

Abstract
We report a compound identification method (SimMR), which simultaneously evaluates the mass spectrum similarity and the retention index distance using an empirical mixture score function, for the analysis of GC-MS data. The performance of the developed SimMR method was compared to that of two existing compound identification strategies. One is mass spectrum matching method without incorporation of retention index information (SM). The other is the method that sequentially evaluates the mass spectrum similarity and retention index distance (SeqMR). For the comparison purpose, we used the NIST/EPA/NIH Mass Spectral Library 2005. Our study demonstrates that SimMR performs the best among the three compound identification methods, by improving the overall identification accuracy up to 1.53% and 4.81% compared to SeqMR and SM, respectively.
Introduction
Compound identification in gas chromatography mass spectrometry (GC–MS) is currently achieved by comparing a query mass spectrum with reference mass spectra in a library via spectrum matching. Several mass spectral libraries have been generated,1-4 and various mass spectral similarity measures have been developed, including composite similarity,5 probability-based matching system,6 Hertz similarity index,7 normalized Euclidean and absolute value distance,8 wavelet and Fourier transform-based composite similarity,9 partial and semi-partial correlations-based composite similarity.10 Most recently, Koo et al.11 compared the performance of several spectral similarity measures and concluded that the performance compound identification depends on multiple factors including the mass spectrum library, spectral similarity measure and weight factors. They further discussed that the compound identification based on mass spectra only has limited accuracy and the high accuracy compound identification can be achieved by incorporating compound separation information into mass spectrum matching.
Since retention time in GC depends on experiment condition dependent, retention index was introduced to reduce such dependency.12 A few approaches using both mass spectrum and retention index have been used for compound identification.13,14 For example, our group developed a method iMatch for compound identification using retention index.15 All of the existing methods employ retention index as a filter to remove the potential false-positive identifications generated by mass spectrum matching. Such an analysis strategy uses the retention index and mass spectrum in two separate analysis steps. The sequential nature of the two-step analysis strategy increases the risk of introducing errors from each independent stage since there is no way to correct the errors caused by the previous step.
The objective of this work was to develop a compound identification method entitled SimMR that simultaneously evaluates the mass spectrum similarity and the retention index distance. An empirical mixture score function was developed to perform the simultaneous evaluation of the mass spectral similarity and the difference of retention index between the experimental data and the data recorded in reference libraries. The performance of the proposed SimMR method was evaluated using the data recorded in the NIST/EPA/NIH Mass Spectral Library 2005 (NIST05).
Materials and Methods
Datasets of mass spectra and retention index
The NIST05 library contains two electron ionization (EI) mass spectrum libraries: the main EI MS library and the replicate EI MS library. A total of 163,198 and 28,234 mass spectra were extracted from the main EI MS library and the replicate EI MS library, respectively. The NIST retention index library is a part of the NIST05 library, from which a total of 242,116 retention index values for 14,878 compounds were extracted. The NIST retention index library characterizes retention index by a set of experimental conditions, including column type, column class, data type, program type, etc. Based on our previous study, the magnitude of retention index on capillary columns can be significantly affected by column class and program type.15
In this study, the query datasets are the replicate EI MS spectra that are present in the retention index library, while the reference mass spectral library is the main EI MS library of the NIST05 library. In detail, the replicate EI MS library and the retention index library are filtered as follows: the compounds with retention index values acquired under ramp condition on the capillary columns are extracted from the retention index library; these extracted retention index values are further split into three sub-libraries based on column class, i.e., standard non-polar, semi non-polar and standard polar; the interception of the replicate EI MS library and each of the three sub-retention index libraries is calculated based on compound's Chemical Abstract Service (CAS) registry numbers, respectively; the retention index value(s) and mass spectrum (spectra) of the compounds in each of the three interceptions forms three query datasets. By doing so, the first query dataset has a total of 7,791 compounds with linear retention index on semi non-polar column and mass spectra in query library; the second query dataset has 8,517 compounds with linear retention index on standard non-polar column and mass spectra in the query library, and the third query dataset has 4,781 compounds with linear retention index on standard polar column and mass spectra in the query library.
During the study, each mass spectrum in a query dataset is first used to search the entire main EI MS library (reference library) for compound identification via mass spectrum matching. Any candidate compounds are removed from the matching list if it does not have a retention index value in the query dataset. Then, the top 10 ranked compounds are used for further analysis to incorporate the retention index value for identification.
