

Abstract
In order to understand how chromatin complexes function in the nucleus, it is important to obtain a comprehensive picture of their protein, DNA, and RNA components and their mutual interactions. Here we present a chromatin cross-linking approach (BioTAP-XL) which utilizes mass-spectrometry to identify protein complex components, together with high-throughput sequencing to identify RNA components and DNA binding sites. We describe full protocols for Drosophila cells and for human cells in culture, along with an additional protocol for Drosophila embryos as the source material. A key element of our approach in all cases is the generation of control data from input chromatin samples.
Keywords: chromatin, formaldehyde crosslinking, LC-MS/MS, next-generation sequencing
INTRODUCTION
Traditional biochemical analyses of multi-component chromatin complexes have been invaluable for the discoveries of enzymatic activities, specific binding properties, and strong subunit interactions of complexes. However, these approaches also involve key compromises. For example, it would be ideal to recover information about all protein components, whether they are strongly or weakly associated. Traditional approaches require the release of the complex from the DNA in order to solubilize it for the characterization of its enzymatic and binding properties. However, each complex requires specific conditions for release from chromatin and it is hard to predict this property in advance. Most importantly, the conditions of release might be incompatible with keeping a given complex intact. In solubilizing the complex to analyze the proteins by traditional methods, genomic positional information is lost (DNA) and, typically, any involved nucleic acids (RNAs) are stripped away. Therefore, different methods must be used to investigate the composition of the complex, its localization on the DNA, and any potential RNA components.
Given these considerations, we reasoned that establishing chemical crosslinks prior to the first step of affinity purification might allow the simultaneous preservation and recovery of the protein and RNA composition of any given complex in addition to its DNA targets.
Here we describe BioTAP-XL, a cross-linking approach which utilizes a special BioTAP tagged transgenic protein bait. The tag includes two epitopes: a Protein A moiety (Rigaut et al., 1999) and a biotinylation targeting signal consisting of a 75-amino acid sequence derived from a P. shermanii transcarboxylase (Guerrero et al., 2006). Endogenous biotin ligases in both prokaryotic and eukaryotic cells catalyze biotinylation of this target sequence in vivo, eliminating the need for co-expression of an exogenous biotin ligase. The transgenic fusion protein can be expressed from a variety of promoters in tissue culture systems and in Drosophila embryos.
Strategic Planning
The BioTAP-XL protocol employs cell culture lines or Drosophila stocks expressing a BioTAP-tagged transgene. A separate transgenic line is required for each protein of interest. These can be established by various selection protocols and Western Blot verified using peroxidase-antiperoxidase soluble complex antibody (PAP) detection before starting the BioTAP-XL protocol. Typically stable cell culture lines require several weeks to establish and expand to the requisite cell counts.
For work with human cells, such as 293T-REx cells, pHAGETRE-DEST-NBioTAP and pHAGE-TRE-DEST-CBioTAP lentiviral viral vectors may be used. They are available from Addgene (http://www.addgene.org/53568/ and http://www.addgene.org/53569/, also see (Alekseyenko et al., 2014a)). Lentiviral transduction and selection with Puromycin has been an effective method for establishing these lines.
For work with Drosophila cell culture or with fly stocks, the BioTAP tags may be cloned from pCAndy-1xProteinA-Bio or pCAndy-2xProteinA-Bio, available from Addgene (http://www.addgene.org/53570/ and http://www.addgene.org/53571, also see (Alekseyenko et al., 2014b)). These vectors may be used to make genomic or cDNA plasmids of BioTAP tagged proteins. Introduction of final BioTAP-tagged constructs along with a HygromycinB-resistance selection plasmid to cell culture by calcium chloride-mediated transfection has been an effective method. For fly lines, microinjection of a mini-white-marked BioTAP-tagged plasmid into pre-blastoderm embryos carrying a site-specific integrase system, and screening for founders that yield red-eyed progeny is an established procedure (Venken et al., 2006).
Here we describe steps to identify protein-protein, protein-RNA, and protein-DNA interactions for a tagged target bait protein from eukaryotic tissue culture cells (see Basic Protocol). We also describe steps to process transgenic Drosophila embryos for the same analysis (see Alternate Protocol 1). Protocols for generating control data from input chromatin and an alternative RNA analysis pipeline method are also provided (see Support Protocols 1 and 2, and Alternate Protocol 2, respectively).
BASIC PROTOCOL: BioTAP-XL for Drosophila and Human cell culture
This protocol is used to identify protein-protein, protein-DNA, and protein-RNA interactions for a specific BioTAP-tagged bait protein from one common starting material. It consists of an initial crude isolation of nuclei, followed by extensive chemical crosslinking by formaldehyde. The resulting material is subjected to sonication to achieve shearing and solubilization of the crosslinked chromatin. Next, tandem affinity purification using the BioTAP tag is performed. After utilizing the ProteinA part of the tag, bound chromatin complexes are eluted with denaturing conditions and subsequently rebound using the biotin part of the tag. The sample can then be split for isolation of peptides by on-bead trypsinization and identification by LC-MS/MS and/or for isolation of nucleic acids (DNA or RNA) and analysis by high-throughput sequencing.
Materials
-
BioTAP-tagged protein-expressing cultured cells:
1–2 × 1010 for S2 cells
1.5–3 × 109 for human cell lines
These numbers depend on the level of expression of the target protein.
Highly expressed targets will require fewer cells.
37% (w/v) formaldehyde (Fisher Scientific, cat. no. BP531-500)
1× PBS pH 7.4 (Boston BioProducts, cat. no. BM-220)
0.1 mM PMSF (Sigma, cat. no. 78830-5G)
1× Dulbecco’s phosphate buffered saline (Thermo Scientific, cat. no. SH300028.02)
NEB buffer (see recipe)
NSucrose buffer (see recipe)
NGlycerol buffer (see recipe)
Protein inhibitor cocktail tablets (Roche, cat. No. 11873580001)
Liquid nitrogen
RIPA buffer (see recipe)
TEN 140 buffer (see recipe)
TE buffer (see recipe)
10% (w/v) Sodium dodecyl sulfate solution (Sigma, cat. no. L4522-100ML)
IgG agarose beads (Sigma, cat. no. A2909)
Streptavidin–agarose beads (Thermo Scientific, cat. no. 20349)
IgG Elution buffer (see recipe)
Reverse Crosslinking buffer (see recipe)
Peroxidase anti-peroxidase soluble antibody complex produced in rabbit (PAP) (Sigma, cat. no. P1291)
Trypsin sequencing-grade (Promega, cat. no. V5111)
50 mM ammonium bicarbonate (Fluka, cat. no. 40867-50G-F)
Pierce Acetonitrile LC-MS grade (Thermo Scientific, cat. no. 51101)
Formic Acid Optima LC/MS (Fisher Scientific, cat. no. A117-10X1AMP)
Trifluoroacetic Acid Optima LC/MS (Fisher Scientific, cat. no. A116-10X1AMP)
HPLC Buffer A (see recipe)
HPLC Buffer B (see recipe)
Trypsin-digested BSA MS standard (New England Biolabs, cat. no. P8108S) (Optional)
TURBO DNase (Invitrogen, cat. no. AM2238)
SUPERase• In (20 U/μl) RNase Inhibitor (Invitrogen, cat. no. AM2694)
5M NaCl solution (Boston BioProducts, cat. no. BM-244)
10% Triton X-100 (Roche, cat. no.10789704001)
10mg/ml DNase-free RNaseA (Roche, cat. no. 11579681001)
CutSmart Buffer (New England BioLabs Inc, cat. no. B7204S)
20 mg/ml proteinase K (Roche, cat. no. 03115879001)
25:24:1 (v/v/v) UltraPure phenol/chloroform/isoamyl alcohol (Invitrogen, cat. no. 15593-031)
24:1 (v/v) chloroform/isoamyl alcohol (Sigma, cat. no. C0549)
3 M sodium acetate, pH 5.5 (Applied Biosystems, cat. no. AM9740)
Ethanol 200 proof (absolute) (Sigma, cat. no. E7023-500ML)
5 mg/ml glycogen (Applied Biosystems, cat. no. AM9510)
75% ethanol (diluted from 95% ethanol), ice cold
70% ethanol (diluted from 95% ethanol), ice cold
UltraPure distilled water (Invitrogen, cat. no.10977-015)
HyClone CCM3 serum-free medium (Thermo Scientific, cat. no.SH30065)
FBS, heat-inactivated (HyClone, Thermo Scientific, cat. no. SH30396.03)
10% (v/v) FBS(HI)-supplemented Schneider’s Drosophila medium (Invitrogen, cat. no. 11720)
10% (v/v) FBS(HI)-supplemented DMEM medium (Invitrogen, cat. no. 11965)
0.25% Trypsin-EDTA (1X), phenol red (Invitrogen, cat. no. 25200056)
Penicillin/Streptomycin (HyClone, Thermo Scientific, cat. no. SV30010)
Antibiotic/Antimycotic (Gibco/Life Technologies, cat. no. 15240-062)
15- and 50-ml Falcon conical tubes
250ml conical centrifuge tubes (Corning, cat. no. 430776)
1L polycarbonate centrifuge bottles (Beckman, cat. no. 355675)
Phase Lock Gel, Heavy 2ml tubes (5Prime, cat. no. 2302830)
1.7-ml Maxymum Recovery microcentrifuge tubes (Axygen INC, cat. no. 311-05-051)
Swinging bucket centrifuge with variable temperature (Beckman J6-MI with JS-4.2 rotor)
Sonicator (Misonix Sonicator 3000 equipped with microtip)
+37°C and +65°C incubators
SpeedVac
NanoDrop ND-3300
Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen, cat. no. P11496)
Quant-iT RiboGreen RNA Assay Kit (Invitrogen, cat. no. R11490)
150mm cell culture dishes (Corning, cat. no.430597)
225cm2 T-flasks with Phenolic-Style cap (Corning, cat. no. 3000)
2.8L Fernbach glass flasks (Bellco, Glass Inc., cat. no. 2552-02800)
40 or 100 ml Dounce homogenizer, with both A and B pestles (Bellco, Glass Inc., cat. no. 1984-10040 or 1984-10100)
10K Amicon Ultra-15 centrifugal filters (Millipore, cat. no. UFC901008)
Pierce C18 Spin Tips (Thermo Scientific, cat. no. 84850)
RotoFlex (Denville Scientific Inc., H5700)
Gyrotory Shaker Model G2 (Brunswick Scientific)
Multitron II ATR Shaker with cooling (INFORS-HT)
Autosampler vial (Fisher Scientific, cat. no. 05-704-225)
Additional materials for DNA and RNA high-throughput sequencing methods
NEBNext ChIP-Seq Library Prep Master Mix Set for Illumina (New England BioLabs Inc, cat. no. E6240S)
NEBNext Ultra Directional RNA Library Prep Kit for Illumina (New England BioLabs Inc, cat. no. E7420S)
CAUTION: Formaldehyde is toxic by inhalation or if swallowed; is irritating to the skin, eyes, and respiratory system; and may be carcinogenic. Formaldehyde should be used with appropriate safety measures such as protective gloves, glasses, clothing, and sufficient ventilation. All waste should be handled according to hazardous waste regulations.
