

Abstract
We report a case of alveolar soft part sarcoma (ASPS) presenting as an isolated frontal lobe metastasis. The tumor demonstrated little or no immunoreactivity for a broad panel of antibodies yet strong, diffuse immunoreactivity with CD68. On electron microscopy, characteristic rectangular to rhomboid crystalline inclusions were not present. Electron-dense granules resembling peroxisomes were present, sometimes in association with elongated granular structures having a periodic, lattice-like arrangement. Metastatic ASPS was confirmed by demonstration of an ASPSCR1-TFE3 fusion and imaging studies that excluded metastatic Xp11.2 translocation renal cell carcinoma. The primary site was subsequently identified in the lower extremity.
Keywords: Alveolar soft part sarcoma, brain metastasis, CD68
Introduction
Alveolar soft part sarcoma (ASPS) is a malignant neoplasm of unknown histogenesis that typically arises in the soft tissues of children and young adults 1. ASPS was likely recognized as (malignant) granular cell myoblastoma2,3 until Christopherson and colleagues provided the currently accepted definition in a series of twelve cases 4. In that original study, the patients studied were predominantly female and were young (median age was 22 years at diagnosis), both of which remain characteristic features in ASPS 5. Characteristic for ASPS, most tumors in that original study arose within (or near) the skeletal muscle of the extremity, or less commonly in the buttock, tongue, or abdominal wall 4. Less common primary sites also reported include other locations in the head and neck, such as the orbit 1,6 and within many viscera and the female genital tract 5. Exceedingly rare are reports of primary ASPS of the central nervous system (CNS) 7.
The characteristic histologic features of ASPS include an organoid arrangement of dyscohesive epithelioid tumor cells, wherein nests of cells are separated by fibrovascular septae 8. Also characteristic of “classic” ASPS are PAS-positive, diastase-resistant needle-shaped crystals in tumor cell cytoplasm and PAS-positive cytoplasmic granules. ASPS most frequently reacts with muscle-associated proteins, including desmin, muscle-specific and smooth muscle actins, and β-enolase, rarely reacting for S-100, and almost never for markers of epithelial, neuroendocrine, or melanocytic differentiation.
Shipkey and colleagues provided the first electron microscopic description of these crystals, noting that they were membrane bound, variable in shape, contained electron-dense granules, and were composed of alternating dark and light lines with a periodicity of 100 angstroms 9, or 10 nm 5. These electron-dense granules were recently found to be immunoreactive to antibodies targeting a ubiquitous proton-linked transporter for monocarboxylates such as lactate, termed monocarboxylate transporter 1 (MCT1), and the MCT-associated protein CD147 10. The significance, if any, of cytoplasmic MCT1 accumulation in ASPS regarding pathogenesis or histogenesis remains unknown.
Critical for diagnostic confirmation, ASPS typically demonstrates a der(17) t(X; 17)(p11.2; q25.3) translocation between ASPSCR1 (17q25.3) and TFE3 (Xp11.2) 6,11,12. This unbalanced translocation results in loss of genetic material telomeric to the ASPSCR1 locus with a gain of material telomeric to TFE3 at Xp11, leading to an ASPSCR1-TFE3 fusion protein that probably induces tumorigenesis through transcriptional deregulation 6. The resulting ASPSCR1-TFE3 fusion may also arise from a balanced translocation, most often observed in a subset of Xp11.2 translocation renal cell carcinomas, which occurs in similarly aged patients 6,12-14. Thus, Xp11.2 translocation renal cell carcinoma is among the critical entities in the differential diagnosis of ASPS, including in the setting of presumed metastasis.
An additional feature of ASPS, fairly unique among sarcomas, is its propensity for metastasis. In the pediatric population, sarcomas metastatic to the CNS include osteosarcoma, Ewing sarcoma/peripheral primitive neuroectodermal tumor, rhabdomyosarcoma, clear cell sarcoma of soft tissue, and ASPS 15. The frequency of ASPS metastases among this group is somewhat remarkable since these tumors represent less than 1% of all soft tissue sarcomas 1. Large series have estimated that up to one-third of patients with ASPS will develop CNS metastases (three times that seen for other sarcomas) at a median interval of 48 months following diagnosis of an extracranial primary16. In several large series, nearly all patients with CNS metastases of ASPS have preceding lung metastases 16-18. Prognosis remains poor in these patients with a median survival after identification of metastasis of 12-27 months and therapeutic strategies including attempted gross total resection, chemoradiation, and/or stereotactic radiosurgery15,16,19.