Spectrum matching-based identification (SM)
To test the performance of the mixture similarity for the three query datasets, four mass spectral similarity methods were used, including Stein and Scott's composite similarity,5 Discrete Fourier- and wavelet-transform (DFT) composite similarity,9 and weighted cosine.16 Following lists the definitions of the four mass spectral similarity measures given a query spectrum X = (x1,…,xn) and a reference spectrum Y = (y1,…y2), where xi and yi are the intensities of the ith fragment ion in X and Y, respectively.
Weighted cosine measure (WC)
Cosine correlation is defined as follows:
| (1) |
where the inner product and the norm . Stein and Scott demonstrated the importance of weight for intensity and m/z value.17 The weighted spectra X, Y are considered as follows:
and
| (2) |
where mi, i = 1,…,n is m/z value of the ith fragment ion, and a, b are the weight factors for peak intensity and m/z value, respectively. The weighted cosine similarity SWC (X,Y) is then defined as follows:
| (3) |
The optimal weight factors are set as (a, b) = (0.53, 1.3).16
Stein and Scott's composite similarity (SS)
The Stein and Scott's composite similarity5 is defined as:
| (4) |
where NX is the number of the non-zero peak intensities in the query spectra. SR is the ratio of peak pair defined as:
| (5) |
where n = −1 or 1 if the term in parentheses is less than or greater than unity, respectively, xi, yi are all non-zero intensities having common m/z value, and the value NX∧Y is the number of non-zero peaks in both the reference and the query spectra. The weight factors (a, b) = (0.5, 3).
Discrete Fourier- and wavelet- transform composite similarity
Discrete Fourier transform (DFT) converts an original spectral signal X = (x1,…,xn) into a new signal XF = (x1F,…,xnF as follows:18
| (6) |
where the notation i is the imaginary unit and is a primitive nth root of unity. By Euler's formula, exp(iϕ) = cosϕ + i sinϕ, the original equation becomes
| (7) |
We have a new transformed signal XFR consisting of real part of as follows:
| (8) |
with
| (9) |
where a function Re(·) is the real part of imaginary number or function.
The discrete wavelet transform of a signal X = (x1,…,xn)is calculated by passing it through a low-pass filter g and a high-pass filter h, resulting in two subsets of signals: approximations and details.19 The coefficients of approximations and details are defined as follows:
| (10) |
| (11) |
where g and h are the low-pass filter and the high-pass filter, respectively. This study used Daubechies' scaling functions with an order of 4 as for low-pass filters.19 Then the approximation and detail DWTs of an original signal X are as follows, respectively:
| (12) |
The DFT with real and DWT with detail composite similarity are defined as follows:9
| (13) |
and
| (14) |
Sequential usage of mass spectrum and retention index for compound identification (SeqMR)
The mass spectrum matching algorithm first ranks compound candidates from the reference library based on their mass spectral similarity to the unknown compound that given rise to the query mass spectrum. A large matching score refers to a high degree of mass spectrum similarity. The retention index information is then employed as a filter to recognize the reference compound that has a large retention index difference with the retention index calculated from the experimental data, by setting a retention index deviation window as follows:13,14,15
| (15) |
where Iexp is the experimental retention index value, Iref denotes the median of the retention index values of a reference compound, ΔI is the threshold of retention index deviation.
Simultaneous evaluation of mass spectrum and retention index similarity (SimMR)
In order to achieve a high degree of accuracy for compound identification, we propose a compound identification method where a list of top ranked reference compounds generated by mass spectrum matching are first selected as the potential identification results, c={c1,c2,…,ck}, where ci is the ith top ranked compound and k is the number of reference compounds selected; the retention index difference between each selected reference compound and query data is computed, respectively; the mass spectral similarity and the retention index difference between a reference compound and the query data are then simultaneously evaluated via an empirical mixture score function defined as follows:
| (16) |
where w is weight factor and 0 ≤ w ≤ 1, fi is a function of retention index, is a function of mass spectral similarity. fi and are defined as follows:
| (17) |
| (18) |
where Ii is the retention index of the compound ci, Imin is the minimum value of retention index in the list of top ranked compounds c, and Imin2 is the second minimum value in c. s̄i is the dissimilarity of the mass spectrum matching of compound ci and , where si is the mass spectral similarity of compound ci, and s̄max and s̄min are the maximum and the minimum of the dissimilarity in compound candidates c, respectively. a and b are two constant numbers that control the scalability of two functions, and usually set as a = 0.05, b = 30.
