

Abstract
Animal and emerging clinical studies have demonstrated that increased ventricular fibrosis in a setting of reduced repolarization reserve promotes early afterdepolarizations (EADs) and triggered activity that can initiate ventricular tachycardia and ventricular fibrillation (VT/VF). Increased ventricular fibrosis plays a key facilitatory role in allowing oxidative and metabolic stress-induced EADs to manifest as triggered activity causing VT/VF. The lack of such an arrhythmogenic effect by the same stressors in normal non-fibrotic hearts highlights the importance of fibrosis in the initiation of VT/VF. These findings suggest that antifibrotic therapy combined with therapy designed to increase ventricular repolarization reserve may act synergistically to reduce the risk of sudden cardiac death.
Keywords: Cardiac fibrosis, ventricular fibrillation, repolarization reserve, oxidative stress, anti-fibrotic therapy
1. Introduction
Classically, increased cardiac fibrosis has been shown to be associated with altered cardiac conduction, causing conduction slowing, block, and reentry in studies on isolated animal and diseased human cardiac tissues [1–3]. Interestingly, similar findings were also demonstrated in isolated Langendorff-perfused explanted human hearts with dilated cardiomyopathy [4,5]. While alterations of cardiac conduction [6,7] and the resulting reentrant wavefront of excitation [8] are uniformly accepted as arrhythmic consequences of increased cardiac fibrosis, recent experimental and computational studies indicate that fibrosis may also importantly modulate the formation of cardiac afterpotentials, notably early afterdepolarizations (EADs),that lead to triggered activity causing atrial fibrillation (AF) [9] and ventricular fibrillation (VF) [10–12]. Taken together, these findings indicate that increased cardiac fibrosis promotes tachyarrhythmias not only by the mechanism of reentry but also by the mechanism of triggered activity, potentially making cardiac fibrosis a highly effective antiarrhythmic target.
In this review, we demonstrate how the interaction of fibrotic ventricles with oxidative or metabolic stress leads to the emergence of EADs, triggered activity, and VF. Specifically, we describe the dynamic scenario starting from cellular EADs that promote triggered activity causing focal ventricular tachycardia (VT), which then degenerates to VF. We also discuss recent experimental and clinical studies that show the potential antiarrhythmic benefits of drug-induced prevention and/or reduction of ventricular fibrosis [13–17].
2.1 The pathology of fibrosis
Cardiac fibrosis develops when the body’s natural wound-healing process becomes altered, causing abnormally elevated fibrosis by mechanisms that still remain poorly defined. Under normal (adaptive) conditions of wound healing, specialized cells known as fibroblasts become activated and transform into myofibroblasts. The myofibroblasts then undergo proliferation, causing increased synthesis of collagen protein in the extracellular matrix composed predominantly of type I collagen and to a lesser extent type III collagen (normal wound healing process). What is initially an adaptive process, perhaps meant to enhance tensile strength, can progress to maladaptive (pathologic) conditions when the “healing” process persists with the development of excessive myocardial fibrosis [15,18–20]. While resident cardiac fibroblasts may be activated and transformed into myofibroblasts, there is also the potential of participation by fibroblasts originating from either endothelial cells via endothelial-mesenchymal transition (EndMT) or from the bone marrow [21,22] and the spleen [23]. For example, it has been shown that transforming growth factor-beta 1 (TGF-β1) induces endothelial cells to undergo EndMT, whereas bone morphogenic protein 7 (BMP-7) preserves the endothelial phenotype. The demonstration that the systemic administration of recombinant human BMP-7 (rhBMP-7) significantly inhibits EndMT and the progression of cardiac fibrosis in mouse models of pressure overload provides new insights into the progression of pathological (maladaptive) cardiac fibrosis [24].
2.2 Aged heart animal model of fibrosis
Atrial and ventricular fibrosis may indeed increase with aging, but fibrosis per se does not promote cardiac arrhythmia [25–28]. Instead, fibrosis provides a substrate that when coupled to a mild form of stress (oxidative or metabolic), which is of no arrhythmic consequence in non-fibrotic hearts, promotes EADs and triggered activity causing VT and VF in fibrotic hearts, as shown in Figure 1. We describe the key role played by increased ventricular fibrosis using the aged rat model exposed to either oxidative stress caused by eitherhydrogen peroxide (H2O2) [10] or glycolytic inhibition (GI) induced by replacing glucose with pyruvate [12]. This substitution deprives the sarcoplasmic reticulum (SR) of high-energy phosphate (ATP) needed for proper reuptake of intracellular calcium from the cytoplasm [29,30].
