

Summary
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder characterized by progressive motor neuron degeneration. The disease pathogenesis is multifaceted in that multiple cellular and molecular pathways have been identified as contributors to the disease progression. Consequently, numerous therapeutic targets have been pursued for clinical development, unfortunately with little success. The recent discovery of mutations in RNA modulating genes such as TARDBP/TDP-43, FUS/TLS or C9ORF72 changed our understanding of neurodegenerative mechanisms in ALS and introduced the role of dysfunctional RNA processing as a significant contributor to disease pathogenesis. This article discusses the latest findings on such RNA toxicity pathways in ALS and potential novel therapeutic approaches.
Keywords: ALS, ALS therapeutics, C9ORF72, repeat expansion, RNA toxicity, TDP-43
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig's disease, is a chronic, incurable and invariably fatal neurodegenerative disease with an annual worldwide incidence of 2–5 per 100,000 people [1,2]. While ALS is rather heterogeneous with regards to age and anatomic site of onset, it is clinically uniformly characterized by the progressive degeneration of motor neurons and interneurons in the brain and spinal cord. This loss leads to continuous diminished motor function such as limb weakness. Most patients die within 3–5 years after diagnosis due to the loss of respiratory muscle capacity [2]. ALS can occur sporadically, without any family history (sALS; 90–95% of patients), while a small percentage of ALS cases are considered familial or hereditary (fALS; 5–10%) [2]. Due to increased genome wide sequencing projects, more mutations are being discovered, adding more heterogeneity to the upstream disease mechanisms. Until recently, the most common genetic mutations of fALS (<20%) were mutations found in SOD1 [3]. Recently, a hexanucleotide repeat expansion in the noncoding region of C9ORF72 has been identified as the cause of approximately 36–46% of familial and up to 8% of sporadic ALS cases (combining Europe and the US), making this the most common genetic cause of ALS to date while also raising questions about the classification of sporadic ALS patients [4–6]. Additional important genetic causes of rare subsets of fALS include mutations in TARDBP/TDP-43 and FUS/TLS [7,8]. Interestingly, these recently discovered mutations are found in DNA/RNA regulation proteins, providing for the first time strong evidence for the existence of a dysregulation of RNA processing in ALS. Aberrant RNA metabolism is known to induce abnormal RNA splicing and editing as well as dysfunctional RNA transport, translation and miRNA biogenesis. This dysfunctional RNA processing has been ascribed to other neurological disorders in the past [9–12], but has only recently come to the forefront of thorough molecular and cellular scientific investigation of ALS pathogenesis. With increased understanding of the specific mechanisms of dysfunctional RNA processing, novel therapeutic targets are being searched for. At the same time, pharmacodynamic biomarker assays aimed to validate drug-target activity are being explored with the hope of better success for future clinical trials. This article highlights the difficulties of past therapeutic targets and failed clinical trials in ALS and will speculate on the potential of targeting different steps along the RNA-mediated disease pathways.
Past & current therapeutic trials for ALS
The only approved drug for ALS to date is Riluzole, which has been on the market since 1995 [13,14]. The mechanism of action for Riluzole is through antiglutamatergic pathways, ranging from blocking presynaptic glutamate release to increasing glutamate uptake via astrocytic glutamate transporters [15]. Despite numerous clinical trials over the last two decades testing over 30 different therapeutic agents, Riluzole remains the only approved therapeutic providing neuro-protection and subsequent increased survival (<3–6 months; see reviews [16–18]).
Many explanations can be given to address the lack of better ALS therapeutics, and one that seems to be discussed frequently is the lack of an appropriate preclinical animal model to test potential new drugs that have been discovered over the last 20+ years. The most common animal model used to date for preclinical testing of ALS therapies is the SOD1mut mouse, an mouse model, which overexpresses the human SOD1 gene with varying mutations [19]. Mutations in SOD1 were the first genetic mutations to be identified in a small percentage of fALS patients (<20%) over 20 years ago. Based on this discovery, transgenic mice overexpressing the human mutated SOD1 protein have been created and closely resemble ALS disease symptoms, including loss of motor neuron function and progressive paralysis, followed by diminished pulmonary function and eventual death within 150–300 days, depending on the specific mutation [19,20].
While this animal model has been extremely helpful in studying ALS disease mechanisms, it has not been very predictive of the efficacy of drug candidates. Independent of the therapeutic target, almost every compound that tested positive in this animal model and which was subsequently advanced toward clinical trials, failed to prove efficacy in ALS patients [16,17]. The pathways targeted in these trials include glutamate excitotoxicity, mitochondrial dysfunction, free radical oxidative injury, immunomodulatory dysfunction (including neuroinflammation and cytokine activity) and protein aggregation, all of which were identified using the SOD1mut mouse models. While most of these mechanisms were subsequently confirmed in human postmortem patient autopsy tissue, the question remains whether any mechanisms identified using this specific mutation-based animal model are representative for the non-SOD1mut ALS patient populations, which generally represent a large percentage of patients participating in ALS clinical trials. Perhaps stratifying the patient population and selecting only mutant SOD1 ALS patients for these trials would have provided more positive outcomes? Perhaps designing more rigorous pre-clinical trials where compounds are only tested when animals start showing disease symptoms, similar to the real patient situation, would give more accurate and predictive datasets on drug efficacy? Given the multifaceted pathogenesis of ALS, perhaps combinatorial drug treatments should be considered instead of targeting individual disease pathways? Alternatively, with the technological advancements of patient-derived induced pluripotent stem cell (iPSC) culture systems one could speculate whether there still is a need of a preclinical animal model at all. Maybe the use of patient- and/or mutation-specific iPSC cultures to test drug candidates provides a more promising and more predictive readout for decision making with regards to the advancement of a novel compound into the clinic. This approach also supports the growing interest in a personalized medicine approach for CNS disorders, similar to what has been developed and successfully implemented for cancer patients (e.g., see reviews [21,22]). Whether the dynamics of ALS disease progression will influence the choice of certain drug candidate is still not fully explored, but should be considered particularly for the treatment of familial ALS patients with a known genetic mutation, where early intervention before disease onset is of great interest.
Finally, no matter which way a drug has been validated preclinically, the need for biomarkers to accompany every trial has finally been recognized and efforts are ongoing to develop pharmacodynamic biomarker assays in conjunction with every newly developed drug candidate. A biomarker measures the pharmacological or phenotypic target response to a therapeutic intervention, independent of a primary outcome measure such as increased survival rate. Biomarker readouts can be obtained very early after initiation of drug treatment and can help select patients who respond positively to the therapy while removing nonresponders early on from the trial. This would make nonresponders available to other trials with the hope of a better response.
Mutations of RNA processing proteins & toxic RNAs in ALS
RNA-mediated toxicity has been gaining increasing importance in contributing to the pathogenesis of neurological diseases. This can be observed in several repeat expansion disorders such as spinocerebellar ataxia or Huntington's disease but also in diseases in which RNA-binding proteins (RBPs) such as SMN, TDP-43 or FUS/TLS are mutated and consequently lead to dysfunctional RNA processing [9]. Aberrant RNA processing has only recently been implicated in ALS, and while these novel pathways of RNA-mediated pathogenesis have added more complexity to the disease, they offer at the same time more opportunities for future therapeutic intervention.
Advances in genomics combined with GWAS analyses of fALS families have led to the identification of numerous ALS-associated gene mutations. Interestingly, mutation(s) in seven RBPs have been identified to date (Table 1; reviewed by [23,24]). Genetic mutations in RBPs and RNA-processing proteins are rare and either comprise approximately 1–5% of fALS cases (TDP-43, ANG, FUS/TLS, MATR3) [24–29] or have unknown prevalence (EWSR1, SETX, TAF15) [23,30–32]. Nevertheless, dysfunctional RNA processing is thought to be a common mechanism underlying neurodegeneration in ALS since pathologic ubiquitinated and hyperphosphorylated inclusions of TDP-43, a RBP, are present in the brain and spinal cord of most (∼97%) fALS and sALS patients [7,8]. A similar TDP-43 pathology has also been found in approximately 45% of frontotemporal dementia (FTD) cases, a disease that has recently been linked to ALS based on shared genetic mutations as well as pathological and mechanistic overlap [7–8,33].
Table 1.
Summary of RNA dysregulation processes in amyotrophic lateral sclerosis.
| RNA process | Gene mutations | Function/amyotrophic lateral sclerosis affected pathway | Ref. |
|---|---|---|---|
| mRNA splicing | TARDB P | Interacts with >6000 RNA and preferentially binds to UG-rich sequences of long introns (>100 kb) knockdown alters splicing of >950 RNAs; silences spicing of CFTR, ApoAII, RXRG and ETF1 mRNAs; promotes splicing of BRCAmut and SKAR mRNAs | [34–39] |
| FUS | Interacts with >5500 RNAs and preferentially binds GUGGU-motifs of long introns (>100 kb) knockdown alters splicing of >370 RNAs | [34,35] | |
| TA F15 | Interacts with hnRNP M, which is involved in splicing | [40] | |
| EWSR1 | Shown to regulate splicing following DNA damage | [41] | |
| SETX | Depletion results in altered splice site selection of SRp20 (SFRS3) splicing factor | [42] | |
| C9ORF72 | GGGGCC expanded RNA colocalize and interact with splicing factors SRSF2, hnRNP H1/F exogenous G4C2 transcripts bind directly to hnRNP H and indirectly to SC35 and SF2 G4C2 expression reduces exon 7 retention of TARBP2 RNA | [43,44] | |
|
| |||
| RNA translational machinery | ANG | Regulates rRNA transcription depleted from nucleus in ALS | [45] |
| C9ORF72 | Sequesters nucleolin (site of rRNA biogenesis) 45S rRNA maturation impaired in motor cortex of C9ORF72 ALS patients | [46] | |
|
| |||
| RNA transport | TARDBP | Localizes axons and dendrites; represents a component of RNP transport granule that localize NEFL mRNA to axons TDP-43 ALS mutations impair trafficking | [47–49] |
| FUS | Localizes to dendritic spines | [50] | |
| MATR3 | Promotes the nuclear retention of inosine containing RNAs | [51] | |
|
| |||
| miRNAs | TARDBP | Associates with Drosha and Dicer Knockdown alters mRNA levels including downregulation of let-7b and upregulation of miR663 | [52] |
| FUS | Binds Drosha and promotes biogenesis of neuronal miRNAs such as miR-9, miR-125b and miR-132 | [53] | |
| Unknown (sALS) | mirR146a*, miR-524–5P, miR-582–3P which target NEFL mRNA are dysregulated in sALS | [54] | |
|
| |||
| RNA editing | Unknown (sALS) | Downregulation of ADAR2 in sALS motor neurons leads to reduced GluA2Q/R editing, thereby increasing Ca2+ permeability and potentially leading to glutamate induced neuronal toxicity | [55–57] |
| C9ORF72 | GGGGCC RNA binds to ADARB2 knockdown of ADARB2 enhanced glutamate sensitivity of iPS neurons | [58] | |
| MATR3 | Represents part of a complex that binds edited A-to-I RNAs | [51] | |
|
| |||
| RNA toxicity | C9ORF72 | GGGGCC RNA sequesters RBPs GGGGCC RNA expression is toxic to cells antisense oligonucleotides targeting the toxic RNA mitigate phenotypes and neuronal sensitivity to extracellular stressors | [43,46,58–60] |
ALS: Amyotrophic lateral sclerosis; NEFL: Neurofilament light polypeptide; sALS: Sporadic ALS.
Adding to the mounting evidence of an RNA-based neurodegenerative mechanism in ALS, two 2011 studies from independent laboratories simultaneously published the discovery of a GGGGCC repeat expansion in the first intron of the C9ORF72 gene in families with ALS/FTD [4,5]. While in healthy patients this sequence is repeated less than 20-times, in C9ORF72 ALS/FTD patients, the sequence is repeated more than 30-times (as determined by repeat primed PCR methodologies) [5] and typically found to be repeated between 400-and 2000-times (as determined by Southern blot analysis) [4–5,61]. One previously identified pathogenic mechanism elucidated in other repeat expansion mutations is the generation of toxic RNA molecules (reviewed by [62]) and early studies of C9ORF72 ALS patient cells and tissue pathology similarly suggest the presence of a neurotoxic RNA species (discussed in depth below) [4,58–60]. As mentioned above, the C9ORF72 repeat expansion is the most prevalent ALS-associated mutation identified to date comprising approximately 36–46% of fALS and up to approximately 8% of sALS [6]. Interestingly, this mutation is also the most common known cause of familial and sporadic FTD cases [23,33,63]. Moreover, the C9ORF72 repeat expansion is the most common genetic cause of Huntington disease phenocopies [64], has been found in some Alzheimer's patients and intermediate length repeats are a risk factor for Parkinson's disease [65–68].
Taken together, the overlap of TDP-43 pathology in ALS and FTD patients as well as the prevalence of the C9ORF72 hexanucleotide repeat expansions across CNS diseases indicate that dysfunctional RNA homeostasis is likely to be a common neurodegenerative pathway for a number of neurological disorders.