CAUTION: Phenol/chloroform/isoamyl-alcohol is harmful if swallowed or in contact with skin, causes severe skin burns and eye damage, is fatal if inhaled, and is potentially carcinogenic. It should be used with appropriate safety measures such as protective gloves, glasses, clothing, and sufficient ventilation. All waste should be handled according to hazardous waste regulations.
CAUTION: Chloroform/isoamyl-alcohol is harmful if swallowed, causes skin and eye irritation, and is potentially carcinogenic. It should be used with appropriate safety measures such as protective gloves, glasses, clothing, and sufficient ventilation. All waste should be handled according to hazardous waste regulations.
CAUTION: Formic acid (FA) is harmful if inhaled or swallowed or in contact with skin; is irritating to the skin, eyes, respiratory system, and gastrointestinal tract; and causes severe burns by all exposure routes. It should be used with appropriate safety measures such as protective gloves, glasses, clothing, and sufficient ventilation. All waste should be handled according to hazardous waste regulations.
CAUTION: Trifluoroacetic Acid (TFA) is harmful if inhaled or swallowed or in contact with skin; inhalation may cause central nervous system effects; and causes severe burns by all exposure routes. It should be used with appropriate safety measures such as protective gloves, glasses, clothing, and sufficient ventilation. All waste should be handled according to hazardous waste regulations.
Growing human 293 T-REx cells for BioTAP-XL
Grow 1.5×109 cells in 150mm cell culture dishes in DMEM medium supplemented with 10% FBS, 1% penicillin/streptomycin (50 to 100 plates).
Add 3ml of 0.25% Trypsin-EDTA to each plate and incubate for 7 min. at room temperature
Add 7ml DMEM supplemented with 10% FBS to inactivate trypsin.
Combine trypsinized cells in 250ml conical centrifuge tubes
-
Centrifuge 5 min at 280 x g, +4°C to pellet the cells. Decant and discard the supernatant.
Note the cell pellet volume. Resuspend the cells in 250 ml ice cold PBS.
Centrifuge 5 min at 280 x g, +4°C, to pellet the cells. Decant and discard the supernatant.
Repeat steps 6 and 7 two additional times, for 3 total washes. Proceed directly with Basic Protocol, step 9.
Growing Drosophila S2 cells for BioTAP-XL
-
1
Grow cells in four 225cm2 T-flasks in Schneider’s Drosophila medium supplemented with 10% FBS, 1% antibiotic/antimycotic to a density of ~5×106 cells/ml.
-
2
Split cells in half by adding an equal volume of HyClone CCM3 serum-free medium (grown in eight new 225cm2 T-flasks). Grow to a density of ~5×106 cells/ml.
-
3
Transfer cells into four 2.8L Fernbach glass flasks. For each flask, mix 100 ml of cells and 400 ml of HyClone CCM3.
-
4
Grow cells at 26.5 °C and 90 rpm to a density of ~1×107 cells/ml in Multitron II shaker.
-
5
Centrifuge 10 min at 600 x g, +4°C to pellet 1×1010 cells. Decant and discard the supernatant.
Note the cell pellet volume. -
6
Resuspend the cells in 500 ml ice cold PBS.
-
7
Centrifuge 10 min at 600 x g, +4°C, to pellet the cells. Decant and discard the supernatant.
-
8
Repeat steps 5 and 6 two additional times, for 3 total washes. Proceed directly with Basic Protocol, step 9.
Formaldehyde crosslinking of crude nuclear extracts
-
9
Resuspend cells in NEB buffer+0.1 mM PMSF, pre-chilled on ice. Add 100 ml of buffer for every 4–5 ml of cell pellet of harvested 293 T-REx or S2 cells.
-
10
Transfer cell suspension to a 100 ml Dounce homogenizer.
-
11
Lyse cells with 5 strokes of pestle A.
-
12
Follow immediately with 5 strokes of pestle B.
-
13
Immediately pour 100 ml of cell/nuclear homogenate into a T-225 flask containing a room-temperature mixture of 360 ml of PBS and 40 ml of 37% formaldehyde.
Prepare the formaldehyde solution fresh. -
14
Incubate 30 min at 25°C on an orbital shaker platform with gentle mixing (100rpm).
-
15
Centrifuge 10 min at 4000 x g, +4°C, to collect fixed nuclei. Decant supernatant into formaldehyde waste.
-
16
Resuspend nuclear pellet in 100 ml of ice-cold PBS with 0.1 mM PMSF.
-
17
Repeat steps 15 and 16 three additional times, for a total of four washes. The waste need not be decanted into formaldehyde waste beyond the second wash. Decant the final wash.
-
18
Resuspend nuclear pellet in 100 ml of ice-cold NSucrose buffer with 0.1 mM PMSF.
-
19
Centrifuge 10 min at 4000 x g, +4°C, to pellet fixed nuclei.
-
20
Resuspend nuclear pellet in 100 ml of ice-cold Nglycerol buffer buffer with 0.1 mM PMSF.
-
21
Centrifuge 10 min at 4000 x g, +4°C, to pellet fixed nuclei.
-
22
Resuspend nuclear pellet in 25 ml of ice-cold Nglycerol buffer supplied with protein inhibitor cocktail.
-
23
Transfer fixed nuclear suspension to a 50 ml falcon tube on ice.
-
24
Proceed directly to Basic Protocol, step 26, or flash-freeze fixed nuclear suspension in liquid nitrogen and store at −80°C.
Once material has been frozen, it can be stored up to 1 year at 80°C.
Shearing and solubilization of cross-linked chromatin by sonication
-
25
If frozen, thaw fixed nuclear suspension at room temperature, then put on ice.
-
26
Centrifuge 10 min at 4000 x g, +4°C, to pellet fixed nuclei.
Note the nuclear pellet volume. -
27
Resuspend the nuclear pellet in 10–20 volumes of TE buffer with 0.1 mM PMSF.
-
28
Centrifuge 10 min at 4000 x g, +4°C, to collect the fixed nuclei.
-
29
Resuspend pellet with 20 volumes of TE buffer with 0.1 mM PMSF by pipetting up and down.
-
30
Add 10% SDS to the suspension to a final concentration of 1% and invert mixture in the tubes 10 times at room temperature.
-
31
Centrifuge 10 min at 4000 x g, +4°C, to pellet the nuclear material. Decant or pipet supernatant into waste.
Remove the supernatant carefully, as the pellet may be quite loose. -
32
Resuspend pellet with 20 volumes of TE buffer with 0.1 mM PMSF by gently pipetting up and down up to 5 times.
-
33
Centrifuge 10 min at 4000 x g, +4°C, to collect nuclear material. Decant or pipet supernatant into waste.
-
34
Repeat steps 32 and 33 two additional times for a total of three washes.
-
35
Resuspend pellet by gently pipetting up and down with 1.5–2 volumes of TE buffer supplied with protein inhibitor cocktail.
Thorough resuspension is required. Avoid proceeding if large visible clumps still exist. -
36
Add 10% SDS to the suspension to make a final concentration of 0.1%.
-
37
Mix viscous suspension on a rotating wheel for 30 min at +4C.
-
38
Split mixture into 5ml aliquots in 15ml polystyrene tubes.
-
39
Sonicate each 5ml aliquot. For a Misonix sonicator, set a total processing time of 3.5 minutes (14 cycles) for S2 cells and 5 minutes (20 cycles) for 293T-REx cells. Set each cycle as a 15″ ON pulse followed by a 45″ OFF pulse, with the power level set to 7.0 (39–42W output). Sonicate the samples in an ice water bath to prevent heating the sample.
These settings may need to be adjusted according to the cell or tissue type, and the model of sonicator used. Avoid foaming, as this likely decreases sonication efficiency. If foaming occurs, centrifuge briefly and gently resuspend all material. In general DNA fragments after sonication should be in the range of 300–3000 bp (see Support Protocol 1 for details). -
40
Add 550μl of 10% Triton X-100 to each tube with 5ml sonicated lysate, and mix the suspension on a rotating wheel for 5 min at +4C.
-
41
Add 166μl of 5M NaCl and mix suspension on a rotating wheel for 5 min at +4C.
-
42
Pool the ~5.5ml aliquots in one 50 ml Falcon tube. Centrifuge 10 min at 12000 x g, +4°C, to pellet nuclear debris.
If the sonication was successful, there will only be a tiny pellet at this point. A large pellet suggests the sonication did not adequately solubilize the chromatin. If this is the case, resuspend the pellet and do additional sonication pulses. -
43
Transfer supernatant containing soluble chromatin to a fresh 50ml Falcon tube.
-
44
Take a minimum of two 0.5 ml aliquot before proceeding with the next step as reference for chromatin input composition and quality (for protein, DNA, and RNA). Flash-freeze the input aliquots in liquid nitrogen and store at −80°C.
See also Support Protocols 1 and 2 for input sample processing. -
45
Use the rest of the chromatin (generally 15–40ml) for tandem affinity purification.
Tandem affinity purification with IgG and streptavidin-agarose
-
46
Thoroughly resuspend IgG agarose beads (50% aqueous slurry).
-
47
Transfer 2ml of beads slurry into a 50ml Falcon tube.
-
48
Add 40ml of RIPA buffer to the beads and mix them on a rotating wheel for 10 min at +4C.
-
49
Centrifuge 5 min at 1000 x g, +4°C, to collect beads. Pipet supernatant into waste.
-
50
Add 15–40ml of the sonicated chromatin from step 45 to the washed beads and rotate end-over-end for 12–16 hours at +4°C.
-
51
Centrifuge 5 min at 1000 x g, +4°C, to collect beads. Pipet supernatant into waste.
-
52
Resuspend beads in 14 ml of ice-cold RIPA buffer and transfer them to a new 15ml Falcon tube.
-
53
Centrifuge 5 min at 1000 x g, +4°C, to collect beads. Pipet supernatant into waste.
-
54
Resuspend beads in 14 ml of ice-cold RIPA buffer and mix them on a rotating wheel for 10 min at +4C.
-
55
Repeat steps 53 and 54 two additional times for a total of three washes. Pipet final wash into waste.
-
56
Resuspend beads in 14 ml of TEN 140 buffer at +25°C.
-
57
Centrifuge 5 min at 1000 x g, +25°C. Pipet supernatant into waste.
-
58
Add 10ml of IgG Elution buffer to the beads and mix slurry by end-over-end rotation for 1 hour at +25°C.
-
59
Centrifuge 5 min at 1000 x g, +25°C, and collect the supernatant with eluted complexes.
-
60
Repeat steps 58 and 59 one additional time for two total elutions.
-
61
Load resulting 20ml eluate into two 10K Amicon Ultra-15 columns (10ml in each).
-
62
Centrifuge 10–15 min at 3000 x g, +25°C, to concentrate eluate from 10ml to 1ml.
-
63
Transfer and pool both 1ml concentrates from each column into one new concentrator column.
-
64
Add 12ml of TEN 140 buffer with 0.1% Triton X-100 to 2ml of samples in the column.
-
65
Centrifuge 10–15 min at 3000 x g, +25°C, to concentrate the samples to 1ml.
-
66
Repeat steps 64 and 65 three additional times for a total of four washes/buffer exchanges.