Here, we report the clinical, histopathologic, and ultrastructural features of a case of ASPS presenting as an isolated CNS parenchymal metastasis in the absence of a known primary site or other metastases. The ultrastructural features of the case are emphasized, as these demonstrate well the spectrum of variant electron microscopic features in ASPS. Recognition of the considerable ultrastructural variation that may be present in ASPS increases the likelihood of an accurate diagnosis, even in the absence of a known primary site.
Materials and Methods
Clinical history
The patient was a 17 year-old male who presented with several episodes of numbness, tingling, and weakness in the right hand, arm, and face over the course of a month. The patient's medical history was significant for prematurity without subsequent complications. Surgical history included open reduction and internal fixation of a right proximal femur fracture that preceded his presentation with a cerebral mass by approximately three months. The pathologic diagnosis on tissue obtained at the fracture site was benign skeletal muscle with crush artifact and rare foci of acute inflammation without evidence of fracture or bone (specimen entirely submitted for histologic examination and serially sectioned). Family history was noncontributory.
Radiologic studies
The patient received a computed tomography (CT) scan without contrast as well as magnetic resonance imaging (MRI) studies with T1-weighted (with and without contrast), T2-weighted, gradient echo, and diffusion-weighted sequences. CT scan revealed an ill-defined area of low-attenuation within the left posterior frontoparietal region, involving both cortical gray matter and subcortical white matter. In the region of the left precentral gyrus, a 1.0 cm area of increased density was observed. There was no midline shift, hemorrhage, hydrocephalus, or herniation. Post-contrast T1-weighted and T2-weighted MR images demonstrated a 1.4 cm avid enhancing lesion in the left superior frontal and precentral gyri and extensive perilesional vasogenic edema (Figure 1). Vascularity was increased in perilesional tissue and no diffusion restriction was identified within the lesion. The radiologic impression was high-grade glioma. Abdominal ultrasound, MRI of the spine, and ophthalmologic examination were performed and the last of these demonstrated borderline elevation of intra-ocular pressure.
Figure 1.
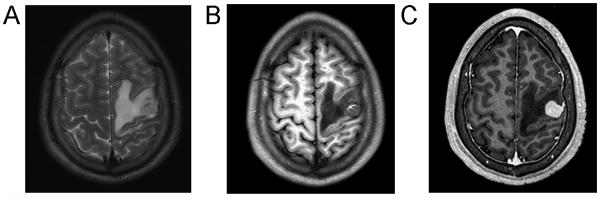
Imaging studies demonstrated a superficial, cortical-based tumor within the left precentral gyrus. T2-FLAIR MRI (A) demonstrated hyperintensity within the tumor and adjacent vasogenic edema. T1-weighted imaging (without contrast) (B) demonstrated a rim of T1 hyperintensity in the anteromedial aspect of the tumor, consistent with intratumoral hemorrhage. T1-weighted MRI with gadolinium (C) demonstrated avid, even enhancement and encroachment of tumor upon the dura.
Surgical procedure and tissue handling
Left frontal craniotomy was performed using Stealth frameless stereotaxy. A biopsy was submitted intraoperatively to the pathology department for frozen section. The completely resected tumor was submitted to the pathology department (1.5 × 1.0 × 0.3 cm in aggregate) and fixed in neutral buffered formalin for light microscopy as well as in glutaraldehyde for electron microscopy.
Immunohistochemistry
Immunohistochemical studies were performed for GFAP (Ventana, prediluted, EP672Y), S100 (Biocare, 1:400, DR96+BC96), synaptophysin (Ventana, prediluted, SP11), CD68 (Ventana, prediluted, KP1), anti-macrophage antibody/MAC387, factor XIII (Ventana, prediluted, AC-1A1), smooth muscle actin (Ventana, prediluted, 1A4), myogenin (Ventana, prediluted, F5D), desmin (Ventana, prediluted, DE-R-11), muscle specific actin (Ventana, prediluted, HHF35), Ki-67 (Ventana, prediluted, 30-9), INI-1 gene product (BAF-47)(Abcam, 1:200, ab58209), p53 (Ventana, prediluted, BP-53-11), pan-keratin (Dako, 1:100, AE1/AE3), CK7 (Ventana, prediluted, SP52), RCC (Ventana, prediluted, PN-15), HMB-45 (Ventana, prediluted, mouse monoclonal), and CD10 (Leica, prediluted, 56C6). Special stains performed included PAS with diastase.