Figure 1. Simultaneous microelectrode and ECG recordings at the onset of VT/VF in an aged rat heart exposed to 0.1 mM H2O2.
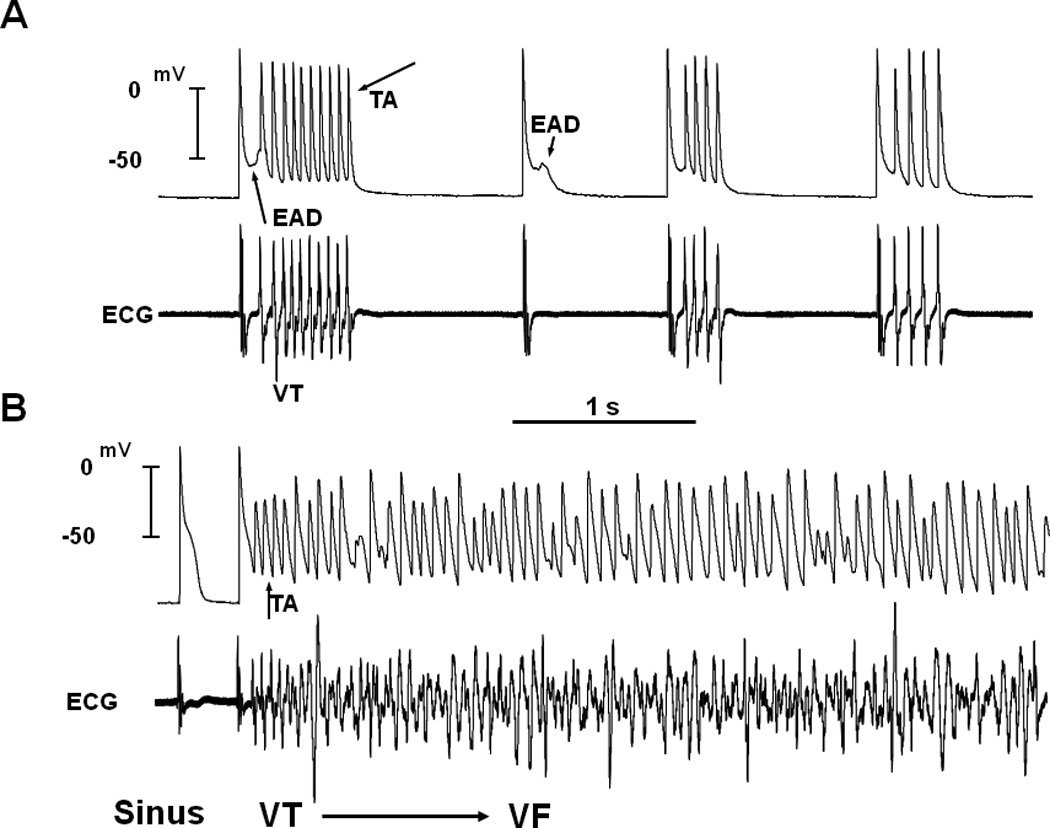
Panel A, onset of early afterdepolarization (EAD)-mediated triggered activity (TA) causing ventricular tachycardia (VT) 5 min after H2O2 exposure. Note the smooth emergence of EAD (upward-pointing arrow) during the isoelectric interval on the ECG followed by a run of 10 TA (downward-pointing arrow) causing non-sustained VT on the ECG. The onset of the EAD precedes the QRS complex of the VT by 8 ms, indicating absence of electrical activity elsewhere in the heart. Two additional short runs of VT with 4 beats each are also shown that follow a single subthreshold EAD (downward-pointing small arrow) with no TA. Panel B shows the degeneration of the TA to VF 15 min after H2O2 exposure. (From reference number 10, Morita et al)
3. Oxidative stress
Hydrogen peroxide (H2O2) is shown to readily promote EADs and triggered activity in isolated rat and rabbit ventricular myocytes by increasing both the L-type Ca channel (ICa-L) and late sodium currents (INa-L) [31–35]. However, this same stress fails to cause EADs in normal non-fibrotic cardiac tissue [10,12]. The discrepancy between non-fibrotic and fibrotic heart tissues in the genesis of EAD-mediated triggered activity either in isolated single myocytes or at the tissue level may result from the “source-to-sink” mismatch [36]. This phenomenon occurs as follows: at a ventricular site, some cells that become greatly stressed and generate EADs (i.e., “source”) may be electrically well coupled to adjoining cells that do not generate EADs and instead repolarize normally back to the resting potential. In this case, such adjoining cells exert a repolarizing influence on the cells that are prone to generate EADs, i.e., “sink effect,” thus suppressing the EADs. Stated otherwise, EAD as the source becomes incapable of overriding the repolarizing sink effect exerted by the electrically coupled normally repolarizing cells, which results in EAD suppression. The importance of the diffusive electrotonic cellular coupling on the modulation of EADs was systematically studied using simulation. For example, when 2 isolated ventricular myocytes were artificially well coupled via a variable electrical resistor, with 1 cell generating EADs and the other cell devoid of EADs, EADs in the first cell were suppressed if the second cell had normal repolarization. However, if the same cell with EADs was uncoupled from the cell with normal repolarization by applying an infinite electrical resistance between the 2 cells (i.e., complete cellular uncoupling, mimicking an isolated single myocyte) the EADs emerged again [37,38]. These single-cell studies were later expanded in a thorough systematic study using 1-, 2-, and 3-dimensionally arranged (1D, 2D, and 3D) tissues to determine the number of contiguous cells with EADs required to override the sink effect and allow the EAD-mediated triggered beats to propagate to the surrounding tissue. For example, using a realistic cardiac cell model, it was shown that in 3D normally-coupled tissue, one needs approximately 700,000 contiguous cells to synchronously produce EADs in order to overcome the source-to-sink mismatch and allow a triggered premature ventricular contraction (PVC) to propagate [36]. In 2D tissue, this number decreases to approximately 7,000, and in a 1D cable to approximately 70. Interestingly, and as expected, the minimum number of cells needed to produce a propagated EAD-induced PVC in 3D tissue decreased by as much as 40-fold when cell-to-cell gap junctional coupling is reduced [36]. Fibrosis, which imposes insulating collagen bands between strands of myocytes (essentially converting 3D tissue into a network of 1D cables), is even more potent at reducing the number of myocytes required for PVC formation [36]. These experimental and simulation studies strongly suggest that the observed discrepancy in EAD production between isolated myocytes and normally coupled cardiac tissue may indeed result from the source-to-sink mismatch. In well-coupled cardiac tissue, the source-to-sink mismatch provides a powerful protective effect for suppression of EAD formation by delinquent cells, preventing the emergence of cardiac arrhythmias. Figure 2 describes schematically the relationships between fibrosis and repolarization reserve reduction under 3 different conditions: the isolated single myocyte, normal tissue, and fibrotic tissue. Based on the findings above, it could be surmised that cardiac diseases that cause myocyte decoupling should facilitate the emergence of EADs when the repolarization reserve is reduced. A number of factors reduce cellular coupling in diseased heart, including gap junction remodeling and fibrosis. Fibrosis forms insulating barriers between cells and groups of cells through the interstitial tissue deposit of collagenous filaments by the proliferating cardiac myofibroblasts [19,20]. We have shown that aging in rats and rabbits manifests not only increased cardiac fibrosis but also down-regulation of the gap junctional connexins43 (C×43) [12,39] that effectively reduce the coupling conductance between cardiac myocytes.
Figure 2. Schematic drawing of the relationship between increased fibrosis and reduced repolarization reserve in the initiation of EADs under 3 different conditions.

Notice that a similar level of stress (0.1 mM H2O2) promotes EADs only at the isolated single cell level and in fibrotic hearts but not in normal hearts.
4. Metabolic stress
Glycolysis accounts for less than 5% of total cellular ATP production in the beating heart under aerobic conditions. However, studies of isolated cardiac myocytes have shown that cardiac stress induced by selectively inhibiting glycolytic ATP production can lead to electromechanical alternans and triggered action potentials, without affecting mitochondrial ATP production or redox state [30,40]. We have shown that fibrotic aged hearts but not non-fibrotic adult hearts are highly vulnerable to EAD-mediated triggered activity causing VT/VF during GI. With GI, simultaneous shortening of the action potential duration (APD) and slowing of intracellular calcium ([Cai2+]) decline rate results in elevation of [Cai+2] during phase 3 repolarization, a condition shown to promote EADs in fibrotic rabbit ventricles [41] and in canine pulmonary veins [42] and atria [43]. Maintained elevation of [Cai+2] enhances the forward mode of the Na+-Ca2+ exchanger (NCX), generating a net depolarizing inward current [42,44]. Inhibition of the NCX prevents GI-mediated EADs, which supports this scenario [12].