Mutations in RNA-binding & -processing proteins
Mutations of RBPs
The discovery of cytoplasmic TDP-43 inclusions in postmortem CNS tissue of both sALS and fALS patients led to the identification of ALS-causing mutations in TARDBP, the gene encoding TDP-43. TDP-43 is a predominantly nuclear RBP with 2 RNA recognition motifs (RRM) and a C-terminal glycine-rich prion-like domain [23,69–71]. Missense mutations in the region of TARDBP that encodes the protein's glycine-rich domain cause a rare but dominant form of ALS [26–27,72]. Interestingly, neuronal and glial pathological TDP-43 inclusions can be found in nearly all ALS patients, except the approximately 2% ALS patients that harbor a SOD1 mutation [73,74]. The cytoplasmic TDP-43 inclusions are often accompanied by a loss of nuclear TDP-43 suggesting that the loss of TDP-43 function might play a role in ALS pathogenesis.
Another recently identified mutant RBP causing an aggressive form of ALS is the FUS/TLS protein [28], which is encoded by the FUS gene and is a member of the FET (FUS/TLS, EWSR1 and TAF15) gene family [23,29,75–86]. FUS/TLS is an RBP with a single RRM and an N-terminal QGSY-rich region and RRM glycine-rich region that, together, comprise a prion-like domain [31,87]. Similar to TDP-43, FUS/TLS is predominantly nuclear but binds RNAs and shuttles between the nucleus and cytoplasm [88]. ALS-causing missense FUS mutations are typically found within FUS' C-terminal NLS [89]; however, some have also been identified in its glycine-rich domain [72]. Notably, CNS tissues from patients with FUS mutations exhibit FUS/TLS inclusions and mutations in the NLS region result in enhanced cytoplasmic localization of the FUS/TLS protein, similar to those seen with TDP-43 in most ALS cases [28–29,89].
RNA splicing in ALS
In addition to their commonalities in ALS pathology, protein structure and localization, TDP-43 and FUS/TLS share a number of cellular RNA regulatory functions. Both TDP-43 and FUS/TLS have been shown to play a role in transcription. TDP-43 was initially identified as a transcriptional suppressor of the HIV virus [69] and FUS/TLS associates with RNA polymerase II [90] as well as nuclear hormone factors [91]. FUS/TLS is also a general repressor of RNA polymerase III-mediated transcription in non-neuronal cultured cells [92]. Both TDP-43 and FUS/TLS are also potent RBPs that participate in and coordinate pre-mRNA splicing events [33]. TDP-43 has been shown to bind both single and double stranded TG-rich DNA sequences [93,94]. Much effort has been directed at determining the RNA targets of these two RBPs and several thousand RNAs have been found to bind TDP-43 or FUS/TLS [34,35]. TDP-43 preferentially binds intronic GU-rich regions of long introns far from exon sequences [34,95]. Studies have also shown that five UG repeat sequences are required for TDP-43:RNA interaction through its RRM1 domain [71]. TDP-43 also promotes skipping of exon 9 of cystic fibrosis transmembrane regulator (CFTR) mRNA [36], exon 3 of apolipoprotein AII (ApoAII) mRNA [37], retinoid × receptor gamma (RXRG) mRNA and eukaryotic termination factor 1 (ETF1) [38]. In contrast, TDP-43 has been shown to promote splicing of mutated breast cancer 1 (BRCA1mut)mRNA [38] and Polymerase (DNA-directed), Delta Interacting protein 3 mRNA (SKAR) [39]. While the targets of FUS are less well known, FUS does seem to preferentially bind with AU-rich sequences in a sawtooth-like pattern along long introns [96,97]
To recapitulate the ALS condition (i.e., loss of nuclear TDP-43 or FUS/TLS and therefore loss of function), studies have employed antisense mediated knockdown of TDP-43 or FUS/TLS. Depletion of TDP-43 and FUS/TLS resulted in the downregulation of numerous genes as well as in aberrant RNA splicing [33–35]. Interestingly, TDP-43 knockdown alters the splicing or RNA levels of a number of genes involved in other neurodegenerative diseases, specifically synaptic proteins with long introns (>100 kb) such as parkin2 and neuroligin (reviewed by [33]). TDP-43 depletion also reduced levels of EAAT2, an astroglial glutamate transporter responsible for buffering extracellular synaptic glutamate to prevent neuronal excitotoxicity [98]. Intriguingly, reduced EAAT2 protein levels are commonly seen in ALS patient tissue [99]. Due to the large number of RNA targets for TDP-43 and FUS, it is easy to imagine how their dysfunction, via mutation or mislocalization as seen in ALS, could disrupt upstream RNA metabolism and exert devastating effects on downstream cellular processes ultimately leading to neurodegeneration.
RNA trafficking & ALS
Although it predominantly resides in the nucleus, TDP-43 contains nuclear localization sequence (NLS) and nuclear export sequence motifs and is trafficked into the cytoplasm under normal physiological conditions [23,100]. Consequently, TDP-43 and FUS/TLS have been identified as components of cytoplasmic RNA granules including ribonucleoprotein (RNP) transport granules, and stress granules [33,47,101–105] and therefore implicating cyptoplasmic RNA transport/localization and translation as important functions of normal TDP-43 and FUS. Supporting this hypothesis, TDP-43 is translocated to dendritic spines in mouse hippocampal neurons in response to neuronal depolarization where it co-localizes with synaptic proteins such as PSD95 [48]. TDP-43 has also been found in the axonal compartments of mouse motor neurons where it is trafficked in response to neurotrophic factor stimulation. Axonal TDP-43 co-localizes with various known axonal RBPs involved in the transport and translation of axonal RNAs [49] suggesting that perturbations in TDP-43 function might alter axonal protein synthesis. Further supporting its role in RNA trafficking, 34% of cytoplasmic TDP-43 targets are located in 3′UTR regions of mRNAs [95], which are known to contain cis localization elements required for mRNA transport into neuronal processes [106]. This was confirmed in a recent study that found TDP-43 moves bidirectionally in Drosophila, mouse and human iPS-derived motor axons and is a component of the mRNP (messenger ribonucleoprotein particle) that antereogradely trafficks neurofilament light polypeptide (NEFL) mRNA. Importantly, ALS-causing TDP-43 mutations (TDP-43A315T, TDP-43G298S and TDP-43M337V) impair the anterograde axonal transport of endogenous NEFL mRNA [47]. Since axonal transport and localized protein synthesis play an important role in growth cone mobility, axonal regeneration and dictating the axonal growth program [106–108], it is likely that deficits in this process might negatively affect axon health in ALS. Moreover, these TDP-43 mutations might also affect synapse formation since TDP-43 is transported with mRNP components into dendrites and dendritic protein synthesis is essential for synaptic plasticity [48,109]. Similarly, FUS/TLS localizes to dendritic spines of mouse hippocampal neurons, is actively transported to the synapses in response to mGluR5 activation, and FUS/TLS null mice exhibit reduced dendritic spine number and abnormal spine morphology [50].
While more work is required to elucidate the specific role of TDP-43 and FUS/TLS in mRNA transport and localized protein synthesis in neurons, these early studies strongly suggest that loss of function of TDP-43 or FUS/TLS via ALS-causing mutations or inclusion formation (as seen in ALS patient tissue) negatively affect dendritic and axonal health and functionality.
Stress-granules & cytoplasmic aggregation
Stress granules are cytoplasmic RNP structures that form in response to environmental stressors (e.g., heat shock or oxidative stress) to prevent the translation of any sequestered mRNAs until the stressor is ameliorated and they are disassembled. Many RBPs that comprise these cytoplasmic RNP structures, including TDP-43 and FUS/TLS, contain prion-like domains that allow for self-aggregation and the formation of these RNP structures [110]. Interestingly, there are a number of ALS mutations found in the prion-like domain of TDP-43 and FUS/TLS, and for TDP-43, these mutations promote its cytoplasmic aggregation and promote cellular toxicity [111]. One gain-of-function hypothesis is that stress granules form under normal conditions, thus recruiting TDP-43 and FUS/TLS; however, the excessive recruitment of these proteins might lead to the formation of toxic cytoplasmic aggregates that cannot be disassembled thus perturbing normal RNP functions and cellular toxicity [110]. Since it is unclear if these cytoplasmic aggregates are neurotoxic, another hypothesis is that cytoplasmic aggregation of TDP-43 or FUS/TLS might also induce a loss-of-function phenotype for TDP-43 disrupting many nuclear RNA processing and transport events [110].
Mutations in other RNA processing proteins associated with ALS
The FET (FUS/TLS, EWSR1 and TAF15) protein family is a group of DNA-binding proteins/RBPs that are predominantly nuclear and are involved in various aspects of normal RNA metabolism [112]. In addition to FUS/TLS, mutations in TAF15 and EWSR1 are also associated with ALS and similar to TDP-43 and FUS/TLS, TAF15 and contain prion-like domains making them aggregation-prone [31,110,113]. Consistent with this, cytoplasmic mislocalization and inclusions of TAF15 and EWSR1 have been found in neurons of sporadic ALS tissue [31,113]. While the pathogenic mechanisms are unknown, EWSR1 has been shown to regulate splicing following DNA damage [41] and TAF15 has been shown to interact with the splicing factor hnRNP M [40], strongly implicating aberrant RNA processing in protein dysfunction.
Angiogenin (ANG) is a member of the ribonuclease A (RNase A) family and missense mutations in this gene have been found in less than 1–5% of ALS patients [23,25]. Angiogenin is involved in ribosomal RNA transcription and regulating levels of cellular tRNAs through its RNase activity [45,114]. Sentaxin (SETX) is a DNA/RNA helicase and mutations in this gene causes an autosomal dominant form of juvenile ALS [30]. While the mechanisms of SETX-mediated pathogenesis remain unclear, helicases are involved in many processes required for RNA metabolism including transcription, splicing, transport, translation and editing [42,114]. Finally, missense mutations in the MATR3 gene that encodes for the Matrin 3 protein were recently identified as a cause of a small percentage of fALS [24]. Although the precise function of Matrin-3 is unknown, it is a DNA/RNA binding protein [115] that is implicated in various RNA processes including RNA editing [51] and Argonaut-mediated gene silencing [116].
miRNAs
miRNAs are small noncoding RNAs that post-transcriptionally regulate gene expression by targeting its complementary RNA sequence for degradation. Analysis of sALS spinal cord tissue showed dysregulation of miR-146a*, miR-524-5P and miR-582-3P, all of which target the 3′UTR of neuroflament light chain mRNA [54]. Another miRNA, miR-206, which is enriched in skeletal muscle, is upregulated in ALS patient muscle tissue [117]. It is believed that miR-206 plays a role in nerve-muscle communication and loss of miR-206 in transgenic SOD1 mouse accelerates the disease phenotype [118]. Interestingly, TDP-43 can bind small noncoding RNAs and associates with the Drosha and Dicer proteins suggesting that TDP-43 dysfunction alters miRNA processing [33]. Consistent with this, TDP-43 knockdown results in the downregulation of let-7b and the upregulation of miR-663 [52]. Similarly, FUS has been shown to interact with Drosha and bind pri-miRNA sequences and plays a role in the biogenesis of some neuronal miRNAs including miR-9, miR-125b and miR-132 [53]. Although the relationship between miRNA regulation and ALS pathology requires further study, these molecules might also be employed as pharmocodynamic biomarkers since miRNAs have been detected in extracellular fluids such as cerebrospinal fluid [119]. The extracellular presence of miRNAs is likely due to leaky membranes of injured cells, release upon cell death, or the active exocytosis of miRNA-containing vesicles [120].
Aberrant RNA editing
RNA editing is a post-transcriptional process in which RNAs (typically pre-mRNAs) are modified or ‘edited’ resulting in a RNA transcript with bases not complementary to their genomic sequence. Two known types of RNA editing are uridine-to-cytodine (C-to-U) and the more common adenosine-to-inosine (A-to-I) [121]. The ‘adenosine deaminases acting on RNA’ or ADAR proteins are enzymes responsible for catalyzing adenosine to inosine via their deaminase activity. During translation, inosine is read as a guanine, therefore, RNA editing of any exonic region can have profound effects on protein structure and function [121]. One target of ADARs is GluR2 (=GluA2), a neuronal specific α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptor, and a subtype of the ionotropic glutamate receptors. Approximately 95–100% of GluR2 pre-mRNA transcripts are edited in the second membrane domain (M2). The unedited transcripts encode for a glutamine (Q), the edited transcripts encode for an arginine (R) and this site of editing is called the Q/R site [122]. Unedited GluR2 transcripts encode for AMPA receptors (GluR2Q) with high Ca2+ permeability compared with their edited counterparts (GluR2R); therefore, if GluR2 is unedited it enhances a cell's susceptibility to Ca2+ dependent glutamate-induced excitotoxicity [123]. Interestingly, motor neurons isolated from the spinal cords of sALS patients show incomplete editing at the GluR2Q/R-site suggesting they exhibit increased susceptibility to Ca2+ dependent excitotoxicity [55]. In support of this, ADAR2, the RNA enzyme responsible for GluR2Q/R-site editing was significantly reduced in spinal motor neurons from sALS patients [56]. Pathological analysis revealed that spinal motor neurons from sALS patients that exhibited cytoplasmic TDP-43 inclusions also showed downregulation of ADAR2 [57]; however, it is unclear how these two phenotypes are related [124]. These studies suggest that the downregulation of ADAR2 in spinal motor neurons of sALS patients may mitigate GluR2 editing and result in a greater proportion of Ca2+ permeable GluR2 receptors leading to neurodegeneration via excitotoxicity.