Electron microscopy
Tissue was fixed overnight in 3% glutaraldehyde, rinsed in Millonig's buffer, and post-fixed in 2% osmium tetroxide for 60 minutes at 4° C. The sample was rinsed in deionized water and dehydrated through a series of alcohols and 100% propylene oxide. The tissue was infiltrated with epoxy resin and propylene oxide, embedded in 100% resin, and allowed to polymerize overnight. 500 nm-thick sections were cut with a glass knife using a Leica Ultracut R ultramicrotome. The sections were heat-fixed to glass slides and stained with toluidine blue for selection of appropriate areas for further imaging. The selected tissue block was trimmed and 120 nm-thin sections were prepared on a Leica UC6 ultramicrotome with a diamond knife (Diatome, U.S.) and stained with 2% uranyl acetate and lead citrate. Fifty-one images were acquired using a JEOL 1200EX transmission electron microscope at 60 kV accelerating voltage (JEOL Ltd., Tokyo, Japan) equipped with a Gatan Orius® 830 Digital Imaging System.
Molecular studies
Fluorescence in situ hybridization (FISH) studies were performed on interphase nuclei using a custom-designed dual color TFE3 break-apart probe set and a three-color probe set composed of BAC clones telomeric to 3′TFE3 (in green) and centromeric to 5′TFE3 (in orange) and a commercially available X centromere probe, DXZ1 (in aqua) (Abbott Molecular Inc., Abbott Park, IL) 14. In addition, FISH analysis was performed using a probe to the 3′ region of TFE3 (Xp11.2) and a probe spanning ASPSCR1 (17q25.3) with DXZ1 as a control probe. Confirmation for the presence of additional X chromosomes was performed using an X centromere probe, DXZ1, and a Y centromere probe, DYZ3.
Results
Operative findings
Left frontal craniotomy demonstrated a tumor whose posterior aspect was in close relation to primary motor cortex and whose medial aspect was adherent to a longitudinal cerebral vein. Grossly, the lesion had several small vessels emanating from it such that it closely resembled hemangioblastoma. Frozen section diagnosis was returned consistent with this intraoperative impression and gross total resection of tumor was performed. Post-operative MRI demonstrated gross total resection of the left frontoparietal tumor with residual blood products in the surgical cavity.
Microscopic findings with H&E and special stains
Microscopic examination revealed a partially encapsulated and hemorrhagic tumor that was sharply demarcated from gliotic brain parenchyma. Adjacent parenchyma showed a perivascular mononuclear inflammatory infiltrate. The tumor consisted of an organoid arrangement of epithelioid tumor cells in loosely cohesive nests with intervening delicate capillaries and ectatic vessels (Figure 2). Intermediate and high-power magnification revealed the tumor cells to have abundant, vacuolated to granular cytoplasm and large nuclei with vesicular chromatin and prominent nucleoli. Mitoses, including atypical forms, were conspicuous. Necrosis and endothelial proliferation were not present and there was no intracellular pigment. PAS with diastase treatment demonstrated occasional cells with abundant diastase-resistant cytoplasmic granules. Only very rare cells contained needle- and rod-shaped crystals.
Figure 2.

Low-power magnification of tumor demonstrating organoid arrangement of tumor cells (A) with abundant capillaries, ectatic vascular channels, and intratumoral hemorrhage (H&E stain). (B, C) At higher-power magnification (PAS with diastase special stain), tumor cells demonstrated vesicular nuclear chromatin, prominent nucleoli, mitoses (B), and diastase-resistant granular cytoplasmic material (C). Very rare cells contained needle- and rod-shaped crystalline material (D). Strong, diffuse immunoreactivity with CD68 (KP1 clone) (E) and strong, patch immunoreactivity with desmin (F) were among the rare immunopositive markers in a comprehensive panel.
Immunohistochemical findings
The most striking immunohistochemical feature was strong, granular cytoplasmic immunoreactivity for CD68 (KP1) (Figure 2). However, additional histiocytic markers, including Factor XIIIa and MAC387, were negative. Membranous and cytoplasmic CD10 immunoreactivity was seen in scattered tumor cells. CK7 and RCC were negative, however. Desmin was strongly reactive in isolated cells although other myogenic markers, including muscle specific actin, myogenin, and smooth muscle actin were negative in tumor cells. Epithelial, melanocytic, and neuroectodermal/glial markers were negative. Strong, diffuse nuclear expression of BAF-47 was retained.
Molecular findings
FISH studies demonstrated a rearrangement of the TFE3 locus in 84.5% of 200 interphase cells analyzed using the two- and three-color probe sets as well as an ASPSCR1-TFE3 fusion in 93.0% of 100 interphase cells examined (Figure 3A, 3B, and 3C, respectively).
Figure 3.