5.1 From EADs to focal VT in intact hearts
To gain insight into the cellular mechanisms of focal VT, we performed high-resolution optical mapping of the left ventricular (LV) epicardial surface during GI or oxidative stress. Consistent with the theory of source-to-sink mismatch, the focal VT at the onset of VF originated from the LV region with an intermediate degree (“critical degree”) of fibrosis that was located at the base of the left ventricle. We performed continuous recording of single cell action potentials with a glass microelectrode from the epicardial surface of the LV base, where the focal VT frequently originated. This showed that the focal VT was initiated by an EAD-mediated triggered activity that arose from a mean take-off potential of −51 ± 16 mV (Figure 1). The mean cycle length(CL) of the triggered activity was 66 ± 10 ms and was not significantly different from the mean CL of the VT (70 ± 18 ms). The EAD preceded the QRS complex of a simultaneously recorded ECG by a mean of 8 ± 4 ms and occurred during an isoelectric interval of the ECG. This indicated absence of electrical activity elsewhere in the heart during the EAD formation. The EADs arose when [Cai2+] remained high relative to the diastolic level, activating the NCX, which then provided a net inward depolarizing current promoting EADs [12].
5.2 Transition from focal VT to VF
The EAD-mediated focal VT degenerates within 3 seconds of the VT onset into sustained VF. The focal VT initially propagated as single wavefront from the base of the heart. This promoted spatially discordant APD alternans, causing wavebreak and leading to reentry and subsequent disorganization of the wavefront to multiple irregular wavelets that signal the onset of VF [12]. This dynamic scenario is shown in Figure 3.
Figure 3. Spontaneous initiation of VT/VF in an aged rat heart exposed to 0.1 mM H2O2.

Panel A is an ECG showing the last 4 sinus beats before the sudden onset of VT leading to VF. Panel B shows voltage snapshots of the last beat of the VT (beat #1) and of the first 2 beats of the VF (beats #2 & #3). In each snapshot, activation time in milliseconds is shown at the bottom right with time 0 (arbitrary) coinciding with the onset of beat #1. The red color in the snap shots represents depolarization (DEP) and the blue repolarization (REP) as shown in panel C. The yellow arrows in the snap shots represent the direction of the wavefront propagation with double horizontal lines denoting the site of conduction block. The VT originates from a focal site at the LV base and propagates as a single wavefront towards the apex and undergoes functional conduction block at site 3. The 2 lateral edges of the front, however, continue to propagate laterally (snap shot 98 ms) forming figure-8 reentry (snapshot 108 ms). During the second reentrant wavefront another wavefront emerges from the apical site of the LV (snapshot 122 ms), disrupting the activation pattern and signaling the onset of VF. Panel D shows 3 optical action potentials (labeled 1, 2, and 3) recorded from sites identified on the heart silhouette (Panel E). The 2 downward-pointing blue arrows indicate the direction of propagation from site 1 to site 3 with the red downward-pointing arrow showing block at site 3 followed by retrograde activation (upward-pointing arrow). Notice the emergence of spatially discordant action potential duration (APD) alternans preceding conduction block at site 3 when the front with short APD (S) at site 1 encroaches at a site (site 3) with long APD (L). S indicates short and L long APD. (From reference number 10, Morita et al)
6. Do myocardial cells in the aged heart undergo electrical remodeling?
Although disruption of normal cell-to-cell coupling by fibrosis provides a plausible explanation for the increased susceptibility of aged hearts to EADs and EAD-mediated arrhythmias, there remains the possibility that aged ventricular myocytes, unlike adult myocytes, become exquisitely sensitive to stress-mediated EADs and triggered activity. To test whether this happens, we studied aged and adult isolated myocytes under patch-clamp conditions and subjected them to similar degrees of stresses. No difference was found between the aged and adult isolated myocytes in their abilities to generate EADs [45]. This indicates lack of electrical remodeling at the single-cell level, making a strong case for enhanced fibrosis in the genesis of EAD at the whole-heart level.