-
67
Take the final 1ml concentrated sample out of the column and split it evenly between two 1.7ml maximum recovery Eppendorf tubes (0.5ml for each).
-
68
Add 1ml of RIPA buffer at +25°C to each tube.
Sample is now ready for streptavidin affinity purification steps. -
69
Thoroughly resuspend streptavidin agarose beads (50% aqueous slurry).
-
70
Transfer 800μl of bead slurry into 15ml Falcon tube.
-
71
Add 12ml of IgG Elution buffer to the beads and mix them on a rotating wheel for 5 min at +25C.
-
72
Centrifuge 5 min at 1000 x g, +25°C, to collect the beads. Pipet supernatant into waste.
-
73
Resuspend beads in 14 ml TEN 140 buffer (no Triton X-100) and mix them on a rotating wheel for 5 min at +25C.
-
74
Centrifuge 5 min at 1000 x g, +25°C, to collect the beads. Pipet supernatant into waste.
-
75
Repeat steps 73 and 74 three additional times except with TEN 140 buffer with 0.1% Triton X-100.
-
76
Resuspend beads in 2ml of TEN 140 buffer with 0.1% Triton X-100 and split between two 1.7ml maximum recovery Eppendorf tubes.
-
77
Centrifuge 5 min at 1000 x g, +25°C, to collect beads. Pipet supernatant into waste.
Beads are now ready for incubation with sample from step 68. -
78
Add 1.5 ml of the samples from step 68 to each tube with washed streptavidin-agarose beads.
-
79
Incubate 12–16 hours at +16°Cwith end-over-end rotation to bind complexes.
-
80
Centrifuge 5 min at 1000 x g, +25°C, to collect beads. Pipet supernatant into waste.
-
81
Resuspend beads in each tube with 1ml of RIPA buffer +25°C. Transfer and pool them to one 15ml Falcon tube.
-
82
Add an additional 12ml of RIPA buffer +25°C, mix for 10 min at +25C on a rotating wheel.
-
83
Centrifuge 5 min at 1000 x g, +25°C, to collect beads. Pipet supernatant into waste.
-
84
Resuspend beads in 12 ml TEN 140 buffer with 0.1% Triton X100 and mix them on a rotating wheel for 5 min at +25C.
-
85
Centrifuge 5 min at 1000 x g, +25°C, to collect beads. Pipet supernatant into waste.
-
86
Repeat washing steps 84 and 85 five additional times as follows:
two washes with 12ml IgG Elution buffer
two washes with 12ml IgG Elution buffer without SDS
one wash with 12ml TEN 140 buffer.
-
87
Split beads equally between two 15ml tubes (each would be ~6ml beads in TEN 140 buffer).
-
88
Centrifuge 5 min at 1000 x g, +25°C, to collect beads. Pipet supernatant into waste.
Use one half for protein recovery steps and the other for DNA and RNA recovery steps.For protein work, proceed to step 89; for DNA and RNA, proceed to step 119.
Protein recovery steps
On-bead trypsin digestion
-
89
Resuspend beads for protein recovery in 12 ml of 50 mM ammonium bicarbonate and mix for 5 min at +25°C on a rotating wheel.
-
90
Centrifuge 5 min at 1000 x g, +25°C, to collect beads. Pipet supernatant into waste.
-
91
Repeat washing steps 89 and 90 for a total of seven washes in order to remove detergent contaminants from the samples which may interfere with mass spectrometry analysis.
In order to check the bait protein integrity and quality of the pulldown, a small aliquot, about 1/20 of bead volume, may be taken for Western-blot analysis. Prior to loading the sample for SDS-PAGE, the beads were incubated with 1.5 bead volumes of Reverse Crosslinking buffer for 25 min at +100°C. -
92
Resuspend remaining beads in 800μl of 50 mM ammonium bicarbonate with 20μl of sequencing-grade Trypsin and incubate 12–16 hours at +37°C on a rotating wheel.
-
93
Add 1μl of 100% formic acid to the bead suspension to stop the trypsinization reaction.
-
94
Centrifuge 5 min at 1000 x g, +25°C. Transfer the supernatant (containing the trypsinized peptides) into one 1.7ml maximum recovery Eppendorf tube.
-
95
To maximize peptide recovery, add 75μl of 25% acetonitrile with 0.1% formic acid to the beads, vortex 5 min at 700 rpm.
-
96
Centrifuge 5 min at 1000 x g, +25°C. Transfer the supernatant to same 1.7ml tube (as step 94).
-
97
Repeat steps 95 and 96 two additional times and pool all eluates in one tube (~1025μL).
-
98
Concentrate samples for 1.5 to 3 hours on a Speedvac to a final volume of 10μl.
Set the Speedvac to the low drying rate setting. Do not use any heat settings. As samples will freeze, thaw the samples at least every 30 min by removing the sample tube from the Speedvac chamber and incubating 10min at room temperature. -
99
Add 50μl of 0.1% trifluoroacetic acid +25°C to the 10μl concentrated sample, gently mix and recollect by brief centrifugation.
C18 column peptide purification
-
100
Prepare a C18 Spin tip: Insert C18 spin tip into spin adapter seated in a microcentrifuge tube. Wet the tip by adding 50μl of 100% acetonitrile and centrifuging 1 min at 1000 x g, +25°C. Repeat washing step one more time. Equilibrate tip by adding 50μl of 0.1% trifluoroacetic acid (in Ultrapure water) and centrifuging 1 min at 1000 x g, +25°C. Repeat equilibration step one more time. Once equilibrated, transfer C18 spin tip and adapter to a new microcentrifuge tube.
-
101
Add the 60μl of sample (from step 99) to the C18 tip. Centrifuge 2 min at 400 x g, +25°C, and collect flow through.
-
102
To maximize peptide binding to the C18 resin, add the flow through to the same C18 spin tip and centrifuge 1 min at 1000 x g, +25°C.
-
103
Wash the tip by adding 50μl of 0.1% trifluoroacetic acid. Centrifuge 1 min at 1000 x g, +25°C.
-
104
Repeat step 103 one additional time.
-
105
Transfer C18 spin tip and adapter to new 1.7ml maximum recovery microcentrifuge tube.
-
106
To elute the peptides from the resin, add 30μl of 50 % acetonitrile and centrifuge 2 min at 400 x g, +25°C.
-
107
Without changing tubes, add 30μl of 100 % acetonitrile and centrifuge 1 min at 1000 x g, +25°C, to take a second peptide elution.
-
108
Dry sample completely on a Speed vac. Store dried peptides at 80°C for up to a month or proceed directly to LC-MS/MS analysis.
LC-MS/MS
Below are recommendations for LC-MS/MS of peptides based on the authors’ experiences with various mass spectrometry facilities. The user is encouraged to adopt the specific protocols used by the mass spectrometry facility actually analyzing the peptides, even if their protocols differ from these written instructions below.
-
109
Re-suspend the dried peptides in at least 20μl of HPLC solvent A and transfer to an autosampler vial.
A 30 minute incubation of the re-suspended peptide solution in a bath sonicator may be required to completely solubilize the peptides prior to transferring to the autosampler vial. -
110
Equilibrate the HPLC column by running a reverse phase gradient either with no sample loaded or with peptide standards such as a BSA tryptic digest mixture.
Running peptide standards is recommended if the column has not been used within 2 – 3 days. -
111
Load 1 – 2 μl of the peptide sample (or 1/10th – 1/20th of sample volume) by autosampler onto the HPLC column.
-
112
Elute peptides with a reverse phase gradient, starting with 2% percent composition of HPLC Buffer B to 30% HPLC Buffer B over a 57 minute gradient and electrospray into the mass spectrometer.
Ensure that electrospray conditions are stable prior to running the sample. -
113
Operate mass spectrometer under data dependent mode by collecting MS/MS generated from collisional induced dissociation of the top 10 – 20 most abundant peptide precursor ions in the MS scan.
-
114
As a reference for overall sample quality, inspect the extracted ion chromatograms of various autocatalytic trypsin peptides after the LC-MS run. Peak shapes should be approximately symmetrical.
Examples of trypsin peptides include, in relative elution order starting with the most hydrophilic, the 98-107 peptide LSSPATLNSR, the 108-115 peptide VATVSLPR, and the 58 – 77 peptide LGEHNIDVLEGNEQFINAAK (see Figure 2). -
115
If the extracted ion chromatograms of the trypsin peptides appear fine (see Figure 2), search the data with the SEQUEST algorithm (http://www.ncbi.nlm.nih.gov/pubmed/10404161) against the appropriate reference proteome database downloaded from the UniProt website (www.uniprot.org). For LTQ Velos ion-trap data, set the precursor and product ion mass tolerances to 2.0 and 1.0 Da respectively and variable modifications to methionine oxidation. Maximum missed cleavage sites should be set to 1 – 2 for searching unmodified peptides or to 2 – 3 for searching peptides containing lysine modifications.
-
116
If the overall run quality is fine, load the same sample again for technical replicates (optional). Three technical replicates are recommended to assess the variability between LC-MS runs.
In the authors’ experience, technical replicates have been highly consistent; the authors thus favor biological replicates (ie, different chromatin preps and/or amino vs carboxy- terminal tags) to check for consistency of interactions. -
117
If the overall run quality is poor, diagnose the problem prior to continuing with additional runs (see Troubleshooting for details).
-
118
To determine candidate proteins enriched by the immunoprecipitation, compare the total and unique peptide counts of the identified proteins between the IP sample and the input sample (see Support Protocol 1 for details). The bait protein should have one of the highest ratios of peptides recovered in the IP relative to input.
Figure 2.

Extracted ion chromatograms (XICs) of tandem mass spectra (MS/MS) of precursor ions corresponding to trypsin autolysis peptides. The top XIC (red) depicts MS/MS of the 523.59 m/z precursor ion corresponding to the doubly charged 98–107 LSSPATLNSR peptide. The middle XIC (green) depicts MS/MS of the 422.01 m/z precursor ion corresponding to the doubly charged 108–115 VATVSLPR peptide. The bottom XIC (blue) represents MS/MS of the 738.15 m/z precursor ion corresponding to the triply charged 58–77 LGEHNIDVLEGNEQFINAAK peptide. Expected average precursor m/z and total number of MS/MS spanned by the main peak are provided for each XIC. A run of good quality will display this relative elution order of these three peptides in reverse phase chromatography, with narrow and symmetrical peak widths (spanning approximately 1–2 minutes).
DNA and RNA recovery steps
-
119
Resuspend beads for DNA/RNA recovery from step 88 in 1ml TE buffer and transfer to a 1.7ml maximum recovery tube.
-
120
Centrifuge 5 min at 1000 x g, +25°C, to collect beads.
-
121
Repeat steps 119 and 120 two additional times.
-
122
Resuspend the beads in 220μl TE. Add 18ul of 10% SDS and 40ul of Proteinase K (20mg/ml).
-
123
Incubate sample overnight at +37°C.
-
124
Transfer sample to +65°C and incubate 6h.
-
125
Add 9μl of 5M NaCl (140mM final) and mix by vortexing.
-
126
Add 300μl of phenol/chloroform/isoamyl alcohol to the samples and mix by vortexing for 30 sec.
-
127
Centrifuge 15 min at 20000 x g, +25°C.