Performance measure
We use the identification accuracy to measure the performance of each identification method. The accuracy is the proportion of the spectra identified correctly in query data, and is defined as follows:
| (19) |
If a spectrum in reference library and its corresponding retention index in the retention index library having the same CAS registry index number with a query mass spectrum, it would be considered as a correct identification. Otherwise, it is incorrect.
Results and Discussion
A total of three compound identification methods are investigated in this study, including mass spectrum matching, sequential evaluation of mass spectrum and retention index similarity (SeqMR), and simultaneous evaluation of mass spectrum and retention index similarity (SimMR). Due to the dependency of the magnitude of the retention index on the experimental conditions,15 we only focused on the compounds that have linear retention index values on the capillary columns with different stationary phases. Therefore, the query data were split into three datasets based on the values of column class defined by NIST, i.e., standard non-polar, semi non-polar and standard polar.
Mass spectrum matching-based identification
The mass spectrum matching is the widely used approach for compound identification in GC-MS. Koo et al. studied the performance of five mass spectral similarity measures for compound identification using all mass spectra extracted from both the replicate library and the main library of the NIST/EPA/NIH Mass Spectral Library 2011 (NIST11) library.11 In this study, we perform compound identification using both mass spectrum and retention index information. Therefore, we first studied the performance of the four mass spectral similarity measures in identifying compounds for each of the three query datasets constructed using the method described in the section of Materials and Methods.
Figure 1 depicts the relation of identification accuracy and the number of top ranked compounds as the result of identification. If a number of top ranked compounds are considered as the identification result, the identification is correct if the true compound is one of the top ranked compounds. For the semi non-polar column (Figure 1A), the compound identification accuracy increases with the increase of the number of top ranked compounds, and such increasing trend levels off at about the top 10 ranked compounds. If the best ranked compound is considered as the identification result, the SS method can correctly identify 71.99% of compounds while DFTR, DWTD and WC achieve an accuracy of 78.57%, 78.76%, and 80.31%, respectively. When the top three ranked compounds are considered as the identification result, the accuracy of SS is increased to 89.48%, while the accuracy of DFTR, DWTD and WC is increased to 93.26%, 93.39%, and 94.19%, respectively. Similar results are observed in Figure 1B and 1C for the standard non-polar column and standard polar column dataset, respectively. Overall, DFTR and DWTD have similar performance and the WC method performs the best at any number of the top ranked compounds. The difference of identification accuracy among the four methods decreases with the increase of the number of top ranked compounds. These results are consistent with the results reported by Koo's using the NIST11 library, even though the magnitude of the identification accuracy varies a little bit due to the difference of the dataset and reference libraries used.11
Fig. 1.
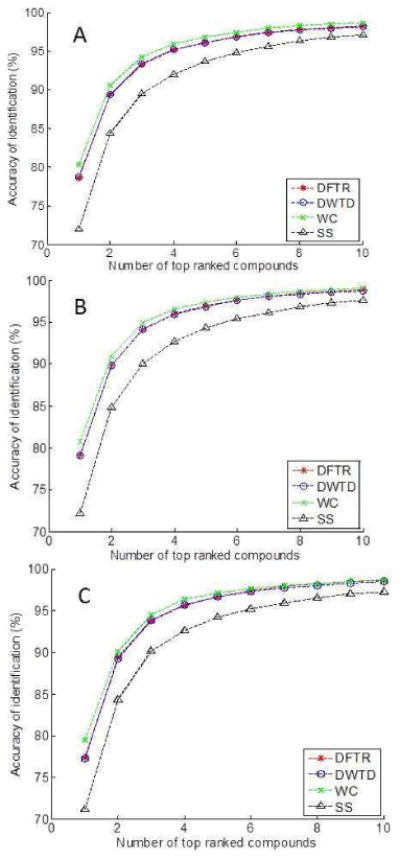
The relation of identification accuracy and the number of top ranked compounds that are considered as the identification results for analysis of three different datasets extracted from the NIST05 library. (A) Query dataset containing retention indices acquired on semi non-polar column, (B) Query dataset containing retention indices acquired on standard non-polar column, and (C) Query dataset with retention index acquired on standard polar column.