7. The role of anti-fibrotic therapy against VT/VF
The link between ventricular fibrosis and the risk of ventricular arrhythmia in the setting of reduced repolarization reserve suggests that targeting both fibrosis and reversal of stress-induced repolarization reserve reduction may be important strategies to prevent VT/VF. We recently have shown that increase in the repolarization reserve by blocking late inward Na+ current with ranolazine suppressed and prevented oxidative stress-induced EADs, triggered activity, and VT/VF in aged fibrotic rat hearts [11]. This pharmacological strategy to suppress EAD-mediated VF appears very promising, as it does not interfere with the normal physiological cardiac ion channel function but instead normalizes the delayed inactivation of the Na current (late INa) while preserving normal excitation-contraction coupling [11]. In addition to increasing the repolarization reserve, new antifibrotic strategies are being developed that hold promise for future effective prevention of sudden cardiac death caused by VF. For example, a recent study found that in patients with hypertrophic cardiomyopathy, myocardial fibrosis as measured by late gadolinium enhancement cardiovascular magnetic resonance (CMR) was an independent predictor of adverse outcome [46]. Interestingly, these investigators found that the extent of myocardial fibrosis was an independent predictor for arrhythmias including sustained VT and VF [46].
In another study Lopez and associates found that patients with chronic heart failure receiving torsemide, a loop diuretic that inhibits the enzyme involved in the myocardial extracellular generation of collagen type I molecules (i.e., procollagen type I carboxy-terminal proteinase or PCP), had reduced myocardial collagen volume fraction (CVF) as assessed in right septal endocardial biopsies [47]. Interestingly, changes in serum PCP activation were positively correlated with changes in CVF in the patients receiving torsemide [47]. These initial clinical studies suggest that ventricular fibrosis may indeed be decreased using antifibrotic therapy; however, its antiarrhythmic significance awaits confirmation in larger patient populations. The emergence of new cardiac imaging techniques for myocardial fibrosis with reasonable degrees of accuracy [48] may help assess the efficacy of antifibrotic therapy for the associated cardiac arrhythmias. Antiarrhythmic therapy would thus be targeted to prevent disproportionate collagen accumulation and myofibroblast proliferation. Such measures will not only prevent arrhythmias, but also help in the management of heart failure. Collectively, there are a wide range of possible preventive antifibrotic treatment options that target TGF-β, endothelin-1 (ET-1), connective tissue growth factor (CTGF), angiotensin II, and platelet-derived growth factor (PDGF) networks [15]. The experimental findings are encouraging, and suggest that reduction of cardiac fibrosis is possible and may indeed reduce the risk of cardiac arrhythmias. For example, studies in rats have shown that pirfenidone mitigates left ventricular fibrosis and dysfunction after myocardial infarction and reduces arrhythmias [49,50] Another animal study showed that the agent relaxin-1 reverses cardiac fibrosis and related cardiac dysfunction [51]. Relaxin is a potent antifibrotic peptide hormone that inhibits fibroblast activation (indicated by suppressed expression of α-smooth muscle actin) and collagen synthesis stimulated by angiotensin II or TGF-β [51]
8. Conclusions
It is hoped that these basic research findings will be translated into treatments for patients at risk for developing cardiac fibrosis-related arrhythmias. To the extent that such a translation will be successful, it is anticipated that a more rational and effective management of patients at risk for VT/VF may be developed. Sudden cardiac death prematurely claims some 300,000 lives each year in the US and 50,000–100,000 lives in Japan, and it remains a global public health problem. New treatment modalities such as reduction of cardiac fibrosis and reversal of repolarization reserve reduction without interfering with excitation-contraction coupling hold future promise.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
This study was supported by AHA Western State Affiliate Research Fellowship Award (0725218Y) and a Grant-in-Aid (0555057Y), and National Institutes of Health Grants P01 HL-78931.