-
128
Transfer 240μl of the aqueous (top) phase to a new 1.7ml maximum recovery tube.
-
129
To maximize nucleic acid recovery, add 300μl of TEN 140 buffer to the organic phase in the original tube to back extract additional material. Mix by vortexing for 30 sec and centrifuge 15 min at 20000 x g, +25°C.
-
130
Recover and combine the aqueous (top) phase with the the material from step 128 in one tube (~540μL total; first extraction 240μl and second 300μl).
-
131
Add one volume (540μl) of chloroform/isoamyl alcohol, mix by vortexing for 30 sec, centrifuge 15 min at 20000 x g, +25°C.
A phase-lock tube may be used here to separate the organic phase from the aqueous phase. -
132
Transfer about 500μl of the upper aqueous phase into two new 1.7ml maximum recovery tube. Split samples evenly (250μl) between tubes.
-
133
Add 25μl of 3M NaAc pH 5.0, 10μl of glycogen (5mg/ml), and 1ml of ethanol (200 proof) to each tube, mix by vortexing for 30 sec, and re-collect by brief centrifugation.
-
134
Incubate at least 12 hours at −80°C.
One tube is for DNA recovery (step 135) and the other is for RNA recovery (step 141).DNA and RNA samples may be stored up to 1 month at −80°C.For RNA samples we strongly recommend proceeding to step 141 as quickly as possible.
DNA preparation for qPCR or Solexa library construction
-
135
Centrifuge 30 min at 16000 x g, +4°C, to pellet DNA recovered after the purification. Pipet supernatant into waste.
A small white pellet should be visible at the bottom of the tube. -
136
Add 1ml of ice cold 70% ethanol to the pellet, mix briefly and centrifuge 1 hour at 16000 x g, +4°C. Pipet supernatant into waste. Briefly centrifuge tube again and remove residual ethanol.
-
137
Air-dry the pellet 15 min at +25°C.
-
138
Dissolve the pellet in 41μl Ultrapure water.
-
139
Using the Quant iT PicoGreen dsDNA Assay kit and a NanoDrop ND-3300, quantify the concentration using 1μl of the purified DNA.
Follow NanoDrop instrument manufacturer’s instructions for accurate quantification. -
140
DNA is now ready for Quantitative Real-Time PCR (qPCR) applications or ChIP-Seq Solexa/Illumina library preparation.
We recommend using NEBNext ChIP-Seq Library Prep Master Mix Set for Illumina, following manufacturer’s instructions, for high-throughput sequencing methods.
RNA preparation for qRT-PCR or for Solexa library construction
-
141
Centrifuge 30 min at 16000 x g, +4°C, to pellet RNA recovered after the purification. Pipet supernatant into waste.
A small white pellet should be visible at the bottom of the tube. -
142
Add 1ml of ice cold 75% ethanol to the pellet, mix briefly and centrifuge 30min at 16000 x g, +4°C. Pipet supernatant into waste.
-
143
Repeat step 142 once more. After discarding supernatant, briefly centrifuge tube again and remove residual ethanol. 144. Air-dry the pellet 15 min at +25°C.
-
145
Dissolve the pellet in 230μl Ultrapure water (RNase-free).
-
146
Add 10μl of SUPERase• In RNase Inhibitor (20 U/μl), 30ul of 10X TURBO DNase Buffer and 30ul of TURBO DNase (2 U/μl). Mix gently by pipetting.
-
147
Incubate 30 min at +37°C.
-
148
To stop the reaction, add 20μl of Proteinase K (20mg/ml), 250μl TEN 140 buffer and 30μl 10% SDS. Mix by vortexing and centrifuge briefly.
-
149
Incubate 5 min at +37°C.
-
150
Add 600μl of phenol/chloroform/isoamyl alcohol to the samples and mix by vortexing for 30 sec.
-
151
Centrifuge 15 min at 20000 x g, +25°C.
-
152
Transfer 550μl of the aqueous (top) phase to a new 1.7ml maximum recovery tube.
-
153
Perform chloroform/isoamyl alcohol extraction and RNA precipitation as before (steps 131–134).
-
154
After −80°C incubation, repeat steps 141–144.
-
155
After air drying the RNA pellet, dissolve it in 6μl Ultrapure water (Invitrogen).
-
156
Using the Quant iT RiboGreen RNA Assay kit and a NanoDrop ND-3300, quantify the concentration using 0.1–1μl of the purified RNA.
Follow NanoDrop instrument manufacturer’s instructions for accurate quantification. -
157
RNA is now ready for Reverse Transcription Quantitative-Real-Time PCR (rt-qPCR) applications or Solexa/Illumina RNA library preparation.
We recommend using NEBNext Ultra Directional RNA Library Prep Kit for Illumina following manufacturer’s instructions for high-throughput sequencing methods.If residual DNA contamination in the RNA sample is a major concern, repeat steps 145–155 once more.
ALTERNATE PROTOCOL 1: BioTAP-XL from Drosophila embryos as starting material
This protocol was developed to generate material from Drosophila embryos for use in the BioTAP-XL Basic Protocol. Note that while the protocol described here is specific to Drosophila embryos, it suggests that the BioTAP-XL protocol could be used with any animal tissue that is expressing a transgenic BioTAP-tagged bait protein.
This protocol outlines the methods to grow and collect embryos, lyse them via mechanical means, and formaldehyde fix the nuclear extract. The resulting crosslinked extract can be fed into the Basic Protocol, step14, for subsequent sonication, tandem-affinity purification, and analysis steps.
Additional Materials (also see Basic Protocol)
Molasses agar plates (see recipe)
Embryo wash solution (see recipe)
Sodium hypochlorite (Sigma Aldrich cat. no. 239305)
Embryo Collection Cage (Flystuff, cat. no. 59-101)
Paint brush, 13mm/½″ (Winsor&Newton, cat. no. 094376864021)
425 micrometer metal sieve (W.S. Tyler, incorporated, No.40)
106 micrometer metal sieve (W.S. Tyler, incorporated, No.140)
100 micrometer Nylon Woven Net Filter (Fisher Scientific, cat. no. NY1H00010)
120 Vac Overhead Stirrer (Wheaton, cat. no. 903475)
Potter-ELV tissue grinder 55ml mortar (Wheaton, cat. no. 358048)
Potter-ELV tissue grinder 55ml teflon pestle (Wheaton, cat. no. 358051)
Miracloth (Calbiochem cat. no. 475855)
Büchner funnel (Fisher, cat. no. FB-966-H)
Side arm filtering flask (Fisher, cat. no. 10-181E)
Fly embryo preparation
Grow BioTAP transgenic flies at 25°C with 65% humidity in Embryo Collection Cages for large scale (50~100g) collection of 12~24 hour-old embryos.
Collect embryo on molasses agar plates covered with yeast paste.
-
Stack the 425 micrometer metal sieve on the 106 micrometer metal sieve and rinse embryos from the molasses agar plates with embryo wash solution in the stacked metal sieves.
The coarse sieve allows embryos to pass through to the fine sieve, but prevents passage of any large debris, adult carcasses, and pupal cases. Clear the surface of molasses agar plates with a paint brush and additional embryo wash solution to collect the residual embryos.
Transfer the embryos collected on the fine sieve (106 um) metal sieve to a Nylon mesh sieve.
-
Immerse the mesh cloth sieve for 3min in 50% bleach (Sodium hypochlorite) to dechorionate the embryos.
Typically one change of 50% bleach solution after 1.5min is used. -
Rinse under running distilled water to remove bleach completely.
This may take several minutes. Blot-dry the embryos in the mesh cloth sieve by wicking away moisture through the cloth with paper towels, and transfer embryos to a weigh boat.
Measure total weight, and divide the embryos into 10 gram aliquots; place on ice.
Resuspend a 10 gram aliquot of embryos in 30ml of chilled Nuclear Extraction Buffer (NEB) with 0.1mM PMSF.
Homogenize with 5 strokes using a Potter-ELV tissue grinder (glass mortar and motorized teflon pestle) using the maximum speed setting of a 120 Vac Overhead Stirrer at 4°C.
Establish an apparatus for suction filtration using a Büchner funnel and side arm filtering flask on ice.
-
Place a single layer of Miracloth in the Büchner funnel and filter the homogenate through under vacuum suction.
This removes unlysed embryos and other debris. Rinse the Miracloth filter with additional 70ml of Nuclear Extraction Buffer (NEB), and pool this wash with the original filtrate for 100ml total volume.
Immediately transfer 100ml of filtered homogenate from the side arm flask to a 225ml T-flask filled with 360ml of room-temperature PBS and 40ml of 37% formaldehyde.
Proceed with fix and subsequent processing and purification steps starting from Basic Protocol, step 14.
ALTERNATE PROTOCOL 2: RNA pull-down sample recovery by using on-bead ligation and library preparation for Solexa/Illumina RNA-seq
This protocol was developed as alternative method for recovering RNA after BioTAP XL pulldown. RNA recovered in the Basic Protocol utilizes a random-priming approach for cDNA synthesis and Illumina RNA-seq. While this is a commonly used approach for RNA reverse transcription prior to expression analysis or other RNA-quantification techniques, the random-priming approach might introduce biases against certain RNA classes (eg. repetitive or short RNA less than 100nt). To attempt to mitigate such biases, an on-bead ligation technique was modified for use with the BioTAP-XL protocol. This alternative method is more laborious and expensive, but may result in increased recovery of specific RNA.
This on-bead ligation approach was developed on the basis of the previously published CRAC technique (Granneman et al., 2009). It allows ligation of Solexa 3′ and 5′ linkers directly to the RNA while still bound to the streptavidin-agarose beads as a part of the crosslinked RNA-protein-DNA complexes recovered from the BioTAP-XL tandem purification.
Additional Materials (also see Basic Protocol)
CRAC-TN150 buffer (see recipe)
CRAC-Wash buffer 1 (see recipe)
1 x CRAC-PNK buffer (see recipe)
5 x CRAC-PNK buffer (see recipe)
TruSeq ChIP Sample Prep Kit for ChIP-Seq (Illumina, IP-202-1012)
RNace-It Ribonuclease Cocktail 6000U (Agilent Technologies Inc, cat. no. 400720)
TSAP Thermosensitive Alkaline Phosphatase (Promega, cat. no. M9910)
RNasin Recombinant Ribonuclease Inhibitor (Promega, cat. no. N2515)
10mM dNTP Mix (Invitrogen, cat. no. 18427-088)
SuperScript III First-Strand Synthesis kit (Invitrogen, cat. no. 18080-051)
Phusion Hot Start II DNA Polymerase 2 U/μL (Thermo Scientific, cat. no. F-549S)
T4 RNA ligase 2 truncated K227Q and 50% PEG 8000 (New England BioLabs Inc, cat. no. M0351L)
T4 RNA Ligase 1 (New England BioLabs Inc, cat. no. M0204L)
T4 PNK-Polynucleotide Kinase from T4-infected Escherichia coli (Sigma, cat. no. P4390-500UN).