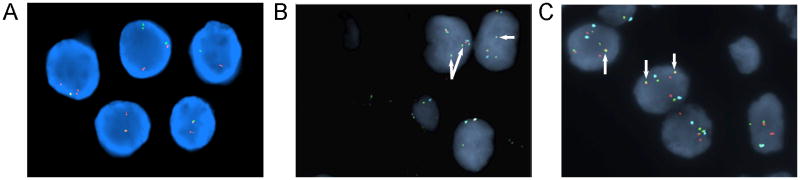
FISH analysis performed on a representative FFPE tissue section using a custom-designed dual color TFE3 break-apart probe set demonstrated separation of the green (centromeric to the 5′TFE3 gene locus at Xp11.2) and orange (telomeric to the 3′TFE3 locus) signals with loss of the 5′TFE3 green signal indicative of an unbalanced rearrangement of TFE3 (A). Similarly, separation of the 5′TFE3 (orange) and 3′TFE3 (green) signals with loss of the 5′TFE3 orange signals was observed with the three-color probe set (B). The three-color probe set also demonstrated the presence of additional copies of the X chromosome (aqua signals) and extra 3′TFE3 signals (arrows) (B). A fusion of the TFE3 and ASPSCR1 (17q25.3) loci was confirmed (arrows) using a probe to the 3′ region of TFE3 (green) and a probe spanning ASPSCR1 (orange) with DXZ1 as an X chromosome control probe (aqua) (C).
Electron microscopic findings
Semi-thin sections demonstrated well-preserved tumor cells with abundant cytoplasm. Cells were arranged in loosely cohesive packets and were surrounded by a well-defined basal lamina (Figure 4). Cytoplasmic glycogen ranged from abundant to sparse and there were droplets of neutral lipid within tumor cells. A notable feature in many cells was the presence of numerous mitochondria and electron dense granules resembling peroxisomes. Some cells additionally demonstrated dilated sacs of endoplasmic reticulum, as well as rough endoplasmic reticulum and Golgi apparatuses. Nuclei were large, containing abundant euchromatin and prominent nucleoli. Very rare membrane-bound crystalloid inclusions were found only after an exhaustive search (Figure 5). They demonstrate a vague lattice-like architecture but classic rhomboid crystals with lattice-like architecture, often illustrated in ASPS, were lacking. Plasma membranes were invaginated and closely associated between tumor cells but intercellular junctions were not present. No intermediate filaments, dense-core vesicles, synaptic structures, cilia, microvilli, premelanosomes, and melanosomes were identified.
Figure 4.
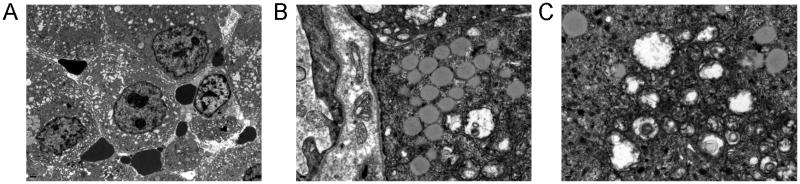
Tumor cells had numerous cytoplasmic organelles, abundant euchromatin, and prominent nucleoli (A). Cells had a well-defined basal lamina and contained cytoplasmic glycogen and droplets of neutral lipid (B). Cells also had numerous mitochondria and electron dense granules resembling peroxisomes (C).
Figure 5.

The tumor was generally crystal-deficient and only very rare, slightly elongated membrane-bound crystalloid inclusions with a vague lattice-like architecture were found. The spectrum of these crystalloid inclusions is demonstrated here in panels A-E. As shown in A and C these inclusions were sometimes seen in close association with electron dense granules. The “classic” rhomboid crystals with lattice-like architecture were not identified (illustrated from a different case in F for comparison to A-E).
Patient status 3 months after initial presentation
Subsequent to the work-up described above the patient was found to have a primary tumor in the deep right thigh. The patient had no complications post-operatively and as of the last available visit he is neurologically intact. The patient had an additional small intracranial metastasis at a site distant from his prior surgical site. This additional focus was treated with gamma-knife radiosurgery.
Discussion
This report demonstrates a case of ASPS metastatic to the frontal lobe without a known primary site or evidence of other metastases. Although the patient's frontal lobe tumor demonstrated characteristic histologic features of ASPS, the diagnosis was challenging due to limited reactivity for a broad-spectrum panel of immunohistochemical markers, and electron microscopic findings were important in providing a definitive diagnosis. Among the few immunopositive markers was strong, diffuse cytoplasmic reactivity for CD68 (KP1). Some ultrastructural findings typical for ASPS were present, including pericellular basal laminae, cytoplasmic glycogen and lipid, and numerous mitochondria. However, the “classic” membrane-bound, rhomboid-shaped crystalline material, often illustrated in ASPS, was not present. Instead, rare membrane-bound inclusions were elongated to round, and these lacked the characteristic lattice-like arrangement of conventional ASPS inclusions. These represent a “variant” of classic rhomboid ASPS inclusions. With the ultrastructural findings, molecular testing for ASPS was undertaken. The diagnosis of ASPS was confirmed by genetic studies demonstrating the ASPSCR1-TFE3 fusion, and most critically, by clinical exclusion of the major differential diagnosis: Xp11.2 translocation renal cell carcinoma. Subsequent to the complete work-up of the metastasis, the patient's primary site was identified in the thigh.