References
- 1.Spach MS, Boineau JP. Microfibrosis produces electrical load variations due to loss of side-to-side cell connections: a major mechanism of structural heart disease arrhythmias. Pacing Clin Electrophysiol. 1997;20:397–413. doi: 10.1111/j.1540-8159.1997.tb06199.x. [DOI] [PubMed] [Google Scholar]
- 2.Spach MS, Dolber PC. Relating extracellular potentials and their derivatives to anisotropic propagation at a microscopic level in human cardiac muscle. Evidence for electrical uncoupling of side-to-side fiber connections with increasing age. Circ Res. 1986;58:356–371. doi: 10.1161/01.res.58.3.356. [DOI] [PubMed] [Google Scholar]
- 3.de Bakker JM, van Capelle FJ, Janse MJ, et al. Slow conduction in the infarcted human heart. 'Zigzag' course of activation. Circulation. 1993;88:915–926. doi: 10.1161/01.cir.88.3.915. [DOI] [PubMed] [Google Scholar]
- 4.Wu TJ, Ong JJ, Hwang C, et al. Characteristics of wave fronts during ventricular fibrillation in human hearts with dilated cardiomyopathy: role of increased fibrosis in the generation of reentry. J Am Coll Cardiol. 1998;32:187–196. doi: 10.1016/s0735-1097(98)00184-3. [DOI] [PubMed] [Google Scholar]
- 5.de Bakker JMT, Coronel R, Tasseron S, et al. Ventricular tachycardia in the infarcted, Langendorff-perfused human heart: role of the arrangement of surviving cardiac fibers. J Am Coll Cardiol. 1990;15:1594–1607. doi: 10.1016/0735-1097(90)92832-m. [DOI] [PubMed] [Google Scholar]
- 6.Spach MS, Heidlage F, Dolber PC, et al. Mechanism of origin of conduction disturbances in aging human atrial bundles: experimental and model study. Heart Rhythm. 2007;4:175–185. doi: 10.1016/j.hrthm.2006.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gaudesius G, Miragoli M, Thomas SP, et al. Coupling of cardiac electrical activity over extended distances by fibroblasts of cardiac origin. Circ Res. 2003;93:421–428. doi: 10.1161/01.RES.0000089258.40661.0C. [DOI] [PubMed] [Google Scholar]
- 8.Spach MS, Dolber PC, Heidlage JF. Interaction of inhomogeneities of repolarization with anisotropic propagation in dog atria. A mechanism for both preventing and initiating reentry. Circ Res. 1989;65:1612–1631. doi: 10.1161/01.res.65.6.1612. [DOI] [PubMed] [Google Scholar]
- 9.Ono N, Hayashi H, Kawase A, et al. Spontaneous atrial fibrillation initiated by triggered activity near the pulmonary veins in aged rats subjected to glycolytic inhibition. Am J Physiol Heart Circ Physiol. 2007;292:639–648. doi: 10.1152/ajpheart.00445.2006. [DOI] [PubMed] [Google Scholar]
- 10.Morita N, Sovari AA, Xie Y, et al. Increased susceptibility of aged hearts to ventricular fibrillation during oxidative stress. Am J Physiol Heart Circ Physiol. 2009;297:H1594–H605. doi: 10.1152/ajpheart.00579.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morita N, Lee JH, Xie Y, et al. Suppression of re-entrant and multifocal ventricular fibrillation by the late sodium current blocker ranolazine. J Am Coll Cardiol. 2011;57:366–375. doi: 10.1016/j.jacc.2010.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morita N, Lee JH, Bapat A, et al. Glycolytic inhibition causes spontaneous ventricular fibrillation in aged hearts. Am J Physiol Heart Circ Physiol. 2011;301:H180–H191. doi: 10.1152/ajpheart.00128.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin CS, Pan CH. Regulatory mechanisms of atrial fibrotic remodeling in atrial fibrillation. Cell Mol Life Sci. 2008;65:1489–1508. doi: 10.1007/s00018-008-7408-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Mello WC. Beneficial effect of eplerenone on cardiac remodelling and electrical properties of the failing heart. J Renin Angiotensin Aldosterone Syst. 2006;7:40–46. doi: 10.3317/jraas.2006.005. [DOI] [PubMed] [Google Scholar]
- 15.Leask A. Potential therapeutic targets for cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ Res. 2010;106:1675–1680. doi: 10.1161/CIRCRESAHA.110.217737. [DOI] [PubMed] [Google Scholar]
- 16.Ehrlich JR, Biliczki P, Hohnloser SH, et al. Atrial-selective approaches for the treatment of atrial fibrillation. J Am Coll Cardiol. 2008;51:787–792. doi: 10.1016/j.jacc.2007.08.067. [DOI] [PubMed] [Google Scholar]