RNA toxicity from repeat expansions
Repeat expansions are the cause of over 20 known neurological disorders including myotonic dystrophy type 1 and 2 (DM1 and 2) and Fragile X ataxia (FXTAS) [125]. Early attempts to identify the pathogenic mechanisms behind the repeat expansion in C9ORF72 revealed a similar pathology as that observed in myotonic dystrophy, specifically the presence of nuclear RNA foci [4]. In both DM1 and 2, the repeat RNA sequences generate ‘toxic’ RNA molecules that sequester the RBP muscleblind-like protein (MBNL1) suggesting that the proposed gain-of-function for toxic RNA may cause a loss of function for interacting RBPs such as MBNL1. Since MBNL1 is important for pre-mRNA splicing, DM1 and 2 cells exhibit aberrant splicing of mature mRNAs [62]. In tissue from DM1 and patients, toxic RNA molecules that sequester nuclear RBPs can be visualized as nuclear RNA foci using RNA fluorescent in situ hybridization (RNA FISH) targeting the repeat RNA sequence [126]. Similarly, initial studies observed GGGGCC RNA foci in the spinal cord from C9ORF72 ALS/FTD patients [4]. In addition, these studies found that C9ORF72 RNA levels were reduced in C9ORF72 ALS/FTD patient tissue [5,61]. In various repeat expansion disorders, the repetitive RNA sequences have been shown to translate and accumulate in the cytoplasm despite the lack of an ATG-start codon, a remarkable event now called repeat associated non-ATG translation (RANT) [127]. Notably, RANT dipeptides accumulate in C9ORF72 patient tissue [128,129]. Based on these initial studies of C9ORF72 ALS pathology, three pathogenic mechanisms have been proposed: toxic gain-of-function RNA, loss of function C9ORF72 and accumulation of gain-of-function RANT dipeptides.
Since C9ORF72 animal models are still being developed, C9ORF72 ALS patient-derived fibroblasts and iPS motor neurons have proven critical in elucidating the neurotoxicity of the GGGGCC expansion. C9ORF72 ALS fibroblasts and iPS neurons all contain both nuclear and cytoplasmic RNA foci [58–60]. iPS neurons from C9ORF72 ALS patients also exhibit increased sensitivity to glutamate-induced excitotoxicity and ER-stress [46,58]. In addition, these neurons exhibit aberrant gene expression of some neuronal and secreted transcripts [58,60]. To date, a number of GGGGCC interactors have been identified via biochemical and immunohistochemical means [130]. For example, ADARB2, an RNA-editing enzyme, has been shown to co-localize and biochemically interact with the GGGGCC repeat, and its depletion has been shown to enhance glutamate toxicity in control iPS neurons. This suggests that ADARB2's loss of function might play a role in glutamate susceptibility in C9ORF72 iPS neurons [58]. Nucleolin, a eukaryotic nucleolar phosphoprotein, has been shown to exhibit a strong biochemical affinity for the GGGGCC RNA G-quardruplex structure and its interaction with the repeat RNA results in disruption of the nucleolus [46]. Inducible tissue-specific expression of an exonic GGGGCCx30 RNA results in a neurodegenerative eye phenotype in Drosophila, which can be rescued by enhancing levels of Purα, a GGGGCC RNA interactor identified by a pull down assay [131]. Expression of 38-and 72mer GGGGCC RNA in neuronal cell lines can also induce the formation of RNA foci that co-localize with SF2, SC35 and hnRNP. Of these, however, only hnRNP-H directly interacts with the GGGGCC RNA sequence [43]. hnRNP-H is required for exon 7 inclusion of TARBP2 RNA, and when TARBP2 RNA splicing was analyzed in cell lines expressing 72 GGGGCC RNA repeats they found a modest reduction in TARBP2 exon 7 inclusion. This supports the toxic RNA hypothesis and suggests that the GGGGCC RNA acts to sequester hnRNP-H resulting in its loss of function [43]. A recent study has also shown the C9ORF72 RNA foci can co-localize and interact with SRSF2, hnRNP H1/F and ALYREF in motor neurons [44]. Although a number of interactors have been identified to date, it is unlikely that a single candidate will be responsible for all of the observed C9ORF72 phenotypes. If this protein does exist, it has not yet been identified. Targeting the GGGGCCexp-containing RNA pharmacologically does ameliorate a number of C9ORF72 phenotypes. For example, antisense oligonucleotides (ASOs) targeting different regions (coding and noncoding) of C9ORF72 RNA have reduced the prevalence of RNA foci in C9ORF72 ALS fibroblasts and iPS neurons [58–60] and rescued aberrant gene expression and glutamate sensitivity of C9ORF72 iPS neurons [58,60]. These studies suggest that RNA toxicity represents a significant pathogenic mechanism in C9ORF72 neurotoxicity. Notably, the GGGGCC antisense transcript (CCCCGG) has also been detected in C9ORF72 patient derived cells and tissue [59] and found to be translated into RANT dipeptides [132].
While the above-mentioned studies implicate RNA toxicity characterized by the sequestration of RBPs as a major cause of C9ORF72 neurodegeneration, it does not rule out C9ORF72 loss of function or RANT dipeptide toxicity as a pathogenic mechanism. Three recent publications overexpressing varying di-peptides in vitro and in vivo support toxicity due to the cytoplasmic accumulation of RANT dipeptides [133–135], although the disadvantages of overexpression models have been noted in all three reports as a caution in regards to the overinterpretation of their datasets. At least one of the five possible RANT dipeptides, glycine-proline (GP) [136,137], has been detected in C9ORF72 iPS neurons, suggesting that the use of iPS neurons could aid in the future investigation of disease-relevant levels of dipeptide accumulations. Interestingly, GP RANT proteins were still detected following acute short-term ASO treatment, which rescued other C9ORF72 phenotypes, including susceptibility to extracellular glutamate [58]. Long-term treatments are needed to fully support a potential ASO rescue of RANT dipeptides. Notably, sense targeting ASOs do not affect the antisense RNA foci [59]; therefore, any mitigated C9ORF72 phenotypes with sense ASO treatment can be attributed to toxicity of the sense strand RNA. Future studies are still required to elucidate the neurotoxicity of the CCCCGGexp antisense RNA.
The function of C9ORF72 protein is not known and, to complicate matters, there is no consensus on a suitable antibody for protein detection. However, a LacZ insertion C9orf72 reporter mouse model revealed C9orf72 expression is enriched in neurons [138]. Support for the haploinsufficiency pathological mechanism emerged when knockdown of the zebrafish C9ORF72 ortholog via morpholinos developed behavioral and cellular deficits, including axonal degeneration of motor neurons [139]. Conversely, acute ASO treatment targeting C9ORF72 exon 2 and reducing global C9ORF72 RNA levels nevertheless rescued susceptibility to glutamate induced excitotoxicity [58]. This suggests that acute knockdown of C9ORF72 is not toxic to neurons in vitro. Moreover, sustained ASO-mediated knockdown of the endogenous mouse C9orf72 for 17-weeks did not induce any significant pathological or behavioral deficits [59].
Potential therapies targeting aberrant RNA homeostasis
As summarized above, abnormal RNA processing has been accepted as a contributor to ALS pathogenesis, even though not all mechanisms of altered RNA metabolism are fully understood and examined to date. Nevertheless, efforts are underway to identify novel therapeutic targets based on these newly discovered disease pathways. Some of these efforts are guided by studies of RNA toxicity in other neurodegenerative disorders and are therefore aggressively moved towards preclinical testing and clinical trials.
Oligonucleotide-based therapeutic strategies
Potential therapeutics for RNA-dominant diseases include the development of oligonucleotides that bind to mRNA in a sequence-specific manner. The end result of this complementary base pairing could either be degradation of the targeted mRNA or steric blockade of the cellular machinery without degradation of the RNA with both mechanisms resulting in decreased gene expression of the target protein [140]. This approach is certainly advantageous when the mutant target protein leads to a disease mechanism involving a ‘protein gain of function,’ but has to be approached carefully when haploinsufficiency and loss of protein function is of concern. ASO approaches have recently been shown to move rapidly into the clinic due to the development of more effective delivery strategies and higher stabilities of ASOs by chemical modifications (for review, see [141]). For example, ASOs targeted to SOD1 have been developed and successfully tried in a Phase I safety trial [142], strongly suggesting that ASO therapeutics are a viable and safe option for gene silencing in ALS patients.
In the case of repeat expansion disorders, where RNA toxicity is often caused by the sequestration of critical RBPs (see above), blocking the binding of these RBPs with the use of repeat-specific ASOs represents another therapeutic approach for these aberrant RNA-disorders (Figure 1B, ‘5’). This approach has been successfully applied in myotonic dystrophy 1 (DM1) to prevent the sequestration of MBNL1 by the (CTG)n repeat expansion in the DMPK gene and thereby eliminating the formation of CTG RNA foci and splicing abnormalities, which are caused due to the loss of function of sequestered MBNL1 protein [143–146].
Figure 1. Schematic of dysfunctional RNA processing events in amyotrophic lateral sclerosis and potential targets for therapeutic intervention (see facing page).
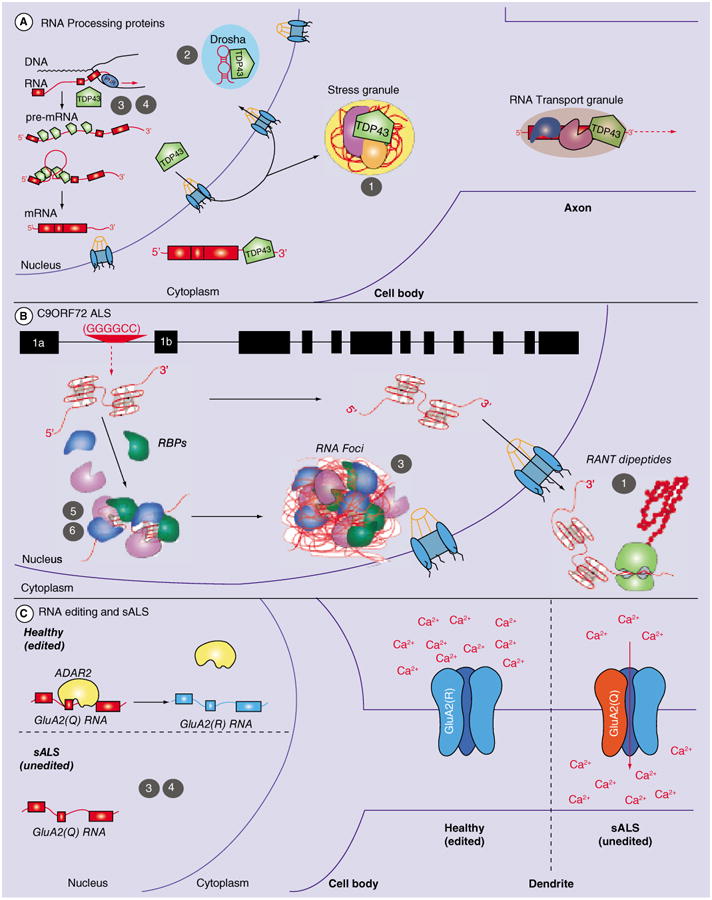
(A) Mutations in RNA processing proteins, such as TARDBP/TDP-43, can lead to dysregulation of RNA processing events at different levels, offering numerous therapeutic targets: (1) Activation of autophagy with small molecule compounds; (2) Targeting of miRNAs with antagomirs; (3) Increasing transcriptional activation of wild-type RNA procession protein with small molecule compounds and (4) Increasing transcriptional activation of wild-type RNA procession protein with gene therapy. (B) Repeat expansions, such as the hexanucleotide expansion in C9ORF72, can sequester RBPs, leading to the formation of RNA foci and a loss of function of RBPs. In addition, RANT of the hexanucleotide repeat can lead to the formation and aggregation of dipeptides. This dipeptide aggregation could potentially be diminished with the use of autophagy activators (1) as described in (A) for accumulation of cytoplasmic RNA binding proteins, such as TDP-43. In addition, transcriptional activation of RBPs with small molecule compounds could overcome their loss of function (3), similar to what has been suggested in (A). Sequestration of RBPs to the repeat foci could be prevented with either antisense oligonucleotides blocking the repeat sequence (5) or with small molecule compounds that bind and thereby block the RBP binding site (6). (C) Yet unknown mechanisms can lead to the dysfunction of RNA editing proteins, such as ADAR2. ADAR2 is responsible for the editing of the GluA2 receptor, which if unedited, shows increased calcium (Ca2+) permeability and can therefore trigger excitotoxicity in amyotrophic lateral sclerosis. Therapeutic approaches as discussed in (A & B) can also be applied to the rescue of RNA editing phenotypes, for example, increased activation and expression of ADAR could be achieved with small molecule transcriptional activators (3) or via gene therapy (4). RANT: Repeat-associated non-ATG translation; RBP: RNA-binding proteins; sALS: Sporadic amyotrophic lateral sclerosis.