Shipkey and colleagues performed the initial and definitive ultrastructural studies on ASPS 9. The most critical finding in their study was the identification of membrane bound, rhomboid, spiked, or polygonal cytoplasmic crystals with a lattice arrangement of alternating light and dark lines (having a periodicity of 100 angstroms or 10 nm) and associated dense granules. Several reports since that time, however, have noted that ASPS may be deficient in cytoplasmic crystals on both electron microscopy and light microscopy. Ladanyi and colleagues demonstrated that these granules and crystals are immunoreactive for the monocarboxylate transport MCT1 and its chaperone protein CD147 10. This provides an immunohistochemical (and possibly pathophysiologic) correlate to this ultrastructural feature. However, Ordóñez described the number of crystals being highly variable between cases and limited to “occasional cells” in some tumors 5. Tucker described two cases of ASPS that were “crystal-deficient” but also noted that these tumors had electron-dense granules, some of which were elongated and had a lattice-like structure internally 20, similar to those seen in the present case. A separate study of fourteen cases by Ordóñez identified “dense bodies”, described as electron-dense structures with a size between large cytoplasmic granules and fully developed crystals 21. An important characteristic of these “dense bodies” was the recognition of a “fine periodicity” within them that permitted the recognition of these tumors as ultrastructural variants of ASPS 21. The case presented here further highlights the presence of these “dense bodies” in an otherwise crystal-deficient form of ASPS. As noted by previous authors, the structures were along a spectrum of small to larger sized granules, to “dense bodies”, to fully developed rhomboid crystals. Not surprisingly then, many authors have described an apparent spatial association between dense cytoplasmic granules and either larger “dense bodies” or rhomboid crystals 8,20,21. The entire spectrum of crystalline material, including associated dense granules and dense bodies, are generally found in association with the Golgi apparatus of tumor cells 9,21. Additional electron microscopic features recognized in ASPS are the presence of a discontinuous basal lamina and numerous mitochondria 1. The present case demonstrated these findings, as well as abundant glycogen and lipid and conspicuous endoplasmic reticulum and Golgi membranes. These intracytoplasmic structures are present in most, if not all, cases of ASPS 5,8. We did not observe intermediate filaments or structures resembling tight junctions between cells, although rare cases have demonstrated both 21.
A surprising immunohistochemical finding in this case was the presence of diffuse, cytoplasmic immunoreactivity for CD68 (KP-1 clone). More commonly emphasized findings include cytoplasmic staining with MyoD1, S-100, desmin 22, nuclear positivity for a polyclonal antibody to the C-terminal portion of TFE3 6, and immunonegativity for myogenin, neuronal markers (e.g., synaptophysin), and various keratins and epithelial membrane antigen (EMA) 1. The finding of CD68 positivity, however, is not commonly emphasized in reports and/or studies of ASPS, so we discuss it further here. The CD68-KP-1 clone utilized in this case report is a mouse monoclonal antibody that targets a 110 kD intracellular glycoprotein found in the cell membrane of macrophages and myeloid cells 23. In light of this, this immunoreactivity can be presumed to be non-specific. Electron microscopy studies, as described above, did not demonstrate features of phagocytic cells, such as primary and secondary lysosomes. Further, additional studies on this patient's tumor with anti-macrophage antibody (MAC387) did not demonstrate reactivity. Along similar lines, Wu and colleagues reported a case of CD68-positive ASPS with “xanthomatous features” arising as a primary breast mass 24. In fact, the immunohistochemical profile of the tumor in their report, including diffuse, cytoplasmic labeling for CD68 (clone not specified), only focal labeling with desmin, and negative reactions for S-100 and epithelial markers, is nearly identical to the findings reported in the present case. Also similar to the present case, those authors briefly entertained the possibility of a histiocytic lesion until arriving at the correct diagnosis via the demonstration of characteristic electron microscopic features and nuclear TFE3 immunoreactivity 24. The authors concluded that CD68 immunoreactivity might represent either the presence of abundant lysosomes within a tumor or the presence of admixed macrophages. However, as noted above, there is not evidence of either in the present case to explain the non-specific immunoreactivity for CD68 (KP-1). To our knowledge, the basis of this immunohistochemical finding remains unexplained in ASPS. In a comprehensive review of numerous immunohistochemical studies on ASPSs, Ordóñez notes only two cases of ASPS in the literature with application of CD68 – both being positive for this marker 5. Thus, all four cases of ASPS tested with anti-CD68 antibody, including the present one, have demonstrated immunoreactivity. As such, CD68 positivity in ASPS may be of diagnostic importance in isolated cases, particularly if the clinical presentation is unusual and there are few other positive markers on a broad-spectrum immunohistochemical panel (both features of the present report).