SeqMR identification
In the SeqMR approach, a query mass spectrum is first searched against all mass spectra in the reference library; the top ranked compound is recognized based on the magnitude of spectral similarity score; the retention index of the compound given rise to the query mass spectrum is compared to the retention index value of the top ranked compound; the identification is considered as correct if the difference between these two retention index values is smaller than a user defined variation window ΔI; otherwise the identification is considered as a false identification. Therefore, the performance of compound identification using retention index matching in the SeqMR approach is heavily dependent on the size of ΔI. A large value of ΔI will reduce the effectiveness of retention index for compound identification, while a small value of ΔI can introduce a high degree of false-negative identification. In order to find the optimal value of ΔI, we studied the identification accuracy in the SeqMR approach by screening ΔI values from 1 to 500 retention index unites (i.u.) for each of the four spectral similarity methods. The optimal value of ΔI is the one that generates the maximum identification accuracy.
In this study, the top 10 ranked candidates for each query spectrum were selected for retention index matching after mass spectrum matching. A compound can have multiple retention index values in the NIST retention index library acquired under the same experimental conditions. For example, the number of unique retention index value for the 7,791 compounds that have linear retention index on semi non-polar column and mass spectra in the replicate MS library ranges from 1 to 77. Considering these multiple choices of retention index for each compound, we randomly selected one retention index value for a compound from these multiple values for the calculation of the retention time difference. For each of the three query dataset, the above mentioned method, i.e., performing mass spectrum matching followed by selecting the top 10 ranked compounds and randomly selecting one retention index value for the calculation of retention index difference, was repeated 100 times. After 100 iterations, the retention index threshold that generates the maximum median identification accuracy for each spectral similarity measure is chosen. Finally, the optimal threshold is used to represent the identification results of the SeqMR approach.
Figure 2 depicts the identification results of using the SeqMR approach with four spectral similarity measures, where the retention index variation window is varied from 1 to 500 i.u. and the median value of the identification accuracy of the 100 iterations are calculated as the final identification accuracy. It can be seen that using the retention index information can improve the accuracy of compound identification. However, such contribution is heavily dependent on the size of retention index variation window ΔI. Both a very large or very small value of ΔI reduces the effectiveness of retention index information. For the semi non-polar column data (Figure 2A), the identification accuracy initially increases with the increase of ΔI. After the value of ΔI reaches to an optimal value, the identification accuracy begins to decrease and levels off at the identification accuracy of mass spectral matching. The optimal ΔI for the four spectral similarity measures SS, DFTR, DWTR, and WC are 10, 11, 11, and 11 i.u., respectively. The corresponding identification accuracy is 74.42%, 81.34%, 81.41%, and 82.60%, respectively. Comparing with the performance of mass spectrum matching (Figure 1A), the SeqMR approach improves the identification accuracy about 2.42%, 2.77%, 2.65%, and 2.29% for the SS, DFTR, DWTD and WC method, respectively.
Fig. 2.
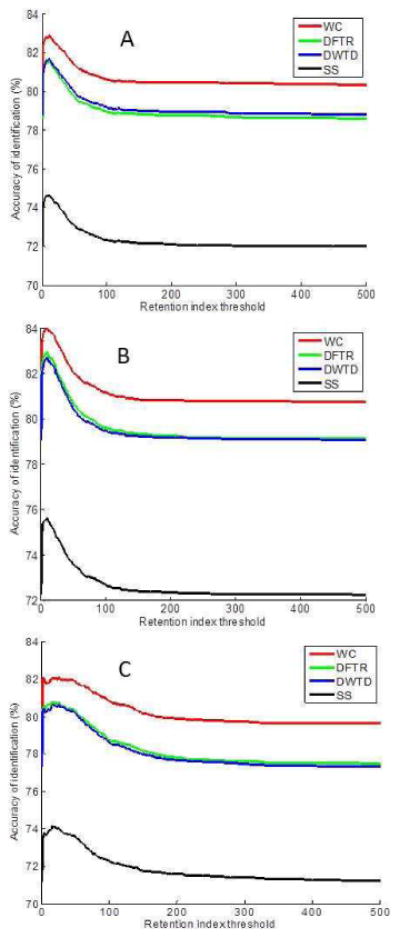
The retention index threshold vs. the accuracy of compound identification using four different similarity methods for analysis of the three query datasets: (A) Query dataset containing retention indices acquired on semi non-polar column, (B) Query dataset containing retention indices acquired on standard non-polar column, and (C) Query dataset with retention index acquired on standard polar column.