Based on this, preliminary studies working with human ALS C9ORF72 patient-derived fibroblasts and iPS neurons confirmed that C9ORF72-specific ASO treatment rescued in vitro disease phenotypes, suggesting that an ASO-based therapeutic approach presents great promise for clinical trials for C9ORF72 patients [58,147–148]. Interestingly, these studies employed ASO targeted to either the repeat expansion, upstream of the repeat or an exonic coding region of C9ORF72 mRNA, the latter leading to a further downregulation of the C9ORF72 gene. Despite the enhanced silencing of C9ORF72, neuroprotection was observed. At the same time, in vivo knock down studies in zebrafish, Caenorhabditis elegans and mice showed conflicting results with regards to confirming or ruling out a role of reduced C9ORF72 gene expression as a disease mechanism [139,147,149]. The observed differences may be explained by either species-related susceptibility to loss of C9ORF72 homologs or due to the different approaches used to reduce the levels of C9ORF72 (genetic knock-out vs use of ASO or morpholinos). Interestingly, one C9ORF72 patient carries the repeat expansion in both alleles without showing any increased severity of clinical symptoms, suggesting that C9ORF72 loss of function may not be a dominant mechanism in C9 pathogenesis [150]. As mentioned earlier, C9ORF72 protein function is still unknown, but the potential loss of protein using an ASO therapeutic approach should nevertheless be addressed during preclinical drug validations. Furthermore, while validating ASO therapeutics for repeat expansion mutations such as C9ORF72, one should be aware of the possibility of bidirectional transcription, which would suggest that ASO might have to be designed for both sense and antisense transcripts [137,147,151].
RNA interference (RNAi) represents another possible oligonucleotide based therapeutic approach targeting RNA toxicity pathways [12]. Similar to the ASO therapeutic, RNAi achieves gene silencing with the use of short interference RNAs (siRNAs), short hairpin RNAs (shRNAs) or artificial miRNAS [12,152–153]) generally without discriminating between the mutant and the normal allele. Therefore, similar to the ASO approach for gene silencing, sufficient knowledge on the cellular function of the target protein is important in order to decide whether a loss of protein function due to the therapeutic knock down is acceptable during treatment, as these approaches would be most suitable to gain-of-function mutations. To overcome the loss of wild-type protein, allele-specific siRNA strategies have been developed and successfully described for Parkinson's disease, Alzheimer's disease and Huntington's disease [154–159]. For ALS, mutant SOD1 and TDP-43 have been targeted using this strategy [160–164]. The hope is that knocking down mutant TDP-43 will not only prevent the formation of cytoplasmic inclusions, but also TDP-43-dependent axonal mRNA trafficking, dysfunction of which has recently been proposed to be a likely contributor to ALS pathogenesis in motor neurons [47]. Delivering these RNAi therapeutics to the cell type of interest in an appropriate vehicle is yet one of the biggest hurdles to take for clinical trial development of these therapeutic strategies [165]. Nevertheless, over 20 siRNA/shRNA therapeutic candidates have been tested in clinical trials to treat more than 16 different diseases [165]. Unfortunately, none of those diseases are CNS diseases, but these statistics encourage the design and development of iRNA therapeutics targeting RNA toxicity pathways in ALS, in particular for dominant, gain-of-function ALS mutations.
If the loss of target protein is not acceptable as a consequence of gene silencing, one could consider the so-called replacement strategy where the normal allele is replaced using nonallele-selective RNAi silencing (see also ‘Gene therapy approaches’) [166–170]. While this therapeutic approach has been successfully applied in vitro, preclinical validation is needed to address potential concerns regarding overexpression of the target protein above endogenous levels as well as safety and efficacy of viral delivery. A novel approach to overcome potential off-target toxicity using viral delivery methods is the use of lipid nanoparticles [171,172], which have been shown recently to efficiently deliver siRNAs into the brain with little apparent toxicity [173].
As mentioned above, RNA toxicity can lead to aberrant alternative splicing of downstream genes. Splicing deficits can lead to the production of truncated, low-functioning proteins, as seen in spinal muscular atrophy (SMA), where splicing of the SMN2 gene is disrupted, leading to a truncated form of the SMN protein. This dysfunction has been targeted with ASOs that either block the binding of splicing suppressors or promote the binding of splice activators and consequently rescue disease phenotypes in SMA animal models [174]. Chemically optimized ASOs have been validated in mouse and nonhuman primates and clinical safety trials are ongoing to advance this therapy to SMA patients [175]. Splicing abnormalities have been observed in response to RNA toxicity in ALS, particularly in mutant TDP-43 and mutant FUS/TLS ALS [176–178]. Whether targeting mutant TPD-43 to prevent splicing abnormalities using ASOs will be appropriate is not known, but one could consider targeting downstream transcripts that are altered due to the loss of function of TDP-43. While it is unlikely that the rescue of only one single downstream transcript will lead to neuroprotection of mutant TDP-43, these pathways have not been explored thoroughly and may therefore harbor suitable drug targets. Although no specific splicing abnormalities have been identified in C9ORF72 mutant cells or tissue, the sequestration of RBPs strongly suggests that splicing abnormalities are highly likely and are hypothesized to contribute to C9ORF72 disease pathogenesis [58]. Hence, targeting these splicing abnormalities could certainly provide rescue of disease phenotypes and subsequent neuroprotection. Notably, by targeting the mutant allele of any gain-of-function RBP mutation, it may be possible to reduce any mutation-dependent splicing deficits.
ASOs can also be designed to specifically silence endogenous miRNAs and these are called antagomirs [179]. An increasing number of miRNAs has been identified to be upregulated in neurodegenerative diseases, including ALS (see above and [180–182]). Initial studies have tested antagomirs to miRNA-155 in the SOD1G93A mouse model leading to extended survival and increased disease duration, suggesting that the use of antagomirs is a promising new therapeutic approach for ALS [183] (Figure 1A, ‘2’). Aberrant miRNA expression has been linked to a list of mutant proteins known to cause cytoplasmic aggregation (TDP-43, FUS/TLS) or altered autophagy regulation (CHMP2B, VCP, Ubiquilin-2) in ALS and/or FTD [181]. While it is yet to be determined whether the repeat expansion in C9ORF72 is causing altered miRNA expression, targeting miRNAs for therapeutic intervention certainly represents a valid approach to intervene with RNA toxicity in ALS. Antagomirs can also be designed to bind to the target RNA of a specific miRNA, while allowing the miRNA to bind to all of its other targets, and are sometimes referred to as blockmirs [184]. If it is known which target mRNA of a miRNA is participating in the disease pathogenesis, blockmirs can be designed to sterically block the miR-NA's binding site on the target RNA to prevent miRNA binding and subsequent gene silencing of their complimentary RNA. This allows for very specific rescue of aberrant miRNA function and reduces off-target effects more frequently to be expected with antagomirs, given the fact that miRNAs often have multiple target RNAs. miRNA therapeutics have been developed for non-CNS diseases and reached clinical trials, once again supporting the design and validation of equivalent therapeutic interventions for CNS diseases including ALS [185].
Yet another experimental paradigm to block miRNA function is the use of so-called miRNA sponges or decoys [185–187]. These sponges are RNA transcripts composed of miRNA binding sites, which upon cell delivery, compete with endogenous miRNA binding sites, similar to peptide block experiments at the protein level [187]. While this miRNA sponge technology is currently mostly used to study the function of individual miRNAs, its use for therapeutic applications could be of great potential.
Small molecule-based therapeutic strategies
The classic pharmacological approach of an inhibitory small molecule compound therapy is certainly applicable to target varying RNA toxicity pathways. For example, there has been great success in DM1 by applying small molecule inhibitors that prevent the interaction of repeat RNA with MBNL1 and thereby rescue disease pathogenesis [188–190]. A similar approach is therefore sought after for the C9ORF72 repeat expansion (Figure 1B, ‘6’). While several repeat-binding RBPs have been identified over the last 2 years [44,58,147–148,191–193], none of those RBPs have been assigned similar unique disease-modifying properties like MBNL1. However, preliminary studies are ongoing to design screening assays with the goal of identifying small molecule compounds that bind to the repeat expansion, or more specifically, to the secondary structure of the RNA and DNA repeat nucleotides, and thereby inhibit the binding of any potential RBPs. It has been shown that the sense strand of the hexanucleotide repeat of C9ORF72 preferentially forms so-called G-quardruplexes [191,194–195] and proof of concept studies in vitro showed that nucleic acid binding small molecules, such as the cationic porphyrin, inhibits the interaction of some RBPs with a (GGGGCC)8 repeat by binding and stabilizing the repeat structure [196]. Successful attempts in targeting RNA structures with small molecules for therapeutic benefits have been made in fragile X-associated tremor ataxia syndrome (FXTAS), DM1 and DM2 [197–200], suggesting that this approach could lead to similar success for C9ORF72-mediated disease progression (ALS and/or FTD). If a MBNL1-like RBP is identified as an interactor with the C9ORF72 hexanucleotide repeat, then more targeted screening efforts can be undertaken in the search for neuroprotective small molecule compounds that inhibit the specific RNA–protein interaction. The alternative is to target not only the interaction of one RBP, but of multiple RBPs at the same time, which could likely occur using the nonspecific repeat blocking approach described above.
Small molecule compounds can also be designed to inhibit protein aggregations, such as those found for mutant TDP-43 [201]. As described above, the RBP TDP-43 forms cytoplasmic aggregates when mutated and thereby loses it nuclear localization and function as a transcriptional regulator of RNA processing such as splicing or trafficking. Screening a library of about 75,000 compounds in an assay that detected stress-induced GFP-labeled TDP-43 aggregation in vitro using a high content image analysis system, 16 compounds were found to inhibit the aggregation, some of which also showed neuroprotection in a mutant TDP-43 C. elegans model [201]. One can imagine that this approach could also be used to screen for RBPs that are sequestered at the C9ORF72 repeat expansion. Almost all of those RBPs form aggregates due to the sequestration and are not only found in the cell nucleus, but also in the cytoplasm, where small molecule inhibitors should gain easy access.
Another way to protect against TDP-43 aggregation (or any other protein aggregation) is to increase autophagy, and thereby enhancing the removal of aggregated protein (Figure 1A, ‘1’). This idea was tested in GFP-TDP-43 overexpression cell lines using automated fluorescence microscopy [202]. The authors screened over one million compounds for increased autophagy stimulation and then tested the positive hits in their TDP-43 screening assay measuring TDP-43 clearance. Compounds that cleared TDP-43 more efficiently also prevented mutant TDP-43-induced cell death, suggesting that therapeutic targeting of these autophagy pathways can prevent RNA toxicity. Clearly, this concept can be applied for any other cellular protein aggregation known to play a role in disease propagation and pathogenesis.
Finally, small molecule compounds can also be used to overcome loss of protein function due to repeat RNA sequestration. Pharmacological upregulation of target proteins has been described in the past showing precedence for this approach. For instance, Rothstein and colleagues discovered ceftriaxone as an upregulator for excitatory amino acid transporter 2 (EAAT2) [203]. This transcriptional upregulation leads to neuroprotection of motor neuron cell death both in vitro and in vivo as well as in several other models of neurodegenerative diseases [204–207]. At the same time, Lin and colleagues discovered translational activators of EAAT2, which have also been shown to provide neuroprotection in an animal model of ALS [208,209]. These studies support the idea of screening for small molecule compounds that can selectively upregulate proteins that are sequestered by repeat expansions such as C9ORF72 and consequently rescue the loss of function of these RBPs, similar to what has been shown for Purα using a genetic approach (see ‘Gene therapy approaches’ Figure 1A & C, 3′) [210]. This approach can certainly also be applied to any other protein loss of function pathway that is known in ALS, for example, the above mentioned downregulation of ADAR2, which leads to editing deficits in ALS. As it is known that dysfunctional RNA processing does lead to general aberrant transcriptome signatures, the approach of upregulating proteins to make up for their loss of function is certainly a strategy that could be applied therapeutically after thorough target validation, similar to what was done with EAAT2.