The histologic differential diagnosis of ASPS is of critical importance in the setting of a metastatic tumor to brain parenchyma. Tumors with similar histologic findings (i.e., an organoid arrangement of polygonal, epithelioid tumor cells with dilated capillaries, inconspicuous mitotic activity, and no evident necrosis), include Xp11.2 translocation renal cell carcinoma, metastatic adrenocortical (ACC), renal (RCC), and hepatocellular carcinomas (HCC), metastatic perivascular epithelioid cell tumors (PEComas), including epithelioid angiomyolipoma, metastatic melanoma, and at the skull base, intracranial extension of a jugulotympanic paraganglioma (JTP). Most of these entities can safely be excluded by consideration of patient age and history and radiologic studies of the chest, abdomen, and pelvis. In an unusual clinical setting, immunohistochemical studies can further narrow the differential diagnosis. This includes exclusion of ASPS by demonstration of immunoreactivity of tumor cells for keratins and/or epithelial membrane antigen (e.g., ACC, HCC, and RCC), for melanocytic markers (e.g., melanoma and PEComa), and for synaptophysin and chromogranin (JTP). Although metastatic malignant PEComa to the brain is presumed to be an exceptionally rare event, there are isolated reports of this 25. These reports are notable in the context of metastatic ASPS because PEComas may share with ASPS immunoreactivity for desmin, as well as nuclear reactivity for TFE3 26. Further, some PEComas (i.e., renal angiomyolipoma) may demonstrate PAS-positive, diastase-resistant cytoplasmic granules with classic rhomboid crystalline architecture on ultrastructural examination, which furthers the overlap with ASPS 10. In this setting, immunoreactivity for HMB-45 and/or microphthalmia-associated transcription factor (MiTF) in tumor cells would be consistent with PEComa and exclude a diagnosis of metastatic ASPS 27. Similarly, these markers should distinguish metastatic amelanotic melanoma to CNS from cases of metastatic ASPS with unusually aggressive histologic features, including a sheet-like arrangement, multinucleation, and cytologic atypia 1.
For pediatric and young adult patients, however, the differential may include alveolar rhabdomyosarcoma 22, clear cell sarcoma 28, and Xp11.2 translocation renal cell carcinoma (TRCC). The latter is the most critical differential diagnosis for a CNS metastasis of ASPS, as a subset of TRCCs result from the formation of an ASPSCR1-TFE3 fusion oncogene 1, as in ASPS. Unlike ASPS this is most commonly observed as a balanced X;17 balanced translocation der(17) t(X; 17) 6,28. Nevertheless, both Xp11.2 TRCCs and ASPSs can exhibit diffuse, strong nuclear immunoreactivity for the TFE3 antibody. Further, Xp11.2 translocation renal cell carcinomas can behave aggressively and demonstrate systemic metastases 29, as well as CNS metastases 30. For these reasons, in the present case, the final diagnosis of ASPS was not conveyed to the patient's treating physicians until high-resolution radiologic studies had excluded the presence of a renal mass. In the absence of such investigations, it may be challenging to distinguish between these entities. Molecular demonstration of a balanced translocation is strongly suggestive of an Xp11.2 TRCC 6 as the reciprocal fusion protein TFE3-ASPSCR1 is only rarely present in ASPS 12,14.
In summary, this case illustrates a particularly diagnostic challenging example of metastatic ASPS to brain parenchyma. The case was unique for the absence of two commonly identified clinical and/or radiologic findings, which are a known primary tumor site and lung metastases, respectively. As described above the histopathologic findings raise a broad differential in this scenario, the most critical of which is Xp11.2 translocation renal cell carcinoma in pediatric and young adult patients. Immunoreactivity for CD68 was also seen, which may be dismissed and/or misinterpreted, but which appears to be a fairly reliable immunohistochemical finding in these tumors, even though the basis for this reactivity is unknown. Finally, by electron microscopy, this case illustrates an example of what has been termed “crystal-deficient” ASPS. In these cases, the presence of electron-dense granules in association with larger, elongated “dense bodies” containing a periodic arrangement of light and dark lines, similar to fully formed rhomboid crystals, are important diagnostic features. These EM structures should be recognized as a variant of classic ASPS rhomboid crystals so that molecular confirmation of the ASPSCR1-TFE3 fusion can be performed.