Figure 2B depicts the analysis results of SeqMR approach using the retention index value acquired on standard non-polar column, while Figure 2C is on the standard polar column. Figure 2C has a broad peak than the curves displayed in Figure 2A and Figure 2B, respectively. The optimal ΔI of the linear retention index acquired on the standard non-polar column for the four spectral similarity measures SS, DFTR, DWTR, and WC are 10, 10, 10, and 10 i.u., respectively, while the optimal ΔI of the linear retention index acquired on the standard polar column for the four spectral similarity measures SS, DFTR, DWTR, and WC are 16, 18, 18, and 26 i.u., respectively. The large values of optimal ΔI in Figure 2C were caused by the large deviation of linear retention index values of each compound acquired on the standard polar columns in the NIST library.
SimMR identification
The SimMR approach proposed in this study evaluates the similarity of mass spectrum and retention index of the query data and the reference data simultaneously using a mixture score defined in equation (11). The analysis was performed as follows: the top 10 ranked candidates for each of query mass spectrum are selected after mass spectrum matching; for each candidate with retention index information, the retention index difference between this candidate and the query data is computed; to determine the parameters (a, b, and w) in the mixture score function, a training set is used to obtain the optimal parameters by minimizing the training identification error.
To evaluate the performance of the mixture score function for compound identification, k-fold cross validation was employed with k = 5. A query dataset were first equally split into five parts in a random manner and a total of five tests were performed. In each test, 80% of query data, i.e., a collect of the four parts of the split query data, were used as training data to obtain the optimal values for three parameters (a, b, and w) while the remaining 20% of data were used to verify the effectiveness of the mixture score function. During the training step, a greedy search algorithm was used to find the optimal values for the three parameters by maximizing identification accuracy of the training data. The boundary of each parameter was set as {a, w} ∈ [0, 1], b ∈ [10, 35], where a was changed with a step of 0.001, w with a step of 0.05, and b with a step of 1. During each of the testing steps, the optimized parameters were applied to equation (16) for the identification of the testing data. The above mentioned 5-fold cross validation was repeated five times. As the results, a total of 25 training accuracies and 25 testing accuracies are obtained for each query dataset, and the final testing identification accuracy is represented as the average of these 25 testing accuracies.
Table 1 lists the training results and the corresponding testing results for the query dataset with retention index values acquired on semi non-polar column. Each datum is the average of the results of a 5-fold cross validation with its standard deviation. Among the five times of 5-fold cross validations, the best testing error is 12.3%, with a training error of 14.2% and trained optimal parameters a = 0.05, b = 30, and w = 0.65, respectively. The testing error is the ratio of the number of testing query data that do not correctly identified divided by the total number of testing query data, while the trained error is the ratio of the number of training query data that do not correctly identified divided by the total number of training query data. The average of the trained optimal parameters of all cross validations are a = 0.05 ± 0.06, b = 23 ± 6, and w = 0.65 ± 0.05, respectively, while the average training error is 13.8% ± 0.3% and the average testing error is 14.2% ± 1.1%. Note that each of the three parameters (a, b, and w) has a large relative standard deviation among the five times of 5-fold cross validation, while the averages of the training error and the testing error remain stable with small relative standard deviation. This indicates that the mixture score function in equation (16) is robust and not sensitive to the values of the three parameters. The results of the five times 5-fold cross validation for the query dataset of standard non-polar column and the query dataset of standard polar column are listed in Table S1 and S2 in the supplementary material, respectively.
Table 1.
Results of the five times of 5-fold cross validation for analysis of the query dataset containing retention indices acquired on semi non-polar column. Each datum is the average of the results of a 5-fold cross validation with its standard deviation.