Gene therapy approaches
One way to overcome haploinsufficiency or to gain back function of repeat expansion-sequestered RBPs is to replace these proteins via gene therapy, in other words, replacing the defective or dysfunctional gene with a healthy and functional one (Figure 1A & C, ‘4’). This approach has been tried for DM1 by overexpressing MBLN1 in a DM1 mouse model [211] as well as for FXTAS and C9ORF72 ALS/FTD by overexpressing Purα in Drosophila and primary mouse cultures, respectively [210,212]. Other targets for ALS could be any of the currently identified GGGGCC repeat binding RBPs, including ADARB2, nucleolin or hnRPs. This therapeutic avenue could certainly also be applied to the above mentioned replacement strategy of mutant alleles, especially in combination with gene silencing, in order to avoid loss of function of the target gene (C9ORF72, TDP-43, FUS/TLS). Proper delivery of the gene of interest to the cell type of interest poses once again the biggest problem for safe, nontoxic therapeutic intervention using gene therapy. Vehicles of choice are viral deliveries using, for example, adeno associated virus (AAV) delivery, similar to siRNA therapeutics discussed above. However, nanoparticles are equally suited for this purpose and have been tried successfully in preclinical settings for brain deliveries [172].
Conclusion & future perspective
Overall, it has become more and more obvious over the last years that the search for therapeutic targets for ALS patients is no longer seen as ‘one-size-fits-all’ approach. Instead, it has become apparent that similar to what we have learned from cancer research, ALS manifests itself in different subtypes of ALS and therefore, therapeutic intervention needs to be developed according to those subtypes. The discovery of dysfunctional RNA processing as a contributor to ALS pathogenesis seems to represent such an ALS subtype and has therefore greatly revolutionized our search for therapeutic targets in ALS. While representing a seemingly large patient population, it is likely that ALS patients characterized by RNA processing deficits will be classified into even more defined subpopulations. However, the fact that cytoplasmic inclusions of TDP-43 are present in the majority of sporadic and familial ALS cases suggests that disrupted RNA homeostasis via RNA binding/processing protein mutations and RNA toxicity likely represent diseases pathway that if targeted therapeutically, could be beneficial to this larger patient population. Similarly, therapeutic development targeted to the highly prevalent C9ORF72 mutation is suggested to provide therapeutic benefits to another large subpopulation of ALS patients. Developing therapeutics for specific subtypes of ALS patients leads the way to a personalized medicine approach in ALS. This strategy is supported by the use of human adult patient-derived induced pluripotent stem cells, which allow for the screening of patient-specific and mutation-specific therapeutics as well as accompanying biomarker assays. The result of these novel screening opportunities are expedited clinical trial development and hopefully better clinical trial outcomes, performed on select patient populations.
Future genome wide sequencing projects may discover other novel mutations in ALS, maybe similar in prevalence to C9ORF72 or even higher. These discoveries may lead to the classification of all ALS patients into familial ALS patients and thereby eliminate the presence of sporadic ALS. Meanwhile, due to the multifaceted nature of ALS disease pathogenesis, perhaps treatment regimens combining different therapeutic approaches, such as small molecule inhibitors or ASOs together with gene therapy, will provide a better and more effective therapeutic strategy.
Practice point.
Amyotrophic lateral sclerosis (ALS) is a chronic and universally fatal neurodegenerative disease, for which there is currently no cure and only one approved therapeutic that leads to a moderate increase in survival.
A total of 90–95% of ALS cases are of sporadic nature, while only 5–10% are familial, with a family history of gene mutations.
An increasing number of mutations are identified in RNA/DNA-regulating genes, supporting an essential role of dysfunctional RNA processing in ALS pathogenesis.
Altered RNA processing includes changes in RNA editing, splicing, stabilization and transport, as well as aberrant miRNA profiles.
The increasing knowledge of RNA toxicity in ALS pathogenesis has led to the discovery of novel therapeutic targets, and in combination with new ways of patient stratification, it provides hope for more efficacious treatments for ALS patients.
Acknowledgments
R Sattler and CJ Donnelly are co-inventors of a pending patent for ‘Composition of modulating C9ORF72.’ This work was supported by the Brain Science Institute; NINDS RO1 NS085207 (Rita Sattler); The Judith and Jean Pape Adams Charitable Foundation (Rita Sattler), ALS Association (Rita Sattler); Maryland Stem Cell Research Fund (Christopher J Donnelly) and the William and Ella Owens Medical Research Foundation (Rita Sattler). JCG is the recipient of a National Science Foundation Graduate Research Fellowship Award.
Footnotes
Financial & competing interests disclosure: The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
• of interest;
•• of considerable interest
- 1.Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. 2004;27:723–749. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- 2.Strong M, Rosenfeld J. Amyotrophic lateral sclerosis: a review of current concepts. Amyotroph Lateral Scler Other Motor Neuron Disord. 2003;4(3):136–143. doi: 10.1080/14660820310011250. [DOI] [PubMed] [Google Scholar]
- 3.Rosen DR, Siddique T, Patterson D, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 4••.Dejesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–256. doi: 10.1016/j.neuron.2011.09.011. Co-published independent report of the discovery of C9ORF72 repeat expansion in ALS/FTD patients and first reports of GGGGCC RNA foci in spinal cord of C9 Amyotrophic lateral sclerosis (ALS)/frontotemporal dementia patients (FTD) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5••.Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–268. doi: 10.1016/j.neuron.2011.09.010. Co-published independent study identifying C9ORF72 repeat expansion as a cause of ALS/FTD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Majounie E, Renton AE, Mok K, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 2012;11(4):323–330. doi: 10.1016/S1474-4422(12)70043-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arai T, Hasegawa M, Akiyama H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351(3):602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- 8•.Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. doi: 10.1126/science.1134108. This paper was the first to link TDP-43 as an ALS/FTD disease protein and to show commonalities between ALS and FTD via TDP-43 accumulation in postmortem CNS tissue. [DOI] [PubMed] [Google Scholar]
- 9.Belzil VV, Gendron TF, Petrucelli L. RNA-mediated toxicity in neurodegenerative disease. Mol Cell Neurosci. 2013;56:406–419. doi: 10.1016/j.mcn.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Renoux AJ, Todd PK. Neurodegeneration the RNA way. Prog Neurobiol. 2012;97(2):173–189. doi: 10.1016/j.pneurobio.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cooper TA. Molecular biology: neutralizing toxic RNA. Science. 2009;325(5938):272–273. doi: 10.1126/science.1177452. [DOI] [PubMed] [Google Scholar]
- 12.Cooper TA, Wan L, Dreyfuss G. RNA and disease. Cell. 2009;136(4):777–793. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis: ALS/Riluzole Study Group. N Engl J Med. 1994;330(9):585–591. doi: 10.1056/NEJM199403033300901. [DOI] [PubMed] [Google Scholar]
- 14.Lacomblez L, Bensimon G, Leigh PN, Guillet P, Meininger V. Dose-ranging study of riluzole in amyotrophic lateral sclerosis: Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet. 1996;347(9013):1425–1431. doi: 10.1016/s0140-6736(96)91680-3. [DOI] [PubMed] [Google Scholar]
- 15.Bellingham MC. A review of the neural mechanisms of action and clinical efficiency of riluzole in treating amyotrophic lateral sclerosis: what have we learned in the last decade? CNS Neurosci Ther. 2011;17(1):4–31. doi: 10.1111/j.1755-5949.2009.00116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morren JA, Galvez-Jimenez N. Current and prospective disease-modifying therapies for amyotrophic lateral sclerosis. Expert Opin Investig Drugs. 2012;21(3):297–320. doi: 10.1517/13543784.2012.657303. [DOI] [PubMed] [Google Scholar]
- 17.Pandya RS, Zhu H, Li W, Bowser R, Friedlander RM, Wang X. Therapeutic neuroprotective agents for amyotrophic lateral sclerosis. Cell Mol Life Sci. 2013;70(24):4729–4745. doi: 10.1007/s00018-013-1415-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vucic S, Rothstein JD, Kiernan MC. Advances in treating amyotrophic lateral sclerosis: insights from pathophysiological studies. Trends Neurosci. 2014;37(8):433–442. doi: 10.1016/j.tins.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 19.Gurney ME, Pu H, Chiu AY, et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264(5166):1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 20.Gurney ME. The use of transgenic mouse models of amyotrophic lateral sclerosis in preclinical drug studies. J Neurol Sci. 1997;152(Suppl. 1):S67–S73. doi: 10.1016/s0022-510x(97)00247-5. [DOI] [PubMed] [Google Scholar]
- 21.Sabatier R, Goncalves A, Bertucci F. Personalized medicine: present and future of breast cancer management. Crit Rev Oncol Hematol. 2014;91(3):223–233. doi: 10.1016/j.critrevonc.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 22.Kraus S, Nabiochtchikov I, Shapira S, Arber N. Recent advances in personalized colorectal cancer research. Cancer Lett. 2014;347(1):15–21. doi: 10.1016/j.canlet.2014.01.025. [DOI] [PubMed] [Google Scholar]
- 23.Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci. 2013;14(4):248–264. doi: 10.1038/nrn3430. [DOI] [PubMed] [Google Scholar]
- 24.Johnson JO, Pioro EP, Boehringer A, et al. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat Neurosci. 2014;17(5):664–666. doi: 10.1038/nn.3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Greenway MJ, Andersen PM, Russ C, et al. ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat Genet. 2006;38(4):411–413. doi: 10.1038/ng1742. [DOI] [PubMed] [Google Scholar]
- 26.Sreedharan J, Blair IP, Tripathi VB, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319(5870):1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rutherford NJ, Zhang YJ, Baker M, et al. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 2008;4(9):e1000193. doi: 10.1371/journal.pgen.1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323(5918):1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 29.Vance C, Rogelj B, Hortobagyi T, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323(5918):1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen YZ, Bennett CL, Huynh HM, et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4) Am J Hum Genet. 2004;74(6):1128–1135. doi: 10.1086/421054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Couthouis J, Hart MP, Shorter J, et al. A yeast functional screen predicts new candidate ALS disease genes. Proc Natl Acad Sci USA. 2011;108(52):20881–20890. doi: 10.1073/pnas.1109434108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Couthouis J, Hart MP, Erion R, et al. Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis. Hum Mol Genet. 2012;21(13):2899–2911. doi: 10.1093/hmg/dds116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79(3):416–438. doi: 10.1016/j.neuron.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Polymenidou M, Lagier-Tourenne C, Hutt KR, et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci. 2011;14(4):459–468. doi: 10.1038/nn.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lagier-Tourenne C, Polymenidou M, Hutt KR, et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci. 2012;15(11):1488–1497. doi: 10.1038/nn.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buratti E, Dork T, Zuccato E, Pagani F, Romano M, Baralle FE. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 2001;20(7):1774–1784. doi: 10.1093/emboj/20.7.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mercado PA, Ayala YM, Romano M, Buratti E, Baralle FE. Depletion of TDP 43 overrides the need for exonic and intronic splicing enhancers in the human apoA-II gene. Nucleic Acids Res. 2005;33(18):6000–6010. doi: 10.1093/nar/gki897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Passoni M, De Conti L, Baralle M, Buratti E. UG repeats/TDP-43 interactions near 5′ splice sites exert unpredictable effects on splicing modulation. J Mol Biol. 2012;415(1):46–60. doi: 10.1016/j.jmb.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 39.Fiesel FC, Weber SS, Supper J, Zell A, Kahle PJ. TDP-43 regulates global translational yield by splicing of exon junction complex component SKAR. Nucleic Acids Res. 2012;40(6):2668–2682. doi: 10.1093/nar/gkr1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marko M, Leichter M, Patrinou-Georgoula M, Guialis A. Selective interactions of hnRNP M isoforms with the TET proteins TAF15 and TLS/FUS. Mol Biol Rep. 2014;41(4):2687–2695. doi: 10.1007/s11033-014-3128-3. [DOI] [PubMed] [Google Scholar]