100 mM ATP (Adenosine 5′-Triphosphate) solution (GE Healthcare Life Sciences, cat. no. 27-2056-01)
3′-BioTAP-Solexa linker-/5rApp/rUrCrGrUrArUrGrCrCrGrUrCrUrUrCrUrGrCrUrUrGrUr/ddC/
This RNA oligo has a blocked 3′ (ddC) end and an activated adenosine at the 5′ end, and may be ordered from BiooScientific.
5′-BioTAP-Solexa linker-InvddT/GrGrUrUrCrArGrArGrUrUrCrUrArCrArGrUrCrCrGrArCrGrArUrC
This DNA-RNA hybrid oligo has an inverted ddT at the 5′ end to block multimerization of the oligo during the ligation reactions, and may be ordered from IDT.
CRAC-RT primer: 5′-CAAGCAGAAGACGGCATAC-3′
CRAC-forward primer: 5′-AATGATACTGCGACCACCGACAGGTTCAGAGTTCTACAGTCCGA-3′.
CRAC -reverse primer: 5′-CAAGCAGAAGACGGCATACGA-3′
Start with beads from after step 119 of the Basic Protocol. Centrifuge 5 min at 1000 x g, +25°C, to pellet the streptavidin beads with bound crosslinked RNA-protein-DNA complexes.
Resuspend beads in 1ml TE buffer, transfer one half of the beads to a new 1.7ml maximum recovery tube. Each tube should have proximally 100μl of packed beads (200μl of initial 50% aqueous slurry). Process one of these tubes for DNA recovery, starting from step 120 of the Basic Protocol. Process the other as follows.
Resuspend beads in 1ml TE buffer, mix on a rotating wheel for 5 min at +25C.
Centrifuge 5 min at 1000 x g, +25°C, to pellet beads.
Repeat wash steps 3 and 4 two additional times with TE buffer, followed by 3 times with TN150 buffer.
Resuspend beads in 1ml TN150 buffer and split equally between 4 new 1.7ml maximum recovery tubes.
Add differing amounts of RNace-It Ribonuclease Cocktail to each tube. If the composition of RNA sizes is unknown, as a starting point, add 0, 0.1, 1, and 10 units.
-
Incubate 5min at +37°C.
Variation of the amount of Ribonuclease Cocktail in each reaction is required to partially digest the long RNA (more than 1000nt) and creates a broad size range of RNA fragments available for ligation in the following steps. Add 1.2ml of CRAC-Wash buffer 1 to each tube to stop the reaction.
Centrifuge 5 min at 1000 x g, +25°C, to pellet beads.
Resuspend beads with 250μl of CRAC-Wash buffer 1 per tube. Combine beads together in one tube for ~1ml total volume.
Wash beads twice (as in steps 3 and 4) with 1ml of CRAC-Wash buffer 1 and three times with 1ml of 1x CRAC-PNK buffer.
Pipet supernatant into waste. After discarding the final wash, briefly centrifuge tube again and remove remaining buffer.
-
Add 96μl of 5 x CRAC-PNK buffer, 48μl of TSAP, 12μl of RNasin and 324μl of Ultrapure water to the beads.
Alkaline phosphatase removes the 3′ phosphates left by the RNace-It cleavage. Incubate 30 minutes at 37°C
Wash beads (as in steps 3 and 4) one time with 1ml of CRAC-Wash buffer 1 and three times with 1ml of 1x CRAC-PNK buffer.
Pipet supernatant into waste. After discarding the final wash, briefly centrifuge tube again and remove remaining buffer.
-
Add 96μl of 5 x CRAC-PNK buffer, 20μl of 3′-BioTAP-Solexa linker (10 μM), 12μl of RNasin, 100μl of 50% PEG 8000, 228μl of Ultrapure water, and 24 μl of T4 RNA ligase 2 truncated K227Q to the beads
At this step, 3′ linker is ligated to the RNA. Incubate 12–16 hours at 16°C.
Repeat wash steps 16 and 17 to stop the reaction and remove free linker.
-
Add 96μl of 5 x CRAC-PNK buffer, 9μl of 10mM ATP, 12μl of RNasin, 339μl of Ultrapure water, and 24μl T4 PNK.
At this step 5′ ends of the RNA gets phosphorylated to allow for subsequent linker ligation. Incubate 1 hour at 37ºC.
Repeat wash steps 16 and 17 to stop the reaction.
-
Add 96μl of 5 x CRAC-PNK buffer, 48μl of 10mM ATP, 12μl of RNasin, and 284μl of Ultrapure water, 20μl of 5′-BioTAP-Solexa linker (10 μM) and 20μl T4 RNA ligase 1.
At this step, 5′ linker is ligated to the RNA. Incubate 12–16 hours at 16ºC.
Repeat wash steps 16 and 17 to stop the reaction and remove free linker.
Resuspend beads in 1ml of TEN 140 buffer with 0.1% Triton X-100 and mix on a rotating wheel for 10 min at +25C.
Centrifuge 5 min at 1000 x g, +4°C., to pellet beads Pipet supernatant into waste.
Repeat steps 27 and 28 two additional times, followed by one time with RIPA buffer
Recover RNA with ligated oligos by the following steps:
Resuspend beads from step 29 in 1ml TE buffer and transfer to 1.7ml maximum recovery tube.
Centrifuge 5 min at 1000 x g, +25°C, to collect beads.
Repeat steps 119 and 120 two additional times.
Resuspend the beads in 220μl TE. Add 18μl of 10% SDS and 40μl of Proteinase K (20mg/ml).
Incubate sample overnight at +37°C.
Transfer sample to +65°C and incubate 6h.
Add 9μl of 5M NaCl (140mM final) and mix by vortexing.
Add 300μl of phenol/chloroform/isoamyl alcohol to the samples and mix by vortexing for 30 sec.
Centrifuge 15 min at 20000 x g, +25°C.
Transfer 240μl of the aqueous (top) phase to a new 1.7ml maximum recovery tube.
To maximize RNA recovery, add 300μl of TEN 140 buffer back to the organic phase in the original tube to back extract additional material. Mix by vortexing for 30 sec and centrifuge 15 min at 20000 x g, +25°C.
Recover and combine the aqueous (top) phase with the the material from step 40 in one tube (~540μL total; first extraction 240μl and second 300μl).
-
Add one volume (540μl) of chloroform/isoamyl alcohol, mix by vortexing for 30 sec, and centrifuge 15 min at 20000 x g, +25°C.
A phase-lock tube may be used here to separate the organic phase from the aqueous phase. Transfer about 500μl of the upper aqueous phase into two new 1.7ml maximum recovery tube. Split samples evenly (250μl) between tubes.
Add 25μl of 3M NaAc pH 5.0, 10μl of glycogen (5mg/ml), and 1ml of ethanol (200 proof) to each tube, mix by vortexing for 30 sec, and re-collect by brief centrifugation.
-
Incubate at least 12 hours at −80°C.
RNA samples may be stored up to 1 month at −80°C, but we strongly recommend proceeding to step 47 as quickly as possible. Because of volume constraints, two tubes are needed for precipitation; however, proceed with both tubes to the next step. Remove DNA from the RNA samples by the following steps:
-
Centrifuge 30 min at 16000 x g, +4°C, to pellet RNA recovered after the purification. Pipet supernatant into waste.
A small white pellet should be visible at the bottom of the tube. Add 1ml of ice cold 75% ethanol to the pellet, mix briefly and centrifuge 30min at 16000 x g, +4°C. Pipet supernatant into waste.
Repeat step 49 once more. After discarding supernatant, briefly centrifuge tube again and remove residual ethanol.
Air-dry the pellet 15 min at +25°C.
Dissolve each pellet in 115μl Ultrapure water (RNase-free), and pool for 230μl total volume.
Add 10μl of SUPERase• In RNase Inhibitor (20 U/μl), 30μl of 10X TURBO DNase Buffer, and 30μl of TURBO DNase (2 U/μl). Mix gently by pipetting.
Incubate 30 min at +37°C.
To stop the reaction, add 20μl of Proteinase K (20mg/ml), 250μl TEN 140 buffer and 30μl 10% SDS. Mix by vortexing and centrifuge briefly.
Incubate 5 min at +37°C.
Add 600μl of phenol/chloroform/isoamyl alcohol to the samples and mix by vortexing for 30 sec.
Centrifuge 15 min at 20000 x g, +25°C.
Transfer 550μl of the aqueous (top) phase to a new 1.7ml maximum recovery tube.
-
Perform chloroform/isoamyl alcohol extraction and RNA precipitation as before (steps 43–46).
Because of volume constraints, two tubes are needed for precipitation; however, proceed with both tubes to the next step. After −80°C incubation, repeat steps 48–51.
Dissolve each pellet in 4μl Ultrapure water and pool for 8μl total volume.
Proceed with reverse transcription reaction by using SuperScript III First-Strand Synthesis kit, following manufacturer’s instructions. It is critical to use the CRAC-RT primer in this reaction. Use 1μl of 2μM stock of CRAC-RT primer for a 20μl RT-reaction.
-
Proceed with Solexa library PCR enrichment. Combine the following in a PCR tube:
10μl of the RT reaction,
26.5μl Ultrapure water,
10μl of 5xPhusion HF buffer,
1μl of each 10 μM CRAC-forward and -reverse primer,
1μl of 10mMdNTP, and
0.5μl of Phusion Hot Start II DNA Polymerase 2 U/μL.
-
Use the following PCR cycling condition:
-
Initial denaturation +98 ºC for 30sec; follow with
20–30 cycles: [+98 ºC for 10sec, [+58 ºC for 30sec, [+72 ºC for 30sec; -
Final extension cycle +72 ºC for 5min.
Number of PCR cycles needs to be carefully considered in order to avoid over amplification of the library.
-
Follow the TruSeq ChIP Sample Preparation Kit instructions (Illumina) for purification of PCR fragments ranging from 200bp to 900bp.
SUPPORT PROTOCOL 1: Peptide recovery from Input Chromatin
While great pains have been taken to ensure high stringency of interactions, affinity-purification after crosslinking may still recover certain proteins based solely on their abundance and general localization to chromatin in the proximity of the bait protein. There may also be other non-specific artifacts from the pulldown procedures. It is therefore important to proceed to candidate validation with caution.
As in any experiment, it is important to perform negative controls. Non-specific contaminants can be identified by performing the BioTAP-XL Basic Protocol using chromatin derived from the original untagged cell line. However, often these purifications result in very little or no protein recovery (reflecting the stringency of the washing steps) and many of these surviving contaminants are among the most abundant chromatin proteins (ie, histones) and other endogenously biotinylated proteins (which may be recovered because of the strength of the biotin-streptavidin interaction). Therefore, if multiple BioTAP-XL experiments have been performed with unrelated proteins, the results from such unrelated BioTAP-tagged chromatin proteins can be very useful to provide additional evidence for the specificity of the interactions with a particular bait of interest.
In addition to these negative controls, we have found that it is very informative to document the relative abundance of candidate interacting proteins in the input sample. The simple enrichment over input ratio is one of the most valuable parameters to identify candidate interactions with the highest specificity.
Here we describe a technique which allows recovery of proteins from the input samples. It is of paramount importance to remove SDS from the samples, as detergents can easily interfere with LC-MS/MS. In the Basic Protocol, SDS can be removed thoroughly from the IP sample when the RNA-protein-DNA complexes are bound to the beads: since the beads have gravity and can be recovered by centrifugation, SDS can be washed away. Since the input sample is entirely soluble, a different method is required for SDS removal. Following SDS removal, a portion of the input sample is treated with trypsin to generate peptides, and these peptides are processed for LC-MS/MS analysis according to the Basic Protocol, steps 98–118.