- 17.Madrid AH, Escobar C, Rebollo JM, et al. Angiotensin receptor blocker as adjunctive therapy for rhythm control in atrial fibrillation: results of the irbesartan-amiodarone trial. Card Electrophysiol Rev. 2003;7:243–246. doi: 10.1023/B:CEPR.0000012391.95928.d2. [DOI] [PubMed] [Google Scholar]
- 18.Baudino TA, Carver W, Giles W, et al. Cardiac fibroblasts: friend or foe? Am J Physiol Heart Circ Physiol. 2006;291:H1015–H1026. doi: 10.1152/ajpheart.00023.2006. [DOI] [PubMed] [Google Scholar]
- 19.Weber KT, Brilla CG. Pathological hypertrophy and cardiac interstitium. Fibrosis and renin-angiotensin-aldosterone system. Circulation. 1991;83:1849–1865. doi: 10.1161/01.cir.83.6.1849. [DOI] [PubMed] [Google Scholar]
- 20.Wynn TA. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest. 2007;117:524–529. doi: 10.1172/JCI31487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takahashi T, Kalka C, Masuda H, et al. Ischemia- and cytokine-induced mobilization of bone marrow-derived endothelial progenitor cells for neovascularization. Nat Med. 1999;5:434–438. doi: 10.1038/7434. [DOI] [PubMed] [Google Scholar]
- 22.Orlic D, Kajstura J, Chimenti S, et al. Bone marrow cells regenerate infarcted myocardium. Nature. 2001;410:701–705. doi: 10.1038/35070587. [DOI] [PubMed] [Google Scholar]
- 23.Yamamoto M, Sobue G, Yamamoto K, et al. Expression of mRNAs for neurotrophic factors (NGF, BDNF, NT-3, and GDNF) and their receptors (p75NGFR, trkA, trkB, and trkC) in the adult human peripheral nervous system and nonneural tissues. Neurochem Res. 1996;21:929–938. doi: 10.1007/BF02532343. [DOI] [PubMed] [Google Scholar]
- 24.Zeisberg EM, Tarnavski O, Zeisberg M, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 25.Boyle AJ, Shih H, Hwang J, et al. Cardiomyopathy of aging in the mammalian heart is characterized by myocardial hypertrophy, fibrosis and a predisposition towards cardiomyocyte apoptosis and autophagy. Exp Gerontol. 2011;46:549–559. doi: 10.1016/j.exger.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eghbali M, Eghbali M, Robinson TF, et al. Collagen accumulation in heart ventricles as a function of growth and aging. Cardiovasc Res. 1989;23:723–729. doi: 10.1093/cvr/23.8.723. [DOI] [PubMed] [Google Scholar]
- 27.de Souza RR. Aging of myocardial collagen. Biogerontology. 2002;3:325–335. doi: 10.1023/a:1021312027486. [DOI] [PubMed] [Google Scholar]
- 28.Hacker TA, McKiernan SH, Douglas PS, et al. Age-related changes in cardiac structure and function in Fischer 344×Brown Norway hybrid rats. Am J Physiol Heart Circ Physiol. 2006;290:H304–H311. doi: 10.1152/ajpheart.00290.2005. [DOI] [PubMed] [Google Scholar]
- 29.Huser J, Wang YG, Sheehan KA, et al. Functional coupling between glycolysis and excitation-contraction coupling underlies alternans in cat heart cells. J Physiol. 2000;524:795–806. doi: 10.1111/j.1469-7793.2000.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kockskamper J, Zima AV, Blatter LA. Modulation of sarcoplasmic reticulum Ca2+ release by glycolysis in cat atrial myocytes. J Physiol. 2005;564:697–714. doi: 10.1113/jphysiol.2004.078782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ward CA, Giles WR. Ionic mechanism of the effects of hydrogen peroxide in rat ventricular myocytes. J Physiol. 1997;500:631–642. doi: 10.1113/jphysiol.1997.sp022048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wagner S, Dybkova N, Rasenack EC, et al. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest. 2006;116:3127–3138. doi: 10.1172/JCI26620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song Y, Shryock JC, Wagner S, et al. Blocking late sodium current reduces hydrogen peroxide-induced arrhythmogenic activity and contractile dysfunction. J Pharmacol Exp Ther. 2006;318:214–222. doi: 10.1124/jpet.106.101832. [DOI] [PubMed] [Google Scholar]