- 41.Paronetto MP, Minana B, Valcarcel J. The Ewing sarcoma protein regulates DNA damage-induced alternative splicing. Mol Cell. 2011;43(3):353–368. doi: 10.1016/j.molcel.2011.05.035. [DOI] [PubMed] [Google Scholar]
- 42.Suraweera A, Lim Y, Woods R, et al. Functional role for senataxin, defective in ataxia oculomotor apraxia type 2, in transcriptional regulation. Hum Mol Genet. 2009;18(18):3384–3396. doi: 10.1093/hmg/ddp278. [DOI] [PubMed] [Google Scholar]
- 43.Lee YB, Chen HJ, Peres JN, et al. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 2013;5(5):1178–1186. doi: 10.1016/j.celrep.2013.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cooper-Knock J, Walsh MJ, Higginbottom A, et al. Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain. 2014;137(Pt 7):2040–2051. doi: 10.1093/brain/awu120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu ZP, Tsuji T, Riordan JF, Hu GF. The nuclear function of angiogenin in endothelial cells is related to rRNA production. Biochem Biophys Res Commun. 2002;294(2):287–292. doi: 10.1016/S0006-291X(02)00479-5. [DOI] [PubMed] [Google Scholar]
- 46.Haeusler AR, Donnelly CJ, Periz G, et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature. 2014;507(7491):195–200. doi: 10.1038/nature13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alami NH, Smith RB, Carrasco MA, et al. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron. 2014;81(3):536–543. doi: 10.1016/j.neuron.2013.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang IF, Wu LS, Chang HY, Shen CK. TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J Neurochem. 2008;105(3):797–806. doi: 10.1111/j.1471-4159.2007.05190.x. [DOI] [PubMed] [Google Scholar]
- 49.Fallini C, Bassell GJ, Rossoll W. The ALS disease protein TDP-43 is actively transported in motor neuron axons and regulates axon outgrowth. Hum Mol Genet. 2012;21(16):3703–3718. doi: 10.1093/hmg/dds205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fujii R, Okabe S, Urushido T, et al. The RNA binding protein TLS is translocated to dendritic spines by mGluR5 activation and regulates spine morphology. Curr Biol. 2005;15(6):587–593. doi: 10.1016/j.cub.2005.01.058. [DOI] [PubMed] [Google Scholar]
- 51.Zhang Z, Carmichael GG. The fate of dsRNA in the nucleus: a p54(nrb)-containing complex mediates the nuclear retention of promiscuously A-to-I edited RNAs. Cell. 2001;106(4):465–475. doi: 10.1016/s0092-8674(01)00466-4. [DOI] [PubMed] [Google Scholar]
- 52.Buratti E, De Conti L, Stuani C, Romano M, Baralle M, Baralle F. Nuclear factor TDP-43 can affect selected microRNA levels. FEBS J. 2010;277(10):2268–2281. doi: 10.1111/j.1742-4658.2010.07643.x. [DOI] [PubMed] [Google Scholar]
- 53.Morlando M, Dini Modigliani S, Torrelli G, et al. FUS stimulates microRNA biogenesis by facilitating co-transcriptional Drosha recruitment. EMBO J. 2012;31(24):4502–4510. doi: 10.1038/emboj.2012.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Campos-Melo D, Droppelmann CA, He Z, Volkening K, Strong MJ. Altered microRNA expression profile in amyotrophic lateral sclerosis: a role in the regulation of NFL mRNA levels. Mol Brain. 2013;6:26. doi: 10.1186/1756-6606-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kawahara Y, Ito K, Sun H, Aizawa H, Kanazawa I, Kwak S. Glutamate receptors: RNA editing and death of motor neurons. Nature. 2004;427(6977):801. doi: 10.1038/427801a. [DOI] [PubMed] [Google Scholar]
- 56.Hideyama T, Yamashita T, Aizawa H, et al. Profound downregulation of the RNA editing enzyme ADAR2 in ALS spinal motor neurons. Neurobiol Dis. 2012;45(3):1121–1128. doi: 10.1016/j.nbd.2011.12.033. [DOI] [PubMed] [Google Scholar]
- 57.Aizawa H, Sawada J, Hideyama T, et al. TDP-43 pathology in sporadic ALS occurs in motor neurons lacking the RNA editing enzyme ADAR2. Acta Neuropathol. 2010;120(1):75–84. doi: 10.1007/s00401-010-0678-x. [DOI] [PubMed] [Google Scholar]
- 58.Donnelly CJ, Zhang PW, Pham JT, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80(2):415–428. doi: 10.1016/j.neuron.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59••.Lagier-Tourenne C, Baughn M, Rigo F, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci USA. 2013;110(47):e4530–e4539. doi: 10.1073/pnas.1318835110. Describes RNA foci pathology in fibroblasts from C9ORF72 ALS patients and shows that antisense oligonucleotides to multiple regions of the C9ORF72 can mitigate the antisense prevalence. Antisense oligonucleotides administered in vivo were also shown to reduce C9ORF72 levels for up to 17 weeks in adult mice with no overt phenotype. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sareen D, O'rourke JG, Meera P, et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med. 2013;5(208):208ra149. doi: 10.1126/scitranslmed.3007529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gijselinck I, Van Langenhove T, Van Der Zee J, et al. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 2012;11(1):54–65. doi: 10.1016/S1474-4422(11)70261-7. [DOI] [PubMed] [Google Scholar]
- 62.Belzil VV, Gendron TF, Petrucelli L. RNA-mediated toxicity in neurodegenerative disease. Mol Cell Neurosci. 2013;56:406–419. doi: 10.1016/j.mcn.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Majounie E, Renton AE, Mok K, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 2012;11(4):323–330. doi: 10.1016/S1474-4422(12)70043-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hensman Moss DJ, Poulter M, Beck J, et al. C9orf72 expansions are the most common genetic cause of Huntington disease phenocopies. Neurology. 2014;82(4):292–299. doi: 10.1212/WNL.0000000000000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cacace R, Van Cauwenberghe C, Bettens K, et al. C9orf72 G4C2 repeat expansions in Alzheimer's disease and mild cognitive impairment. Neurobiol Aging. 2013;34(6):1712 e1711–e1717. doi: 10.1016/j.neurobiolaging.2012.12.019. [DOI] [PubMed] [Google Scholar]
- 66.Kohli MA, John-Williams K, Rajbhandary R, et al. Repeat expansions in the C9ORF72 gene contribute to Alzheimer's disease in Caucasians. Neurobiol Aging. 2013;34(5):1519. doi: 10.1016/j.neurobiolaging.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Majounie E, Abramzon Y, Renton AE, et al. Repeat expansion in C9ORF72 in Alzheimer's disease. N Engl J Med. 2012;366(3):283–284. doi: 10.1056/NEJMc1113592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nuytemans K, Bademci G, Kohli MM, et al. C9ORF72 intermediate repeat copies are a significant risk factor for Parkinson disease. Ann Hum Genet. 2013;77(5):351–363. doi: 10.1111/ahg.12033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ou SH, Wu F, Harrich D, Garcia-Martinez LF, Gaynor RB. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol. 1995;69(6):3584–3596. doi: 10.1128/jvi.69.6.3584-3596.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang HY, Wang IF, Bose J, Shen CK. Structural diversity and functional implications of the eukaryotic TDP gene family. Genomics. 2004;83(1):130–139. doi: 10.1016/s0888-7543(03)00214-3. [DOI] [PubMed] [Google Scholar]
- 71.Ayala YM, Pantano S, D'ambrogio A, et al. Human, Drosophila, and C. elegans TDP43: nucleic acid binding properties and splicing regulatory function. J Mol Biol. 2005;348(3):575–588. doi: 10.1016/j.jmb.2005.02.038. [DOI] [PubMed] [Google Scholar]
- 72.Lattante S, Rouleau GA, Kabashi E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: summary and update. Hum Mutat. 2013;34(6):812–826. doi: 10.1002/humu.22319. [DOI] [PubMed] [Google Scholar]
- 73.Mackenzie IR, Bigio EH, Ince PG, et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol. 2007;61(5):427–434. doi: 10.1002/ana.21147. [DOI] [PubMed] [Google Scholar]
- 74.Tan CF, Eguchi H, Tagawa A, et al. TDP-43 immunoreactivity in neuronal inclusions in familial amyotrophic lateral sclerosis with or without SOD1 gene mutation. Acta Neuropathol. 2007;113(5):535–542. doi: 10.1007/s00401-007-0206-9. [DOI] [PubMed] [Google Scholar]
- 75.Chio A, Calvo A, Moglia C, et al. A de novo missense mutation of the FUS gene in a ‘true’ sporadic ALS case. Neurobiol Aging. 2011;32(3):553 e523–556. doi: 10.1016/j.neurobiolaging.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Damme PV, Goris A, Race V, et al. The occurrence of mutations in FUS in a Belgian cohort of patients with familial ALS. Eur J Neurol. 2010;17(5):754–756. doi: 10.1111/j.1468-1331.2009.02859.x. [DOI] [PubMed] [Google Scholar]
- 77.Dejesus-Hernandez M, Kocerha J, Finch N, et al. De novo truncating FUS gene mutation as a cause of sporadic amyotrophic lateral sclerosis. Hum Mutat. 2010;31(5):e1377–e1389. doi: 10.1002/humu.21241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hewitt C, Kirby J, Highley JR, et al. Novel FUS/TLS mutations and pathology in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2010;67(4):455–461. doi: 10.1001/archneurol.2010.52. [DOI] [PubMed] [Google Scholar]
- 79.Lai SL, Abramzon Y, Schymick JC, et al. FUS mutations in sporadic amyotrophic lateral sclerosis. Neurobiol Aging. 2011;32(3):550 e551–e554. doi: 10.1016/j.neurobiolaging.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rademakers R, Stewart H, Dejesus-Hernandez M, et al. Fus gene mutations in familial and sporadic amyotrophic lateral sclerosis. Muscle Nerve. 2010;42(2):170–176. doi: 10.1002/mus.21665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tsai CP, Soong BW, Lin KP, Tu PH, Lin JL, Lee YC. FUS, TARDBP, and SOD1 mutations in a Taiwanese cohort with familial ALS. Neurobiol. Aging. 2011;32(3):553 e513–e521. doi: 10.1016/j.neurobiolaging.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 82.Waibel S, Neumann M, Rabe M, Meyer T, Ludolph AC. Novel missense and truncating mutations in FUS/TLS in familial ALS. Neurology. 2010;75(9):815–817. doi: 10.1212/WNL.0b013e3181f07e26. [DOI] [PubMed] [Google Scholar]
- 83.Yamamoto-Watanabe Y, Watanabe M, Okamoto K, et al. A Japanese ALS6 family with mutation R521C in the FUS/TLS gene: a clinical, pathological and genetic report. J Neurol Sci. 2010;296(1–2):59–63. doi: 10.1016/j.jns.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 84.Ticozzi N, Silani V, Leclerc AL, et al. Analysis of FUS gene mutation in familial amyotrophic lateral sclerosis within an Italian cohort. Neurology. 2009;73(15):1180–1185. doi: 10.1212/WNL.0b013e3181bbff05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Corrado L, Del Bo R, Castellotti B, et al. Mutations of FUS gene in sporadic amyotrophic lateral sclerosis. J Med Genet. 2010;47(3):190–194. doi: 10.1136/jmg.2009.071027. [DOI] [PubMed] [Google Scholar]
- 86.Belzil VV, Valdmanis PN, Dion PA, et al. Mutations in FUS cause FALS and SALS in French and French Canadian populations. Neurology. 2009;73(15):1176–1179. doi: 10.1212/WNL.0b013e3181bbfeef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sun Z, Diaz Z, Fang X, et al. Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol. 2011;9(4):e1000614. doi: 10.1371/journal.pbio.1000614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zinszner H, Sok J, Immanuel D, Yin Y, Ron D. TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J Cell Sci. 1997;110(Pt 15):1741–1750. doi: 10.1242/jcs.110.15.1741. [DOI] [PubMed] [Google Scholar]
- 89.Dormann D, Rodde R, Edbauer D, et al. ALS-associated fused in sarcoma (FUS) mutations disrupt transportin-mediated nuclear import. EMBO J. 2010;29(16):2841–2857. doi: 10.1038/emboj.2010.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bertolotti A, Lutz Y, Heard DJ, Chambon P, Tora L. hTAF(II)68, a novel RNA/ssDNA-binding protein with homology to the pro-oncoproteins TLS/FUS and EWS is associated with both TFIID and RNA polymerase II. EMBO J. 1996;15(18):5022–5031. [PMC free article] [PubMed] [Google Scholar]
- 91.Powers CA, Mathur M, Raaka BM, Ron D, Samuels HH. TLS (translocated-in-liposarcoma) is a high-affinity interactor for steroid, thyroid hormone, and retinoid receptors. Mol Endocrinol. 1998;12(1):4–18. doi: 10.1210/mend.12.1.0043. [DOI] [PubMed] [Google Scholar]
- 92.Tan AY, Manley JL. TLS inhibits RNA polymerase III transcription. Mol Cell Biol. 2010;30(1):186–196. doi: 10.1128/MCB.00884-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Acharya KK, Govind CK, Shore AN, Stoler MH, Reddi PP. Cis-requirement for the maintenance of round spermatid-specific transcription. Dev Biol. 2006;295(2):781–790. doi: 10.1016/j.ydbio.2006.04.443. [DOI] [PubMed] [Google Scholar]
- 94.Kuo PH, Doudeva LG, Wang YT, Shen CK, Yuan HS. Structural insights into TDP-43 in nucleic-acid binding and domain interactions. Nucleic Acids Res. 2009;37(6):1799–1808. doi: 10.1093/nar/gkp013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tollervey JR, Curk T, Rogelj B, et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci. 2011;14(4):452–458. doi: 10.1038/nn.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hoell JI, Larsson E, Runge S, et al. RNA targets of wild-type and mutant FET family proteins. Nat Struct Mol Biol. 2011;18(12):1428–1431. doi: 10.1038/nsmb.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rogelj B, Easton LE, Bogu GK, et al. Widespread binding of FUS along nascent RNA regulates alternative splicing in the brain. Sci Rep. 2012;2:603. doi: 10.1038/srep00603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rothstein JD, Dykes-Hoberg M, Pardo CA, et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16(3):675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 99.Bristol LA, Rothstein JD. Glutamate transporter gene expression in amyotrophic lateral sclerosis motor cortex. Ann Neurol. 1996;39(5):676–679. doi: 10.1002/ana.410390519. [DOI] [PubMed] [Google Scholar]