Acknowledgments
We wish to thank Dr. Alyaa Al-Ibraheemi, both of the Department of Pathology and Laboratory Medicine at the University of Texas Health Science Center at Houston, for their assistance. Thanks to Drs. Irina Vaysertreyger, Yuanyuan Zhang, and Xiao-qiong Liu of the University of Nebraska Medical Center for their technical assistance. Dr. Olar was supported by the National Institutes of Health/National Cancer Institute (Training Grant No. 5T32CA163185).
References
- 1.Ordonez N, Ladanyi M. Alveolar soft part sarcoma. In: Fletcher CDM, Unni KK, Mertens F, editors. World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon, France: IARC Press; 2002. p. 427. [Google Scholar]
- 2.Horn RC, Stout AP. Granular cell myoblastoma. Surg, Gynec, and Obst. 1943;76:315–318. [Google Scholar]
- 3.Ackerman LV, Phelps CR. Malignant granular cell myoblastoma of the gluteal region. Surgery. 1946;20:511–519. [PubMed] [Google Scholar]
- 4.Christopherson WM, Foote FW, Jr, Stewart FW. Alveolar soft-part sarcomas; structurally characteristic tumors of uncertain histogenesis. Cancer. 1952 Jan;5(1):100–111. doi: 10.1002/1097-0142(195201)5:1<100::aid-cncr2820050112>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 5.Ordonez NG. Alveolar soft part sarcoma: a review and update. Adv Anat Pathol. 1999 May;6(3):125–139. doi: 10.1097/00125480-199905000-00001. [DOI] [PubMed] [Google Scholar]
- 6.Pfeifer JD. Molecular Genetic Testing in Surgical Pathology. Philadelphia: Lippincott Williams and Wilkins; 2005. [Google Scholar]
- 7.Ahn SH, Lee JY, Wang KC, et al. Primary alveolar soft part sarcoma arising from the cerebellopontine angle. Childs Nerv Syst. 2014 Feb;30(2):345–350. doi: 10.1007/s00381-013-2193-6. [DOI] [PubMed] [Google Scholar]
- 8.Folpe AL, Deyrup AT. Alveolar soft-part sarcoma: a review and update. J Clin Pathol. 2006 Nov;59(11):1127–1132. doi: 10.1136/jcp.2005.031120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shipkey FH, Lieberman PH, Foote FW, Jr, Stewart FW. Ultrastructure of Alveolar Soft Part Sarcoma. Cancer. 1964 Jul;17:821–830. doi: 10.1002/1097-0142(196407)17:7<821::aid-cncr2820170702>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 10.Ladanyi M, Antonescu CR, Drobnjak M, et al. The precrystalline cytoplasmic granules of alveolar soft part sarcoma contain monocarboxylate transporter 1 and CD147. Am J Pathol. 2002 Apr;160(4):1215–1221. doi: 10.1016/S0002-9440(10)62548-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joyama S, Ueda T, Shimizu K, et al. Chromosome rearrangement at 17q25 and xp11.2 in alveolar soft-part sarcoma: A case report and review of the literature. Cancer. 1999 Oct 1;86(7):1246–1250. [PubMed] [Google Scholar]
- 12.Ladanyi M, Lui MY, Antonescu CR, et al. The der(17)t(X;17)(p11;q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25. Oncogene. 2001 Jan 4;20(1):48–57. doi: 10.1038/sj.onc.1204074. [DOI] [PubMed] [Google Scholar]
- 13.Zhong M, De Angelo P, Osborne L, et al. Dual-color, break-apart FISH assay on paraffin-embedded tissues as an adjunct to diagnosis of Xp11 translocation renal cell carcinoma and alveolar soft part sarcoma. Am J Surg Pathol. 2010 Jun;34(6):757–766. doi: 10.1097/PAS.0b013e3181dd577e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hodge JC, Pearce KE, Wang X, Wiktor AE, Oliveira AM, Greipp PT. Molecular cytogenetic analysis for TFE3 rearrangement in Xp11.2 renal cell carcinoma and alveolar soft part sarcoma: validation and clinical experience with 75 cases. Mod Pathol. 2014 Jan;27(1):113–127. doi: 10.1038/modpathol.2013.83. [DOI] [PubMed] [Google Scholar]