| CV 1 | CV 2 | CV 3 | CV 4 | CV 5 | |
|---|---|---|---|---|---|
| a | 0.05±0.03 | 0.08±0.09 | 0.03±0.02 | 0.03±0.02 | 0.08±0.09 |
| b | 21±8 | 24±6 | 24±5 | 24±6 | 24±6 |
| w | 0.61±0.05 | 0.66±0.02 | 0.69±0.08 | 0.66±0.02 | 0.66±0.02 |
| Training error (%) | 13.8±0.2 | 13.8±0.3 | 13.8±0.3 | 13.8±0.3 | 13.8±0.3 |
| Testing error (%) | 14.2±1.2 | 14.1±1.3 | 14.3±0.9 | 14.1±1.3 | 14.1±1.3 |
After the five times of 5-fold cross validation using each query dataset, all data in each of the three query datasets were then used for compound identification using the trained optimal parameters, respectively. Table 2 summaries the identification results of SimMR as well as the results of mass spectrum matching and SeqMR, where ΔIo is the optimal retention index variation window used during SeqMR analysis. The value of ΔIo was derived from the curves displayed in Figure 2. At each ΔIo, the SeqMR generates the best identification accuracy. For the query dataset of semi non-polar column data, the identification accuracy is improved to 75.16% by mixture score with SS as the spectral similarity measure when the top ranked compound is considered as the identification result, while the identification accuracy of mixture score for DFTR, DWTR, and WC as the spectral similarity measures is improved to 82.08%, 82.23%, and 83.50%, respectively. These identification accuracies are 0.74%, 0.74%, 0.82%, and 0.92% increase compared to the accuracy acquired by the SeqMR approach, and 3.17%, 3.52%, 3.47%, and 3.19% improvement by mass spectrum matching.
Table 2.
The compounds identification results for the analysis of three query datasets using different analysis strategies, including mass spectrum matching (SM), sequential evaluation of the similarity of mass spectrum and retention index (SeqMR), and the simultaneous evaluation of the similarity of mass spectrum and retention index (SimMR). For each analysis strategy, a total of four mass spectral similarity measures were employed.
| Spectral similarity measure | Column Class | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Semi non-polar column | Standard non-polar column | Standard polar column | ||||||||||
| SM | ΔIo | SeqMR | SimMR | SM | ΔIo | SeqMR | SimMR | SM | ΔIo | SeqMR | SimMR | |
| SS | 71.99 | 10 | 74.42 | 75.16 | 72.23 | 10 | 75.41 | 76.16 | 71.18 | 16 | 74.17 | 75.21 |
| DFTR | 78.57 | 11 | 81.34 | 82.08 | 79.14 | 10 | 82.79 | 83.89 | 77.41 | 18 | 80.69 | 82.22 |
| DWTD | 78.76 | 11 | 81.41 | 82.23 | 79.07 | 10 | 82.54 | 83.58 | 77.29 | 18 | 80.46 | 81.86 |
| WC | 80.31 | 11 | 82.59 | 83.50 | 80.74 | 10 | 83.85 | 84.84 | 79.52 | 26 | 82.07 | 83.06 |
Table 2 shows that the optimal linear retention index difference ΔIo between the experimental value and the database value for the semi-non polar column is 11 i.u. To an EI mass spectrum of compound beta-Estradiol (CAS 50-28-2) in the NIST replicate database, the SM approach ranked compound 17-alpha-Estradiol (CAS 57-91-0) as the top candidate with a spectral similarity score S=0.9564 using the WC measure. The true compound beta-Estradiol (CAS 50-28-2) was ranked as the second with a spectral similarity score S=0.9465. If the top ranked compound is selected as the identification result, the SM approach will generate a false positive identification for the testing mass spectrum. In case of the SeqMR approach, the top 10 matched results of SM approach were first sorted in descending order based on their spectral similarity scores. Starting from the top ranked compound, the first compound with retention index value difference less than the optional retention index difference ΔIo was considered as the identification result. Therefore, compound 17-alpha-Estradiol (CAS 57-91-0) was selected as the identification result because its retention index difference between the experimental data and the database value (ΔI =9.1) is less than the retention index threshold ΔIo = 11 iu, showing that SeqMR also generated a false positive identification for the testing spectrum. In case of SimMR approach, the mixture scores SM for the top 10 SM ranked compounds were calculated, respectively. Compound beta-Estradiol (CAS 50-28-2) has the largest mixture score SM = 0.9101, while the SM of compound 17-alpha-Estradiol (CAS 57-91-0) was ranked as the second (SM = 0.8260). For this reason, beta-Estradiol (CAS=50-28-2) is considered as the identification result by SimMR, which is a true positive identification.