- 34.Xie LH, Chen F, Karagueuzian HS, et al. Oxidative stress-induced afterdepolarizations and calmodulin kinase II signaling. Circ Res. 2009;104:79–86. doi: 10.1161/CIRCRESAHA.108.183475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wagner S, Ruff HM, Weber SL, et al. Reactive oxygen species-activated Ca/calmodulin kinase IIdelta is required for late I(Na) augmentation leading to cellular Na and Ca overload. Circ Res. 2011;108:555–565. doi: 10.1161/CIRCRESAHA.110.221911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xie Y, Sato D, Garfinkel A, et al. So little source, so much sink: requirements for afterdepolarizations to propagate in tissue. Biophys J. 2010;99:1408–1415. doi: 10.1016/j.bpj.2010.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huelsing DJ, Spitzer KW, Pollard AE. Electrotonic suppression of early afterdepolarizations in isolated rabbit Purkinje myocytes 1. Am J Physiol Heart Circ Physiol. 2000;279:H250–H259. doi: 10.1152/ajpheart.2000.279.1.H250. [DOI] [PubMed] [Google Scholar]
- 38.Saiz J, Ferrero JM, Jr, Monserrat M, et al. Influence of electrical coupling on early afterdepolarizations in ventricular myocytes. IEEE Trans Biomed Eng. 1999;46:138–147. doi: 10.1109/10.740876. [DOI] [PubMed] [Google Scholar]
- 39.Sato D, Xie LH, Sovari AA, et al. Synchronization of chaotic early afterdepolarizations in the genesis of cardiac arrhythmias. Proc Natl Acad Sci U S A. 2009;106:2983–2988. doi: 10.1073/pnas.0809148106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu KY, Zweier JL, Becker LC. Functional coupling between glycolysis and sarcoplasmic reticulum Ca2+ transport. Circ Res. 1995;77:88–97. doi: 10.1161/01.res.77.1.88. [DOI] [PubMed] [Google Scholar]
- 41.Ogawa M, Morita N, Tang L, et al. Mechanisms of recurrent ventricular fibrillation in a rabbit model of pacing-induced heart failure. Heart Rhythm. 2009;6:784–792. doi: 10.1016/j.hrthm.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Patterson E, Lazzara R, Szabo B, et al. Sodium-calcium exchange initiated by the Ca2+ transient: an arrhythmia trigger within pulmonary veins 1. J Am Coll Cardiol. 2006;47:1196–1206. doi: 10.1016/j.jacc.2005.12.023. [DOI] [PubMed] [Google Scholar]
- 43.Burashnikov A, Antzelevitch C. Reinduction of atrial fibrillation immediately after termination of the arrhythmia is mediated by late phase 3 early afterdepolarization-induced triggered activity. Circulation. 2003;107:2355–2360. doi: 10.1161/01.CIR.0000065578.00869.7C. [DOI] [PubMed] [Google Scholar]
- 44.Milberg P, Pott C, Fink M, et al. Inhibition of the Na+/Ca2+ exchanger suppresses torsades de pointes in an intact heart model of long QT syndrome-2 and long QT syndrome-3. Heart Rhythm. 2008;5:1444–1452. doi: 10.1016/j.hrthm.2008.06.017. [DOI] [PubMed] [Google Scholar]
- 45.Bapat A, Nguyen TP, Lee JH, et al. Enhanced sensitivity of aged fibrotic hearts to angiotensin II- and hypokalemia-induced early afterdepolarizations-mediated ventricular arrhythmias. Am J Physiol Heart Circ Physiol. 2012;302:H2331–H2340. doi: 10.1152/ajpheart.00094.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O'Hanlon R, Grasso A, Roughton M, et al. Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2010;56:867–874. doi: 10.1016/j.jacc.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 47.Lopez B, Gonzalez A, Beaumont J, et al. Identification of a potential cardiac antifibrotic mechanism of torasemide in patients with chronic heart failure. J Am Coll Cardiol. 2007;50:859–867. doi: 10.1016/j.jacc.2007.04.080. [DOI] [PubMed] [Google Scholar]
- 48.Patel R, Nagueh SF, Tsybouleva N, et al. Simvastatin induces regression of cardiac hypertrophy and fibrosis and improves cardiac function in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circulation. 2001;104:317–324. doi: 10.1161/hc2801.094031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nguyen DT, Ding C, Wilson E, et al. Pirfenidone mitigates left ventricular fibrosis and dysfunction after myocardial infarction and reduces arrhythmias. Heart Rhythm. 2010;7:1438–1445. doi: 10.1016/j.hrthm.2010.04.030. [DOI] [PubMed] [Google Scholar]
- 50.Lee KW, Everett TH, Rahmutula D, et al. Pirfenidone prevents the development of a vulnerable substrate for atrial fibrillation in a canine model of heart failure. Circulation. 2006;114:1703–1712. doi: 10.1161/CIRCULATIONAHA.106.624320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Du XJ, Xu Q, Lekgabe E, et al. Reversal of cardiac fibrosis and related dysfunction by relaxin. Ann N Y Acad Sci. 2009;1160:278–284. doi: 10.1111/j.1749-6632.2008.03780.x. [DOI] [PubMed] [Google Scholar]