Materials
See also Basic Protocol
Pierce Detergent Removal Spin Columns 2ml (Thermo Scientific, cat. no. 87778)
Quick Start Bradford Protein Assay Kit 1 (Bio-Rad, cat. no. 500-0201EDU)
Input protein recovery
Thaw one 0.5 ml aliquot of the sheared chromatin (from Basic protocol, step 44).
In a new microcentrifuge tube, mix 100μl of input chromatin with 400μl of Ultrapure water.
Remove detergent by loading the 500μl sample to a 2ml Pierce Detergent Removal Spin Column. Follow the manufacturer’s instructions. It is critical to use 50 mM ammonium bicarbonate to wash and equilibrate columns.
Collect detergent-free samples as per the manufacturer’s instructions.
Quantify protein concentration using the Quick Start Bradford Protein Assay Kit 1, following manufacturer’s instructions, and using BSA as a reference.
Dilute a 5–10μg aliquot of the detergent-free sample up to 800μl with 50 mM ammonium bicarbonate.
Add 20μl of sequencing-grade Trypsin and mix the suspension on a rotating wheel for 12–16 hours at +37°C.
Recover and prepare the resulting peptides for LC-MS/MS analysis by following the Basic Protocol, steps 98–118.
SUPPORT PROTOCOL 2: DNA/RNA recovery from Input Chromatin
A ubiquitous analysis scheme for chromatin immunoprecipitation (ChIP) and RNA immunoprecipitation (RIP) methods is to compare the amount of immunoprecipitated nucleic acid to the amount found in a reference input sample. This controls for any potential underlying biases of the input chromatin (enrichment for open/active chromatin, copy-number differences, etc) which may arise from processing techniques in generating chromatin or from biological variation. Similarly it is important to isolate the DNA and RNA content from the starting input chromatin in the BioTAP-XL technique to help interpret the immunoprecipitation data.
The isolation of DNA from the input sample also reflects the extent of chromatin sonication. A typical sonication aims for fragmentation of the input chromatin to 300–3000bp. It is advisable to check that the chromatin is fragmented to this size by running a small aliquot of DNA (after step 15) on an agarose gel and visualizing with a DNA stain.
The steps presented here describe how to isolate nucleic acids from the input sample, and then more specifically how to process DNA and RNA samples separately for applications such as (reverse transcription)-quantitative real-time PCR (RT-qPCR or qPCR) or high-throughput sequencing. The final DNA or RNA isolated in this protocol may be processed in parallel to the material purified through the Basic Protocol for high-throughput sequencing library construction.
Materials
See also Basic Protocol
NanoDrop ND-1000
Input DNA and RNA isolation
-
1
Thaw one 0.5 ml aliquot of the sheared chromatin (from Basic protocol, step 44).
-
2
Add 40μl of Proteinase K (20mg/ml), 30μl of 10% SDS and 30μl RIPA buffer. Mix by vortexing and centrifuge briefly.
-
3
Incubate sample overnight at +37°C.
-
4
Transfer sample to +65°C and incubate 6h.
-
5
Add 600μl of phenol/chloroform/isoamyl alcohol to the sample, mix by vortexing for 30 sec, and centrifuge 15 min at 20000 x g, +25°C.
-
6
Transfer 550μl of aqueous (top) phase to a new 1.7ml maximum recovery tube.
-
7
Add 550μl of chloroform/isoamyl alcohol and mix by vortexing for 30 sec.
-
8
Centrifuge 5 min at 20000 x g, +25°C.
-
9
Transfer about 500μl of the aqueous (top) phase into two new 1.7ml maximum recovery tube. Split samples evenly (250μl) between tubes.
-
10
Add 25μl of 3M NaAc pH 5.0, 10μl of glycogen (5mg/ml), and 1ml of ethanol (200 proof) to each tube. Mix by vortexing for 30 sec, and centrifuge briefly.
-
11
Incubate at least 12 hours at −80°C.
One tube is for DNA recovery (step 12) and the other is for RNA recovery (step 26).DNA and RNA samples may be stored up to 1 month at −80°C.For RNA samples we strongly recommend proceed to step 26 as quickly as possible.
Input DNA preparation for qPCR or Solexa library construction
-
12
Centrifuge 30 min at 16000 x g, +4°C, to pellet DNA recovered after the purification. Pipet supernatant into waste.
A small white pellet should be visible at the bottom of the tube. -
13
Add 1ml of ice cold 70% ethanol to the pellet, mix briefly and centrifuge 1 hour at 16000 x g, +4°C. Pipet supernatant into waste. Briefly centrifuge tube again and remove residual ethanol.
-
14
Air-dry the pellet 15min at +25°C.
-
15
Dissolve the pellet in 100μl Ultrapure water.
-
16
Quantify the concentration by UV absorbance using 2μl of input DNA and a NanoDrop ND-1000.
Follow instrument manufacturer’s instructions for accurate quantification. -
17
Dilute a 20μg aliquot of input DNA up to 267μl with Ultrapure water.
-
18
Add 30μl of 10X CutSmart Buffer (New England BioLabs Inc) and 3μl of 10mg/ml DNase-free RNaseA (Roche). Mix by vortexing and centrifuge briefly.
-
19
Incubate 30min at +37°C.
-
20
To stop the reaction, add 20μl of Proteinase K (20mg/ml), 250μl TEN 140 buffer and 30μl 10% SDS. Mix by vortexing and centrifuge briefly.
-
21
Incubate 5min at +37°C.
-
22
Perform phenol/chloroform/isoamyl alcohol extraction as above (steps 5–11).
Due to volume requirements, two tubes are needed for precipitation; proceed with both of them to step 23. -
23
After incubation at −80°C, repeat steps 12–14 to recover the final DNA pellets.
-
24
Dissolve each pellet in 10μl Ultrapure water, and pool for 20μl total volume.
-
25
Quantify the concentration using 2μl of input DNA and a NanoDrop ND-1000.
Follow instrument manufacturer’s instructions for accurate quantification.Input DNA is now ready for Quantitative Real-Time PCR (qPCR) applications or ChIP- Seq Solexa/Illumina library preparation. We recommend using NEBNext ChIP-Seq Library Prep Master Mix Set for Illumina, following manufacturer’s instructions, for high-throughput sequencing methods.
Input RNA preparation for qPCR or Solexa library construction
-
26
Centrifuge 30 min at 16000 x g, +4°C, to pellet DNA recovered after the purification. Pipet supernatant into waste. A small white pellet should be visible at the bottom of the tube.
-
27
Add 1ml of ice cold 70% ethanol to the pellet, mix briefly and centrifuge 1 hour at 16000 x g, +4°C. Pipet supernatant into waste. Briefly centrifuge tube again and remove residual ethanol.
-
28
Air-dry the pellet 15min at +25°C.
-
29
Dissolve the pellet in 100μl Ultrapure water.
-
30
Quantify the concentration by UV absorbance using 2μl of input RNA and a NanoDrop ND-1000.
Follow instrument manufacturer’s instructions for accurate quantification. -
31
Dilute a 20μg aliquot of input RNA up to 230μl with Ultrapure water.
-
32
Add 10μl of SUPERase• In RNase Inhibitor (20 U/μl), 30μl of 10X TURBO DNase Buffer and 30μl of TURBO DNase (2 U/μl). Mix gently by pipetting.
-
33
Incubate 30min at +37°C.
-
34
To stop the reaction, add 20μl of Proteinase K (20mg/ml), 250μl TEN 140 buffer and 30μl 10% SDS. Mix by vortexing and centrifuge briefly.
-
35
Incubate 5min at +37°C.
-
36
Perform phenol/chloroform/isoamyl alcohol extraction as above (steps 5–11).
Due to volume requirements, two tubes are needed for precipitation; proceed with both of them to step 37. -
37
After incubation at −80°C, repeat steps 26–28 to recover the RNA pellet.
-
38
Dissolve each pellet in 115μl Ultrapure water, and pool for 230μl total volume.
-
39
Repeat DNase digestion steps 32–37 once more.
-
40
Dissolve each final RNA pellet in 10μl Ultrapure water, and pool for 20μl total volume.
-
41
Quantify the concentration using 2μl of input RNA and a NanoDrop ND-1000.
Follow instrument manufacturer’s instructions for accurate quantification.Input RNA is now ready for Reverse Transcription Quantitative Real-Time PCR (RT- qPCR) applications or Solexa/Illumina RNA library preparation. We recommend using NEBNext Ultra Directional RNA Library Prep Kit for Illumina, following manufacturer’s instructions, for high-throughput sequencing methods.If residual DNA contamination in the RNA sample is a major concern, repeat steps 32–37 once more.
REAGENTS AND SOLUTIONS
Nuclear extraction buffer (NEB)
20mM HEPES pH7.6 (Boston BioProducts, cat. no. BBH-76)
10% Sucrose (MP Biomedicals, cat. no. 821713)
10mM NaCl (Boston BioProducts, cat. no. BM-244)
3mM MgCl2 (Boston BioProducts, cat. no. MT-200)
0.2% Triton (Roche, cat. no.10789704001)
Store 3 to 6 months at +4°C
NSucrose buffer
10 mM HEPES pH8.0 (Boston BioProducts, cat. no. BBH-80)
300 mM sucrose (MP Biomedicals, cat. no. 821713)
1% Triton X-100 (Roche, cat. no. 10789704001)
2 mM MgOAc (Sigma, cat. no. M5661-250G)
Store 3 to 6 months at +4°C
NGlycerol buffer
10 mM HEPES pH8.0 (Boston BioProducts, cat. no. BBH-80)
25% Glycerol (Fisher Scientific, cat. no. G33-1)
0.1 mM EGTA (Boston BioProducts, cat. no. BM-151)
5 mM MgOAc (Sigma, cat. no. M5661-250G)
Store 3 to 6 months at +4°C
RIPA buffer
140 mM NaCl (Boston BioProducts, cat. no. BM-244)
10 mM Tris-HCl pH8.0 (Boston BioProducts, cat. no. BM-320)
1 mM EDTA pH8.0 (Boston BioProducts, cat. no. BM-150)
1% Triton X-100 (Roche, cat. no. 10789704001)
0.1% SDS (Sigma, cat. no. L4522-100ML)
Store 3 to 6 months at +4°C
TEN 140 buffer
10 mM Tris-HCl pH8.0 (Boston BioProducts, cat. no. BM-320)
1 mM EDTA pH8.0 (Boston BioProducts, cat. no. BM-150)
140 mM NaCl (Boston BioProducts, cat. no. BM-244)
Store 6 to 12 months at +4°C
TE buffer
10 mM Tris-HCl pH8.0 (Boston BioProducts, cat. no. BM-320)
1 mM EDTA pH8.0 (Boston BioProducts, cat. no. BM-150)
Store 6 to 12 months at +4°C
IgG Elution buffer
100 mM Tris-HCl pH8.0 (Boston BioProducts, cat. no. BM-320)
200 mM NaCl (Boston BioProducts, cat. no. BM-244)
6 M urea (Invitrogen, cat. no.15505-035)
0.2% SDS (Sigma, cat. no. L4522-100ML)
Made fresh on day of use
Reverse Crosslinking buffer
250 mM Tris-HCl pH8.8 (Boston BioProducts, cat. no. BM-321)
2% SDS (Sigma, cat. no. L4522-100ML)
0.5 M beta-mercaptoethanol (Sigma, cat. no. M7522)
Made fresh on day of use
TN150 buffer
50 mM Tris-HCl pH 7.8 (Boston BioProducts, cat. no. BM-318)
150 mM NaCl (Boston BioProducts, cat. no. BM-244)
0.1% NP-40 (Roche, cat. no. 11754599001)
5 mM beta-mercaptoethanol (Sigma, cat. no. M7522)
Store 6 to 12 months at +4°C
Beta-mercaptoethanol, add fresh on day of use.