- 100.Ayala YM, Zago P, D'ambrogio A, et al. Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci. 2008;121(Pt 22):3778–3785. doi: 10.1242/jcs.038950. [DOI] [PubMed] [Google Scholar]
- 101.Ling SC, Albuquerque CP, Han JS, et al. ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS. Proc Natl Acad Sci USA. 2010;107(30):13318–13323. doi: 10.1073/pnas.1008227107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dewey CM, Cenik B, Sephton CF, et al. TDP-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Mol Cell Biol. 2011;31(5):1098–1108. doi: 10.1128/MCB.01279-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mcdonald KK, Aulas A, Destroismaisons L, et al. TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum Mol Genet. 2011;20(7):1400–1410. doi: 10.1093/hmg/ddr021. [DOI] [PubMed] [Google Scholar]
- 104.Meyerowitz J, Parker SJ, Vella LJ, et al. C-Jun N-terminal kinase controls TDP-43 accumulation in stress granules induced by oxidative stress. Mol Neurodegener. 2011;6:57. doi: 10.1186/1750-1326-6-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Vance C, Scotter EL, Nishimura AL, et al. ALS mutant FUS disrupts nuclear localization and sequesters wild-type FUS within cytoplasmic stress granules. Hum Mol Genet. 2013;22(13):2676–2688. doi: 10.1093/hmg/ddt117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Donnelly CJ, Fainzilber M, Twiss JL. Subcellular communication through RNA transport and localized protein synthesis. Traffic. 2010;11(12):1498–1505. doi: 10.1111/j.1600-0854.2010.01118.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Donnelly CJ, Willis DE, Xu M, et al. Limited availability of ZBP1 restricts axonal mRNA localization and nerve regeneration capacity. EMBO J. 2011;30(22):4665–4677. doi: 10.1038/emboj.2011.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Donnelly CJ, Park M, Spillane M, et al. Axonally synthesized beta-actin and GAP-43 proteins support distinct modes of axonal growth. J Neurosci. 2013;33(8):3311–3322. doi: 10.1523/JNEUROSCI.1722-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liu-Yesucevitz L, Bassell GJ, Gitler AD, et al. Local RNA translation at the synapse and in disease. J Neurosci. 2011;31(45):16086–16093. doi: 10.1523/JNEUROSCI.4105-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Li YR, King OD, Shorter J, Gitler AD. Stress granules as crucibles of ALS pathogenesis. J Cell Biol. 2013;201(3):361–372. doi: 10.1083/jcb.201302044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Johnson BS, Snead D, Lee JJ, Mccaffery JM, Shorter J, Gitler AD. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem. 2009;284(30):20329–20339. doi: 10.1074/jbc.M109.010264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mackenzie IR, Neumann M. FET proteins in frontotemporal dementia and amyotrophic lateral sclerosis. Brain Res. 2012;1462:40–43. doi: 10.1016/j.brainres.2011.12.010. [DOI] [PubMed] [Google Scholar]
- 113.Neumann M, Bentmann E, Dormann D, et al. FET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from amyotrophic lateral sclerosis with FUS mutations. Brain. 2011;134(Pt 9):2595–2609. doi: 10.1093/brain/awr201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Van Blitterswijk M, Landers JE. RNA processing pathways in amyotrophic lateral sclerosis. Neurogenetics. 2010;11(3):275–290. doi: 10.1007/s10048-010-0239-4. [DOI] [PubMed] [Google Scholar]
- 115.Salton M, Elkon R, Borodina T, et al. Matrin 3 binds and stabilizes mRNA. PLoS ONE. 2011;6(8):e23882. doi: 10.1371/journal.pone.0023882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hock J, Weinmann L, Ender C, et al. Proteomic and functional analysis of Argonaute-containing mRNA-protein complexes in human cells. EMBO Rep. 2007;8(11):1052–1060. doi: 10.1038/sj.embor.7401088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Russell AP, Wada S, Vergani L, et al. Disruption of skeletal muscle mitochondrial network genes and miRNAs in amyotrophic lateral sclerosis. Neurobiol Dis. 2012;49C:107–117. doi: 10.1016/j.nbd.2012.08.015. [DOI] [PubMed] [Google Scholar]
- 118.Williams AH, Valdez G, Moresi V, et al. MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science. 2009;326(5959):1549–1554. doi: 10.1126/science.1181046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chen X, Ba Y, Ma L, et al. Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008;18(10):997–1006. doi: 10.1038/cr.2008.282. [DOI] [PubMed] [Google Scholar]
- 120.Etheridge A, Lee I, Hood L, Galas D, Wang K. Extracellular microRNA: a new source of biomarkers. Mutat Res. 2011;717(1–2):85–90. doi: 10.1016/j.mrfmmm.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Seeburg PH. A-to-I editing: new and old sites, functions and speculations. Neuron. 2002;35(1):17–20. doi: 10.1016/s0896-6273(02)00760-2. [DOI] [PubMed] [Google Scholar]
- 122.Puchalski RB, Louis JC, Brose N, et al. Selective RNA editing and subunit assembly of native glutamate receptors. Neuron. 1994;13(1):131–147. doi: 10.1016/0896-6273(94)90464-2. [DOI] [PubMed] [Google Scholar]
- 123.Brusa R, Zimmermann F, Koh DS, et al. Early-onset epilepsy and postnatal lethality associated with an editing-deficient GluR-B allele in mice. Science. 1995;270(5242):1677–1680. doi: 10.1126/science.270.5242.1677. [DOI] [PubMed] [Google Scholar]
- 124.Yamashita T, Hideyama T, Teramoto S, Kwak S. The abnormal processing of TDP-43 is not an upstream event of reduced ADAR2 activity in ALS motor neurons. Neurosci Res. 2012;73(2):153–160. doi: 10.1016/j.neures.2012.02.015. [DOI] [PubMed] [Google Scholar]
- 125.La Spada AR, Taylor JP. Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat Rev Genet. 2010;11(4):247–258. doi: 10.1038/nrg2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Caillet-Boudin ML, Fernandez-Gomez FJ, Tran H, Dhaenens CM, Buee L, Sergeant N. Brain pathology in myotonic dystrophy: when tauopathy meets spliceopathy and RNAopathy. Front Mol Neurosci. 2014;6:57. doi: 10.3389/fnmol.2013.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zu T, Gibbens B, Doty NS, et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci USA. 2011;108(1):260–265. doi: 10.1073/pnas.1013343108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mori K, Weng SM, Arzberger T, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339(6125):1335–1338. doi: 10.1126/science.1232927. [DOI] [PubMed] [Google Scholar]
- 129.Ash PE, Bieniek KF, Gendron TF, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77(4):639–646. doi: 10.1016/j.neuron.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Vatovec S, Kovanda A, Rogelj B. Unconventional features of C9ORF72 expanded repeat in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Neurobiol Aging. 2014;35(10):e2412. doi: 10.1016/j.neurobiolaging.2014.04.015. [DOI] [PubMed] [Google Scholar]
- 131.Xu Z, Poidevin M, Li X, et al. Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc Natl Acad Sci USA. 2013;110(19):7778–7783. doi: 10.1073/pnas.1219643110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zu T, Liu Y, Banez-Coronel M, et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci USA. 2013;110(51):E4968–E4977. doi: 10.1073/pnas.1315438110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mizielinska S, Gronke S, Niccoli T, et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science. 2014;345(6201):1192–1194. doi: 10.1126/science.1256800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kwon I, Xiang S, Kato M, et al. Poly-dipeptides encoded by the C9ORF72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science. 2014;345(6201):1139–1145. doi: 10.1126/science.1254917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.May S, Hornburg D, Schludi MH, et al. C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol. 2014;128(4):485–503. doi: 10.1007/s00401-014-1329-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gendron TF, Cosio DM, Petrucelli L. c9RAN translation: a potential therapeutic target for the treatment of amyotrophic lateral sclerosis and frontotemporal dementia. Expert Opin Ther Targets. 2013;17(9):991–995. doi: 10.1517/14728222.2013.818659. [DOI] [PubMed] [Google Scholar]
- 137.Zu T, Liu Y, Banez-Coronel M, et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci USA. 2013;110(51):e4968–e4977. doi: 10.1073/pnas.1315438110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Suzuki N, Maroof AM, Merkle FT, et al. The mouse C9ORF72 ortholog is enriched in neurons known to degenerate in ALS and FTD. Nat Neurosci. 2013;16(12):1725–1727. doi: 10.1038/nn.3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ciura S, Lattante S, Le Ber I, et al. Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Ann Neurol. 2013;74(2):180–187. doi: 10.1002/ana.23946. [DOI] [PubMed] [Google Scholar]
- 140.Kole R, Krainer AR, Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov. 2012;11(2):125–140. doi: 10.1038/nrd3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Malik R, Roy I. Making sense of therapeutics using antisense technology. Expert Opin Drug Discov. 2011;6(5):507–526. doi: 10.1517/17460441.2011.565744. [DOI] [PubMed] [Google Scholar]
- 142.Miller TM, Pestronk A, David W, et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a Phase 1, randomised, first-in-man study. Lancet Neurol. 2013;12(5):435–442. doi: 10.1016/S1474-4422(13)70061-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Arambula JF, Ramisetty SR, Baranger AM, Zimmerman SC. A simple ligand that selectively targets CUG trinucleotide repeats and inhibits MBNL protein binding. Proc Natl Acad Sci USA. 2009;106(38):16068–16073. doi: 10.1073/pnas.0901824106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mulders SA, Van Den Broek WJ, Wheeler TM, et al. Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. Proc Natl Acad Sci USA. 2009;106(33):13915–13920. doi: 10.1073/pnas.0905780106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145••.Wheeler TM, Sobczak K, Lueck JD, et al. Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science. 2009;325(5938):336–339. doi: 10.1126/science.1173110. Described the successful use of morpholino DNA antisense oligonucleotides targeted at the CUG exp RNA in a mouse model of DM1. Neutralizing the RNA and releasing MBLN1 from the repeat reversed splicing abnormalities and eliminated CAG RNA foci leading to the loss of myotonia in this mouse model. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Warf MB, Nakamori M, Matthys CM, Thornton CA, Berglund JA. Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proc Natl Acad Sci USA. 2009;106(44):18551–18556. doi: 10.1073/pnas.0903234106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lagier-Tourenne C, Baughn M, Rigo F, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci USA. 2013;110(47):e4530–e4539. doi: 10.1073/pnas.1318835110. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sareen D, O'Rourke JG, Meera P, et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med. 2013;5(208):208ra149. doi: 10.1126/scitranslmed.3007529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Therrien M, Rouleau GA, Dion PA, Parker JA. Deletion of C9ORF72 results in motor neuron degeneration and stress sensitivity in C. elegans. PLoS ONE. 2013;8(12):e83450. doi: 10.1371/journal.pone.0083450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Fratta P, Poulter M, Lashley T, et al. Homozygosity for the C9orf72 GGGGCC repeat expansion in frontotemporal dementia. Acta Neuropathol. 2013;126(3):401–409. doi: 10.1007/s00401-013-1147-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mizielinska S, Lashley T, Norona FE, et al. C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol. 2013;126(6):845–857. doi: 10.1007/s00401-013-1200-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Grimm D, Kay MA. Therapeutic application of RNAi: is mRNA targeting finally ready for prime time? J Clin Invest. 2007;117(12):3633–3641. doi: 10.1172/JCI34129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Grimm D, Kay MA. RNAi and gene therapy: a mutual attraction. Hematology Am Soc Hematol Educ Program. 2007;2007:473–481. doi: 10.1182/asheducation-2007.1.473. [DOI] [PubMed] [Google Scholar]
- 154.De Ynigo-Mojado L, Martin-Ruiz I, Sutherland JD. Efficient allele-specific targeting of LRRK2 R1441 mutations mediated by RNAi. PLoS ONE. 2011;6(6):e21352. doi: 10.1371/journal.pone.0021352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Fiszer A, Mykowska A, Krzyzosiak WJ. Inhibition of mutant huntingtin expression by RNA duplex targeting expanded CAG repeats. Nucleic Acids Res. 2011;39(13):5578–5585. doi: 10.1093/nar/gkr156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lombardi MS, Jaspers L, Spronkmans C, et al. A majority of Huntington's disease patients may be treatable by individualized allele-specific RNA interference. Exp Neurol. 2009;217(2):312–319. doi: 10.1016/j.expneurol.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 157.Miller VM, Gouvion CM, Davidson BL, Paulson HL. Targeting Alzheimer's disease genes with RNA interference: an efficient strategy for silencing mutant alleles. Nucleic Acids Res. 2004;32(2):661–668. doi: 10.1093/nar/gkh208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Rodriguez-Lebron E, Gouvion CM, Moore SA, Davidson BL, Paulson HL. Allele-specific RNAi mitigates phenotypic progression in a transgenic model of Alzheimer's disease. Mol Ther. 2009;17(9):1563–1573. doi: 10.1038/mt.2009.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Van Bilsen PH, Jaspers L, Lombardi MS, Odekerken JC, Burright EN, Kaemmerer WF. Identification and allele-specific silencing of the mutant huntingtin allele in Huntington's disease patient-derived fibroblasts. Hum Gene Ther. 2008;19(7):710–719. doi: 10.1089/hum.2007.116. [DOI] [PubMed] [Google Scholar]