Table 2 also shows that similar improvements are achieved in the other two query datasets. For the query dataset of the standard non-polar column, the SimMR method has 3.93%, 4.75%, 4.51%, and 4.10% improvement compared to the mass spectrum matching method when the SS, DFTR, DWTR, and WC measures are used, respectively. Compared to the SeqMR approach, 0.75%, 1.10%, 1.04%, and 0.99% of improvement are achieved when SS, DFTR, DWTR, and WC are used, respectively. For the query dataset of the standard polar column, the SimMR method has 4.04%, 4.81%, 4.57%, and 3.53% improvement compared to the mass spectrum matching method when the SS, DFTR, DWTR, and WC measures are used, respectively. Compared to the SeqMR approach, 1.05%, 1.53%, 1.39%, and 0.98% of improvement are achieved when SS, DFTR, DWTR, and WC are used, respectively.
The significant improvement of compound identification accuracy by SimMR in analysis of all three query datasets demonstrates that the proposed mixture score method outperforms both the mass spectrum matching and the SeqMR approaches. Mass spectrum of retention index reveals only partial molecular information of a compound. Many experimental conditions and data analysis parameters involved in data acquisition and data reduction affect the accuracy of mass spectrum and retention index. A compound could be removed from the identification list by the SeqMR approach if a large variation is introduced to either mass spectrum or retention index, due to its sequential nature of evaluating the similarity of mass spectrum and retention index in two isolated analysis steps. However, this compound may be kept as the identification result by the SimMR method because the overall variation of mass spectrum and retention index may still be small enough to make the compound have the best mixture score. The nature of simultaneous evaluation of the mass spectrum and retention index similarity in the SimMR approach improves the compound identification accuracy by reducing the chance of removing a true identification as well as increasing the chance of excluding false identification.
The mixture equation proposed in equation (16) is empirical, which may partially contribute to the false positive identification of SimMR approach. For instance, to a query EI mass spectrum of compound oleic acid (CAS 21556-26-3) in the NIST replicate database, the SeqMR approach correctly identified the mass spectrum of compound oleic acid (CAS 21556-26-3) in the NIST main library as the top ranked compound with a WC mass spectral similarity score of S=0.9458 and retention index difference (semi-non polar column) of ΔI =11.0 iu, while SimMR approach ranked compound TMS trans-9-octadecenoate (CAS 96851-47-7) as the top compound withSM = 0.9996, ΔI =11.3 iu, and S=0.9588. Such a false positive identification by SimMR was induced by the small difference between the retention index and spectral similarity score. Therefore, it is still necessary to further improve the mixture score for accurate compound identification.
This study demonstrates that mass spectral similarity measure can affect the performance of the SimMR method. We believe that the accuracy of library information, i.e., mass spectrum and retention index, also affects the identification accuracy of the mixture score function. It is necessary to explore the performance of the SimMR approach using different datasets. The compound identification accuracy of using the proposed SimMR method may be further improved by exploring different forms of mixture score functions as well as incorporating the mixture score function with more accuracy mass spectral similarity measures.
Conclusions
The developed compound identification method, SimMR, simultaneously evaluates the mass spectrum similarity and retention index distance using an empirical mixture score function. Due to the popularity of capillary columns and temperature gradient experiments in GC-MS analysis, the compounds with linear retention indices acquired on different capillary columns were extracted from NIST/EPA/NIH Mass Spectral Library 2005 (NIST05). The intercepts of these compounds and the compounds with EI MS spectra in the replicate EI MS library of NIST05 were used to form three query datasets based on column class defined in NIST05.
The performance of the SimMR method was compared to that of two other compound identification strategies: spectrum matching (SM), and sequential evaluation the similarity of mass spectrum and retention index (SeqMR). By analyzing the three query datasets, the performance of these three identification strategy is as follows in a descending order: SimMR > SeqMR > SM. For the query dataset of semi non-polar column data, the SimMR approach improves the compound identification accuracy 0.74-0.92% compared to the accuracy acquired by the SeqMR approach, and 3.17-3.52% by mass spectrum matching. For the query dataset of the standard non-polar column data, the SimMR method has 3.93-4.75% of improvement compared to the mass spectrum matching method, and 0.75-1.10% of improvement compared to the SeqMR approach. For the query dataset of the standard polar column, the SimMR method has 3.53-4.81% of improvement compared to the mass spectrum matching method, and 0.98-1.53% of improvement compared to the SeqMR approach.