CRAC-Wash buffer 1
50 mM Tris-HCl pH 7.8 (Boston BioProducts, cat. no. BM-318)
300 mM NaCl (Boston BioProducts, cat. no. BM-244)
10 mM Imidazole (Sigma, cat. no. I5513)
6M Guanidine-HCl (Sigma, cat. no. G3272)
0.1% NP-40 (Roche, cat. no. 11754599001)
5 mM beta-mercaptoethanol (Sigma, cat. no. M7522)
Store 1 to 2 months at +25°C
Beta-mercaptoethanol, add fresh on day of use.
1 x CRAC-PNK buffer
50 mM Tris-HCl pH 7.8 (Boston BioProducts, cat. no. BM-318)
10 mM MgCl2 (Boston BioProducts, cat. no. MT-200)
0.1% NP-40 (Roche, cat. no. 11754599001)
5 mM beta-mercaptoethanol (Sigma, cat. no. M7522)
Store 6 to 12 months at +4°C
Beta-mercaptoethanol, add fresh on day of use.
5 x CRAC-PNK buffer
250 mM Tris-HCl pH 7.8 (Boston BioProducts, cat. no. BM-318)
50 mM MgCl2 (Boston BioProducts, cat. no. MT-200)
50 mM beta-mercaptoethanol (Sigma, cat. no. M7522)
Store 6 to 12 months at −20°C
Beta-mercaptoethanol, add fresh on day of use.
HPLC Buffer A
2.5% (vol:vol) Acetonitrile
0.1% Formic acid
97.4% Water
Store up to 1 month at room temperature, protect from light
HPLC Buffer B
97.5% (vol:vol) Acetonitrile
0.1% Formic acid
2.4% Water
Store up to 1 month at room temperature, protect from light
Molasses agar plate (1L)
28g Agar (Genesee Scientific, cat. no. 66-103)
140ml molasses (Genesee Scientific, cat. no. 62-117)
Store up to 1 month at +4°C
Warm to room temperature before use
Embryo wash solution
0.7% NaCl (Fisher cat. no. S271)
0.03% Triton-X100 (Roche, cat. no. 10789704001)
Make fresh
COMMENTARY
Background Information
By tagging multiple unrelated bait proteins in Drosophila and human cell culture, different chromatin complexes have been studied using BioTAP-XL. In each case, previously known protein subunits as well as novel, yet functionally relevant interactors were identified (Alekseyenko et al., 2014a, Alekseyenko et al., 2014b). Similarly, the DNA targeting of the tagged bait was resolved, and agreed strongly with previous experiments. In several cases, ncRNA was also discovered as a specific part of the complex (Alekseyenko et al., 2014b).
Comparison with other methods
The process of purifying chromatin bound complexes presents numerous biochemical challenges. Solutions to these challenges have been addressed previously in various ways. These other methods typically rely on light treatment of chromatin so as not to disrupt existing interactions and/or only mild washing steps.
One such approach, developed in yeast, is to employ light sonication and to wash solubilized chromatin under very mild conditions in order to preserve protein interactions as extensively as possible (Lambert et al., 2010, Lambert et al., 2009). Another approach in yeast is to add light cross-linking prior to sonication and washing under mild conditions (Smart et al., 2009). Recently, an approach for systematic analysis of endogenous protein-protein interactions and protein-DNA binding events was developed (Mohammed et al., 2013). Named rapid immunoprecipitation mass spectrometry of endogenous proteins (RIME), it combines several methods including formaldehyde crosslinking and on-bead digestion purification of endogenous-interacting proteins. The key to successful implementation of this method is having a high quality ChIP-grade antibody to the desired target protein for pulldown.
Another solution, in which the initial idea of robust cross-linking is used to capture protein-protein interactions in the cytoplasm (Guerrero et al., 2006, Tagwerker et al., 2006) was adapted to study chromatin complexes (Tardiff et al., 2007, Wang et al., 2013). A key aspect of this approach is the use of a 75–amino acid sequence from bacteria as an affinity tag which is recognized by endogenous biotinylation enzymes in yeast, flies and humans.
It was previously shown in our lab that one-step affinity-purification using this tag (ChIP-MS) was sufficient to define MSL3 protein interactions in S2 tissue culture cells (Wang et al., 2013). In the course of this study, it was noted that the one-step purification was less effective for more heterogenous samples such as fly embryos due to high levels of contamination from endogenous biotinylated proteins. This led to the development of the two-step purification in the BioTAP-XL procedure (Alekseyenko et al., 2014a). With the modifications to the ChIP-MS protocol to eliminate the problem of endogenously biotinylated protein contamination, it was discovered that, in addition to the higher quality protein recovery results, it was now possible to isolate RNA subunits in the complex and to map the genomic localization (DNA targets).
Critical Parameters
There are several critical parameters for a successful BioTAP-XL experiment.
Initial Cellular Lysis
While typical chromatin-immunoprecipitation experiments do not perform a cell lysis prior to crosslinking, breaking open the cells to obtain the nuclei is important for this procedure. The initial cell lysis and crude nuclear isolation prevents the crosslinking of cytoplasmic proteins which would interfere with subsequent lysis steps and create a higher background signal of contaminants. Furthermore, only starting the crosslinking with crude nuclear extract allows for more extensive crosslinking of the chromatin to be established. Especially in Drosophila embryos, delaying the crosslinking until after the embryos are disrupted greatly improves the amount of soluble chromatin recovered.
Crosslinking concentration, duration, and temperature
The BioTAP-XL procedure requires the chromatin to be stably crosslinked, such that interactions may survive the stringent wash conditions. For this reason, extensive crosslinking of the nuclear material is necessary. A final concentration of formaldehyde of 3% has been highly effective. Incubating at room temperature for 30 minutes allows for the establishment of extensive chemical crosslinks. Reduction of formaldehyde concentration, crosslink duration, and/or incubation temperature may result in insufficiently crosslinked chromatin which may result in less protein and DNA recovered.
Input and untagged controls
An understanding of the protein composition of the input sample is an important parameter for analysis of protein recovery by BioTAP-XL (see Support Protocol 1). Typically one input control per parental (no BioTAP construct) cell line or source tissue will suffice. The authors have found little variation between the parental cell line and BioTAP-expressing lines in terms of input protein composition. As with standard ChIP/RIP experiments, DNA and RNA from input are important to control for the biases introduced due to variability of starting material (see Support Protocol 2).
An untagged control is also an important parameter, as this assesses the background of a particular cell type. Typically one untagged control per parental (no BioTAP construct) cell line or source tissue will suffice.
Replicates
The BioTAP-XL procedure has been highly consistent across biological replicates and across tissues. Replicates may either be different isolates using the same construct, or different isolates using the same bait protein with the BioTAP tag attached to the opposite ends of the protein (ie, amino vs carboxy terminal). Technical replicates of LC-MS/MS may be favorable to explore the variability of mass spectrometry runs, but typically will return nearly identical profiles.
Troubleshooting
| Problem | Potential cause | Solution |
|---|---|---|
| Poor quality LC-MS run | Problem with Sample or with HPLC column | Run a peptide standard; if standard = poor, replace HPLC if standard = fine, problem with sample |
| Poor electrospray stability or overall low signal intensity | Insufficient desalting | More washes of sample during peptide recovery |
| Sudden increase in HPLC pressure | Particulate debris in the sample | Replace HPLC column, lines, junctions to autosampler, and/or the rotor and stator of the autosampler valve; Centrifuge peptide sample at 16000 x g, transfer top 90% of volume |
| Poor chromatography – fronting peaks (peak skews to earlier retention time) | Column capacity overloaded due to overly concentrated peptide sample | Dilute sample |
| Poor chromatography – tailing peaks (peak skews to later retention time) | Residual reactive silanol groups in the stationary phase, or improper formulation of the mobile phases (pH) | Replace HPLC column and/or HPLC buffers |
Anticipated Results
Identification of interacting proteins
The peptides isolated by the BioTAP-XL procedure and identified by LC-MS/MS should be compared to peptides from input and from an untagged experiment (see Table 1). Generally, the proteins will fall into two classes: (1) those observed to be abundant in the BioTAP-XL sample and absent from input, and (2) those observed to have slight nor no enrichment of peptide counts in the BioTAP-XL sample compared to input. The Group 1 proteins are obvious candidates for follow-up. Group 2 candidates may be highly abundant in the BioTAP-XL sample, but given their similar levels in input, may not be the strongest candidates for followup.
Table 1.
Representative results for a BioTAP-XL experiment. Peptide counts for top interactions from EZH2 pulldowns from human 293T-REx cells are listed along with peptide counts recovered in two input replicates, two mock-BioTAP-XL replicates, and from a CBX4-BioTAP experiment for comparison. Ezh2 and CBX4 are subunits in two related, but distinct, chromatin complexes. Group 1 candidates are most highly enriched over input, making them strong candidates for follow-up, whereas Group 2 candidates are abundant in both the pulldown and input. Adapted from (Alekseyenko et al., 2014a).
| Ezh2 | Input | Mock | CBX4 | ||||
|---|---|---|---|---|---|---|---|
| Group 1 | C | N | R1 | R2 | R1 | R2 | C |
| SUZ12 | 124 (26) | 102 (26) | 0 | 0 | 0 | 0 | 2 (2) |
| EED | 81 (14) | 69 (18) | 0 | 0 | 0 | 0 | 0 |
| MTF2 | 75 (22) | 85 (28) | 0 | 0 | 0 | 0 | 0 |
| EZH2-bait | 103(13) | 105(20) | 0 | 1 | 0 | 0 | 0 |
| C10ORF12 | 66 (27) | 64 (30) | 0 | 0 | 0 | 0 | 0 |
| JARID2 | 40 (18) | 52 (27) | 0 | 0 | 0 | 0 | 0 |
| PHF1 | 21 (10) | 24 (14) | 0 | 0 | 0 | 0 | 0 |
| C17ORF96 | 15 (7) | 18 (10) | 0 | 0 | 0 | 0 | 0 |
| AEBP2 | 9 (6) | 16 (9) | 0 | 0 | 0 | 0 | 0 |
| PHF19 | 8 (6) | 12 (10) | 0 | 0 | 0 | 0 | 0 |
| RBBP7 | 16 (4) | 16 (8) | 0 | 1 | 0 | 0 | 5 (2) |
| SKIDA1 | 4 (3) | 4 (4) | 0 | 0 | 0 | 0 | 0 |
| LCOR | 3 (3) | 4 (3) | 0 | 0 | 0 | 0 | 2 (2) |
| RBBP4 | 10 (7) | 12 (5) | 2 (1) | 3 (2) | 0 | 0 | 2 (2) |
| SCML2 | 2 (2) | 3 (3) | 0 | 0 | 0 | 0 | 57 (18) |
| Group 2 | |||||||
| PARP1 | 31 (18) | 51 (30) | 46 (26) | 43 (21) | 0 | 0 | 66 (34) |
| TOP2A | 11 (8) | 16 (14) | 16 (12) | 20 (13) | 0 | 0 | 34 (26) |
| SMC1A | 4 (4) | 13 (12) | 8 (7) | 8 (8) | 0 | 0 | 18 (17) |
| SSRP1 | 8 (8) | 8 (7) | 10 (8) | 10 (7) | 0 | 0 | 14 (11) |
| H2AFY | 14 (7) | 19 (1) | 24 (9) | 16 (8) | 0 | 0 | 9 (6) |
| TOP1 | 5 (4) | 10 (10) | 10 (5) | 8 (6) | 0 | 0 | 8 (5) |
| MKI67 | 13 (12) | 25 (24) | 38 (31) | 30 (24) | 0 | 0 | 33 (30) |
Abbreviations: C = carboxy-terminal BioTAP tag; N = amino-terminal BioTAP tag; R = replicate. Peptide counts are listed in the following format: Total (Unique).