- 160.Nishimura AL, Shum C, Scotter EL, et al. Allele-specific knockdown of ALS-associated mutant TDP-43 in neural stem cells derived from induced pluripotent stem cells. PLoS One. 2014;9(3):e91269. doi: 10.1371/journal.pone.0091269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ding H, Schwarz DS, Keene A, et al. Selective silencing by RNAi of a dominant allele that causes amyotrophic lateral sclerosis. Aging Cell. 2003;2(4):209–217. doi: 10.1046/j.1474-9728.2003.00054.x. [DOI] [PubMed] [Google Scholar]
- 162.Maxwell MM, Pasinelli P, Kazantsev AG, Brown RH., Jr RNA interference-mediated silencing of mutant superoxide dismutase rescues cyclosporin A-induced death in cultured neuroblastoma cells. Proc Natl Acad Sci USA. 2004;101(9):3178–3183. doi: 10.1073/pnas.0308726100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ralph GS, Radcliffe PA, Day DM, et al. Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model. Nat Med. 2005;11(4):429–433. doi: 10.1038/nm1205. [DOI] [PubMed] [Google Scholar]
- 164.Raoul C, Abbas-Terki T, Bensadoun JC, et al. Lentiviral-mediated silencing of SOD1 through RNA interference retards disease onset and progression in a mouse model of ALS. Nat Med. 2005;11(4):423–428. doi: 10.1038/nm1207. [DOI] [PubMed] [Google Scholar]
- 165.Burnett JC, Rossi JJ. RNA-based therapeutics: current progress and future prospects. Chem Biol. 2012;19(1):60–71. doi: 10.1016/j.chembiol.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Fiszer A, Olejniczak M, Switonski PM, et al. An evaluation of oligonucleotide-based therapeutic strategies for polyQ diseases. BMC Mol Biol. 2012;13:6. doi: 10.1186/1471-2199-13-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kiang AS, Palfi A, Ader M, et al. Toward a gene therapy for dominant disease: validation of an RNA interference-based mutation-independent approach. Mol Ther. 2005;12(3):555–561. doi: 10.1016/j.ymthe.2005.03.028. [DOI] [PubMed] [Google Scholar]
- 168.Kubodera T, Yamada H, Anzai M, et al. In vivo application of an RNAi strategy for the selective suppression of a mutant allele. Hum Gene Ther. 2011;22(1):27–34. doi: 10.1089/hum.2010.054. [DOI] [PubMed] [Google Scholar]
- 169.Kubodera T, Yokota T, Ishikawa K, Mizusawa H. New RNAi strategy for selective suppression of a mutant allele in polyglutamine disease. Oligonucleotides. 2005;15(4):298–302. doi: 10.1089/oli.2005.15.298. [DOI] [PubMed] [Google Scholar]
- 170.Xia XG, Zhou H, Zhou S, Yu Y, Wu R, Xu Z. An RNAi strategy for treatment of amyotrophic lateral sclerosis caused by mutant Cu, Zn superoxide dismutase. J Neurochem. 2005;92(2):362–367. doi: 10.1111/j.1471-4159.2004.02860.x. [DOI] [PubMed] [Google Scholar]
- 171.Gastaldi L, Battaglia L, Peira E, et al. Solid lipid nanoparticles as vehicles of drugs to the brain: current state of the art. Eur J Pharm Biopharm. 2014;87(3):433–444. doi: 10.1016/j.ejpb.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 172.Perez-Martinez FC, Carrion B, Cena V. The use of nanoparticles for gene therapy in the nervous system. J Alzheimers Dis. 2012;31(4):697–710. doi: 10.3233/JAD-2012-120661. [DOI] [PubMed] [Google Scholar]
- 173.Rungta RL, Choi HB, Lin PJ, et al. Lipid nanoparticle delivery of siRNA to silence neuronal gene expression in the brain. Mol Ther Nucleic Acids. 2013;2:e136. doi: 10.1038/mtna.2013.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Porensky PN, Burghes AH. Antisense oligonucleotides for the treatment of spinal muscular atrophy. Hum Gene Ther. 2013;24(5):489–498. doi: 10.1089/hum.2012.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Rigo F, Chun SJ, Norris DA, et al. Pharmacology of a central nervous system delivered 2′-o-methoxyethyl-modified survival of motor neuron splicing oligonucleotide in mice and nonhuman primates. J Pharmacol Exp Ther. 2014;350(1):46–55. doi: 10.1124/jpet.113.212407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Arnold ES, Ling SC, Huelga SC, et al. ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc Natl Acad Sci USA. 2013;110(8):e736–e745. doi: 10.1073/pnas.1222809110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Orozco D, Edbauer D. FUS-mediated alternative splicing in the nervous system: consequences for ALS and FTLD. J Mol Med. 2013;91(12):1343–1354. doi: 10.1007/s00109-013-1077-2. [DOI] [PubMed] [Google Scholar]
- 178.Yang C, Wang H, Qiao T, et al. Partial loss of TDP-43 function causes phenotypes of amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 2014;111(12):e1121–e1129. doi: 10.1073/pnas.1322641111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Krutzfeldt J, Rajewsky N, Braich R, et al. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438(7068):685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 180.Tan L, Yu JT, Tan L. Causes and consequences of MicroRNA dysregulation in neurodegenerative diseases. Mol Neurobiol. 2014 doi: 10.1007/s12035-014-8803-9. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 181.Gascon E, Gao FB. The emerging roles of microRNAs in the pathogenesis of frontotemporal dementia-amyotrophic lateral sclerosis (FTD-ALS) spectrum disorders. J Neurogenet. 2014;28(1–2):30–40. doi: 10.3109/01677063.2013.876021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Shinde S, Arora N, Bhadra U. A complex network of MicroRNAs expressed in brain and genes associated with amyotrophic lateral sclerosis. Int J Genomics. 2013;2013:383024. doi: 10.1155/2013/383024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Koval ED, Shaner C, Zhang P, et al. Method for widespread microRNA-155 inhibition prolongs survival in ALS-model mice. Hum Mol Genet. 2013;22(20):4127–4135. doi: 10.1093/hmg/ddt261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Yue J. miRNA and vascular cell movement. Adv Drug Deliv Rev. 2011;63(8):616–622. doi: 10.1016/j.addr.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Broderick JA, Zamore PD. MicroRNA therapeutics. Gene Ther. 2011;18(12):1104–1110. doi: 10.1038/gt.2011.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Care A, Catalucci D, Felicetti F, et al. MicroRNA-133 controls cardiac hypertrophy. Nat Med. 2007;13(5):613–618. doi: 10.1038/nm1582. [DOI] [PubMed] [Google Scholar]
- 187.Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: Competitive inhibitors of small RNAs in mammalian cells. Nat Methods. 2007;4(9):721–726. doi: 10.1038/nmeth1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Gareiss PC, Sobczak K, Mcnaughton BR, Palde PB, Thornton CA, Miller BL. Dynamic combinatorial selection of molecules capable of inhibiting the (CUG) repeat RNA-MBNL1 interaction in vitro: discovery of lead compounds targeting myotonic dystrophy (DM1) J Am Chem Soc. 2008;130(48):16254–16261. doi: 10.1021/ja804398y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Chen CZ, Sobczak K, Hoskins J, et al. Two high-throughput screening assays for aberrant RNA-protein interactions in myotonic dystrophy type 1. Anal Bioanal Chem. 2012;402(5):1889–1898. doi: 10.1007/s00216-011-5604-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Childs-Disney JL, Hoskins J, Rzuczek SG, Thornton CA, Disney MD. Rationally designed small molecules targeting the RNA that causes myotonic dystrophy type 1 are potently bioactive. ACS Chem Biol. 2012;7(5):856–862. doi: 10.1021/cb200408a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Haeusler ARD, CJ Periz G, Simko EAJ, Shaw PG, Kim MS, Maragakis NJ, Troncoso JC, Pandey A, Sattler R, Rothstein JD, Wang J. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature. 2014;507(7491):195–200. doi: 10.1038/nature13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Lee YB, Chen HJ, Peres JN, et al. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 2013;5(5):1178–1186. doi: 10.1016/j.celrep.2013.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Almeida S, Gascon E, Tran H, et al. Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol. 2013;126(3):385–399. doi: 10.1007/s00401-013-1149-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Reddy K, Zamiri B, Stanley SY, Macgregor RB, Jr, Pearson CE. The disease-associated r(GGGGCC)n repeat from the C9orf72 gene forms tract length-dependent uni- and multimolecular RNA G-quadruplex structures. J Biol Chem. 2013;288(14):9860–9866. doi: 10.1074/jbc.C113.452532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Fratta P, Mizielinska S, Nicoll AJ, et al. C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Sci Rep. 2012;2:1016. doi: 10.1038/srep01016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Zamiri B, Reddy K, Macgregor RB, Jr, Pearson CE. TMPyP4 porphyrin distorts RNA G-quadruplex structures of the disease-associated r(GGGGCC)n repeat of the C9orf72 gene and blocks interaction of RNA-binding proteins. J Biol Chem. 2014;289(8):4653–4659. doi: 10.1074/jbc.C113.502336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Childs-Disney JL, Yildirim I, Park H, et al. Structure of the myotonic dystrophy type 2 RNA and designed small molecules that reduce toxicity. ACS Chem Biol. 2014;9(2):53–550. doi: 10.1021/cb4007387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Tran T, Childs-Disney JL, Liu B, Guan L, Rzuczek S, Disney MD. Targeting the r(CGG)repeats that cause FXTAS with modularly assembled small molecules and oligonucleotides. ACS Chem Biol. 2014;9(4):904–912. doi: 10.1021/cb400875u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Guan L, Disney MD. Recent advances in developing small molecules targeting RNA. ACS Chem Biol. 2012;7(1):73–86. doi: 10.1021/cb200447r. [DOI] [PubMed] [Google Scholar]
- 200.Disney MD, Lee MM, Pushechnikov A, Childs-Disney JL. The role of flexibility in the rational design of modularly assembled ligands targeting the RNAs that cause the myotonic dystrophies. ChemBioChem. 2010;(3):375–382. doi: 10.1002/cbic.200900716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Boyd JD, Lee-Armandt JP, Feiler MS, et al. A high-content screen identifies novel compounds that inhibit stress-induced TDP-43 cellular aggregation and associated cytotoxicity. J Biomol Screen. 2014;19(1):44–56. doi: 10.1177/1087057113501553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Barmada SJ, Serio A, Arjun A, et al. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat Chem Biol. 2014;10(8):677–685. doi: 10.1038/nchembio.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Rothstein JD, Patel S, Regan MR, et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433(7021):73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- 204.Lipski J, Wan CK, Bai JZ, Pi R, Li D, Donnelly D. Neuroprotective potential of ceftriaxone in in vitro models of stroke. Neuroscience. 2007;146(2):617–629. doi: 10.1016/j.neuroscience.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 205.Miller BR, Dorner JL, White RD, Sengelaub DR, Rebeck GV. Ceftriaxone elevates GLT1 expression and improves the Huntington's disease phenotype in R6/2 mice. Soc Neurosci Abs. 2006;36(677):671. [Google Scholar]
- 206.Mineur YS, Picciotto MR, Sanacora G. Antidepressant-like effects of ceftriaxone in male C57BL/6J mice. Biol Psychiatry. 2006;61(2):250–252. doi: 10.1016/j.biopsych.2006.04.037. [DOI] [PubMed] [Google Scholar]
- 207.Rumbaugh JA, Li G, Rothstein J, Nath A. Ceftriaxone protects against the neurotoxicity of human immunodeficiency virus proteins. J Neurovirol. 2007;13(2):168–172. doi: 10.1080/13550280601178218. [DOI] [PubMed] [Google Scholar]
- 208.Kong Q, Chang LC, Takahashi K, et al. Small-molecule activator of glutamate transporter EAAT2 translation provides neuroprotection. J Clin Invest. 2014;124(3):1255–1267. doi: 10.1172/JCI66163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Colton CK, Kong Q, Lai L, et al. Identification of translational activators of glial glutamate transporter EAAT2 through cell-based high-throughput screening: an approach to prevent excitotoxicity. J Biomol Screen. 2010;15(6):653–662. doi: 10.1177/1087057110370998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Jin P, Duan R, Qurashi A, et al. Pur alpha binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron. 2007;55(4):556–564. doi: 10.1016/j.neuron.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Kanadia RN, Shin J, Yuan Y, et al. Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc Natl Acad Sci USA. 2006;103(31):11748–11753. doi: 10.1073/pnas.0604970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Xu Z, Poidevin M, Li X, et al. Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc Natl Acad Sci USA. 2013;110(19):7778–7783. doi: 10.1073/pnas.1219643110. [DOI] [PMC free article] [PubMed] [Google Scholar]