- 15.Kebudi R, Ayan I, Gorgun O, Agaoglu FY, Vural S, Darendeliler E. Brain metastasis in pediatric extracranial solid tumors: survey and literature review. J Neurooncol. 2005 Jan;71(1):43–48. doi: 10.1007/s11060-004-4840-y. [DOI] [PubMed] [Google Scholar]
- 16.Reichardt P, Lindner T, Pink D, Thuss-Patience PC, Kretzschmar A, Dorken B. Chemotherapy in alveolar soft part sarcomas. What do we know? Eur J Cancer. 2003 Jul;39(11):1511–1516. doi: 10.1016/s0959-8049(03)00264-8. [DOI] [PubMed] [Google Scholar]
- 17.Portera CA, Jr, Ho V, Patel SR, et al. Alveolar soft part sarcoma: clinical course and patterns of metastasis in 70 patients treated at a single institution. Cancer. 2001 Feb 1;91(3):585–591. doi: 10.1002/1097-0142(20010201)91:3<585::aid-cncr1038>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 18.Sood S, Baheti AD, Shinagare AB, et al. Imaging features of primary and metastatic alveolar soft part sarcoma: single institute experience in 25 patients. Br J Radiol. 2014 Apr;87(1036):20130719. doi: 10.1259/bjr.20130719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fox BD, Patel A, Suki D, Rao G. Surgical management of metastatic sarcoma to the brain. J Neurosurg. 2009 Jan;110(1):181–186. doi: 10.3171/2008.4.17505. [DOI] [PubMed] [Google Scholar]
- 20.Tucker JA. Crystal-deficient alveolar soft part sarcoma. Ultrastruct Pathol. 1993 May-Aug;17(3-4):279–286. doi: 10.3109/01913129309027774. [DOI] [PubMed] [Google Scholar]
- 21.Ordonez NG, Ro JY, Mackay B. Alveolar soft part sarcoma. An ultrastructural and immunocytochemical investigation of its histogenesis. Cancer. 1989 May 1;63(9):1721–1736. [PubMed] [Google Scholar]
- 22.LeGallo RG, Wick MR. Soft Tissue. In: Gattuso P, Reddy VB, Odile D, Spitz DJ, Haber MH, editors. Differential Diagnosis in Surgical Pathology. Philadelphia: Saunders; 2010. pp. 889–948. [Google Scholar]
- 23.Ventana Medical Systems I. CD68 (KP-1) Primary Antibody, CONFIRM. 2013 [Google Scholar]
- 24.Wu J, Brinker DA, Haas M, Montgomery EA, Argani P. Primary alveolar soft part sarcoma (ASPS) of the breast: report of a deceptive case with xanthomatous features confirmed by TFE3 immunohistochemistry and electron microscopy. Int J Surg Pathol. 2005 Jan;13(1):81–85. doi: 10.1177/106689690501300112. [DOI] [PubMed] [Google Scholar]
- 25.Parfitt JR, Keith JL, Megyesi JF, Ang LC. Metastatic PEComa to the brain. Acta Neuropathol. 2006 Sep;112(3):349–351. doi: 10.1007/s00401-006-0105-5. [DOI] [PubMed] [Google Scholar]
- 26.Argani P, Aulmann S, Illei PB, et al. A distinctive subset of PEComas harbors TFE3 gene fusions. Am J Surg Pathol. 2010 Oct;34(10):1395–1406. doi: 10.1097/PAS.0b013e3181f17ac0. [DOI] [PubMed] [Google Scholar]
- 27.Chang KL, Folpe AL. Diagnostic utility of microphthalmia transcription factor in malignant melanoma and other tumors. Adv Anat Pathol. 2001 Sep;8(5):273–275. doi: 10.1097/00125480-200109000-00004. [DOI] [PubMed] [Google Scholar]
- 28.Brooks JSJ. Disorders of Soft Tissue. In: Mills SE, editor. Sternberg's Diagnostic Surgical Pathology. Vol. 1. Philadelphia: Lippincott Williams and Wilkins; 2010. pp. 124–197. [Google Scholar]
- 29.Ross H, Argani P. Xp11 translocation renal cell carcinoma. Pathology. 2010 Jun;42(4):369–373. doi: 10.3109/00313021003767348. [DOI] [PubMed] [Google Scholar]
- 30.Morii A, Fujiuchi Y, Nomoto K, Komiya A, Fuse H. Rapidly progressing renal cell carcinoma associated with Xp11.2 translocations: a case report. J Med Case Rep. 2012;6(1):164. doi: 10.1186/1752-1947-6-164. [DOI] [PMC free article] [PubMed] [Google Scholar]