Identification of genomic targets
Using the DNA recovered from the BioTAP-XL procedure, two main avenues of analysis are available. For small-scale analysis of known sites, qPCR may be used. For genome wide binding profiles, high-throughput sequencing may be used. Data analysis may be performed using standard methods, and binding profiles may be visualized with a genome browser of choice (see Figure 3).
Figure 3.
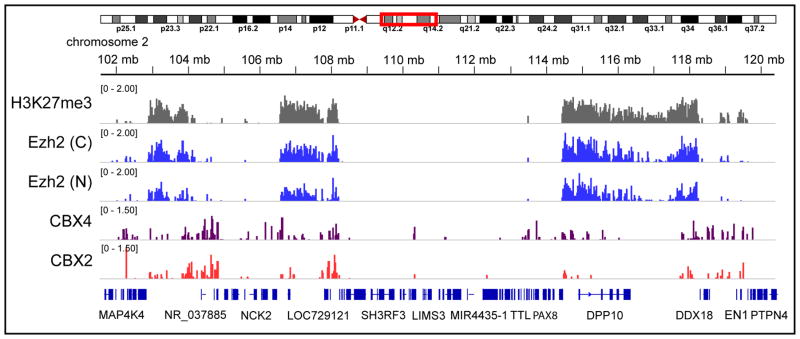
Representative results for a BioTAP-XL experiment. ChIP-seq profiles for BioTAP-tagged Ezh2 (both carboxy- [C] and amino- [N] terminal tagged constructs) from human 293T-REx cells exhibit consistent binding patterns and also track with the PRC2-associated H3K27me3 binding profile. BioTAP-tagged CBX4 and Flag-His-tagged CBX2 display patterns distinct from the PRC2 profiles. Figure from (Alekseyenko et al., 2014a), with H3K27me3 and Flag-His-CBX2 profiles from (Gao et al., 2012). Ezh2 is the catalytic subunit of PRC2 (Polycomb Repressive Complex 2); CBX2 and CBX4 are interchangeable subunits in a related, but distinct complex, PRC1.
Identification of interacting RNAs
Similarly, RNA samples may be interrogated using RT-qPCR or high-throughput sequencing, with subsequent standard analyses.
Time Considerations
The BioTAP-XL protocol takes about 5 days once the proper number of cells are obtained. The first day consists of cell lysis, crosslinking, and extract preparation. The second and third are tandem-affinity purification steps. The fourth and fifth are preparation of peptide samples for LC-MS/MS mass spectrometry and/or library samples for high-throughput sequencing. There is a natural stopping point after nuclei are crosslinked and processed into glycerol buffer. Once committed to sonication, the samples should be processed rapidly through the protocol to peptides or nucleic acid ethanol precipitation. DNA or RNA may be stored during the ethanol precipitation step at −80°C for up to a month and final processing of these samples performed later.
Figure 1.
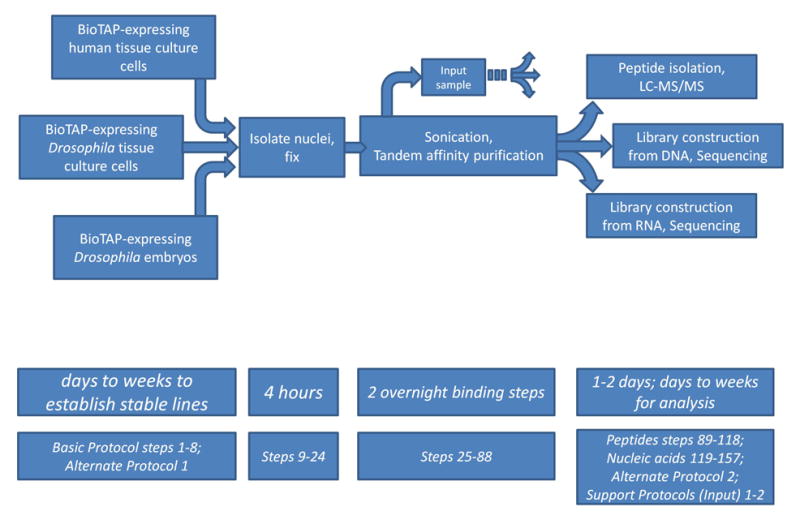
Strategic Planning for BioTAP-XL experiments. This flowchart outlines sample generation and processing stages along with their associated Basic Protocol steps and/or Support or Alternative protocols, and time considerations. Different source materials are processed with the same Basic Protocol. A successful BioTAP-XL experiment takes approximately 5 days of benchwork. There is an optional stopping point after fixing the crude nuclear extract.
Acknowledgments
We thank Ross Tomaino (Taplin Mass Spectrometry Facility, Harvard Medical School) for helpful advice regarding mass spectrometry; Stephen Elledge, Alberto Ciccia, and Natasha Pavlova for sharing reagents and advice regarding human tissue culture; and Peter Kaiser for reagents. Our work on chromatin complexes is supported by NIH grants GM045744 and GM101958.
Footnotes
INTERNET RESOURCES
For an example of a more statistically rigorous evaluation of enrichment of BioTAP-XL pulldowns, see the Bamse method provided at the URL below. Briefly, this approach employs statistical methods that control for multiple sources of bias, including biological replicate variability (IPs and inputs), differential base-level abundance of proteins in different cell types (inputs), variability in peptide ionization or detection rates (technical), and nonspecific effects of pull-down procedures. http://pklab.med.harvard.edu/mass.spec/viewms.html
LITERATURE CITED
- Alekseyenko AA, Gorchakov AA, Kharchenko PV, Kuroda MI. Reciprocal interactions of human C10orf12 and C17orf96 with PRC2 revealed by BioTAP-XL cross-linking and affinity purification. Proc Natl Acad Sci U S A. 2014a;111:2488–2493. doi: 10.1073/pnas.1400648111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseyenko AA, Gorchakov AA, Zee BM, Fuchs SM, Kharchenko PV, Kuroda MI. Heterochromatin-associated interactions of Drosophila HP1a with dADD1, HIPP1, and repetitive RNAs. Genes Dev. 2014b;28:1445–1460. doi: 10.1101/gad.241950.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Zhang J, Bonasio R, Strino F, Sawai A, Parisi F, Kluger Y, Reinberg D. PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol Cell. 2012;45:344–356. doi: 10.1016/j.molcel.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granneman S, Kudla G, Petfalski E, Tollervey D. Identification of protein binding sites on U3 snoRNA and pre-rRNA by UV cross-linking and high-throughput analysis of cDNAs. Proc Natl Acad Sci U S A. 2009;106:9613–9618. doi: 10.1073/pnas.0901997106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero C, Tagwerker C, Kaiser P, Huang L. An integrated mass spectrometry-based proteomic approach: quantitative analysis of tandem affinity-purified in vivo cross-linked protein complexes (QTAX) to decipher the 26 S proteasome-interacting network. Mol Cell Proteomics. 2006;5:366–378. doi: 10.1074/mcp.M500303-MCP200. [DOI] [PubMed] [Google Scholar]
- Lambert JP, Fillingham J, Siahbazi M, Greenblatt J, Baetz K, Figeys D. Defining the budding yeast chromatin-associated interactome. Mol Syst Biol. 2010;6:448. doi: 10.1038/msb.2010.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JP, Mitchell L, Rudner A, Baetz K, Figeys D. A novel proteomics approach for the discovery of chromatin-associated protein networks. Mol Cell Proteomics. 2009;8:870–882. doi: 10.1074/mcp.M800447-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed H, D’Santos C, Serandour AA, Ali HR, Brown GD, Atkins A, Rueda OM, Holmes KA, Theodorou V, Robinson JL, Zwart W, Saadi A, Ross-Innes CS, Chin SF, Menon S, Stingl J, Palmieri C, Caldas C, Carroll JS. Endogenous purification reveals GREB1 as a key estrogen receptor regulatory factor. Cell Rep. 2013;3:342–349. doi: 10.1016/j.celrep.2013.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Seraphin B. A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol. 1999;17:1030–1032. doi: 10.1038/13732. [DOI] [PubMed] [Google Scholar]
- Smart SK, Mackintosh SG, Edmondson RD, Taverna SD, Tackett AJ. Mapping the local protein interactome of the NuA3 histone acetyltransferase. Protein Sci. 2009;18:1987–1997. doi: 10.1002/pro.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagwerker C, Flick K, Cui M, Guerrero C, Dou Y, Auer B, Baldi P, Huang L, Kaiser P. A tandem affinity tag for two-step purification under fully denaturing conditions: application in ubiquitin profiling and protein complex identification combined with in vivocross-linking. Mol Cell Proteomics. 2006;5:737–748. doi: 10.1074/mcp.M500368-MCP200. [DOI] [PubMed] [Google Scholar]
- Tardiff DF, Abruzzi KC, Rosbash M. Protein characterization of Saccharomyces cerevisiae RNA polymerase II after in vivo cross-linking. Proc Natl Acad Sci U S A. 2007;104:19948–19953. doi: 10.1073/pnas.0710179104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venken KJ, He Y, Hoskins RA, Bellen HJ. P[acman]: a BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science. 2006;314:1747–1751. doi: 10.1126/science.1134426. [DOI] [PubMed] [Google Scholar]
- Wang CI, Alekseyenko AA, LeRoy G, Elia AE, Gorchakov AA, Britton LM, Elledge SJ, Kharchenko PV, Garcia BA, Kuroda MI. Chromatin proteins captured by ChIP-mass spectrometry are linked to dosage compensation in Drosophila. Nat Struct Mol Biol. 2013;20:202–209. doi: 10.1038/nsmb.2477. [DOI] [PMC free article] [PubMed] [Google Scholar]