse of acute myeloid leukemia. Blood. 2009;113:1086–1096. doi: 10.1182/blood-2008-01-132316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ono R, Kumagai H, Nakajima H, et al. Mixed-lineage-leukemia (MLL) fusion protein collaborates with Ras to induce acute leukemia through aberrant Hox expression and Raf activation. Leukemia. 2009;23:2197–2209. doi: 10.1038/leu.2009.177. [DOI] [PubMed] [Google Scholar]
- 141.Tamai H, Miyake K, Takatori M, et al. Activated K-Ras protein accelerates human MLL/AF4-induced leukemo-lymphomogenicity in a transgenic mouse model. Leukemia. 2011;25:888–891. doi: 10.1038/leu.2011.15. [DOI] [PubMed] [Google Scholar]
- 142.Ng MH, Ng RK, Kong CT, Jin DY, Chan LC. Activation of Ras-dependent Elk-1 activity by MLL-AF4 family fusion oncoproteins. Exp Hematol. 2010;38:481–488. doi: 10.1016/j.exphem.2010.03.014. [DOI] [PubMed] [Google Scholar]
- 143.Nakanishi H, Nakamura T, Canaani E, Croce CM. ALL1 fusion proteins induce deregulation of EphA7 and ERK phosphorylation in human acute leukemias. Proc Natl Acad Sci U S A. 2007;104:14442–14447. doi: 10.1073/pnas.0703211104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Manara E, Baron E, Tregnago C, et al. MLL-AF6 fusion oncogene sequesters AF6 into the nucleus to trigger RAS activation in myeloid leukemia. Blood. 2014;124:263–272. doi: 10.1182/blood-2013-09-525741. [DOI] [PubMed] [Google Scholar]
- 145.Wang Y, Krivtsov AV, Sinha AU, et al. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science. 2010;327:1650–1653. doi: 10.1126/science.1186624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yeung J, Esposito MT, Gandillet A, et al. beta-Catenin mediates the establishment and drug resistance of MLL leukemic stem cells. Cancer Cell. 2010;18:606–618. doi: 10.1016/j.ccr.2010.10.032. [DOI] [PubMed] [Google Scholar]
- 147.Staal FJ, Luis TC. Wnt signaling in hematopoiesis: crucial factors for self-renewal, proliferation, and cell fate decisions. J Cell Biochem. 2010;109:844–849. doi: 10.1002/jcb.22467. [DOI] [PubMed] [Google Scholar]
- 148.Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149:1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 149.de la Roche M, Rutherford TJ, Gupta D, et al. An intrinsically labile alpha-helix abutting the BCL9-binding site of beta-catenin is required for its inhibition by carnosic acid. Nat Commun. 2012;3:680. doi: 10.1038/ncomms1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wang X, Zhu K, Li S, et al. MLL1, a H3K4 methyltransferase, regulates the TNFalpha-stimulated activation of genes downstream of NF-kappaB. J Cell Sci. 2012;125:4058–4066. doi: 10.1242/jcs.103531. [DOI] [PubMed] [Google Scholar]
- 151.Austenaa L, Barozzi I, Chronowska A, et al. The histone methyltransferase Wbp7 controls macrophage function through GPI glycolipid anchor synthesis. Immunity. 2012;36:572–585. doi: 10.1016/j.immuni.2012.02.016. [DOI] [PubMed] [Google Scholar]
- 152.Kuo HP, Wang Z, Lee DF, et al. Epigenetic roles of MLL oncoproteins are dependent on NF-kappaB. Cancer Cell. 2013;24:423–437. doi: 10.1016/j.ccr.2013.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ohlsson E, Hasemann MS, Willer A, et al. Initiation of MLL-rearranged AML is dependent on C/EBPalpha. J Exp Med. 2014;211:5–13. doi: 10.1084/jem.20130932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zhou J, Wu J, Li B, et al. PU.1 is essential for MLL leukemia partially via crosstalk with the MEIS/HOX pathway. Leukemia. 2014;28:1436–1448. doi: 10.1038/leu.2013.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Mills AA. Throwing the cancer switch: reciprocal roles of polycomb and trithorax proteins. Nat Rev Cancer. 2010;10:669–682. doi: 10.1038/nrc2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Schuettengruber B, Martinez AM, Iovino N, Cavalli G. Trithorax group proteins: switching genes on and keeping them active. Nat Rev Mol Cell Biol. 2011;12:799–814. doi: 10.1038/nrm3230. [DOI] [PubMed] [Google Scholar]
- 157.Tanaka S, Miyagi S, Sashida G, et al. Ezh2 augments leukemogenicity by reinforcing differentiation blockage in acute myeloid leukemia. Blood. 2012;120:1107–1117. doi: 10.1182/blood-2011-11-394932. [DOI] [PubMed] [Google Scholar]