Supplementary Material
Acknowledgments
This work was supported by NIH grant RO1GM087735 through the National Institute of General Medical Sciences (NIGMS), NSF grant DMS-1312603, and NIH grant R21ES021311 through the National Institute of Environmental Health Sciences (NIEHS).
Notes and references
- 1.Horai H, Arita M, Kanaya S, Nihei Y, Ikeda T, Suwa K, Ojima Y, Tanaka K, Tanaka S, Aoshima K, Oda Y, Kakazu Y, Kusano M, Tohge T, Matsuda F, Sawada Y, Hirai MY, Nakanishi H, Ikeda K, Akimoto N, Maoka T, Takahashi H, Ara T, Sakurai N, Suzuki H, Shibata D, Neumann S, Iida T, Tanaka K, Funatsu K, Matsuura F, Soga T, Taguchi R, Saito K, Nishioka T. J Mass Spectrom. 2010;45:703. doi: 10.1002/jms.1777. [DOI] [PubMed] [Google Scholar]
- 2.Schauer N, Steinhauser D, Strelkov S, Schomburg D, Allison G, Moritz T, Lundgren K, Roessner-Tunali U, Forbes MG, Willmitzer L, Fernie AR, Kopka J. Febs Lett. 2005;579:1332. doi: 10.1016/j.febslet.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 3.IST N. Office of the Standard Reference Data Base. National Institute of Standards and Technology; Gaithersburg, Maryland: 2005. [Google Scholar]
- 4.Stein S. Anal Chem. 2012;84:7274. doi: 10.1021/ac301205z. [DOI] [PubMed] [Google Scholar]
- 5.Stein SE, Scott DR. J Am Soc Mass Spectr. 1994;5:859. doi: 10.1016/1044-0305(94)87009-8. [DOI] [PubMed] [Google Scholar]
- 6.McLafferty FW, Zhang MY, Stauffer DB, Loh SY. Journal of the American Society for Mass Spectrometry. 1998;9:92. doi: 10.1016/S1044-0305(97)00235-3. [DOI] [PubMed] [Google Scholar]
- 7.Hertz HS, Hites RA, Biemann K. Analytical chemistry. 1971;43:681. [Google Scholar]
- 8.Visvanathan A. Information-theoretic mass spectral library search for comprehensive two-dimensional gas chromatography with mass spectrometry. ProQuest. 2008 [Google Scholar]
- 9.Koo I, Zhang X, Kim S. Analytical chemistry. 2011;83:5631. doi: 10.1021/ac200740w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim S, Koo I, Jeong J, Wu SW, Shi X, Zhang X. Analytical chemistry. 2012;84:6477. doi: 10.1021/ac301350n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koo I, Kim S, Zhang X. Journal of chromatography A. 2013;1298:132. doi: 10.1016/j.chroma.2013.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kováts E. Helvetica Chimica Acta. 1958;41:1915. [Google Scholar]
- 13.Lisec J, Schauer N, Kopka J, Willmitzer L, Fernie AR. Nat Protoc. 2006;1:387. doi: 10.1038/nprot.2006.59. [DOI] [PubMed] [Google Scholar]
- 14.Dunn WB, Broadhurst D, Begley P, Zelena E, Francis-McIntyre S, Anderson N, Brown M, Knowles JD, Halsall A, Haselden JN, Nicholls AW, Wilson ID, Kell DB, Goodacre R, C HSMH. Nat Protoc. 2011;6:1060. doi: 10.1038/nprot.2011.335. [DOI] [PubMed] [Google Scholar]
- 15.Zhang J, Fang AQ, Wang B, Kim SH, Bogdanov B, Zhou ZX, McClain C, Zhang X. Journal of Chromatography A. 2011;1218:6522. doi: 10.1016/j.chroma.2011.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim S, Koo I, Wei XL, Zhang X. Bioinformatics. 2012;28:1158. doi: 10.1093/bioinformatics/bts083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stein SE, Scott DR. J Am Soc Mass Spectr. 1994;5:859. doi: 10.1016/1044-0305(94)87009-8. [DOI] [PubMed] [Google Scholar]
- 18.Brigham EO. The fast Fourier transform. Prentice-Hall; Englewood Cliffs, N.J.: 1974. [Google Scholar]
- 19.Daubechies I. Ten lectures on wavelets. Society for Industrial and Applied Mathematics; Philadelphia, Pa.: 1992. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.