- 158.Neff T, Sinha AU, Kluk MJ, et al. Polycomb repressive complex 2 is required for MLL-AF9 leukemia. Proc Natl Acad Sci U S A. 2012;109:5028–5033. doi: 10.1073/pnas.1202258109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Xie H, Xu J, Hsu JH, et al. Polycomb repressive complex 2 regulates normal hematopoietic stem cell function in a developmental-stage-specific manner. Cell Stem Cell. 2014;14:68–80. doi: 10.1016/j.stem.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Shi J, Wang E, Zuber J, et al. The Polycomb complex PRC2 supports aberrant self-renewal in a mouse model of MLL-AF9;Nras(G12D) acute myeloid leukemia. Oncogene. 2013;32:930–938. doi: 10.1038/onc.2012.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kim W, Bird GH, Neff T, et al. Targeted disruption of the EZH2-EED complex inhibits EZH2-dependent cancer. Nat Chem Biol. 2013;9:643–650. doi: 10.1038/nchembio.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Yuan J, Takeuchi M, Negishi M, Oguro H, Ichikawa H, Iwama A. Bmi1 is essential for leukemic reprogramming of myeloid progenitor cells. Leukemia. 2011;25:1335–1343. doi: 10.1038/leu.2011.85. [DOI] [PubMed] [Google Scholar]
- 163.Smith LL, Yeung J, Zeisig BB, et al. Functional crosstalk between Bmi1 and MLL/Hoxa9 axis in establishment of normal hematopoietic and leukemic stem cells. Cell Stem Cell. 2011;8:649–662. doi: 10.1016/j.stem.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 164.Tan J, Jones M, Koseki H, et al. CBX8, a polycomb group protein, is essential for MLL-AF9–induced leukemogenesis. Cancer Cell. 2011;20:563–575. doi: 10.1016/j.ccr.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Maethner E, Garcia-Cuellar MP, Breitinger C, et al. MLL-ENL inhibits polycomb repressive complex 1 to achieve efficient transformation of hematopoietic cells. Cell Rep. 2013;3:1553–1566. doi: 10.1016/j.celrep.2013.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Nakamura T, Mori T, Tada S, et al. ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol Cell. 2002;10:1119–1128. doi: 10.1016/s1097-2765(02)00740-2. [DOI] [PubMed] [Google Scholar]
- 167.Harris WJ, Huang X, Lynch JT, et al. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell. 2012;21:473–487. doi: 10.1016/j.ccr.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 168.Kerenyi MA, Shao Z, Hsu YJ, et al. Histone demethylase Lsd1 represses hematopoietic stem and progenitor cell signatures during blood cell maturation. Elife. 2013;2:e00633. doi: 10.7554/eLife.00633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Thiel AT, Blessington P, Zou T, et al. MLL-AF9–induced leukemogenesis requires coexpression of the wild-type MLL allele. Cancer Cell. 2010;17:148–159. doi: 10.1016/j.ccr.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Mishra BP, Zaffuto KM, Artinger EL, et al. The histone methyltransferase activity of MLL1 is dispensable for hematopoiesis and leukemogenesis. Cell Rep. 2014;7:1239–1247. doi: 10.1016/j.celrep.2014.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Osaki H, Walf-Vorderwulbecke V, Mangolini M, et al. The AAA ATPase RUVBL2 is a critical mediator of MLL-AF9 oncogenesis. Leukemia. 2013;27:1461–1468. doi: 10.1038/leu.2013.42. [DOI] [PubMed] [Google Scholar]
- 172.Shi J, Whyte WA, Zepeda-Mendoza CJ, et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 2013;27:2648–2662. doi: 10.1101/gad.232710.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Stumpel DJ, Schneider P, van Roon EH, Pieters R, Stam RW. Absence of global hypomethylation in promoter hypermethylated Mixed Lineage Leukaemia-rearranged infant acute lymphoblastic leukaemia. Eur J Cancer. 2013;49:175–184. doi: 10.1016/j.ejca.2012.07.013. [DOI] [PubMed] [Google Scholar]
- 174.Schafer E, Irizarry R, Negi S, et al. Promoter hypermethylation in MLL-r infant acute lymphoblastic leukemia: biology and therapeutic targeting. Blood. 2010;115:4798–4809. doi: 10.1182/blood-2009-09-243634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Bernot KM, Nemer JS, Santhanam R, et al. Eradicating acute myeloid leukemia in a MLLPTD/wt:Flt3ITD/wt murine model: a path to novel therapeutic approaches for human disease. Blood. 2013;122:3778–3783. doi: 10.1182/blood-2013-06-507426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Trowbridge JJ, Sinha AU, Zhu N, Li M, Armstrong SA, Orkin SH. Haploinsufficiency of Dnmt1 impairs leukemia stem cell function through derepression of bivalent chromatin domains. Genes Dev. 2012;26:344–349. doi: 10.1101/gad.184341.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Broske AM, Vockentanz L, Kharazi S, et al. DNA methylation protects hematopoietic stem cell multipotency from myeloerythroid restriction. Nat Genet. 2009;41:1207–1215. doi: 10.1038/ng.463. [DOI] [PubMed] [Google Scholar]
- 178.Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Huang H, Jiang X, Li Z, et al. TET1 plays an essential oncogenic role in MLL-rearranged leukemia. Proc Natl Acad Sci U S A. 2013;110:11994–11999. doi: 10.1073/pnas.1310656110. [DOI] [PMC free article] [PubMed] [Google Scholar]